
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkyl sulfonate surfactant mediates electroreduction of carbon dioxide to ethylene or ethanol over hydroxide-derived copper catalysts†
Yiding
Wang
ab,
Runyao
Zhao
ab,
Yunpeng
Liu
 c,
Fengtao
Zhang
b,
Yuepeng
Wang
ab,
Zhonghua
Wu
bc,
Buxing
Han
ab and
Zhimin
Liu
*ab
c,
Fengtao
Zhang
b,
Yuepeng
Wang
ab,
Zhonghua
Wu
bc,
Buxing
Han
ab and
Zhimin
Liu
*ab
aBeijing National Laboratory for Molecular Sciences, CAS Laboratory of Colloid and Interface and Thermodynamics, CAS Research/Education Center for Excellence in Molecular Sciences, Center for Carbon Neutral Chemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: liuzm@iccas.ac
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cInstitute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
First published on 8th February 2024
Abstract
For CO2 electroreduction (CO2ER) to C2 compounds, it is generally accepted that the formation of ethylene and ethanol shares the same intermediate, *HCCOH. The majority of studies have achieved high faradaic efficiency (FE) towards ethylene, but faced challenges to get high ethanol FE. Herein, we present an alkyl sulfonate surfactant (e.g., sodium dodecyl sulfonate, SDS) mediated CO2ER to a C2 product over an in situ generated Cu catalyst (Cu@SDS) from SDS-modified Cu(OH)2. It achieves the CO2ER to ethylene as the sole C2 product at low applied voltages with a FE of 55% at −0.6 V vs. RHE and to ethanol as the main product at potentials ≥0.7 V with a maximum FE of 64% and a total C2 FE of 86% at −0.8 V, with a current density of 231 mA cm−2 in a flow cell. Mechanism investigation indicates that SDS modifies the oxidation state of the in situ formed Cu species in Cu@SDS, thus tuning the catalyst activity for CO2ER and lowering the C–C coupling energy barrier; meanwhile, it tunes the adsorption mode of the *HCCOH intermediates on the catalyst, thus mediating the selectivity of CO2ER towards C2 products.
Introduction
The greenhouse effect, primarily driven by excessive emission of carbon dioxide (CO2), has emerged as a widely discussed topic. Interestingly, CO2 can be applied as a valuable C1 synthon, given its abundant availability.1 CO2 electroreduction (CO2ER) has shown promise as a viable method to transform CO2 into a range of valuable chemicals, including CO,2 methane,3 methanol,4 ethylene,5,6 and ethanol.7 Ethylene and ethanol, in particular, have been widely employed in modern industry, making them highly desirable. However, achieving CO2ER selectively either to ethylene or to ethanol presents great challenges due to the involvement of twelve proton–electron-coupled intermediates8 and the competing hydrogen evolution reaction (HER). Copper-based catalysts have demonstrated superior activity in promoting carbon–carbon coupling during CO2ER, benefiting from their moderate adsorption capabilities for CO2 and intermediates.9,10 While extensive research has successfully achieved CO2ER to ethylene with high faradaic efficiency (FE), attaining a high FE of ethanol (over 60%) is seldom reported.11–14It is generally accepted that the formation of ethylene and ethanol shares the same intermediate, *HCCOH, which undergoes hydrogenation and deoxygenation to form ethanol and ethylene, respectively.15,16 Therefore, catalysts that can enhance hydrogenation while suppressing deoxygenation of *HCCOH are highly desirable for CO2ER to ethanol. Several strategies have been reported to prepare such catalysts, for example, control over copper species,11 surface modification17–19 and addition of dopants to catalysts.12,13
Surfactants are commonly employed as protective agents20 or templates21 in catalyst synthesis processes. Recently, it has been reported that the surfactant decorated on the catalyst surface can effectively tune both the selectivity of CO2ER to desired products and current density.22,23 These surfactant-induced effects are mainly attributed to the following aspects: tuning the hydrophilicity and charge distribution of the catalyst,23 enriching reactants on the catalyst surface,24 and modifying electrode–electrolyte interfaces.25,26 Though some progress has been made, surfactant-mediated CO2ER is still seldom reported.
In this work, we present an alkyl sulfonate surfactant (e.g., sodium dodecyl sulfonate, SDS) mediated CO2ER to ethanol, which is achieved over Cu@SDS derived from electroreduction of SDS modified Cu(OH)2 (Cu(OH)2@SDS) in the CO2ER process. The in situ generated Cu@SDS exhibited high performance for CO2-to-ethanol conversion in 1.0 M KOH electrolyte, affording an ethanol faradaic efficiency (FE) of 64% and a total C2 product FE of 86%, with a current density of 231 mA cm−2 in a flow cell. This catalyst showed much higher activity for catalysing CO2ER than the OHDCu catalyst originated from reduction of Cu(OH)2. It was found that the SDS-functionalized Cu species derived from electroreduction of Cu(OH)2@SDS are responsible for the generation of ethanol in the CO2ER process, while the in situ formed Cu species without SDS could afford only ethylene as the C2 compound. From the results of density functional theory (DFT) calculations, not only CO adsorbance is enhanced, but C–C coupling is also facilitated in the presence of SDS. Importantly, the strong hydrogen bonding interaction between the SDS anion and *HCCOH suppresses the deoxygenation of *HCCOH over Cu@SDS, thus producing ethanol.
