
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-stimuli-responsive luminescence enabled by crown ether anchored chiral antimony halide phosphors†
Xiao
Han
a,
Puxin
Cheng
a,
Shanshan
Han
a,
Zhihua
Wang
a,
Junjie
Guan
a,
Wenqing
Han
a,
Rongchao
Shi
a,
Songhua
Chen
b,
Yongshen
Zheng
*a,
Jialiang
Xu
 *a and
Xian-He
Bu
a
*a and
Xian-He
Bu
a
aSchool of Materials Science and Engineering, Smart Sensing Interdisciplinary Science Center, Frontiers Science Center for New Organic Matter, Nankai University, Tongyan Road 38, Tianjin 300350, P. R. China. E-mail: yszheng@nankai.edu.cn; jialiang.xu@nankai.edu.cn
bCollege of Chemistry and Material Science, Longyan University, Longyan 364012, Fujian, P. R. China
First published on 1st February 2024
Abstract
Stimuli-responsive optical materials have provided a powerful impetus for the development of intelligent optoelectronic devices. The family of organic–inorganic hybrid metal halides, distinguished by their structural diversity, presents a prospective platform for the advancement of stimuli-responsive optical materials. Here, we have employed a crown ether to anchor the A-site cation of a chiral antimony halide, enabling convenient control and modulation of its photophysical properties. The chirality-dependent asymmetric lattice distortion of inorganic skeletons assisted by a crown ether promotes the formation of self-trapped excitons (STEs), leading to a high photoluminescence quantum yield of over 85%, concomitant with the effective circularly polarized luminescence. The antimony halide enantiomers showcase highly sensitive stimuli-responsive luminescent behaviours towards excitation wavelength and temperature simultaneously, exhibiting a versatile reversible colour switching capability from blue to white and further to orange. In situ temperature-dependent luminescence spectra, time-resolved luminescence spectra and theoretical calculations reveal that the multi-stimuli-responsive luminescent behaviours stem from distinct STEs within zero-dimensional lattices. By virtue of the inherent flexibility and adaptability, these chiral antimony chlorides have promising prospects for future applications in cutting-edge fields such as multifunctional illumination technologies and intelligent sensing devices.
Introduction
Stimuli-responsive materials have gained considerable attention for their talent to toggle or alter their intrinsic properties upon external stimuli, embracing light,1,2 temperature,3 electric field,4,5 chemical perturbation,6etc. Numerous artificial sensing devices, therefore, have been established by incorporating such stimuli-responsive materials. In particular, stimuli-responsive luminescence materials,7 being able to afford feedback on ambient stimuli through optical outputs in directable manners,8 have emerged as trending on account of their various advantages, encompassing facile detection of luminescent signals, exceptional sensitivity, rapid response speed, and an expansive scope for emission intensity and wavelength modulations.9 Hence, various stimuli-responsive luminescence materials including organic small molecules,10,11 polymers,12 organometallic complexes,13 and hybrid organic–inorganic metal halides (HOMHs)14 have been extensively utilized in prominent domains15–17 such as intelligent optoelectronics,18,19 biosensors,20 and information storage.21–23Among these stimuli-responsive luminescence materials, HOMHs feature rich chemical composition and structural diversity24 along with tailorable bandgaps, long carrier diffusion lengths,25 large oscillator strengths and exceptional optical absorption coefficients.26 Furthermore, compared to pure organic molecular systems, HOMHs furnish a streamlined alternative, circumventing the complexities associated with synthesis procedures,27 leading to the widespread application of various facile, low-temperature, and efficient solution-processing synthesis methods. Besides, the dimensionality of HOMHs is variable, which exerts a significant impact on their luminescent properties. Shrinking the dimensionality of HOMHs from three-dimensional (3D) to low-dimensional (2D, 1D,28 0D) can endow them with unique properties such as large exciton binding energy, multiple quantum wells and confinement effects,29 controllable charge-carrier mobility, and efficient photoluminescence intensity.30–32 Low-dimensional metal halides have thus been witnessed to exhibit high photoluminescence quantum yield (PLQY) and broad emission range33 on account of the formation of self-trapped excitons (STEs).34 Organic cations play a key role in the formation of STEs and the distribution of hydrogen bond interactions within these HOMHs.35 Recently, the integration of crown ethers into organic cations has been demonstrated with the exceptional ability to engage in intricate host–guest interactions with various metal cations and even organic ammonium cations.36–39 The introduction of crown ethers not only imparts a more diverse repertoire of hydrogen bonding interactions to the system, but also enhances the overall stability of the framework.40 Additionally, the adaptable ring-opening diameter also offers the selectivity for distinct cations,41 enabling the modulation of luminescence in HOMHs.42 More importantly, the self-assembly systems of HOMHs, enabled by the presence of weak ionic bonds, low formation energies, flexible crystal nature, and multiple excited states, offer a handy and efficient avenue for facile modulation of the intrinsic optical characteristics exposed to external stimuli.23,26,43,44
Chiral low-dimensional metal halides combine the merits of chiral materials and low-dimensional metal halides.45 Chiral materials can manipulate the circular polarization states of light naturally.46 Applying the strategy of chirality transfer from organic chiral ligands, the chiroptoelectrics of low-dimensional metal halides have been comprehensively explored and developed, including circularly polarized luminescence (CPL), which refers to the emission of light with a specific polarization state, wherein the electric field vector rotates in a circular manner.47 Due to its distinctive optical properties,48 CPL has found extensive applications49 in crucial domains such as 3D imaging and printing, sensing and probing, anti-counterfeiting, quantum computing, etc.49 Consequently, chiral low-dimensional metal halides pave a potential pathway for intelligent modulation of CPL by virtue of the integration of CPL emission and multi-stimuli responsiveness.27
Herein, a pair of new zero-dimensional (0D) chiral antimony halides, i.e., [R-/S-1,2-propanediamine(18-crown-6)2]SbCl6, referred to as R-/S-Sb, have been synthesized via antisolvent vapor-assisted crystallization. Within their crystalline configurations, the self-assembly behaviour of 18-crown-6 with chiral amine cations bestows an abundance of hydrogen bonding interactions and efficaciously segregates the inorganic octahedra, thereby boosting the photoluminescence efficiency (51.6% and 87.2% quantum yield for white and orange emission, respectively). Moreover, the chirality transfer engendered by the asymmetric hydrogen bonding interactions confers the system with effective CPL. More importantly, the chiral R-Sb and S-Sb exhibit multi-stimuli-responsive luminescence, showcasing their capacity to dynamically respond to excitation wavelength and temperature, enabling a back-and-forth emission colour switch from blue to orange emission. This study not only unveils a novel approach for the development of low-dimensional eco-friendly emitters with CPL, but also broadens the scope of candidate materials for optical and thermal sensing optoelectronic devices.
