
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure and polymerization of liquid sulfur across the λ-transition†
Manyi
Yang
 a,
Enrico
Trizio
ab and
Michele
Parrinello
*a
a,
Enrico
Trizio
ab and
Michele
Parrinello
*a
aAtomistic Simulations, Italian Institute of Technology, 16156 Genova, Italy. E-mail: michele.parrinello@iit.it
bDepartment of Materials Science, Università di Milano-Bicocca, 20126 Milano, Italy
First published on 31st January 2024
Abstract
The anomalous λ-transition of liquid sulfur, which is supposed to be related to the transformation of eight-membered sulfur rings into long polymeric chains, has attracted considerable attention. However, a detailed description of the underlying dynamical polymerization process is still missing. Here, we study the structures and the mechanism of the polymerization processes of liquid sulfur across the λ-transition as well as its reverse process of formation of the rings. We do so by performing ab initio-quality molecular dynamics simulations thanks to a combination of machine learning potentials and state-of-the-art enhanced sampling techniques. With our approach, we obtain structural results that are in good agreement with the experiments and we report precious dynamical insights into the mechanisms involved in the process.
Introduction
Elemental sulfur exhibits a very complex phase diagram. This richness derives from the sulfur's propensity to be twofold coordinated. This results in the possibility of either forming ring-like structures (Sπ) or polymeric chains (S∞) of competing energy. The ring-like arrangements dominate the stable solid structures1–3 (orthorhombic α-S, monoclinic β-S and γ-S) with polymeric arrangements reported only for pressures higher than 2.0 GPa.4–6 Among the cyclic polymorphs, small rings are preferred, and the 8-membered crown-shaped rings (S8) are by far the most stable configurations. However, evidence of a minority fraction of rings with sizes ranging from 6 to 32 atoms has also been reported.7,8A mixture of these two structural models has also been proposed to describe the liquid phase, and several liquid–liquid phase transitions, similar to a variety of other systems,9–12 have been reported in a wide temperature and pressure range.1,2,7,13,14 For instance, a transition between a low-density liquid, richer in rings, and a high-density liquid, richer in polymers, has been observed with a transition line that terminates in a critical point at around 1000 K and 2.0 GPa.7
Here, we focus on a small part of the phase diagram, namely the in proximity of the so-called lambda transition2,13,15 that occurs at Tλ = 432 K and ambient pressure and results in an anomalous behavior of physical properties like heat capacity,2 viscosity,15 and density.16 The behavior observed around Tλ has been widely studied and associated with the onset of a living polymerization of the S8 rings.17,18 It is believed that before the λ-transition, where the phase equilibrium is mostly determined by enthalpic contributions, ring-like structures dominate, but as one increases the temperature and entropic driving forces become more relevant, the polymeric fraction of the liquid slowly grows and suddenly increases around Tλ.
Over the years, extensive experimental studies have been conducted to characterize the species involved in the transition.1,19–22 However, they still provide limited insight into the underlying dynamical process and a detailed picture of this phenomenon is still missing. Previous theoretical studies,9,23 including ab initio-based simulations,24–28 have partially filled this gap. However, the computational costs of first principles methods, which are needed to faithfully describe the complex changes in the forming and breaking of chemical bonds, have limited the scope of these simulations. Indeed, such calculations are still computationally too expensive to perform extensive sampling, even on small systems. Thus, all these studies have been limited to relatively short timescales (some 100 ps) and/or simulation cells with only a few hundred atoms. Such limitations are quite severe when studying polymeric systems, which by definition involve a large number of atoms and often exhibit slow dynamics.18,29
In recent years, the combination of machine-learning interatomic potentials (MLPs) of ab initio-quality and enhanced sampling (ES) in an active learning framework has proven effective in overcoming similar difficulties in processes as complex as the liquid–liquid transition in phosphorus,11 the nucleation and phase diagram of gallium,31 the decomposition of urea32 or the dynamical nature of heterogenous catalytic processes.33–35 In our case, such an approach has allowed the limitation of standard molecular dynamics (MD) simulations to be overcome and to simulate systems of thousands of atoms for timescales of the order of nanoseconds.
Indeed, MLPs allow performing ab initio-quality MD simulations at a fraction of the cost of first principles methods. In this approach, following the strategy pioneered by Behler and Parrinello,36 the interatomic interactions are modeled through a neural network (NN), which is trained to faithfully predict the energies and forces obtained with DFT calculations on a set of reference configurations. To be accurate for the study of reactive processes, it is then of the utmost importance that the training dataset does not contain only low-energy configurations coming from the sampling of metastable states but also includes transition state configurations. Unfortunately, in the case of complex systems such as liquid sulfur, due to the presence of large free energy barriers, most reactive events take place on a temporal scale that far exceeds that accessible in standard MD simulations and cannot be sampled.
Fortunately, ES methods are aimed at overcoming this limitation and allowing rare events to be sampled in an affordable computational time. Many such methods are based on the addition of an external bias potential, which is taken to be a function of a small number of collective variables (CVs). The CVs are, in turn, functions of the atomic coordinates s = s(R) and should be wisely chosen to encode the relevant slow modes of the process for a successful simulation. Recently, the advent of machine learning (ML) methods has greatly facilitated the CV determination. In the present case, the complex structural changes that occur close to the λ-transition are difficult to express in simple geometrical terms, and we use instead a more abstract CV that results from a combination of graph theory and ML.
In the first part of the paper, we construct the interaction potential and show that the predictions of our model potential are in agreement with experimental diffraction data and diffusion properties. In the second part, we study the polymerization and depolymerization processes that take place close to the λ-transition. In nanosecond-long reactive simulations, with the help of an analysis of charge distribution, we describe reaction mechanisms that shed light on the puzzling mechanism of formation and breaking of the sulfur polymers.