Experimental
Preparation of catalysts
Cu(OH)2@SDS was synthesized as follows. First, 1 mmol of CuCl2 was added into a flask containing 50 mL of water, and 100 mg of SDS was then added. After being stirred to full dispersion, 2 mL of 1 M KOH was injected into the solution, and a grey suspension was formed within 2 h. The suspension was subsequently washed with ethanol and water twice, respectively. Similarly, Cu(OH)2@SOS was prepared using SOS to replace SDS. Cu(OH)2 was prepared without any surfactant, and blue suspension was formed. Mechanically mixed Cu(OH)2 and SDS were prepared by grinding Cu(OH)2 with SDS. Cu(OH)2@SDS and Cu(OH)2 and were firstly characterized by means of different techniques (see ESI, Fig. S2–S9†). After electroreduction at a constant potential of −0.7 V (vs. RHE, reversible hydrogen electrode) under a CO2 atmosphere, Cu@SDS and OHDCu were obtained from Cu(OH)2@SDS and Cu(OH)2, respectively, which were examined and evaluated for CO2ER.CO2 electroreduction experiments
Typically, 10 mg of the as-prepared catalyst was dispersed in 3 mL of ethanol and mixed with 50 μL of Nafion D-521 solution (5%, Thermo Scientific) to form a uniform catalyst ink after being sonicated for 1 h. The ink (675 μL) was evenly loaded on 1.5 × 1.5 cm2 SIGRACET 29BC (0.2–0.5 mg cm−2) under reduced pressure. The electrocatalytic activity of the catalyst was evaluated in a flow cell with a three-electrode system using 1 M KOH as electrolyte at room temperature. 20 sccm of CO2 was provided to the cathode, and the flowrate of the catholyte and anolyte was controlled at 50 mL min−1. The membrane Fumasep FAB-PK-130 was used to separate the anode and cathode.A LSV test was carried out under the conditions that CO2 was passed at a sweep rate of 10 mV s−1. The products from CO2ER were analyzed by gas chromatography (GC) and 1H NMR analysis over a certain electric amount of 100C at various potentials. The FE of products was calculated as follows:
| FE = (amount of M × n × F/C) × 100% |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485C mol−1), and C is the amount of charge passing through the working electrode. An IR compensation of 3.7 ohm was used in LSV tests and amperometric i–t tests.
485C mol−1), and C is the amount of charge passing through the working electrode. An IR compensation of 3.7 ohm was used in LSV tests and amperometric i–t tests.
Results and discussion
The electrocatalytic performances of the as-synthesized Cu@SDS and OHDCu catalysts were initially evaluated using linear sweep voltammetry (LSV) under a N2 or CO2 atmosphere. It was indicated that both in situ generated Cu@SDS and OHDCu catalysts exhibited a noticeable increase in current density when CO2 was used as the feedstock rather than N2 (Fig. 1a, S12†), indicating that they show activity for CO2ER. Lower current densities are observed on Cu@SDS, which means that the electrochemical process is regulated, caused by a reduced electrochemically active surface area (ECSA) (62 cm2 for Cu@SDS and 89 cm2 for OHDCu) and improved impedance (Fig. S30 and S31†). It is reasonable that SDS could occupy active sites, thus suppressing the total reaction dynamics. Both catalysts exhibited high activity for CO2ER to C2 compounds in the gas and liquid phases, respectively. The OHDCu catalyst derived from as-prepared Cu(OH)2 afforded ethylene