Results and discussion
Structural characterization
The target chiral 0D antimony halides (R-/S-Sb) have been obtained via a facile antisolvent vapor-assisted crystallization (see details in the Experimental section). Their spatial configurations have been confirmed through single crystal X-ray diffraction (SCXRD) analyses. Chiral R-Sb and S-Sb crystallize in tetragonal chiral space groups of P43212 and P41212, respectively, subordinate to the 422 crystal class (Table S1†). In an asymmetric unit, a chiral 1,2-propanediamine cation (PDD2+) is anchored with double 18-crown-6 molecules through multiple N–H⋯OC hydrogen bonding interactions, featuring a host–guest architecture (Fig. 1a). The incorporation of the bulky 18-crown-6 isolates the inorganic [SbCl6]3− octahedra effectively, forming the classical 0D configuration (Fig. 1b). Among the 0D metal halides, the large distances between [MXn]m− units eliminate the electronic interactions and energy transfer between neighboring units, leading to a highly localized exciton in the excited state,50–53 facilitating the formation of highly emissive HOMHs. The steric effect of bulkier organic cations [PPD(18-crown-6)2]2+ enables the enhanced separation of [SbCl6]3− octahedra, showing the shortest Sb⋯Sb distance of 10.11 Å for R-Sb and 9.66 Å for rac-Sb (Fig. S1a†). In comparison, in the absence of the crown, the longest distances between atoms of the chiral [S-PPD]2+ cation along the crystallographic b- and c-axis directions are shortened to 5.26 Å and 3.42 Å (Fig. S2†), respectively. As a result, the shortest Sb⋯Sb distance of [S-1,2-propanediamine]SbCl6 (S-PPD-Sb) shrinks to 6.22 Å. Different from the usual organic–inorganic hybrid antimony halides, the inorganic frameworks combine with organic cations through Sb–Cl⋯H–C hydrogen bonding interactions (Fig. S3†) rather than Sb–Cl⋯H–N interactions. By investigating the number of Sb–Cl⋯H–C hydrogen bonds concerning an inorganic unit and the proportion of H⋯Cl interactions around organic amines in R-Sb and rac-Sb structures by the Hirshfeld surface analysis method, it is revealed that R-Sb exhibits stronger hydrogen bonding interactions than rac-Sb (Fig. S4 and S5†).54 Compared to the non-covalent interactions in S-PPD-Sb, the crown ether in the R-Sb structure obstructs the contact between the chiral amine cation and [SbCl6]3− octahedron, resulting in a decline in the proportion of H⋯Cl interactions and the predominance of H⋯O interactions (Fig. S6†). Moreover, the results of thermogravimetry-differential thermal analyses (TG-DTA) show that R-/S-Sb preserves higher melting temperature than rac-Sb (Fig. S7†), consistent with the fact that the hydrogen bonding interactions are intensified in the chiral antimony chlorides. Under the effect of asymmetric hydrogen bonding interactions, severe octahedral distortion is observed in the chiral antimony chlorides (Fig. 1c). The hydrogen bonding with the axial chlorine atom induces a 13.4° tilt and the equatorial Cl–Sb–Cl bond angle was compressed to 71.1°. The corresponding bond angle variances (σ2), as calculated viaeqn (1):55,56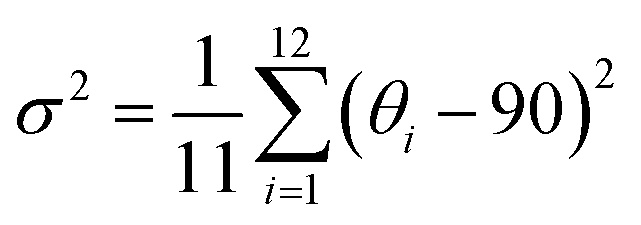 | (1) |
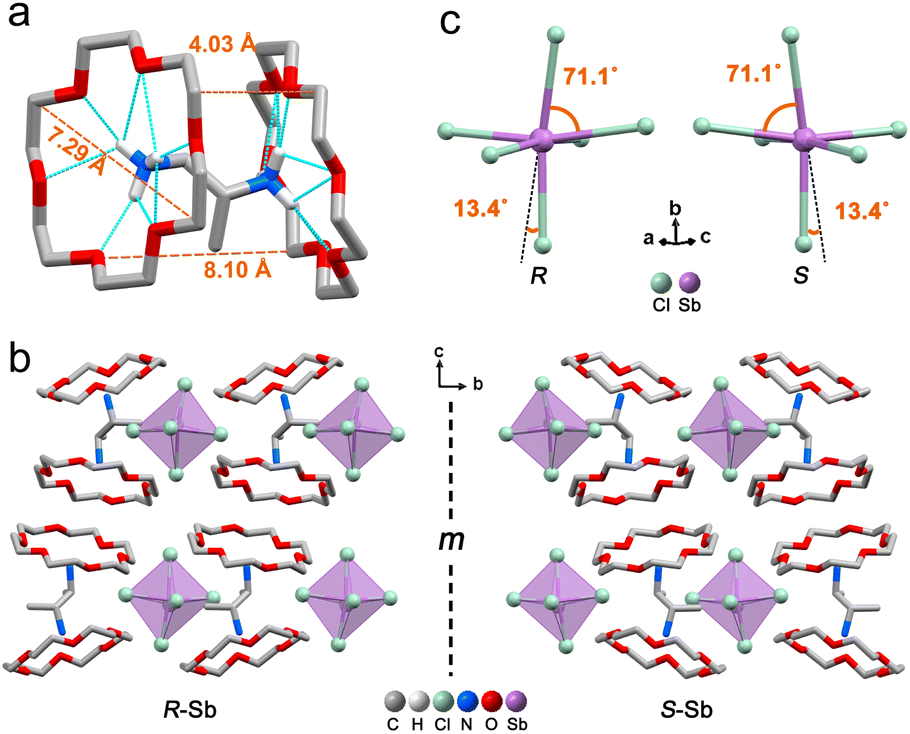 | ||
| Fig. 1 Single-crystal structures of the chiral 0D antimony halides. (a) The structure of the [R-PPD(18-crown-6)2]2+ cation, showing host–guest interactions, size of 18-crown-6 (7.29 Å), longest distance (8.10 Å) and shortest distance (4.03 Å) between the two adjacent 18-crown-6 molecules. (b) Structural models of R-Sb and S-Sb (hydrogen atoms are omitted for clarity, m represents mirror plane). (c) Inorganic scaffold in R-/S-Sb. | ||
reveal a greater extent of distortion of R-/S-Sb compared to rac-Sb. The pronounced distortions in the octahedral geometry can be identified in R- and S-Sb with σ2 of 71.0 deg.2 significantly larger than that of rac-Sb (12.3 deg.2). The powder X-ray diffraction (PXRD) patterns of R-/S-Sb as depicted in Fig. 2a accord well with the simulated results from the corresponding SCXRD data, verifying the high phase purity of the obtained single crystals.