Results
Topological collective variables for enhanced sampling
To build the CV for ES simulations, in this work, we use the Deep Targeted Discriminant Analysis30 (Deep-TDA) method, and the whole protocol of our CV design process is schematically depicted in Fig. 1a. In Deep-TDA, the CV is expressed as the output of a NN that is optimized according to a classification criterion (panels 3 and 4) and takes as input a set of (physical) descriptors, which, in our case, are built from the adjacency matrix of the system (panels 1 and 2). | ||
| Fig. 1 (a) Schematic representation of the construction of the topological collective variable (CV) for polymerization in liquid sulfur. For each configuration, we build the corresponding adjacency matrix and compute its eigenvalues distribution with a continuous histogram. The values of such histograms are fed as inputs of a neural network (NN) that combines them and returns the CV as output, according to the Deep-TDA CV scheme.30 For the training of the NN, we build a dataset of configurations from pure rings (blue) and pure polymer (red) phases. The NN is optimized such that the projection of the training data in the CV-space matches a pre-assigned target distribution in which the states are well-discriminated. (b) Value of the Deep-TDA CV as a function of time in a biased simulation, in which the biasing is applied along the Deep-TDA CV. Starting from a pure S8 phase at 0.0 ns, the system polymerizes to progressively higher polymer fractions. Instantaneous snapshots at times of 0.0 ns, 0.5 ns and 1.5 ns are given. In both panels, ring-related features are colored in blue and polymer-related ones in red, respectively. | ||
As mentioned in the introduction, the use of ML greatly simplified the CV design procedure. However, the effectiveness of such approaches is still affected by the choice of input descriptors, as they should be informative on the slow modes of the process. In this sense, the complex processes that take place at the λ-transition presented us with the new challenge of finding proper descriptors that could describe the ring opening and formation while retaining permutational invariance. Since the system undergoes enormous changes in connectivity (see upper panel in Fig. 1b) we resort to graph theory and, in particular, to the adjacency matrix eigenvalues (panels 1 and 2 in Fig. 1a), which directly relates to the topology of the system. For example, the values  and 0 reflect S8 ring arrangements, and the multiplicity of such eigenvalues is proportional to the number of such rings in the system.
and 0 reflect S8 ring arrangements, and the multiplicity of such eigenvalues is proportional to the number of such rings in the system.
In practice, to train our Deep-TDA CV, we used a two-state model, using unbiased data collected in the pure ring and pure polymeric phases (see Fig. 1 whole). Our choice is motivated by the experimental evidence that suggests that the relevant properties in the λ-transition region depend on the relative fraction of these two phases. We also note that even if the pure polymeric phase is not reported in the experiments, it still can be used to simplify the training of the model by making the relevant polymer-related features more evident and deepen our understanding of the system, providing a measure of the phase composition of the system during our simulations as shown in the bottom of Fig. 1b.
Radial distribution function and structure factor
The radial distribution function g(r) and the structure factor S(k) depend on the structural ordering of the atoms and their features on the relative concentration of the phases in the sample. For this reason, these quantities have been monitored at different temperatures around the λ-transition.37 However, the experimental determination of the S8 fraction is still subject to much uncertainty.13,18,38,39This is the typical scenario in which theoretical modeling can provide helpful insights. Simulations can access the pure phases (i.e., only rings S8 and only polymers S∞), which are never found in the experiments. Despite sounding somehow unphysical, this information can be used to interpret the experimental results. One can then obtain the g(r) and S(k) for all the intermediate compositions through a linear combination of the contribution from the pure S8 and S∞ phases. For example, the radial distribution function gα(r) at a given concentration of rings α is
| gα(r) = αgS8(r) + (1 − α)gS∞(r) | (1) |
| Sα(k) = αSS8(k) + (1 − α)SS∞(k) | (2) |