as the sole C2 compound at all applied voltages, reaching a maximum ethylene FE of 55% at −0.9 V (Fig. 1b). Interestingly, the resultant Cu@SDS could adjust the selectivity of CO2ER towards C2 compounds via changing the applied voltages (Fig. 1c and d). At low voltages of −0.5 and −0.6 V ethylene was obtained as the sole C2 product, with the highest FE of 54% at −0.6 V, while at higher applied voltages of −0.7 to −0.9 V ethanol was formed, reaching the highest ethanol FE of 64% at −0.8 V, with a total C2 compound FE of 86%. The electroreduction of 13CO2 instead of CO2 was performed, and 13C-labeled ethanol was detected (Fig. S19†), suggesting that the carbonaceous products originated from CO2ER and excluding SDS as a reactant.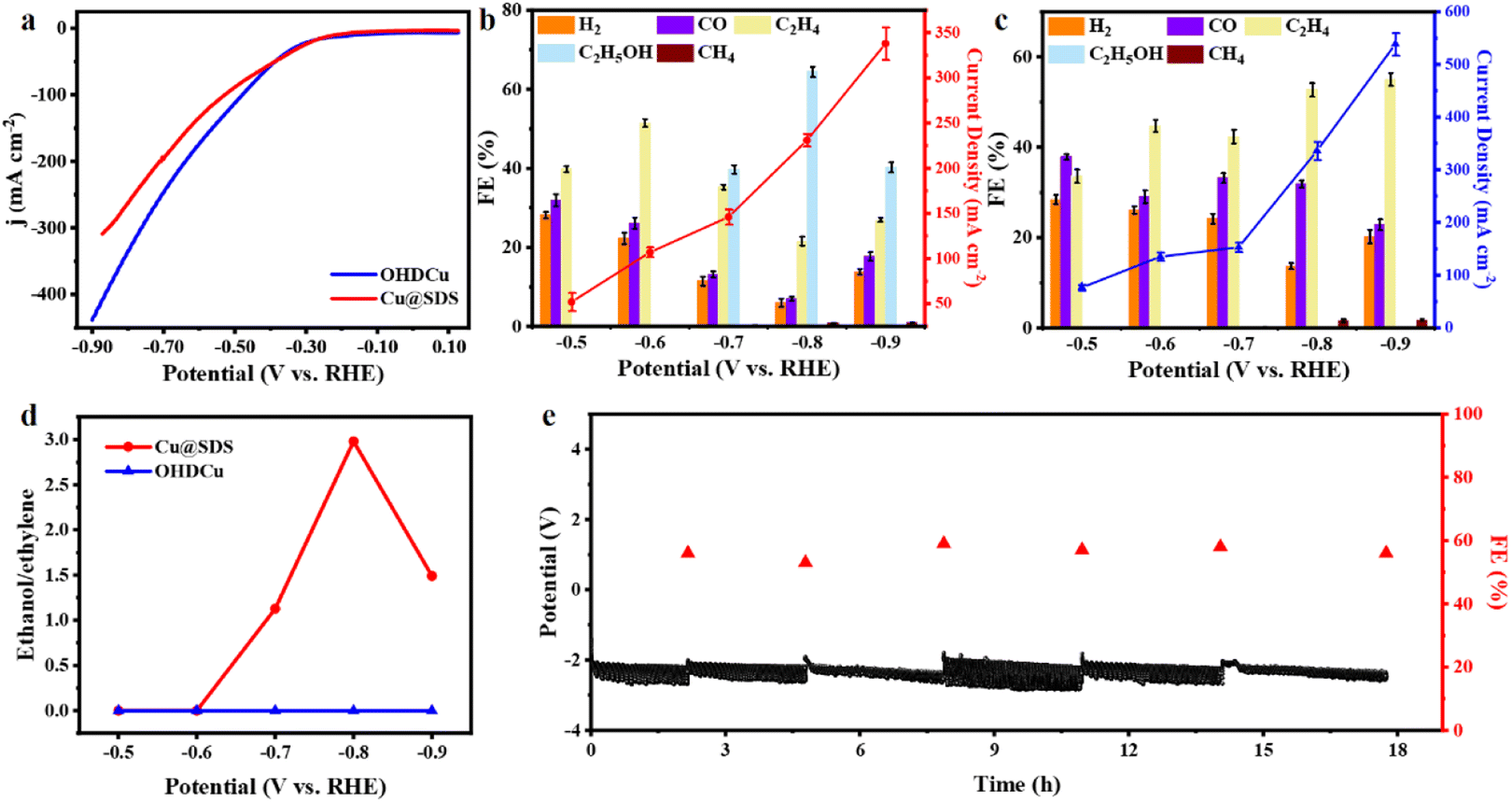 | ||
| Fig. 1 CO2ER performances of catalysts. (a) LSV comparison of Cu@SDS and OHDCu; FEs and current densities under different potentials of Cu@SDS (b) and OHDCu (c). (d) The ratios of ethanol to ethylene derived from CO2ER over Cu@SDS and OHDCu at different potentials. (e) Potentials and FEs of ethanol derived from continuous CO2 reduction at 100 mA cm−2 without iR compensation. 1.0 M KOH was applied as electrolyte for the above tests. | ||
In comparison, Cu@SDS showed much higher FE of C2 chemicals and lower hydrogen FE than OHDCu at the same applied voltages, suggesting that SDS promoted CO2ER and efficiently inhibiting the generation of H2. As mechanically mixed Cu(OH)2 and SDS were tested for CO2ER, no ethanol was obtained (Fig. S14†). From the above findings, it can be deduced that SDS plays a key role in mediating the selectivity of CO2ER and the SDS-functionalized Cu species are responsible for the generation of ethanol.
For comparison, sodium octyl sulfonate (SOS) functionalized Cu(OH)2 was prepared and applied in CO2ER. Ethanol was obtained as expected at the suitable applied voltages, but in a smaller amount compared to the case using Cu@SDS as the catalyst under the same conditions (Fig. S18†). This indicates that SOS plays a similar role to SDS in mediating the production of C2 products from CO2ER. The FE differences induced by these two surfactants may be ascribed to the discrepancy in modification on the Cu catalysts and to the difference in their interactions with the *HCCOH intermediate.
To assess the stability of Cu@SDS, continuous CO2 reduction was conducted at a constant current of 100 mA cm−2. As depicted in Fig. 1e, no significant decrease in ethanol FE or change in potential was observed as the reaction was performed for 18 h, except for minor fluctuations caused by periodic bubble formation. This indicates the excellent stability of Cu@SDS for CO2ER.
From scanning electron microscopy (SEM) observation, it is clear that Cu@SDS (Fig. S20d†) and OHDCu (Fig. S21†) appeared as similar irregular nanoparticles with size around 100 nm, different from the nanorod-like morphology of Cu(OH)2@SDS and OHDCu (Fig. S2 and S3†). The X-ray diffraction (XRD) patterns of Cu@SDS and OHDCu display two peaks at 43.3° and 50.4° (Fig. 2a) ascribed to the Cu (111) and (200) crystal planes, which confirms the formation of metallic Cu in these two samples (Fig. S32†).27 Extended X-ray absorption fine structure (EXAFS) analysis also confirmed the formation of metallic Cu with a Cu–Cu coordination environment in the Cu@SDS sample.