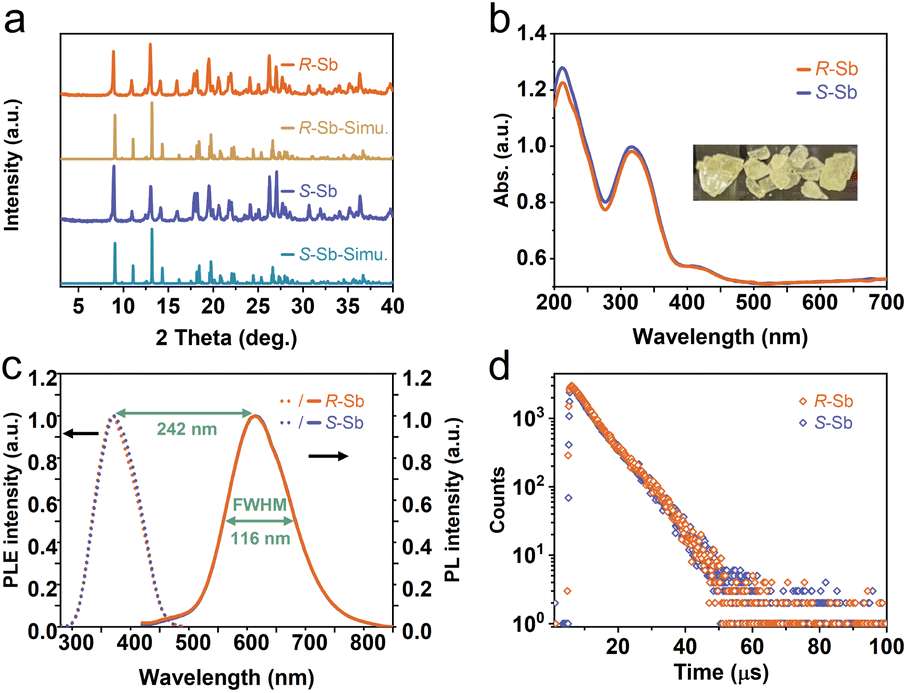 | ||
| Fig. 2 Characterization of chiral 0D antimony halides. (a) PXRD patterns and (b) UV-vis absorption spectra of R-/S-Sb. Inset: optical image of R-Sb crystals under the radiation of natural light. (c) Normalized PL excitation (PLE), PL spectra and (d) time-resolved PL decay spectra of R-/S-Sb at 612 nm. | ||
Photophysical properties of chiral Sb halides
The obtained UV-vis absorption spectra reveal prominent absorption bands of R-/S-Sb in the range of 200–400 nm, accompanied by a minor absorption in the visible range (400–500 nm) (Fig. 2b), consistent with the pale-yellow colour observed in the crystals under ambient light. Due to the excited state splitting behaviours of the 5s2 configuration like Sb3+, the two main absorption peaks around 213 nm and 315 nm can be attributed to characteristic 1S0 → 1P1 and 1S0 → 3P1 electronic transitions of the Sb(III) ion containing 5s2 lone-pair electrons.57,58 Under UV light irradiation at 365 nm, R-Sb exhibits intense orange emission (Fig. S8†). Therefore, steady-state photoluminescence (PL) and photoluminescence excitation (PLE) spectra (Fig. 2c) have been collected to investigate their luminescent behaviours. Upon excitation at the optimal excitation wavelength of 370 nm, R-/S-Sb displays broadband emission centered at 612 nm, with a full width at half maximum (FWHM) of 116 nm (0.40 eV), and a significant Stokes shift of 242 nm (1.32 eV). Such a large Stokes shift signifies low self-absorption effects in the luminescent material, which is highly desirable for high-performance luminescent materials.29,59 Under the optimal excitation wavelength, R-Sb exhibits a high quantum yield of 87.2% (Fig. S9a†). However, the S-PDD-Sb crystal is nonluminous under ambient conditions (Fig. S10†), reflecting the key role of 18-crown-6 in the emission process. The luminescence lifetimes of R-Sb and S-Sb have been monitored by time-resolved decay PL spectra (Fig. 2d). The resulting decay curves could be well fitted with a single exponential function, with a lifetime of approximately 7.0 μs. Based on these aforementioned luminescence features of R-/S-Sb, including a broad FWHM, large Stokes shift, and long luminescence lifetime (microsecond scale), it could be concluded that the emission of R-/S-Sb centered at around 612 nm originates from triplet STE emission.60–62 The STE formation of R-/S-Sb is contributed by its intrinsic low electronic dimensionality and soft lattice nature.63Stimuli-responsive luminescence of chiral Sb halides
For chiral R-Sb halides, a gradual blue-shift in the excitation wavelength engenders the gradual emergence of a high-energy emission peak at 492 nm. In stark contrast to the long-lived luminescence at 612 nm, which exhibits a lifetime of 7.0 μs, time-resolved PL decay spectroscopy reveals a significantly shorter lifetime of 9.3 ns at 492 nm (Fig. S11†), thereby signifying the single STE (1P1 → 1S0) electronic transition process.64,65 The time-resolved emission spectra of R-Sb with excitation at 365 nm were recorded to assign the dual emissions (Fig. 3c and d). The emission intensity at 492 nm rises steadily, reaching its maximum at 19.98 ns, before swiftly decaying and extinguishing within a short duration. Conversely, the emission peak at 612 nm exhibits a much slower decline, taking approximately 0.7 μs to diminish and eventually fade away. The distinct excitation spectra observed at 492 nm and 612 nm thus prove the involvement of different transition mechanisms for the dual emission bands (Fig. S12†). For the low-dimensional halides with the 5s2-configuration cations (such as Sb3+ or Sn2+), the nsnp excited states will be split into 1P (singlet) and 3P (triplet) states under the impact of the Coulomb and exchange interactions.66 Further, the 3P states will be split into non-degenerate states 3P2, 3P1, and 3P0 by the spin–orbit coupling interaction (Fig. S13a†). The STE3 state should be converted from the STE1 state by undergoing an intersystem crossing (ISC) process (Fig. S13b†), resulting to the delay emission process and long lifetime.67 | ||
| Fig. 3 Excitation wavelength-dependent PL properties of R-Sb. (a) Excitation–emission mapping patterns of R-Sb under ambient conditions. (b) The corresponding CIE chromaticity coordinates at different excitation wavelengths. Insets: luminescence photographs at excitation wavelengths of 300 nm (left), 360 nm (medium), and 380 nm (right). (c) and (d) Time-resolved emission spectra for R-Sb at the excitation wavelength of 365 nm. Note that t = 0 μs (t = 6.0 μs) represents the start (end) of the excitation pulse and that the emission intensity reached its maximum at t = 6.0 μs. | ||
As the excitation wavelength transitions from 300 nm to longer wavelengths of up to 400 nm, a pronounced stimulus response is observed in the emission spectra of R-Sb. The emission at 492 nm is negligible for R-Sb when the excitation wavelength is longer than 350 nm, which can be ascribed to the direct 3P1 → 1S0 transition channel. The alteration of excitation wavelength can affect the exciton transition channel readily. Upon excitation at 320 nm, both singlet STE (STE1) and triplet STE (STE3) states can be activated simultaneously, implying the energy transfer channel from STE1 to STE3. Specifically, the high-energy emission intensity (IHE) at 492 nm and the low-energy emission intensity (ILE) at 612 nm initially increase before gradually diminishing upon the change of excitation wavelength from 300 to 400 nm, while the LE emission increasingly overshadows the one in the HE emission (Fig. 3a). This comprehensive variation in the relative intensities of the two peaks brings about a luminescence colour switch of R-Sb from the original yellowish white to yellow, and orange, resulting from the stimuli-responsive behaviours of excitation wavelength emission colours between STE1 and STE3. Therefore, by controlling the excitation wavelengths, one could deftly generate the white emission (Fig. 3b). Blending a pair of complementary wavelengths, the CIE chromaticity coordinates of the white emission of R-Sb at 320 nm excitation were calculated to be (0.35, 0.37), obtaining high colour temperatures (CCT) of 4109 K, along with a high PLQY of 51.6% (Fig. S9b†). Thus, the dynamic emission colour modulation can be realized facilely via targeted excitation of distinct STEs. For the multiexcitonic emission in a single system, the colour kinetic performance offers promising strategies for scheming multicolour light-emitting diodes (LEDs) and multidimensional optical anti-counterfeiting materials.68
To further investigate the photophysical mechanism of R-Sb, temperature-dependent PL spectra were recorded. Under excitation at a higher energy of 320 nm, the emission peak intensity at 492 nm gradually increases with lowering temperature (Fig. 4a). Furthermore, the fluorescence lifetime prolongs significantly from 5.2 ns at 320 K to 28.1 ns at 80 K (Fig. 4b), implying nonradiative recombination is suppressed in the cryogenic environment.69 Additionally, the FWHM has noticeably narrowed down, indicating a weaker electron–phonon coupling interaction in the cryogenic environment.60 Similar variations can be also observed at 612 nm in both luminescence intensity and lifetime (Fig. 4c and d). Under excitation by low-energy photons, the emission intensity at 492 nm becomes inconspicuous, signifying that only the STE3 state is actuated. In contrast, upon excitation by high-energy photons, the ground-state electrons experience excitation to higher energy states and are subsequently trapped by the STE1 state. Then the electrons undergo an intersystem crossing (ISC) process, transiting from the STE1 state to the STE3 state, and exhibiting dual-emission luminescence.