The values of gS8, gS∞, SS8, SS∞ were obtained by analyzing 200 configurations collected over one ns of equilibrated dynamics of 3456 atoms (box size 46.9 Å) in the pure S8 and S∞ phases. We have tested the accuracy of this approach by comparing it with the results from simulations performed at preassigned S8 concentrations, as we report in the ESI.†
In Fig. 2, we compare the experimental data37 from X-ray diffraction (XRD) at ambient pressure and different temperatures with our estimates of the g(r) and Sk (panel a and b, respectively) for different S8 concentrations (95% to 40% S8 rings). The values of α were chosen to best fit the experimental data within the range of concentrations reported in the experiments at the considered temperatures. In the case of the g(r), for a meaningful and direct comparison with the experimental data, we also applied a Gaussian convolution to the simulated results. The non-processed data are available in the ESI.†
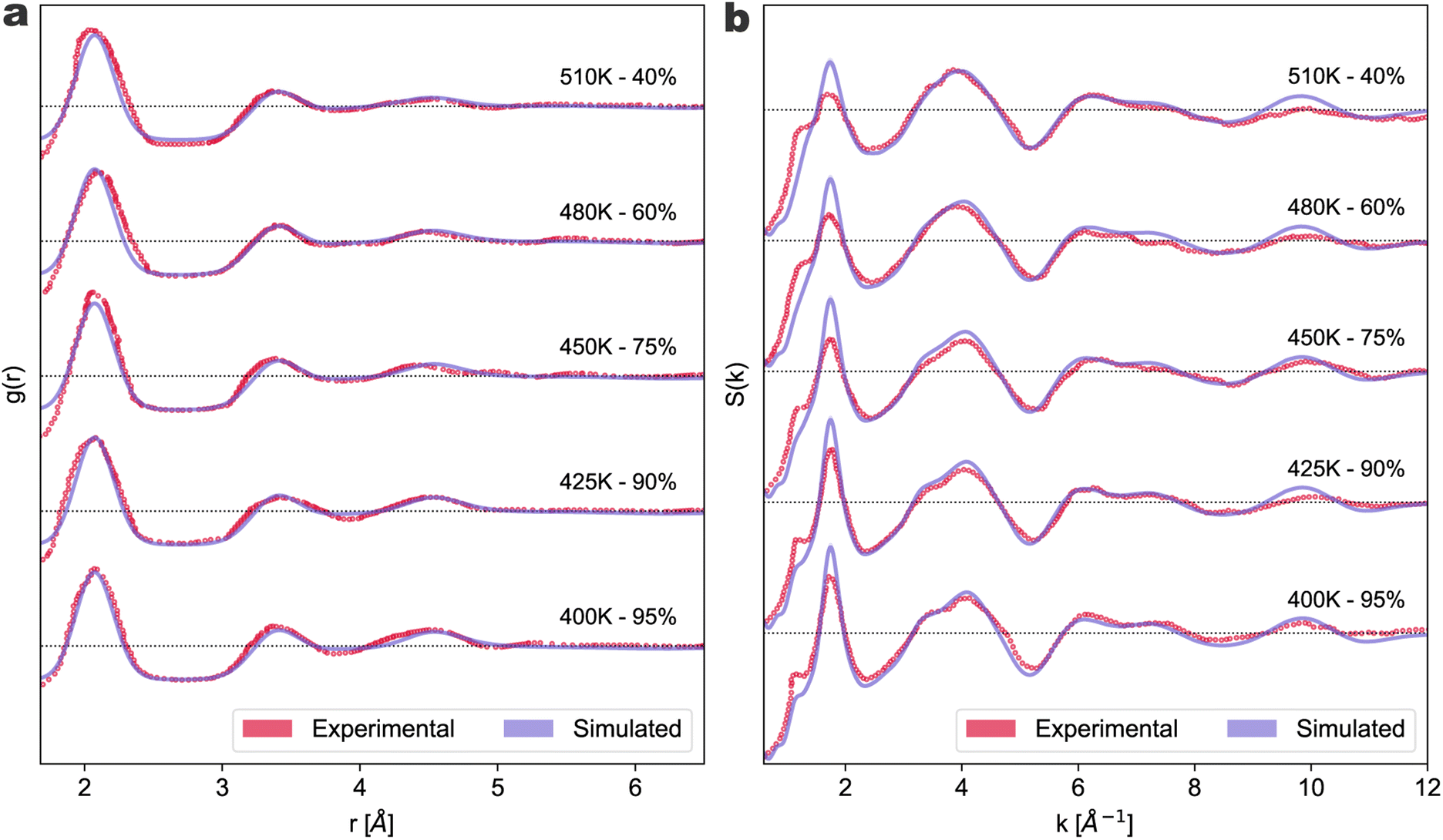 | ||
| Fig. 2 Radial distribution function g(r) (a) and structure factor S(k) (b) for liquid sulfur at temperatures around the Tλ. Simulated results at different ring concentrations (solid purple lines) are compared with experimental data37 at different temperatures (red void dots). Each experimental temperature and the matching percentage of rings are reported close to the corresponding curve. The ±5% interval on the ring concentration for each simulated curve is given as a shaded purple area. The black dotted lines mark the g(r) = 1 and S(k) = 1 value for each couple of curves as they are offset in the vertical direction for visualization purposes by four and one units, respectively. | ||
From Fig. 2, it can be seen that the short-range order of the liquid is well-fitted, giving credibility to our estimation of the S8 fraction. Remarkably, our results reproduce well the evolution with the temperature of the g(r) third peak around 4.5 Å. This elusive signal is associated with third neighboring distances in the S8, thus tends to disappear as the temperature and the polymer fraction increase. The long-range structure is also well reproduced, except for the pre-peak in S(k) at k ∼ 1 Å−1, which is less apparent in our simulations. This is a reflection of the fact that our system is still too small to resolve this peak well. However, in the previous ab initio calculations that out of necessity were performed on smaller cells, both the peak at k ∼ 2 Å−1 and the corresponding pre-peak were not reproduced almost at all.
Atomic mobility: displacement analysis
One of the main features of the λ-transition is its sudden increase in viscosity above Tλ. At the atomic level, this should correspond to a decreased mobility of the atoms.Especially in the polymeric phase, the atomic motions become sluggish and a direct calculation of the viscosity is impossible. However, in order to have an insight into the dynamics, we compute and compare the atomic displacements after 10, 25, 50, and 100 ps for ring concentrations that resemble conditions below Tλ (100% rings concentration), slightly above (75%) and well above (55%).
The results from this analysis are reported in Fig. 3 and clearly show that, on average, atoms in the molecular S8 phase have the highest mobility. On the other hand, as the polymeric content increases, a peak in the distribution starts to appear below the ∼2 Å threshold of the first coordination shell. This comes from the polymer atoms, which mostly oscillate around their positions rather than showing any net drift, thus inducing the rise in the viscosity.
 | ||
| Fig. 3 Histogram of the atomic displacements in liquid sulfur at different ring concentrations, given in the legend, and different lag times, indicated by white labels. | ||
Reaction mechanisms and charge analysis
Having assessed the reliability of our potential's prediction when compared to the experiments, we move to the study of the chemical mechanisms involved in the λ-transition with the crucial help of enhanced sampling simulations and our topological CV. In the following, we propose mechanisms for the polymerization/depolymerization process. We note that we report only those mechanisms that we found to be dominant in our simulations, i.e., they were observed in the majority of several independent simulations, and for all of them, we double-checked the agreement of our potential with DFT calculations to avoid artifacts.For each mechanism, we provide a prototypical example and an analysis of the instantaneous charges involved in the process. This analysis is made with the help of Bader charge distribution40 as obtained from the DFT charge density.