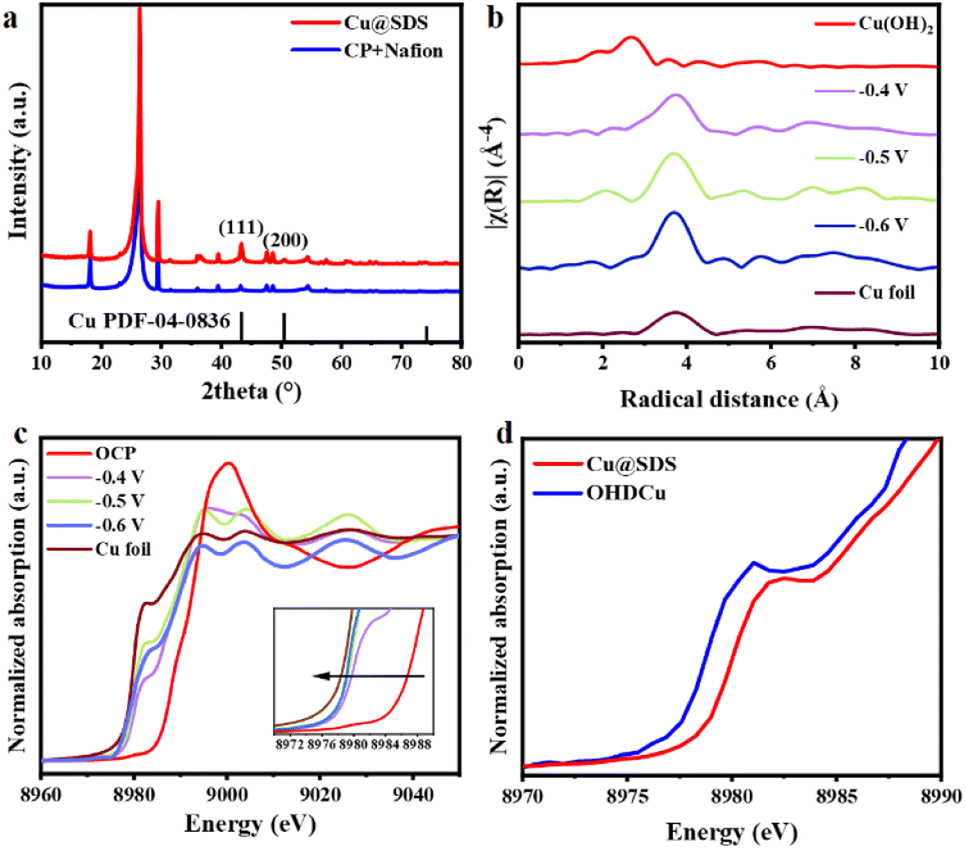 | ||
| Fig. 2 (a) XRD pattern of Cu@SDS and bare Nafion on carbon paper. (b) Fourier transform k3-weighted R space χ EXAFS spectra of Cu@SDS applied in CO2ER at different applied potentials with the comparison of Cu RE (Cu foil reference). (c) In situ XANES of OHDCu at different applied potentials. (d) Comparison of in situ XANES of Cu@SDS and OHDCu at −0.8 V. | ||
To explore the structural changes of Cu(OH)2@SDS and the role of SDS in the CO2ER process, in situ measurements were conducted under the same conditions as the CO2ER tests.28 Considering that the oxidation state of Cu species can significantly affect their performance for CO2ER,29,30in situ X-ray absorption near-edge structure (XANES) analysis was performed to probe the oxidation states of Cu in CO2ER (Fig. 2c and d, S22†).31 From the XANES spectra (Fig. 2c), it is obvious that the oxidation states of Cu species in OHDCu obtained at various potentials were higher than that of Cu foil and gradually came close to that of Cu foil as the applied potential increased. This means that the Cu species in the in situ formed catalysts exhibit a tunable oxidation state related to the applied potentials, meaning that they have tunable activity in the CO2ER process. The presence of SDS can remarkably influence the oxidation state of the reduced Cu species, confirmed by the fact that the Cu species in Cu@SDS showed an even higher oxidation state compared to those in OHDCu at an applied potential of −0.8 V. The higher oxidation state induced by the SDS molecule could enhance CO adsorption and thus facilitate C–C coupling,30 inducing a higher C2/C1 ratio on Cu@SDS than that on OHDCu (Fig. S13†). However, at −0.9 V, the Cu species in Cu@SDS and OHDCu exhibit nearly identical oxidation states (Fig. S22†), suggesting that some SDS molecules may desorb from Cu@SDS under more negative potentials. This may partially explain why the resultant Cu@SDS catalyst showed lower ethanol FE at −0.9 V than at −0.8 V. Furthermore, charge density difference calculations32 demonstrated a clear charge loss of Cu in the presence of the sulfonate anion (Fig. S23†), leading to a higher Cu oxidation state, which supports the XANES analysis results.
The in situ Raman spectroscopy and in situ Fourier transform infrared (FTIR) spectroscopy analyses provide information on the intermediates of CO2ER on the catalysts. In the in situ Raman spectra of CO2ER carried out at −0.8 V (Fig. 3a), apart from the D band and G band induced by the carbon paper substrate, the ν(Cu–CO)33 peak appeared at 354 cm−1 for OHDCu, while it shifted to 362 cm−1 for Cu@SDS, indicating a stronger interaction between Cu@SDS and the CO intermediate, which is also supported by DFT calculations. The calculated bond length of Cu–CO on Cu@SDS (1.82 Å) is shorter than that on OHDCu (1.85 Å) (Fig. 3c and d). The suitable Cu–CO bonding interaction is favorable to carbon–carbon coupling, which can explain why Cu@SDS showed higher C2 FE than OHDCu (Fig. 1b and c), in line with XANES analysis.