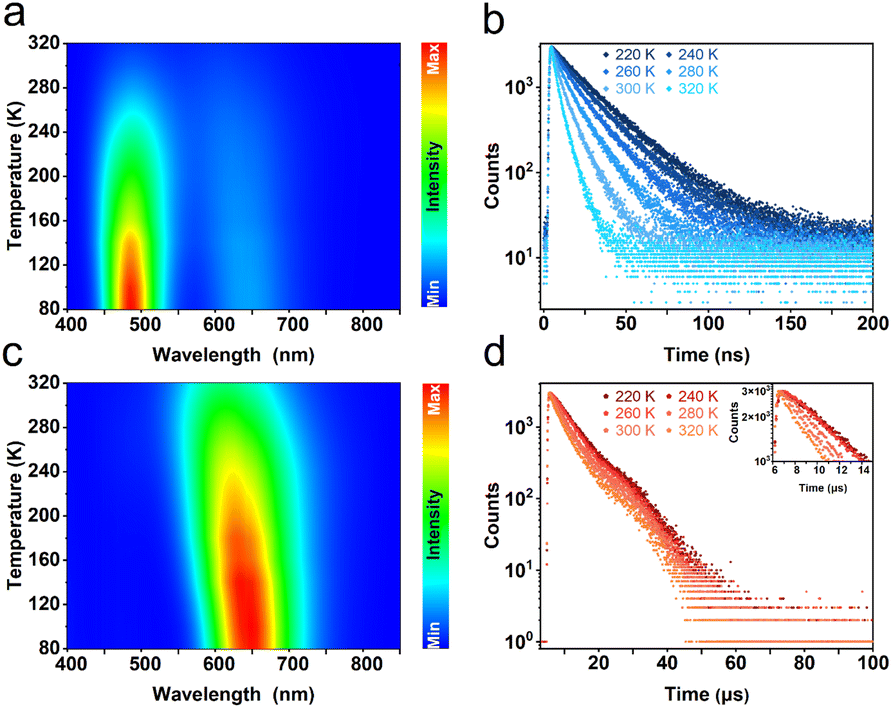 | ||
| Fig. 4 Temperature-dependent PL measurements of R-Sb. (a) Temperature-dependent PL spectra (λex = 320 nm) and (b) decay PL spectra (λex = 300 nm) for R-Sb; (c) temperature-dependent PL spectra (λex = 370 nm) and (d) decay PL spectra (λex = 370 nm) for R-Sb. Inset: its partial amplification diagram from 6 to 14 μs. | ||
The dual-emission behaviour of R-Sb exhibits an exceptional temperature sensitivity, particularly in the vicinity of the 492 nm emission peak. Throughout the progressive cooling process from 320 to 80 K, the emission intensity at 492 nm experienced an obvious amplification by 17.3-fold, while the intensity at 612 nm saw a comparatively modest 2.4-fold increase. The temperature-dependent non-uniformity in the intensity variations of these two bands engenders a captivating colour-switching response in the luminescence (Fig. S14a†). At 80 K, the prevailing emission dominance in the blue emission region, specifically at 492 nm, endows R-Sb with an overall blue-emitting appearance, as evidenced by the CIE coordinates of (0.18, 0.27). However, as the temperature gradually rises, the prominence of the blue emission wanes, prompting a transition in the overall luminescence to a white emission at 320 K, exemplified by the CIE coordinates of (0.35, 0.37).
The Huang–Rhys factor (S) is widely employed to evaluate the strength of electron–phonon coupling interactions in metal halides, which can be estimated via the following equation (eqn (2)).
 | (2) |
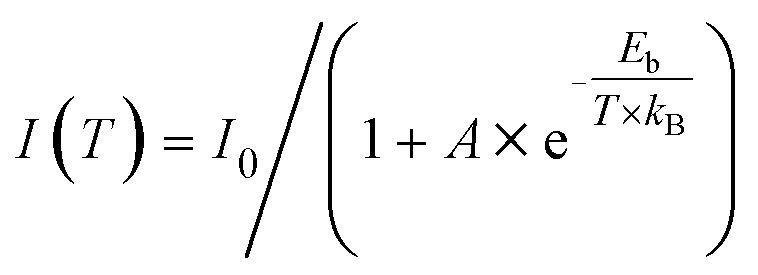 | (3) |
CPL properties of chiral Sb halides
One of the fascinating optical characteristics exhibited by chiral metal halides is their response to and control of circularly polarized light. Through an analysis of the crystal structure presented earlier, it becomes evident that the chirality of organic cations can be effectively transferred to the inorganic framework through asymmetrical hydrogen bonding interactions. This assertion finds validation in the circular dichroism (CD) spectra of the chiral antimony halides. The CD spectra of R- and S-Sb display mirror-image signals at 263 nm, 320 nm, and 356 nm, where all peaks are anterior to the absorption edge (Fig. 5a). This phenomenon is well known as the Cotton effect, associated with the splitting of electronic states under excitation in distinct CPL. The anisotropy factor, gCD, is calculated via the following equation (eqn (4)):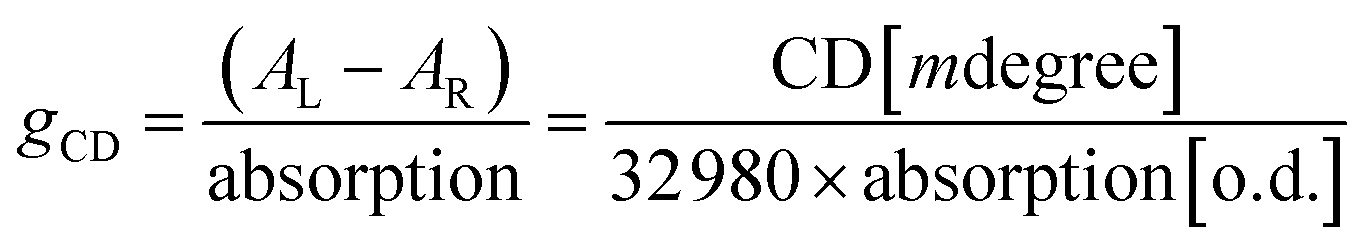 | (4) |
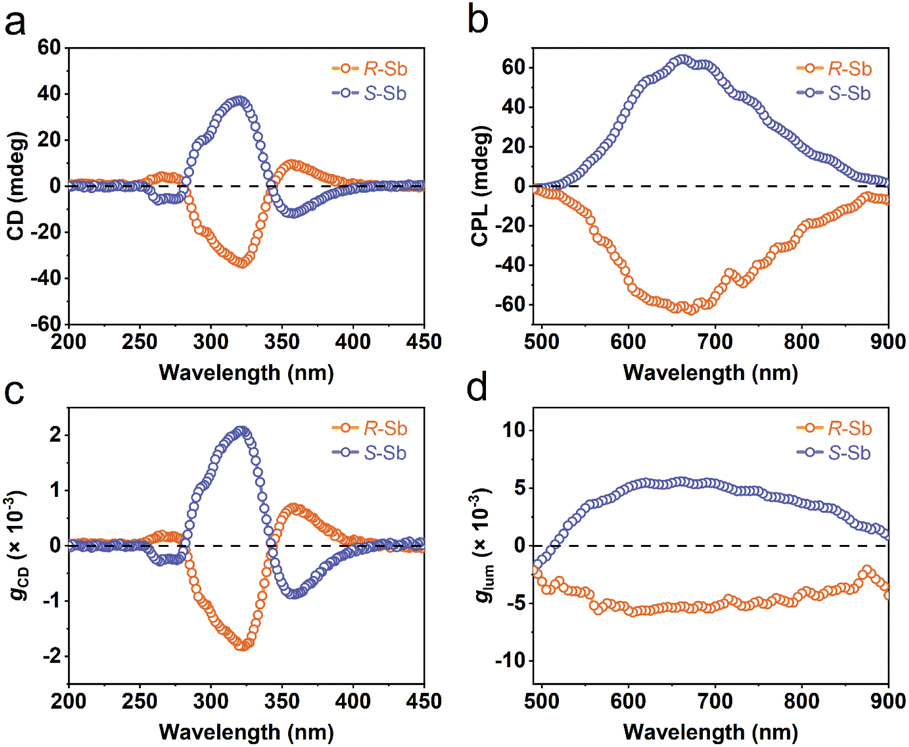 | ||
| Fig. 5 Chiroptical properties of R-/S-Sb. (a) CD spectra, (b) CPL emission spectra, and the corresponding (c) gCD factor and (d) glum factor of R-/S-Sb. | ||
Given their chiral structures and strong photoluminescence properties, it is not surprising that R-/S-Sb displays pronounced CPL responses. The CPL signals of R- and S-Sb crystals manifest as a pair of mirror-symmetric curves in the range of 500–900 nm (Fig. 5b). Similarly, the extent of CPL is quantified and assessed by the luminescence anisotropy factor glum. The glum is defined as the difference in emission intensity of the material under excitation by two different circularly polarized lights, i.e., glum = 2(IL − IR)/(IL + IR),79 where IL and IR are the intensities of left- and right- handed CPL. Fig. 5d implies the glum values of R- and S-Sb at the maximum emission wavelength are −5.7 × 10−3 and 5.2 × 10−3, respectively, showing a high luminescence dissymmetry factor among the previously reported chiral antimony halides (Table S7†).