A schematic chemical diagram for the proposed mechanism is also available in the ESI.†
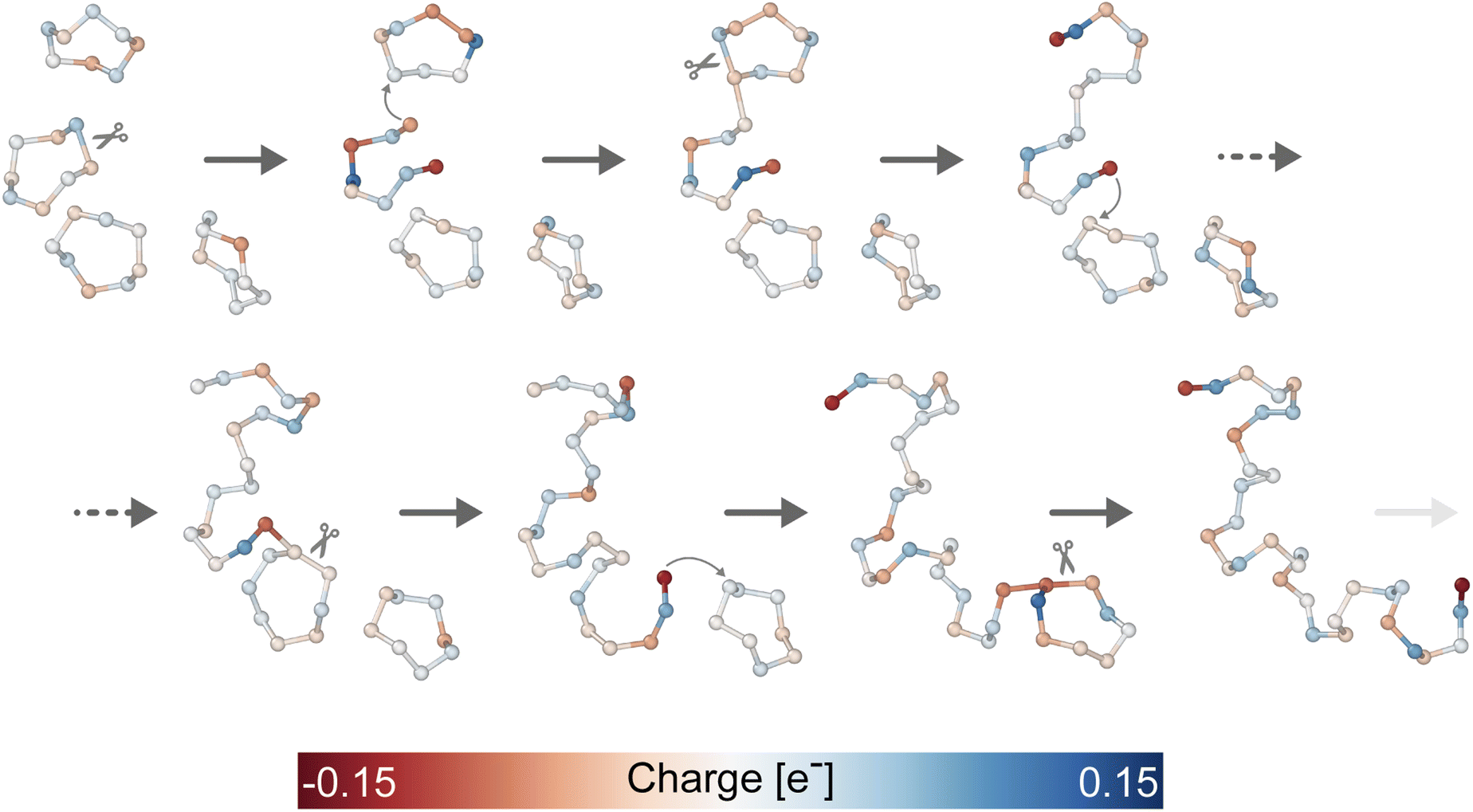 | ||
| Fig. 4 Polymerization mechanism in liquid sulfur starting from four S8 rings. The atoms are colored according to the instantaneous charge obtained by computing the Bader's charge from the DFT electronic density. For visualization purposes, only the relevant atoms are represented from the 512 in the simulation cell. | ||
Even if the ideal crown-shaped S8 rings dominate the liquid phase, our calculations confirmed the experimental evidence1 that other cyclic monomers (Sn, n ≠ 8) and sub-stable isomers of S8 rings (see ESI†) also contribute to this polymerization process.
Overall, from our results, it clearly appears that the under-coordinated nature of the chain tails mainly drives the polymerization. This makes the terminal atoms highly reactive, as indicated by the negative charge localization, and eager to find new partners.
Another key factor is related to the increased stabilization of the charge unbalance in longer polymeric strands. In that case, the average delocalization of the charge is more efficient with respect to the case of short oligomers. In this regard, we also found, in agreement with previous theoretical studies,25 that shorter chains tend to be rather unstable and often revert to rings if left to relax in short unbiased dynamics, at variance with the longer chains, which remain stable.
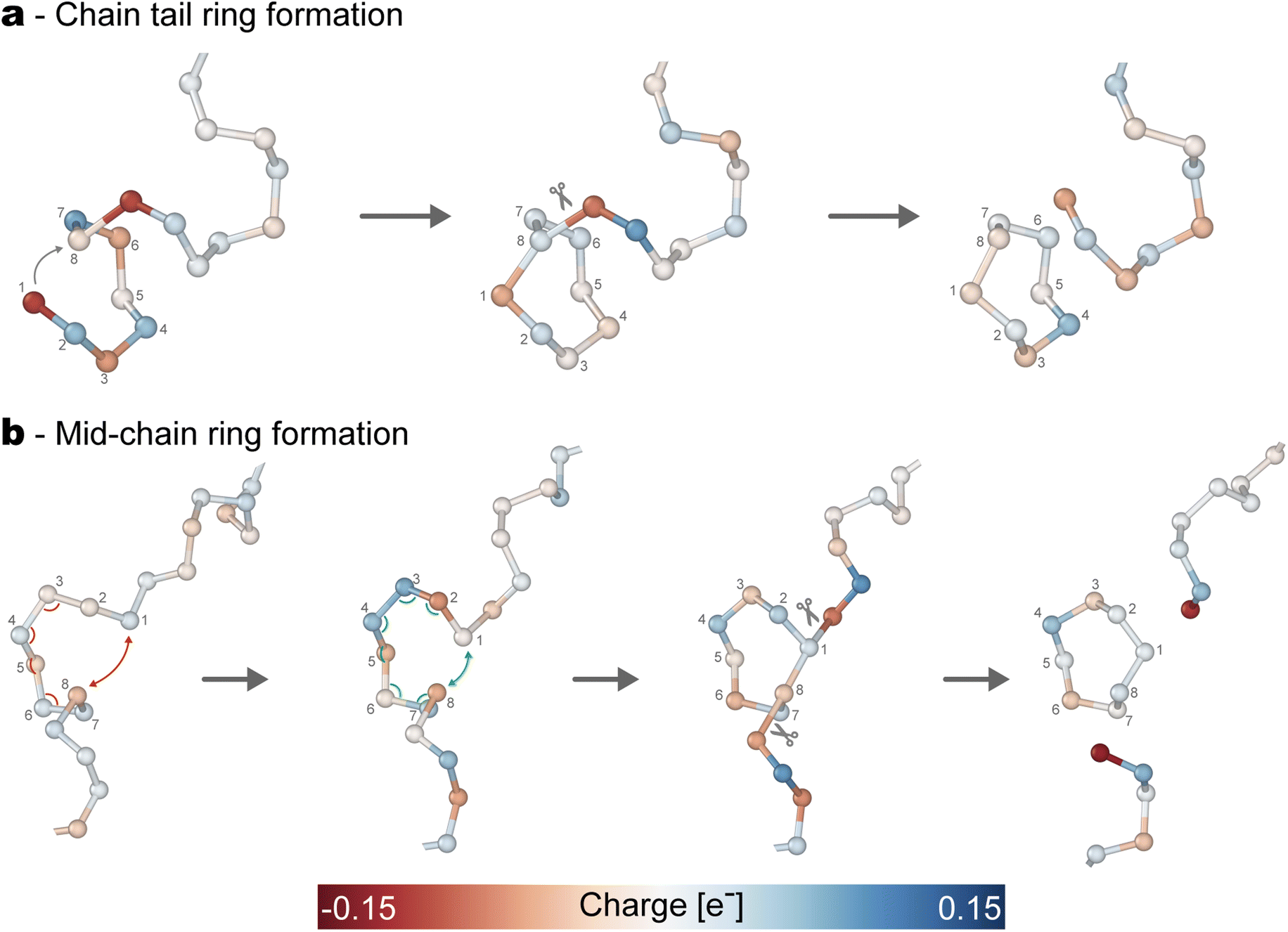 | ||
| Fig. 5 Mechanisms for the formation of rings in liquid sulfur starting from S∞ polymers. (a) Formation of a ring from the tail of the polymeric chain. (b) Formation of a ring in the middle of the chain. The atoms are colored according to the instantaneous charge obtained by computing the Bader's charge from the DFT electronic density. For visualization purposes, only the relevant atoms are represented from the 512 in the simulation cell. | ||
In the first case (see Fig. 5a), the chain tail is characterized by two elements: a charge unbalance and higher mobility with respect to the rest of the chain. The first element is specifically due to the sulfur atoms being under-coordinated, thus leading to their negative polarization. This is the chemical driving force of the reaction, as it makes such atoms eager for new partners and ready to react. However, this is not enough for the reaction to take place, as the reactive tail has to fold onto the chain to form the loop that will eventually lead to the ring. This is made possible thanks to the higher mobility of the terminal atoms, which is typical of polymeric chain ends. Of course, this loop is most stable if the terminal atom folds such that it interacts with its 7th neighbor, thus ensuring the S8 arrangement, but this same mechanism can also lead with less probability to some of the different-sized rings as we report in the ESI.†
In the second mechanism, the scenario is significantly different as the atoms involved in the formations of the loop belong to the middle of the polymeric chain (see Fig. 5b). Such atoms are indeed fully coordinated, meaning that they are much less reactive than in the first case and that any polarization fluctuation is short-lived. We found that to compensate for this weaker reactivity, it is crucial that the arrangement of the atoms resembles as much as possible that of one of the stable Sπ rings. Of course, the choice shall preferably be the ideal crown-shaped S8. Thus, we report this case in Fig. 5b. In the first frame, the eight-membered loop that starts to appear in the middle of the chain still has a wrong combination of angles and distances. On the other hand, in the configuration reported in the second frame, the sequence of angles becomes favorable, and the distance between the 1st and 8th S atoms reduces enough they can interact. As a consequence of this interaction, the adjacent atoms show a weak negative polarization (see third frame), which becomes stronger as the ring finally separates from the original chain.
Discussion