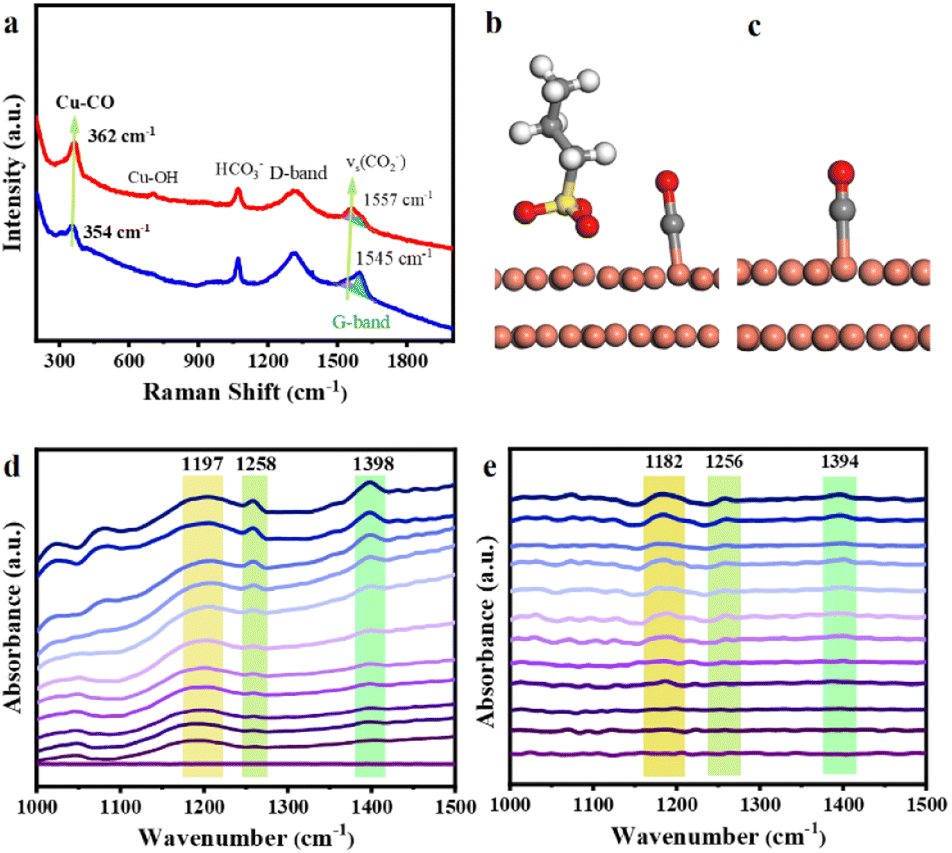 | ||
| Fig. 3 (a) In situ Raman spectrum of Cu@SDS and OHDCu at −0.8 V. Optimized structure of CO adsorbed on Cu(111)@PS (b) and Cu(111) (c). Cu–CO band lengths are 1.82 Å for Cu@PS and 1.85 Å for OHDCu. In situ FTIR spectra of CO2ER intermediates on Cu@SDS (d) and OHDCu (e) at potential applied from OCP, −0.1 to −1.1 V. | ||
In situ FTIR analysis provides information on the adsorption of CO2 and intermediates of CO2ER on the catalysts. In the in situ FTIR spectra of CO2ER over Cu@SDS (Fig. 3d), the peak at 1398 cm−1 corresponds to the chemisorbed *CO2 species.34 In the case of using OHDCu as the catalyst, this peak appears at 1394 cm−1 (Fig. 3f). These findings indicate that the presence of SDS in Cu@SDS may impact the adsorption mode of CO2 on the catalyst. The corresponding in situ Raman spectra (Fig. 2a) show identical information, in which the peak assigned to the adsorbed CO2− species on Cu@SDS shifted to 1557 cm−1 from 1545 cm−1 for those on OHDCu. The gradually increased intensity of the band at 1398 cm−1 in the in situ FTIR spectra indicates that more CO2 molecules are chemically adsorbed and activated as the applied potentials increase, which is favorable to CO2ER.
Another characteristic band corresponding to the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band in the *COOH intermediate35 appears at 1258 and 1256 cm−1 using Cu@SDS and OHDCu catalysts, respectively, which also reflects the influence of SDS on the properties of the catalyst. The peak at 1197 cm−1 is attributed to the stretching vibration of C–OH from the *HCCOH intermediate,36 while it appears at 1182 cm−1 when using OHDCu as the catalyst. This indicates that Cu@SDS provides a microenvironment to make the C–OH bond in *HCCOH become stronger, probably due to the hydrogen bonding interaction between the SDS anion and hydroxyl H of this intermediate. The enhanced C–OH bond strength may inhibit the deoxygenation of the *HCCOH intermediate upon further reduction, thus producing ethanol. The presence of bands at 2960, 2922, and 2853 cm−1 corresponding to the asymmetric stretching vibration of –CH3 and –CH2–, and the symmetric stretching of –CH2–, respectively (Fig. S25†), provides direct evidence of the evolution of ethanol.
O band in the *COOH intermediate35 appears at 1258 and 1256 cm−1 using Cu@SDS and OHDCu catalysts, respectively, which also reflects the influence of SDS on the properties of the catalyst. The peak at 1197 cm−1 is attributed to the stretching vibration of C–OH from the *HCCOH intermediate,36 while it appears at 1182 cm−1 when using OHDCu as the catalyst. This indicates that Cu@SDS provides a microenvironment to make the C–OH bond in *HCCOH become stronger, probably due to the hydrogen bonding interaction between the SDS anion and hydroxyl H of this intermediate. The enhanced C–OH bond strength may inhibit the deoxygenation of the *HCCOH intermediate upon further reduction, thus producing ethanol. The presence of bands at 2960, 2922, and 2853 cm−1 corresponding to the asymmetric stretching vibration of –CH3 and –CH2–, and the symmetric stretching of –CH2–, respectively (Fig. S25†), provides direct evidence of the evolution of ethanol.
To further explore the electrochemical performance differences between Cu@SDS and OHDCu, density functional theory (DFT) calculations were conducted using Cu(111) and propyl sulfonate (PS, Cu(111)@PS) as a substitute for SDS to save computational resources, considering that from the third carbon atom to the tail end, charge distribution is similar (Fig. S11†) and the distance to all intermediates are far enough to ignore the interaction. Firstly, the stability of SDS on the Cu(111) surface was considered by introducing auxiliary atoms. After two auxiliary atoms were introduced, the average bond length of Cu–O coordinated between directly adsorbed O from SDS and the nearest three Cu atoms is not obviously changed (Fig. S24†), proving the stability of the structure. As shown in Fig. 4a and b, once the first CO molecule is adsorbed on Cu(111)@PS, another CO molecule is more preferable to be adsorbed on the surface, than on bare Cu(111), exhibiting a 2.9 eV lower reaction energy (*CO + CO → *CO + *CO). The result gives proof that in the presence of SDS, the surface coverage of CO could be higher, benefitting the C–C coupling procedure. The C–C coupling reaction barrier (*CO + *CO → *COCOH) is also lowered from 1.04 to 0.64 eV, in accordance with the facilitated C2 products shown in Fig. 1b and c.