Density functional theory (DFT) calculations
To understand the PL nature of R-Sb, theoretical calculations based on the first-principles DFT have been performed. As depicted in Fig. 6a, the flat electronic bands indicate a localized electron band structure. Moreover, the band gap of R-Sb is direct, and the corresponding calculated PBE bandgap at M point is determined to be 3.67 eV, slightly lower than the experimental value probably due to the discontinuity of the exchange-correlation function.80 Both the conduction band minimum (CBM) and valence band maximum (VBM) are contributed from the localized electronic states of inorganic parts, which also agree with the calculated projected density of states (PDOS) (Fig. 6b). The CBM mainly consists of a mixture of Sb-p and Cl-p, while the VBM is mainly derived from Sb-s and Cl-p orbitals with dominant Cl-p orbitals. The electronic charge density patterns also unveil the electronic transfer from the halide to metal (Fig. 6c and d), implying luminescence process is primarily determined by the inorganic components.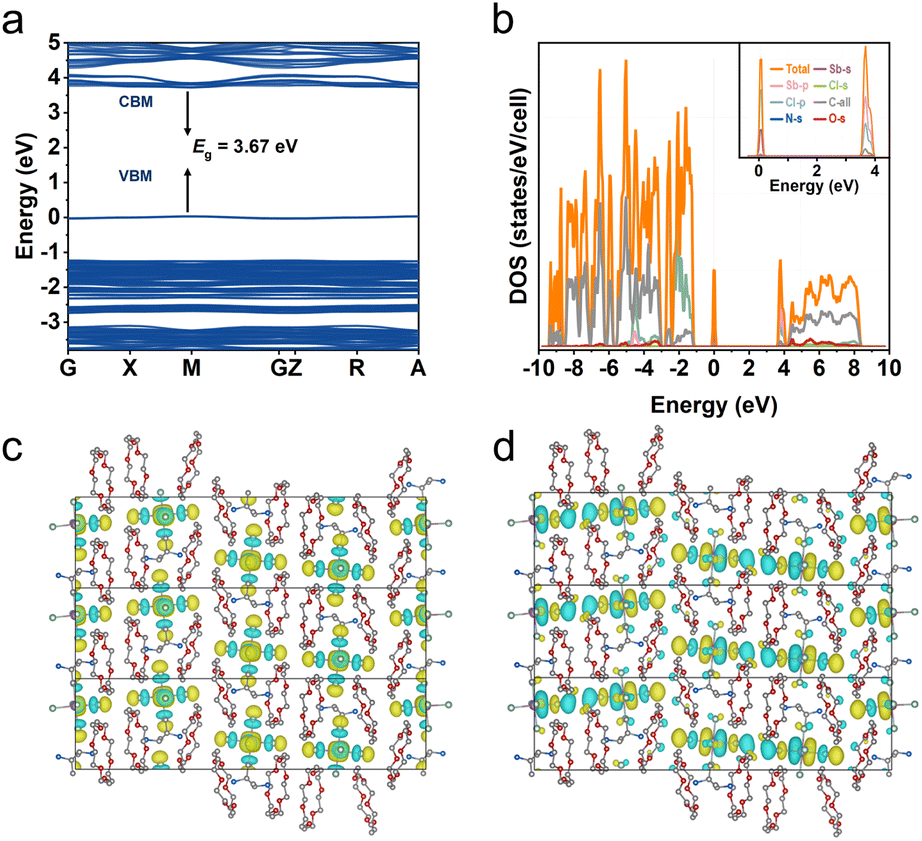 | ||
| Fig. 6 DFT simulation of R-Sb. (a) Calculated band structure and (b) the corresponding PDOS for R-Sb. (c) The conduction band minimum (CBM) and (d) valence band maximum (VBM) of R-Sb. | ||
Photophysical properties of achiral Sb halides
The UV absorption spectra of the achiral Sb halide rac-Sb indicate its similar absorption characteristics to those of R-Sb (Fig. S16†). The photophysical properties of rac-Sb are primarily assessed through its corresponding PL spectra. In the excitation-emission mapping patterns (Fig. S17a†), two emission centers can be observed, located at 490 nm and 626 nm, respectively. Time-resolved decay spectra reveal that the PL lifetimes at 490 nm and 626 nm are measured to be 7.2 ns and 7.4 μs, respectively, suggesting that the luminescent behaviours at the two peaks can be attributed to the 1P1 → 1S0 and 3P1 → 1S0 transition processes, respectively. As shown in Fig. S17a,† switching the excitation wavelength from 300 nm to 400 nm results in non-synergistic alterations in the emission intensities of the two main peaks, with the highest PL intensity at 490 nm significantly higher than that at 626 nm. Similarly, as the excitation wavelength gradually redshifts, rac-Sb undergoes a significant emission transition from blue to white and eventually to orange (Fig. S17b†).Nevertheless, the emission of rac-Sb is much lower than that of the chiral R-Sb. At the optimal excitation wavelengths of 319 nm and 371 nm, the PLQYs of rac-Sb are both below 15%. This can be mainly attributed to the smaller degree of lattice distortion in the inorganic octahedra of rac-Sb, resulting in weaker electron–phonon coupling interactions. Temperature-dependent PL spectra measurements of rac-Sb at excitation wavelengths of 319 nm and 371 nm were conducted (Fig. S18†). By analyzing the relationship between the FWHM at 490 nm (HE) and 626 nm (LE) and temperature, the corresponding S of HE and LE can be determined to be 19.1 and 17.4, respectively (Fig. S19†), both lower than those of R-Sb, verifying the weaker electron–phonon coupling interactions in rac-Sb. Moreover, the denser arrangement of octahedra in rac-Sb also leads to the increased energy transfer between adjacent octahedra, thereby lowering the efficiency of photoluminescence.