In this work, we studied the structures and the polymerization and depolymerization mechanisms across λ-transition in liquid sulfur using a combination of ab initio-quality machine learning potentials and state-of-the-art enhanced sampling techniques. For an effective application of the latter approach, we designed a topological collective variable by combining machine learning and graph theory. This has been crucial for the construction of a proper dataset on which our machine-learning potentials were optimized and for the study of the polymerization and depolymerization mechanisms of liquid sulfur. Our methodology proved powerful and greatly improved over previous DFT-based theoretical results. The calculated static structural quantities, including g(r) and S(k), are in good agreement with the experimental results. Similarly, our calculations reproduce well the trend in the atomic mobility in the different phases that can be associated with the anomalous rise in the viscosity during the sulfur polymerization across the λ-transition. In addition, we proposed dynamic reaction mechanisms for the processes involved in the λ-transition, which also shed light on the critical role that charge localization plays in driving the polymerization and depolymerization process. Moreover, such mechanisms show that specific conformational requirements need to be fulfilled in order for both processes to take place, confirming the well-known complexity of this peculiar system and possibly explaining the presence of residues of the less stable phases at all temperatures that are observed in the experiments.Overall, our calculations allowed us to harvest precious results in close agreement with the experiments and the success of our strategy encourages us to study other complex systems in the future with this approach.
Methods
Code and software
All ab initio molecular dynamics simulations (AIMD) and single-point energies and forces needed for training the neural network (NN) potentials were performed using the CP2K 9.2 code.41 In addition, we double-checked that the quantities computed using the CP2K code are consistent with those obtained with Quantum Espresso code.42 The Bader's charges were obtained by analyzing the DFT electronic densities with the Bader40 code.The DeepMD-kit package43,44 was used for the training of the NN potentials for the atomic interactions and as a plugin in the LAMMPS45 MD engine for performing the simulations.
For the application of enhanced sampling methods, we relied on the PLUMED46 plugin patched with LAMMPS.
The training of the machine learning collective variable (Deep-TDA CV) has been done using the mlcolvar47 library based on PyTorch.48 In particular, we refer to the 4387073 commit49 of such a library, which includes additional preprocessing tools for adjacency-matrix-related calculations. The CVs have been deployed to PLUMED using the interface provided in its PyTorch module.47
The atomic displacement analysis was performed using the visualization and post-processing code Ovito,50 which was also used for the rendering of the molecular snapshots reported in the paper.
AIMD simulations
In AIMD simulations, the energies and forces were computed using the Perdew–Burke–Ernzerhof (PBE) exchange–correlation density functional.51 The Kohn and Sham orbitals were expanded in a m-DZVP Gaussian basis and the plane wave expansion of the electronic density was truncated at an energy cutoff of 300 Ry. The core electrons were treated using the Goedecker–Teter–Hutter (GTH) pseudopotentials52,53 optimized for PBE. To keep the computational cost low, only the Γ-point was used to sample the supercell Brillouin zone.All AIMD simulations were performed in the NPT ensemble with a time step of 2.0 fs. Temperature and pressure were controlled using Nosé–Hoover thermostat54 and a Nosé–Hoover-like barostat55 with coupling constants of 0.05 ps and 0.5 ps, respectively. To mitigate the computational costs, only a smaller cubic simulation cell consisting of 128 atoms was used. Nonetheless, the results discussed in the text were calculated on larger cubic simulation cells.