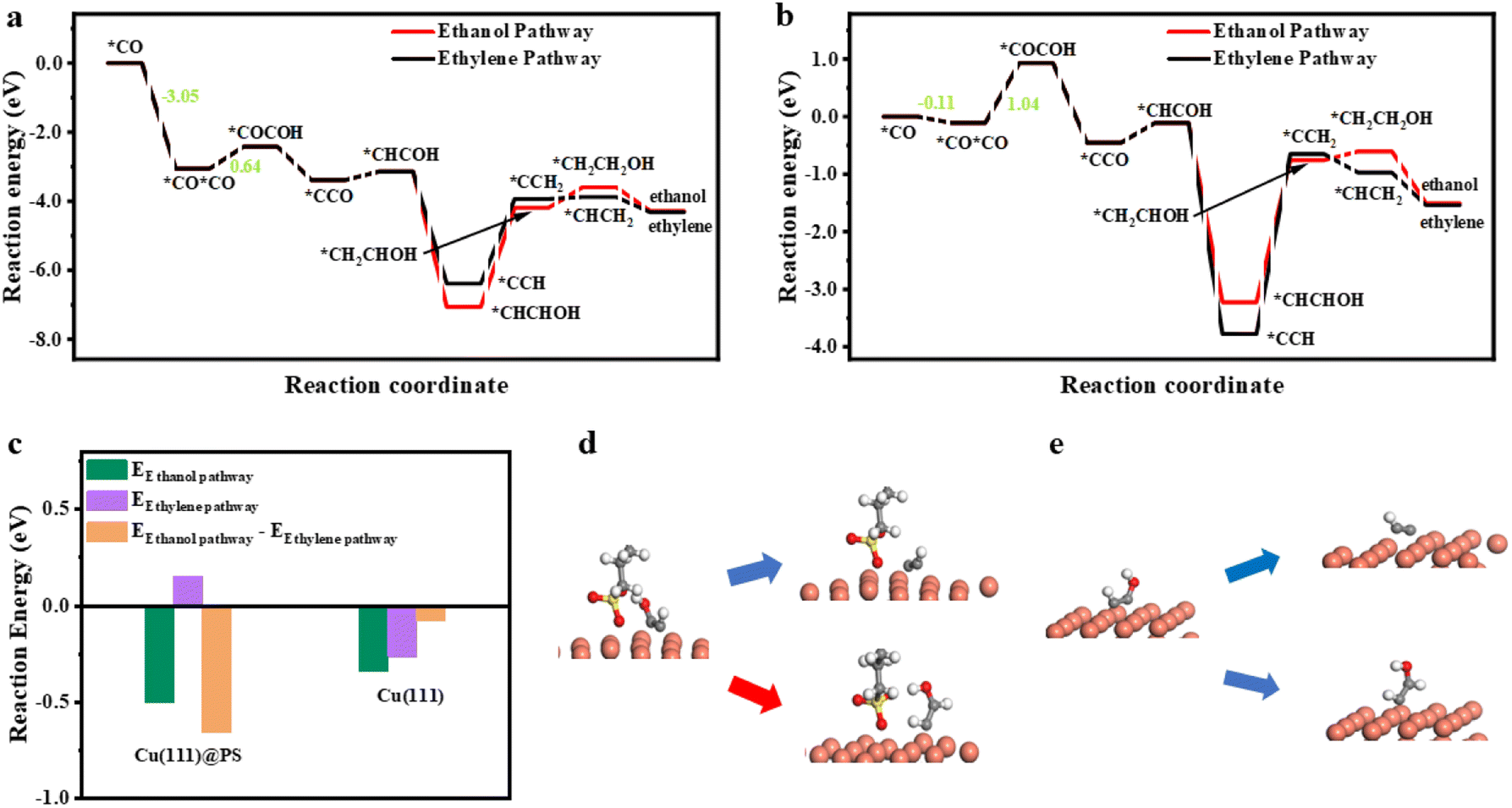 | ||
| Fig. 4 Free energy diagram for ethanol and ethylene on Cu(111)@PS (a) and Cu(111) (b). (c) DFT calculated reaction energies of the ethylene pathway (*HCCOH to *CCH) and the ethanol pathway (*HCCOH to *HCCHOH), and their deviation on Cu(111)@PS and Cu(111). Illustration of calculation models and reaction pathways on Cu(111)@PS (d) and Cu(111) (e). See full version picture of structures in Fig. S25 and S27.† Cu, O, C, S, and H are illustrated as orange, red, gray, yellow and white balls, respectively. | ||
To explain the selectivity difference between ethanol and ethylene, the intermediate *HCCOH was taken as the starting point for the bifurcation towards ethylene and ethanol.37 The reaction energies from *HCCOH to *HCCHOH and to *CCH were calculated and plotted in Fig. 4c. Obviously, the pathway to ethanol through *HCCOH (*HCCOH → *HCCHOH) is more energetically favorable on Cu(111) @PS (−0.5 eV) than on bare Cu(111) (−0.34 eV), which supports that Cu@SDS can achieve CO2ER to ethanol. However, the subsequent step (*CHCHOH → *CH2CHOH or *CCH → *CCH2) is energy demanding, and for Cu@PS, the reaction energy for the former one is higher (2.87 eV over 2.45 eV), resulting in similar production distribution at −0.5 V and −0.6 V. Once the energy demand is satisfied, the ethanol pathway will be unchoked and more preferred. The energy deviation between the ethanol and ethylene formation (energy difference between reactions *HCCOH → *HCCHOH and *HCCOH → *CCH) directly affects CO2ER selectivity to these two compounds. A more negative value of −0.66 eV was obtained for PS@Cu(111), compared to that for Cu(111) (−0.08 eV), which indicates that PS@Cu(111) is more favorable for catalyzing CO2ER towards ethanol. From the optimized geometry of SDS, Cu(111) and *HCCOH (Fig. 4d) via DFT calculations, it is clear that a strong hydrogen bond could be formed between the O atom of the SDS anion and the hydroxyl H of *HCCOH with a length of 1.53 Å (Fig. S28†), which may be responsible for suppressing the deoxygenation of *HCCOH upon further reduction, thus generating ethanol.