Noticeably, rac-Sb can be readily prepared via the solvent-free synthesis. By mixing stoichiometric amounts of precursor materials in this ternary system and dry grinding the precursors in the glovebox, rac-Sb can be rapidly and facilely obtained. After grinding for 10 minutes, the samples can express the orange emission under the 365 nm UV radiation (Fig. S20b†). The PXRD pattern of the ground sample is fitted well with the simulated result from the SCXRD data (Fig. S20c†), testifying to the successful fabrication of rac-Sb. However, grinding the stoichiometric R-/S-Sb precursor materials can only obtain the rac-Sb phase (Fig. S21†), probably due to the racemization of the chiral amine cation during the grinding process.
Conclusion
Adopting the host–guest self-assembly strategy mediated by a crown ether, we have designed and synthesized a pair of zero-dimensional chiral antimony halides (R-/S-Sb). The inclusion of crown ethers not only creates rich hydrogen bonding networks, but also contributes to the formation of bulky chiral cations, segregating the [SbCl6]3− inorganic octahedra and enhancing the PLQY, as high as 87.2% and 51.6% for orange and white emission, respectively. By virtue of the asymmetric hydrogen bonding interactions, the chirality of the organic cations is conveyed to the inorganic framework readily, inducing conspicuous asymmetrical lattice distortion within the octahedral structure. Consequently, R-/S-Sb exhibits strong CPL emission, along with the good luminescence anisotropy factor (|glum| ≥ 5.2 × 10−3), suggesting its potential as a circularly polarized LED (CP-LEDs). Intriguingly, the lattice distortion-induced dual-stimuli STE emission exhibits reversible emission transitions of chiral antimony halides from blue to white to orange colour, demonstrating sensitivity to both excitation wavelength and temperature. Meanwhile, the emission of rac-Sb also shares sensitivity to excitation wavelength, although it manifests a weaker emission than its chiral counterparts on account of the diminished electron–phonon coupling interactions. Moreover, rac-Sb can also be prepared readily via the solvent-free solid-state grinding pathway. This study provides a new protocol for the fabrication of a lead-free, efficient CPL emitter, while also expanding the application of chiral zero-dimensional metal halides in the field of multi-stimuli-responsive luminescence.Experimental section
Materials
18-Crown-6 (99%) and N,N-dimethylformamide (DMF, 99.5%) were purchased from Aladdin. R-/S-/rac-1,2-propanediamine dihydrochloride (R-/S-/rac-PDD·2HCl, 98%) was purchased from Bidepharm. Antimony trichloride (SbCl3, 99%) was purchased from Macklin. Tetrahydrofuran (THF, 99%) and dimethyl sulfoxide (DMSO, 99%) were purchased from Concord Technology (Tianjin). All reagents and solvents were used without further purification.Synthetic procedures
Structural characterization
Single-crystal X-ray diffractions (SCXRD) were conducted on a Rigaku XtaLAB MM007 CCD diffractometer with Cu Kα radiation (λ = 1.5418 Å) or Mo Kα radiation (λ = 0.71073 Å) at 100 K. The structures were solved by SHELXT methods with the Olex2 programs and all non-hydrogen atoms were refined anisotropically by the least-squares technique on weighted F2 using SHELXL. Powder X-ray diffraction (PXRD) spectra were recorded on a Rigaku D/Max-2500 diffractometer at 40 kV, 100 mA with a Cu-target tube and a graphite monochromator. Thermogravimetry analyses (TGA) were carried out using a standard TG-DTA analyser under an Ar flux of 10 mL min−1 in the temperature range 30–800 °C at a heating rate of 10 °C min−1.Computational methods
DFT calculations were implemented in the DS-PAW adopting the projector augmented-wave method (PAW) to deal with the ion–electron interaction.81–83 We selected the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) to approximate the exchange-correlation energy.84 Considering the nonbonding and long-range interaction in the hybrid organic–inorganic metal halides, the van der Waals (vdW) correction (DFT-D3) was adopted in the calculations.85 The plane-wave basis set was defined by the energy cut-off at 450 eV. The energy convergence criterion for electronic iteration was set to be 10−5 eV. The structural relaxation was performed until the Hellmann–Feynman forces on each atom were less than 0.001 eV Å−1. A Γ-centered 5 × 5 × 1 grid was adopted to simulate the k-space integrations.Data availability
The data supporting this study are available within the main text and ESI.†Author contributions
This article was written by X. H. after scientific discussion with all authors; J. X., B. X., and S. C. supervised the research; synthesis of single crystals and characterisations were carried out by X. H. with the help of S. H., Z. W., and J. G.; data treatments were performed by Y. Z., W. H., and R. S.; J. X. and Y. Z. revised the manuscript; theoretical calculations were performed by P. C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (52172045 and 22035003), the National Key R&D Program of China (2022YFA1204500 and 2022YFA1204504), the Fundamental Research Funds for the Central Universities, and Collaborative Innovation Platform Project of Fu-Xia-Quan National Independent Innovation Demonstration Zone (2022-P-021) is gratefully acknowledged. We gratefully acknowledge HZWTECH for providing computation facilities.References
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- Z. Tang, H. Gao, X. Chen, Y. Zhang, A. Li and G. Wang, Nano Energy, 2021, 80, 105454 CrossRef CAS.
- E. Moulin, L. Faour, C. C. Carmona-Vargas and N. Giuseppone, Adv. Mater., 2020, 32, 1906036 CrossRef CAS PubMed.
- X. Xia, C. M. Spadaccini and J. R. Greer, Nat. Rev. Mater., 2022, 7, 683–701 CrossRef PubMed.
- L. Ren, X. Xu, Y. Du, K. Kalantar-Zadeh and S. X. Dou, Mater. Today, 2020, 34, 92–114 CrossRef CAS.
- M. C. Koetting, J. T. Peters, S. D. Steichen and N. A. Peppas, Mater. Sci. Eng., R, 2015, 93, 1–49 CrossRef PubMed.
- J. Zhang, B. He, Y. Hu, P. Alam, H. Zhang, J. W. Y. Lam and B. Z. Tang, Adv. Mater., 2021, 33, 2008071 CrossRef CAS PubMed.
- Z. Li and Y. Yin, Adv. Mater., 2019, 31, 1807061 CrossRef PubMed.