Single-point energies and forces calculations
The energies and forces needed for the NN potential training were computed using the same exchange–correlation density functional (PBE) and pseudopotential (GTH) as that of the AIMD simulations. However, the energy cutoff was increased to 350 Ry and the D3 dispersion corrections56 were included. Single-point calculations have been performed on cells with 128 and 512 atoms. For the smallest cells, we used k-points grids of 2 × 2 × 2. In contrast, we only used the Γ-point for the larger ones as we checked that the accuracy on energies and forces with this setup was almost indistinguishable from the one on smaller cells using the grids.NN potential-based MD simulations
All the results reported in the text were obtained using NN-potential-based MD simulations. Specifically, we performed unbiased and biased simulations with a timestep of 1.0 fs in the NVT ensemble, using the global velocity rescaling thermostat57 with a relaxation time of 0.05 ps and periodic boundary conditions (PBC) on cubic simulation cells with box sizes chosen to be consistent with the experimental densities.The unbiased approach has been used to study the radial distribution function, the structure factor, and the atomic mobility. For this latter analysis, we simulated a system consisting of 512 atoms (box 24.8 Å) starting from configurations generated from biased simulations, whereas, for the others, we simulated a much larger cell with 3456 atoms (box 46.9 Å).
The more expensive enhanced sampling approach has been used to simulate the dynamics of 512 atoms to study the polymerization and depolymerization mechanisms of sulfur.
Enhanced sampling method: OPES
We reverted to enhanced sampling to study the polymerization and depolymerization processes of sulfur that are supposed to take place close to λ-transition. In particular, we used the on-the-fly probability enhanced sampling (OPES) method.58,59 In OPES, the equilibrium probability distribution P(s) in the CV space s is estimated on the fly, and a bias potential Vn(s) is constructed so as to drive P(s) toward a target distribution Ptg(s).Here, we used as target the well-tempered distribution,60 , where γ > 1 is the bias factor. In this case, the bias potential at nth iteration is written as:
, where γ > 1 is the bias factor. In this case, the bias potential at nth iteration is written as:
 | (3) |
Collective variables method: Deep-TDA
The OPES biasing potential is applied along a small set of CVs, which are continuous and derivable functions of the atomic coordinates s = s(R). These are meant to encode the slow modes of the system and, as an additional requirement, they shall be invariant with respect to the symmetries of the system (rotation, translation, and permutation of identical atoms).In the Deep Targeted Discriminant Analysis (Deep-TDA) method, the CV is the output of a feed-forward NN (see panel 3 in Fig. 1a) whose inputs are a set of physical descriptors, such as distances, angels, coordination numbers, collected with short unbiased runs in the metastable basins that are supposed to be visited in the process of interest. The NN is optimized such that the training data, when projected in the CV space, are distributed according to a preassigned target distribution (panel 4 in Fig. 1a). This target is defined as a series of Gaussian with fixed positions and widths, one for each state, such that data from different basins are localized in different regions of the CV space.
Topological collective variable training
In our Deep-TDA CV model, we used the mlcolvar library47 preprocessing tools to compute the NN input descriptors starting from the Cartesian coordinates of all the atoms in the system, which are the inputs of our deployed model in PLUMED.To compute the NN input descriptors, we build the adjacency matrix A in which the Aij element is Aij = 1 if the scalar distance dij between atom i and j is lower than a cutoff distance dcutoff and Aij = 0 otherwise. However, the application of such a sharp cutoff would give a matrix A with discontinuous derivatives that would not be suited for a biasing context. Thus, we applied the cutoff using a sharp switching function S(dij) in the form
 | (4) |
We then computed the full eigenvalues spectrum of A with Pytorch48 tools and computed a histogram of such values with 100 bins in the range (−2.2,2.2) using a Gaussian expansion to ensure continuous derivatives. Finally, we took the values of the histogram in the 100 bins as input for the Deep-TDA NN. In the training, the Deep-TDA targets were chosen to be μA = −25 and μB = 25 for the centers of the distributions and σA = 0.2 and σB = 0.2. The training set was composed of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 configurations for each of the two states for a total of 36000 configurations. Including input and output layers, the architecture was [100, 64, 32, 1] nodes/layer, and we used the rectified linear unit (ReLU) as activation function with a learning rate of 0.001 for the optimization.
000 configurations for each of the two states for a total of 36000 configurations. Including input and output layers, the architecture was [100, 64, 32, 1] nodes/layer, and we used the rectified linear unit (ReLU) as activation function with a learning rate of 0.001 for the optimization.
NN potential for interatomic interactions training
The NN potentials were trained with the DeepMD method61 using the attention-based Deep Potential scheme,62 which, in our case, is able to give a much more accurate reproduction and prediction of DFT energies and atomic forces compared to the standard Deep Potential-Smooth Edition scheme63 (see ESI†). The cutoff radius was set to smoothly decay from 0.5 Å to 7.5 Å. The maximum possible number of neighbors in the cutoff was set to 90, and the number of layers in the attention scheme to 3. We used three hidden layers with [30, 60, 120] nodes/layer for the embedding network and four hidden layers with [240, 240, 240, 240] nodes/layer for the fitting network, whereas the size of the embedding matrix was set to 16. The learning rate was set to decay from 1.0 × 10−3 to 5.0 × 10−8 an we used a batch size of 8. The prefactors of the energy and force terms in the loss function were set to change during the training from 0.01 to 5 and from 1000 to 1, respectively, and the final NN model was trained for 3.0 × 106 steps.Collection of the training set
The key step in the construction of a machine learning potential is the collection of the training data set. This is particularly challenging in the case of liquid sulfur that exhibits a wide range of ring-like and chain-like metastable structures.1 It is thus necessary to include these configurations in the training set as well as those related to the transition state of the interconversion process.To improve and simplify this step, active learning strategies boosted by the use of enhanced sampling methods have been applied to study several complex systems.11,31–34,64 The advantage of this approach is that it allows crucially relevant reactive configurations to be extracted at an affordable computational cost.