Conclusions
In summary, we present a surfactant-mediated strategy for CO2ER to ethanol, which is achieved over an in situ formed catalyst from Cu(OH)2@SDS. A high ethanol FE of 64% with a total C2 FE of 86% was obtained at −0.8 V, with a current density of 231 mA cm−2 using a flow cell. It has been indicated that SDS plays key role in mediating the selectivity of CO2ER to ethanol from the following aspects. SDS could modify the oxidation state of the in situ formed Cu species in the catalyst depending on the applied potentials, thus tuning the catalyst activity for CO2ER. Meanwhile, SDS could interact with the *HCCOH intermediate via hydrogen bonding interaction, thus suppressing the deoxygenation of *HCCOH and generating ethanol preferentially. This work provides a simple and novel way to achieve CO2ER to ethanol in high FE, which may have promising applications in the production of ethanol from CO2ER.Data availability
The data underlying this study are available in the published article and its ESI.†Author contributions
Z. L. conceived and supervised the experiments. Y. W. performed the experiments and all the simulations and F. Z. analysed the calculated data. R. Z. carried out the TEM analysis on the catalysts. Y. L. facilitated the XAS related experiments. All authors discussed the results and wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was financially supported by the National Key Research and Development Program of China (2020YFA0710203) and National Natural Science Foundation of China (22121002). The authors thank the 4B9A and 1W2B beamline stations of the Beijing Synchrotron Radiation Facility.Notes and references
- X. She, Y. Wang, H. Xu, S. Chi Edman Tsang and S. Ping Lau, Angew. Chem., Int. Ed., 2022, 61, e202211396 CrossRef CAS PubMed.
- R. Zhao, Y. Wang, G. Ji, J. Zhong, F. Zhang, M. Chen, S. Tong, P. Wang, Z. Wu, B. Han and Z. Liu, Adv. Mater., 2022, 35, e2205262 CrossRef PubMed.
- L. Xiong, X. Zhang, L. Chen, Z. Deng, S. Han, Y. Chen, J. Zhong, H. Sun, Y. Lian, B. Yang, X. Yuan, H. Yu, Y. Liu, X. Yang, J. Guo, M. H. Rümmeli, Y. Jiao and Y. Peng, Adv. Mater., 2021, 33, e2101741 CrossRef PubMed.
- D. Bagchi, J. Raj, A. K. Singh, A. Cherevotan, S. Roy, K. S. Manoj, C. P. Vinod and S. C. Peter, Adv. Mater., 2022, 34, e2109426 CrossRef PubMed.
- W. Liu, P. B. Zhai, A. W. Li, B. Wei, K. P. Si, Y. Wei, X. G. Wang, G. D. Zhu, Q. Chen, X. K. Gu, R. F. Zhang, W. Zhou and Y. J. Gong, Nat. Commun., 2022, 13, 1877 CrossRef CAS PubMed.
- W. J. Li, L. F. Li, Q. N. Xia, S. Hong, L. J. Wang, Z. B. Yao, T. S. Wu, Y. L. Soo, H. Zhang, T. W. B. Lo, A. W. Robertson, Q. Y. Liu, L. D. Hao and Z. Y. Sun, Appl. Catal., B, 2022, 318, 121823 CrossRef CAS.
- R. K. Miao, Y. Xu, A. Ozden, A. Robb, C. P. O'Brien, C. M. Gabardo, G. Lee, J. P. Edwards, J. E. Huang, M. Y. Fan, X. Wang, S. J. Liu, Y. Yan, E. H. Sargent and D. Sinton, Joule, 2021, 5, 2742–2753 CrossRef CAS.
- Z. Y. Zhang, L. Bian, H. Tian, Y. Liu, Y. Bando, Y. Yamauchi and Z. L. Wang, Small, 2022, 18, e2107450 CrossRef.
- X. Y. Liu, J. P. Xiao, H. J. Peng, X. Hong, K. Chan and J. K. Norskov, Nat. Commun., 2017, 8, 15438 CrossRef CAS.
- L. Y. Q. Xie, Y. J. Jiang, W. L. Zhu, S. C. Ding, Y. Zhou and J. J. Zhu, Chem. Sci., 2023, 14, 13629–13660 RSC.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean, R. E. Winans, D.-J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- W. Sun, P. Wang, Y. Jiang, Z. Jiang, R. Long, Z. Chen, P. Song, T. Sheng, Z. Wu and Y. Xiong, Adv. Mater., 2022, 34, e2207691 CrossRef PubMed.
- L. Ding, N. Zhu, Y. Hu, Z. Chen, P. Song, T. Sheng, Z. Wu and Y. Xiong, Angew. Chem., Int. Ed., 2022, 61, e202209268 CrossRef CAS PubMed.
- C. Guo, Y. Guo, Y. Shi, X. Lan, Y. Wang, Y. Yu and B. Zhang, Angew Chem. Int. Ed. Engl., 2022, 61, e202205909 CrossRef CAS PubMed.
- J. Li, Z. Y. Wang, C. McCallum, Y. Xu, F. W. Li, Y. H. Wang, C. M. Gabardo, C. T. Dinh, T. T. Zhuang, L. Wang, J. Y. Howe, Y. Ren, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 1124–1131 CrossRef CAS.
- H. Xiao, T. Cheng and W. A. Goddard, J. Am. Chem. Soc., 2016, 139, 130–136 CrossRef PubMed.