- G. Guan, M. Wu and M.-Y. Han, Adv. Funct. Mater., 2020, 30, 1903439 CrossRef CAS.
- D. Li, Y. Yang, J. Yang, M. Fang, B. Z. Tang and Z. Li, Nat. Commun., 2022, 13, 347 CrossRef CAS PubMed.
- X. Bi, Y. Shi, T. Peng, S. Yue, F. Wang, L. Zheng and Q.-E. Cao, Adv. Funct. Mater., 2021, 31, 2101312 CrossRef CAS.
- T. P. Russell, Science, 2002, 297, 964–967 CrossRef CAS PubMed.
- J.-J. Pang, Z.-Q. Yao, K. Zhang, Q.-W. Li, Z.-X. Fu, R. Zheng, W. Li, J. Xu and X.-H. Bu, Angew. Chem., Int. Ed., 2023, 62, e202217456 CrossRef CAS PubMed.
- W. Ma, Q. Qian, S. M. H. Qaid, S. Zhao, D. Liang, W. Cai and Z. Zang, Nano Lett., 2023, 23, 8932–8939 CrossRef CAS PubMed.
- X.-Y. Lou, N. Song and Y.-W. Yang, Natl. Sci. Rev., 2021, 8, nwaa281 CrossRef CAS PubMed.
- K. Lou, Z. Hu, H. Zhang, Q. Li and X. Ji, Adv. Funct. Mater., 2022, 32, 2113274 CrossRef CAS.
- J. Li, X. Huang, X. Zhao, L.-J. Chen and X.-P. Yan, Angew. Chem., Int. Ed., 2021, 60, 2398–2405 CrossRef CAS PubMed.
- B. Zhou and D. Yan, Chem. Sci., 2022, 13, 7429–7436 RSC.
- M. Lu, Y. Zhang, S. Wang, J. Guo, W. W. Yu and A. L. Rogach, Adv. Funct. Mater., 2019, 29, 1902008 CrossRef.
- B. Zhou and D. Yan, Adv. Funct. Mater., 2023, 33, 2300735 CrossRef CAS.
- B. Zhou, Z. Qi, M. Dai, C. Xing and D. Yan, Angew. Chem., Int. Ed., 2023, 62, e202309913 CrossRef CAS PubMed.
- Y. Lin, S. Liu and D. Yan, Research, 2023, 6, 0259 CrossRef CAS PubMed.
- C. Xing, B. Zhou, D. Yan and W.-H. Fang, CCS Chem., 2023, 5, 2866–2876 CrossRef.
- J. Xu, X. Li, J. Xiong, C. Yuan, S. Semin, T. Rasing and X.-H. Bu, Adv. Mater., 2020, 32, 1806736 CrossRef CAS PubMed.
- K. P. Goetz, A. D. Taylor, F. Paulus and Y. Vaynzof, Adv. Funct. Mater., 2020, 30, 1910004 CrossRef CAS.
- A. A. Zhumekenov, M. I. Saidaminov, O. F. Mohammed and O. M. Bakr, Joule, 2021, 5, 2027–2046 CrossRef CAS.
- D. Han, J. Wang, T. Zheng, L. Peng and T. Jiao, Cell Rep. Phys. Sci., 2023, 4, 101523 CrossRef CAS.
- D. Duan, C. Ge, M. Z. Rahaman, C.-H. Lin, Y. Shi, H. Lin, H. Hu and T. Wu, NPG Asia Mater., 2023, 15, 8 CrossRef CAS.
- C. Zhou, H. Lin, Q. He, L. Xu, M. Worku, M. Chaaban, S. Lee, X. Shi, M.-H. Du and B. Ma, Mater. Sci. Eng., R, 2019, 137, 38–65 CrossRef.
- M. Li and Z. Xia, Chem. Soc. Rev., 2021, 50, 2626–2662 RSC.
- M. I. Saidaminov, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2017, 2, 889–896 CrossRef CAS.
- K. Han, J. Jin, B. Su and Z. Xia, Trends Chem., 2022, 4, 1034–1044 CrossRef CAS.
- J.-S. Yao, J.-J. Wang, J.-N. Yang and H.-B. Yao, Acc. Chem. Res., 2021, 54, 441–451 CrossRef CAS PubMed.
- X.-K. Liu, W. Xu, S. Bai, Y. Jin, J. Wang, R. H. Friend and F. Gao, Nat. Mater., 2021, 20, 10–21 CrossRef CAS PubMed.
- T. Zhang, C. Zhou, X. Feng, N. Dong, H. Chen, X. Chen, L. Zhang, J. Lin and J. Wang, Nat. Commun., 2022, 13, 60 CrossRef CAS PubMed.
- E. Merzlyakova, S. Wolf, S. Lebedkin, L. Bayarjargal, B. L. Neumeier, D. Bartenbach, C. Holzer, W. Klopper, B. Winkler, M. Kappes and C. Feldmann, J. Am. Chem. Soc., 2021, 143, 798–804 CrossRef CAS PubMed.
- Z. Wang, J.-T. Mo, J.-J. Pan and M. Pan, Adv. Funct. Mater., 2023, 33, 2300021 CrossRef CAS.
- H.-P. Lv, Y.-R. Li, X.-J. Song, N. Zhang, R.-G. Xiong and H.-Y. Zhang, J. Am. Chem. Soc., 2023, 145, 3187–3195 CrossRef CAS PubMed.
- C. Zhu, J. Jin, M. Gao, A. M. Oddo, M. C. Folgueras, Y. Zhang, C.-K. Lin and P. Yang, J. Am. Chem. Soc., 2022, 144, 12450–12458 CrossRef CAS PubMed.
- J. Zhao, T. Zhang, X.-Y. Dong, M.-E. Sun, C. Zhang, X. Li, Y. S. Zhao and S.-Q. Zang, J. Am. Chem. Soc., 2019, 141, 15755–15760 CrossRef CAS PubMed.
- J. Li, D. Yim, W.-D. Jang and J. Yoon, Chem. Soc. Rev., 2017, 46, 2437–2458 RSC.
- V. Morad, S. Yakunin and M. V. Kovalenko, ACS Mater. Lett., 2020, 2, 845–852 CrossRef CAS PubMed.
- L. Fan, K. Liu, S. He, F. Zhao, J. Zhao, Y. Wang and Q. Liu, Adv. Funct. Mater., 2022, 32, 2110771 CrossRef CAS.
- D.-Y. Liu, H.-Y. Li, R.-P. Han, H.-L. Liu and S.-Q. Zang, Angew. Chem., Int. Ed., 2023, 62, e202307875 CrossRef CAS PubMed.
- Y. Zheng, X. Han, P. Cheng, X. Jia, J. Xu and X.-H. Bu, J. Am. Chem. Soc., 2022, 144, 16471–16479 CrossRef CAS PubMed.
- T.-T. Kim, S. S. Oh, H.-D. Kim, H. S. Park, O. Hess, B. Min and S. Zhang, Sci. Adv., 2023, 3, e1701377 CrossRef PubMed.
- H. Lu, Z. V. Vardeny and M. C. Beard, Nat. Rev. Chem., 2022, 6, 470–485 CrossRef PubMed.
- L. E. MacKenzie and R. Pal, Nat. Rev. Chem., 2021, 5, 109–124 CrossRef CAS PubMed.
- Y. Sang, J. Han, T. Zhao, P. Duan and M. Liu, Adv. Mater., 2020, 32, 1900110 CrossRef CAS PubMed.