In our specific case, we applied the on-the-fly probability enhanced sampling58,59 (OPES) method combined with state-of-the-art machine learning collective variables (CVs) (see Topological collective variables for enhanced sampling section). The whole procedure of exploring the relevant atomic configurations in the training set is as follows.
First, we ran a series of unbiased AIMD simulations in the NPT ensemble on systems of 128 atoms for times ranging from 2 to 10 ps. These simulations were performed in the 500–1200 K temperature range and 0.2–3.0 GPa pressure range to collect configurations in both the polymeric and ring phases of liquid sulfur. Indeed, approaching high temperature and pressure, S8 rings are destabilized in favor of the S∞ polymers.
From these simulations, we collected about 7800 atomic configurations to build the initial training set and start our active learning procedure, which alternate cycles of training and sampling according to the following steps:
• Step 1: we train four NN potentials using different initial weights and the previous iteration's updated training set. For the first iteration, we shall use the initial training set of AIMD configurations.
• Step 2: we perform a series of simulations using one of the four NN potentials trained in step 1 to explore new relevant atomic configurations. Not limited to AIMD simulations anymore, we expand our systems from 128 atoms to 512 atoms and run enhanced sampling simulations using OPES combined with our topological CVs. These biased simulations are essential for exploring the active atomic configurations along the polymerization of S8 rings.
During the simulations, we monitor the reliability of the potential on the sampled configurations based on the maximal standard deviation σ65 of the atomic forces predicted by the four NN potentials:
 | (5) |
 is the average force on the atom i over the four NN potentials.
is the average force on the atom i over the four NN potentials.
To minimize the number of new relevant atomic configurations to be added to the training set while ensuring maximum diversity, we follow the same strategy described in ref. 34, with the low (σl) and up (σu) bound values set to 0.15 and 0.4, respectively. We refer the readers to ref. 34 for further details.
• Step 3: we calculate the DFT energies and forces for the configurations selected in step 2 and include them in the training set for the next iteration.
Following our previous work, this active learning process is repeated until less than 10% of the sampled atomic configurations fall into the candidate list of step 2. At the end of the procedure, our dataset included roughly 1.5 × 105 atomic configurations, with almost 90% of them consisting of 512 atoms.
Data availability
All the inputs and files needed to reproduce the results presented in this manuscript are available on GitHub.66Author contributions
M. Y., E. T., and M. P. made substantial contributions to the design and implementation of the work and to the writing of the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
M. Y. and E. T want to thank Ana Maria Borrego Sanchez, Umberto Raucci, Luigi Bonati for useful discussions and support. The authors acknowledge the HPC infrastructure and the Support Team at Fondazione Istituto Italiano di Tecnologia. Computational resources were also provided by the Swiss National Supercomputing Centre (CSCS) under project ID S1134 and S1183.References
- R. Steudel, Elemental sulfur and sulfur-rich compounds I, Springer Science & Business Media, 2003, vol. 2 Search PubMed.
- B. Meyer, Elemental sulfur, Chem. Rev., 1976, 76, 367–388 CrossRef CAS.
- L. Crapanzano, W. A. Crichton, G. Monaco, R. Bellissent and M. Mezouar, Alternating sequence of ring and chain structures in sulphur at high pressure and temperature, Nat. Mater., 2005, 4, 550–552 CrossRef CAS PubMed.
- S. Geller, Pressure-induced phases of sulfur, Science, 1966, 152, 644–646 CrossRef CAS PubMed.
- M. Lind and S. Geller, Structure of pressure-induced fibrous sulfur, J. Chem. Phys., 1969, 51, 348–353 CrossRef CAS.
- W. A. Crichton, G. B. M. Vaughan and M. Mezouar, In situ structure solution of helical sulphur at 3GPa and 400°C, Z. Kristallogr.–Cryst. Mater., 2001, 216, 417–419 CrossRef CAS.
- L. Henry, M. Mezouar, G. Garbarino, D. Sifré, G. Weck and F. Datchi, Liquid–liquid transition and critical point in sulfur, Nature, 2020, 584, 382–386 CrossRef CAS PubMed.
- R. Bellissent, L. Descotes, F. Bouc and P. Pfeuty, Liquid sulfur: Local-order evidence of a polymerization transition, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 2135 CrossRef CAS PubMed.
- M. A. Anisimov, M. Duška, F. Caupin, L. E. Amrhein, A. Rosenbaum and R. J. Sadus, Thermodynamics of fluid polyamorphism, Phys. Rev. X, 2018, 8, 011004 Search PubMed.
- N. R. Fried, T. J. Longo and M. A. Anisimov, Thermodynamic modeling of fluid polyamorphism in hydrogen at extreme conditions, J. Chem. Phys., 2022, 157, 101101, DOI:10.1063/5.0107043/16548562/101101_1_online.pdf.
- M. Yang, T. Karmakar and M. Parrinello, Liquid-liquid critical point in phosphorus, Phys. Rev. Lett., 2021, 127, 080603 CrossRef CAS PubMed.
- H. Tanaka, Liquid–liquid transition and polyamorphism, J. Chem. Phys., 2020, 153, 130901 CrossRef CAS PubMed.
- R. Steudel, Liquid sulfur, Top. Curr. Chem., 2012, 230, 81–116 CrossRef.
- L. Liu, Y. Kono, C. Kenney-Benson, W. Yang, Y. Bi and G. Shen, Chain breakage in liquid sulfur at high pressures and high temperatures, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 174201 CrossRef.
- G. E. Sauer and L. B. Borst, Lambda transition in liquid sulfur, Science, 1967, 158, 1567–1569, DOI:10.1126/science.158.3808.1567.
- K. M. Zheng and S. C. Greer, The density of liquid sulfur near the polymerization temperature, J. Chem. Phys., 1992, 96, 2175–2182 CrossRef CAS ), https://pubs.aip.org/aip/jcp/article-pdf/96/3/2175/11261025/<?pdb_no 2175?>2175<?pdb<?db_id PDB?> END?>_1_online.pdf.
- A. V. Tobolsky and A. Eisenberg, Equilibrium polymerization of sulfur, J. Am. Chem. Soc., 1959, 81, 780–782, DOI:10.1021/ja01513a004.