- X. Wang, Z. Y. Wang, F. P. G. de Arquer, C. T. Dinh, A. Ozden, Y. G. C. Li, D. H. Nam, J. Li, Y. S. Liu, J. Wicks, Z. T. Chen, M. F. Chi, B. Chen, Y. Wang, J. Tam, J. Y. Howe, A. Proppe, P. Todorovic, F. W. Li, T. T. Zhuang, C. M. Gabardo, A. R. Kirmani, C. McCallum, S. F. Hung, Y. W. Lum, M. C. Luo, Y. M. Min, A. N. Xu, C. P. O'Brien, B. Stephen, B. Sun, A. H. Ip, L. J. Richter, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Energy, 2020, 5, 478–486 CrossRef CAS.
- Y. P. Zang, T. F. Liu, P. F. Wei, H. F. Li, Q. Wang, G. X. Wang and X. H. Bao, Angew Chem. Int. Ed. Engl., 2022, 61, e202209629 CrossRef CAS PubMed.
- C. Peng, S. Yang, G. Luo, S. Yan, M. Shakouri, J. Zhang, Y. Chen, W. Li, Z. Wang, T. K. Sham and G. Zheng, Adv. Mater., 2022, 34, e2204476 CrossRef PubMed.
- Y. Moriyama, Y. Kawasaka and K. Takeda, J. Colloid Interface Sci., 2003, 257, 41–46 CrossRef CAS PubMed.
- T. T. Zhuang, Y. J. Pang, Z. Q. Liang, Z. Y. Wang, Y. Li, C. S. Tan, J. Li, C. T. Dinh, P. De Luna, P. L. Hsieh, T. Burdyny, H. H. Li, M. X. Liu, Y. H. Wang, F. W. Li, A. Proppe, A. Johnston, D. H. Nam, Z. Y. Wu, Y. R. Zheng, A. H. Ip, H. R. Tan, L. J. Chen, S. H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS.
- W. X. Ge, Y. X. Chen, Y. Fan, Y. H. Zhu, H. L. Liu, L. Song, Z. Liu, C. Lian, H. L. Jiang and C. Z. Li, J. Am. Chem. Soc., 2022, 144, 6613–6622 CrossRef CAS PubMed.
- T. Kumeda, H. Tajiri, O. Sakata, N. Hoshi and M. Nakamura, Nat. Commun., 2018, 9, 4378 CrossRef.
- W. Ge, Y. Chen, Y. Fan, Y. Zhu, H. Liu, L. Song, Z. Liu, C. Lian, H. Jiang and C. Li, J. Am. Chem. Soc., 2022, 144, 6613–6622 CrossRef CAS PubMed.
- S. Banerjee, Z. Q. Zhang, A. S. Hall and V. S. Thoi, ACS Catal., 2020, 10, 9907–9914 CrossRef CAS.
- K. H. Wu, D. Wang, X. Y. Lu, X. F. Zhang, Z. L. Xie, Y. F. Liu, B. J. Su, J. M. Chen, D. S. Su, W. Qi and S. J. Guo, Chem, 2020, 6, 1443–1458 CAS.
- S. Y. Lee, H. Jung, N.-K. Kim, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2018, 140, 8681–8689 CrossRef CAS PubMed.
- S. L. Zhao, Y. C. Yang and Z. Y. Tang, Angew Chem. Int. Ed. Engl., 2022, 61, e202110186 CrossRef CAS PubMed.
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS.
- Y. S. Zhou, F. L. Che, M. Liu, C. Q. Zou, Z. Q. Liang, P. De Luna, H. F. Yuan, J. Li, Z. Q. Wang, H. P. Xie, H. M. Li, P. N. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- R. O. Yang, J. Y. Duan, P. P. Dong, Q. L. Wen, M. Wu, Y. W. Liu, Y. Liu, H. Q. Li and T. Y. Zhai, Angew Chem. Int. Ed. Engl., 2022, 61, e202116706 CrossRef CAS PubMed.
- R. J. Meng, Q. Y. Deng, C. X. Peng, B. J. Chen, K. X. Liao, L. J. Li, Z. Y. Yang, D. L. Yang, L. Zheng, C. Zhang and J. H. Yang, Nano Today, 2020, 35, 100991 CrossRef CAS.
- Y. Zhao, X.-G. Zhang, N. Bodappa, W.-M. Yang, Q. Liang, P. M. Radjenovica, Y.-H. Wang, Y.-J. Zhang, J.-C. Dong, Z.-Q. Tian and J.-F. Li, Energy Environ. Sci., 2022, 15, 3968–3977 RSC.
- T. Cheng, A. Fortunelli and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7718–7722 CrossRef CAS PubMed.
- Y. Cao, S. Chen, S. Bo, W. Fan, J. Li, C. Jia, Z. Zhou, Q. Liu, L. Zheng and F. Zhang, Angew Chem. Int. Ed. Engl., 2023, 62, e202303048 CrossRef CAS PubMed.
- E. Perez-Gallent, M. C. Figueiredo, F. Calle-Vallejo and M. T. Koper, Angew Chem. Int. Ed. Engl., 2017, 56, 3621–3624 CrossRef CAS PubMed.
- X. Wang, Z. Wang, F. P. García de Arquer, C.-T. Dinh, A. Ozden, Y. C. Li, D.-H. Nam, J. Li, Y.-S. Liu, J. Wicks, Z. Chen, M. Chi, B. Chen, Y. Wang, J. Tam, J. Y. Howe, A. Proppe, P. Todorović, F. Li, T.-T. Zhuang, C. M. Gabardo, A. R. Kirmani, C. McCallum, S.-F. Hung, Y. Lum, M. Luo, Y. Min, A. Xu, C. P. O'Brien, B. Stephen, B. Sun, A. H. Ip, L. J. Richter, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Energy, 2020, 5, 478–486 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of materials, experimental instruments and measurement process, characterization of precatalysts, identification of products (GC and NMR), other electrochemical related characterization, and DFT calculation results. See DOI: https://doi.org/10.1039/d3sc06351h |
| This journal is © The Royal Society of Chemistry 2024 |