- C. Sun, Z. Deng, Z. Li, Z. Chen, X. Zhang, J. Chen, H. Lu, P. Canepa, R. Chen and L. Mao, Angew. Chem., Int. Ed., 2023, 62, e202216720 CrossRef CAS PubMed.
- L. Mao, P. Guo, S. Wang, A. K. Cheetham and R. Seshadri, J. Am. Chem. Soc., 2020, 142, 13582–13589 CrossRef CAS PubMed.
- J.-Q. Zhao, H.-S. Shi, L.-R. Zeng, H. Ge, Y.-H. Hou, X.-M. Wu, C.-Y. Yue and X.-W. Lei, Chem. Eng. J., 2022, 431, 134336 CrossRef CAS.
- B. Li, J. Jin, M. Yin, X. Zhang, M. S. Molokeev, Z. Xia and Y. Xu, Angew. Chem., Int. Ed., 2022, 61, e202212741 CrossRef CAS PubMed.
- M.-E. Sun, Y. Wang, F. Wang, J. Feng, L. Wang, H. Gao, G. Chen, J. Gu, Y. Fu, K. Bu, T. Fu, J. Li, X. Lü, L. Jiang, Y. Wu and S.-Q. Zang, J. Am. Chem. Soc., 2023, 145, 8908–8916 CrossRef CAS PubMed.
- Y. Zhang, Y. Zhang, Y. Zhao, H. Jia, Z. Yang, B. Yin, Y. Wu, Y. Yi, C. Zhang and J. Yao, J. Am. Chem. Soc., 2023, 145, 12360–12369 CrossRef CAS PubMed.
- C.-C. Fan, X.-B. Han, B.-D. Liang, C. Shi, L.-P. Miao, C.-Y. Chai, C.-D. Liu, Q. Ye and W. Zhang, Adv. Mater., 2022, 34, 2204119 CrossRef CAS PubMed.
- Y. Jing, Y. Liu, M. Li and Z. Xia, Adv. Opt. Mater., 2021, 9, 2002213 CrossRef CAS.
- F. Zhang, X. Chen, X. Qi, W. Liang, M. Wang, Z. Ma, X. Ji, D. Yang, M. Jia, D. Wu, X. J. Li, Y. Zhang, Z. Shi and C.-X. Shan, Nano Lett., 2022, 22, 5046–5054 CrossRef CAS PubMed.
- Y. Zhou, J. Chen, O. M. Bakr and O. F. Mohammed, ACS Energy Lett., 2021, 6, 739–768 CrossRef CAS.
- J.-L. Li, Y.-F. Sang, L.-J. Xu, H.-Y. Lu, J.-Y. Wang and Z.-N. Chen, Angew. Chem., Int. Ed., 2022, 61, e202113450 CrossRef CAS PubMed.
- B. Su, S. Geng, Z. Xiao and Z. Xia, Angew. Chem., Int. Ed., 2022, 61, e202208881 CrossRef CAS PubMed.
- F. Jiang, Z. Wu, M. Lu, Y. Gao, X. Li, X. Bai, Y. Ji and Y. Zhang, Adv. Mater., 2023, 35, 2211088 CrossRef CAS PubMed.
- Q. Guo, X. Zhao, B. Song, J. Luo and J. Tang, Adv. Mater., 2022, 34, 2201008 CrossRef CAS PubMed.
- V. Morad, Y. Shynkarenko, S. Yakunin, A. Brumberg, R. D. Schaller and M. V. Kovalenko, J. Am. Chem. Soc., 2019, 141, 9764–9768 CrossRef CAS PubMed.
- J.-H. Wei, J.-F. Liao, L. Zhou, J.-B. Luo, X.-D. Wang and D.-B. Kuang, Sci. Adv., 2021, 7, eabg3989 CrossRef CAS PubMed.
- K. M. McCall, V. Morad, B. M. Benin and M. V. Kovalenko, ACS Mater. Lett., 2020, 2, 1218–1232 CrossRef CAS PubMed.
- L. Zhang, S. Li, H. Sun, Y. Fang, Y. Wang, K. Wang, H. Jiang, L. Sui, G. Wu, K. Yuan and B. Zou, Angew. Chem., Int. Ed., 2023, 62, e202311912 CrossRef CAS PubMed.
- B. Chen, Y. Guo, Y. Wang, Z. Liu, Q. Wei, S. Wang, A. L. Rogach, G. Xing, P. Shi and F. Wang, J. Am. Chem. Soc., 2021, 143, 17599–17606 CrossRef CAS PubMed.
- G. Zhang, D. Wang, B. Lou, C.-G. Ma, A. Meijerink and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202207454 CrossRef CAS PubMed.
- P. Klement, N. Dehnhardt, C.-D. Dong, F. Dobener, S. Bayliff, J. Winkler, D. M. Hofmann, P. J. Klar, S. Schumacher, S. Chatterjee and J. Heine, Adv. Mater., 2021, 33, 2100518 CrossRef CAS PubMed.
- B. Zhang, T. Zheng, J. You, C. Ma, Y. Liu, L. Zhang, J. Xi, G. Dong, M. Liu and S. Liu, Adv. Mater., 2023, 35, 2208875 CrossRef CAS PubMed.
- L. Zhang, Y. Fang, L. Sui, J. Yan, K. Wang, K. Yuan, W. L. Mao and B. Zou, ACS Energy Lett., 2019, 4, 2975–2982 CrossRef CAS.
- P. Cheng, L. Sun, L. Feng, S. Yang, Y. Yang, D. Zheng, Y. Zhao, Y. Sang, R. Zhang, D. Wei, W. Deng and K. Han, Angew. Chem., Int. Ed., 2019, 58, 16087–16091 CrossRef CAS PubMed.
- H. Shao, X. Wu, J. Zhu, W. Xu, L. Xu, B. Dong, J. Hu, B. Dong, X. Bai, H. Cui and H. Song, Chem. Eng. J., 2021, 413, 127415 CrossRef CAS.
- W. Tao, Y. Zhang and H. Zhu, Acc. Chem. Res., 2022, 55, 345–353 CrossRef CAS PubMed.
- B. Yang, F. Hong, J. Chen, Y. Tang, L. Yang, Y. Sang, X. Xia, J. Guo, H. He, S. Yang, W. Deng and K. Han, Angew. Chem., Int. Ed., 2019, 58, 2278–2283 CrossRef CAS PubMed.
- R. Zeng, L. Zhang, Y. Xue, B. Ke, Z. Zhao, D. Huang, Q. Wei, W. Zhou and B. Zou, J. Phys. Chem. Lett., 2020, 11, 2053–2061 CrossRef CAS PubMed.
- L. Zhou, J.-F. Liao, Y. Qin, X.-D. Wang, J.-H. Wei, M. Li, D.-B. Kuang and R. He, Adv. Funct. Mater., 2021, 31, 2102654 CrossRef CAS.
- C. Zhang, S. Li, X.-Y. Dong and S.-Q. Zang, Aggregate, 2021, 2, e48 CrossRef CAS.
- C. Wu, X. Jiang, Z. Wang, H. Sha, Z. Lin, Z. Huang, X. Long, M. G. Humphrey and C. Zhang, Angew. Chem., Int. Ed., 2021, 60, 14806–14810 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 2303422, 2303423, 2303424 and 2303425. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06362c |
| This journal is © The Royal Society of Chemistry 2024 |