- V. F. Kozhevnikov, W. B. Payne, J. K. Olson, C. L. McDonald and C. E. Inglefield, Physical properties of sulfur near the polymerization transition, J. Chem. Phys., 2004, 121, 7379–7386 CrossRef CAS PubMed.
- R. Winter, C. Szornel, W. C. Pilgrim, W. S. Howells, P. A. Egelstaff and T. Bodensteiner, The structural properties of liquid sulphur, J. Phys.: Condens. Matter, 1990, 2, 8427 CrossRef CAS.
- M. Stolz, R. Winter, W. S. Howells, R. L. McGreevy and P. A. Egelstaff, The structural properties of liquid and quenched sulphur II, J. Phys.: Condens. Matter, 1994, 6, 3619 CrossRef CAS.
- C. Biermann, R. Winter, C. Benmore and P. Egelstaff, Structural and dynamic properties of liquid sulfur around the λ-transition, J. Non-Cryst. Solids, 1998, 232–234, 309–313 CrossRef CAS.
- A. G. Kalampounias, K. S. Andrikopoulos and S. N. Yannopoulos, Probing the sulfur polymerization transition in situ with Raman spectroscopy, J. Chem. Phys., 2003, 118, 8460–8467 CrossRef CAS ), https://pubs.aip.org/aip/jcp/article-pdf/118/18/8460/10847574/<?pdb_no 8460?>8460<?pdb END?>_1_online.pdf.
- N. A. Shumovskyi, T. J. Longo, S. V. Buldyrev and M. A. Anisimov, Modeling fluid polyamorphism through a maximum-valence approach, Phys. Rev. E, 2022, 106, 015305 CrossRef CAS PubMed.
- H. Flores-Ruiz and M. Micoulaut, Crucial role of s 8-rings in structural, relaxation, vibrational, and electronic properties of liquid sulfur close to the λ transition, J. Chem. Phys., 2022, 157(5), 054507 CrossRef CAS PubMed.
- R. O. Jones and P. Ballone, Density functional and Monte Carlo studies of sulfur. I. Structure and bonding in Sn rings and chains (n=2−18), J. Chem. Phys., 2003, 118, 9257–9265 CrossRef CAS ), https://pubs.aip.org/aip/jcp/article-pdf/118/20/9257/10847755/<?pdb_no 9257?>9257<?pdb<?db_id PDB?> END?>_1_online.pdf.
- S. Munejiri, F. Shimojo and K. Hoshino, Photo-induced structural change in liquid sulphur, J. Phys.: Condens. Matter, 2000, 12, 7999–8008 CrossRef CAS.
- J. S. Tse and D. D. Klug, Structure and dynamics of liquid sulphur, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 34–37 CrossRef CAS.
- M. W. Wong, Y. Steudel and R. Steudel, Novel species for the sulfur zoo: isomers of S8, Chem. Phys. Lett., 2002, 364, 387–392 CrossRef CAS.
- P. C. Hiemenz and T. P. Lodge, Polymer chemistry, CRC press, 2007 Search PubMed.
- E. Trizio and M. Parrinello, From enhanced sampling to reaction profiles, J. Phys. Chem. Lett., 2021, 12, 8621–8626 CrossRef CAS PubMed.
- H. Niu, L. Bonati, P. M. Piaggi and M. Parrinello, Ab initio phase diagram and nucleation of gallium, Nat. Commun., 2020, 11, 2654 CrossRef CAS PubMed.
- M. Yang, L. Bonati, D. Polino and M. Parrinello, Using metadynamics to build neural network potentials for reactive events: the case of urea decomposition in water, Catal. Today, 2022, 387, 143–149 CrossRef CAS.
- L. Bonati, D. Polino, C. Pizzolitto, P. Biasi, R. Eckert, S. Reitmeier, R. Schlögl and M. Parrinello, The role of dynamics in heterogeneous catalysis: Surface diffusivity and n2 decomposition on fe(111), Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2313023120, DOI:10.1073/pnas.2313023120.
- M. Yang, U. Raucci and M. Parrinello, Reactant-induced dynamics of lithium imide surfaces during the ammonia decomposition process, Nat. Catal., 2023, 6, 829–836 CrossRef CAS.
- S. Tripathi, L. Bonati, S. Perego, and M. Parrinello, How poisoning is avoided in a step of relevance to the haber-bosch catalysis, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-0zzcd.
- J. Behler and M. Parrinello, Generalized neural-network representation of high-dimensional potential-energy surfaces, Phys. Rev. Lett., 2007, 98, 146401 CrossRef PubMed.
- K. S. Vahvaselkä and J. M. Mangs, X-ray diffraction study of liquid sulfur, Phys. Scr., 1988, 38, 737 CrossRef.
- W. J. Klement and J. C. Koh, Polymer content of sulfur quenched rapidly from the melt, J. Phys. Chem., 1970, 74, 4280–4284, DOI:10.1021/j100718a017.
- L. Crapanzano, Polymorphism of sulfur: Structural and Dynamical Aspects, PhD thesis, Université Joseph-Fourier, Grenoble I, 2006.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for bader decomposition of charge density, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- T. D. Kühne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Glöβ, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, CP2K: An electronic structure and molecular dynamics software package - Quickstep: Efficient and accurate electronic structure calculations, J. Chem. Phys., 2020, 152, 194103, DOI:10.1063/5.0007045/16718133/194103_1_online.pdf.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. B. Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. D. Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Kokalj, E. Kúçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H.-V. Nguyen, A. O. de-la Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, Advanced capabilities for materials modelling with Quantum ESPRESSO, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- H. Wang, L. Zhang, J. Han and W. E, DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics, Comput. Phys. Commun., 2018, 228, 178–184 CrossRef CAS.
- J. Zeng, D. Zhang, D. Lu, P. Mo, Z. Li, Y. Chen, M. Rynik, L. Huang, Z. Li, S. Shi, Y. Wang, H. Ye, P. Tuo, J. Yang, Y. Ding, Y. Li, D. Tisi, Q. Zeng, H. Bao, Y. Xia, J. Huang, K. Muraoka, Y. Wang, J. Chang, F. Yuan, S. L. Bore, C. Cai, Y. Lin, B. Wang, J. Xu, J.-X. Zhu, C. Luo, Y. Zhang, R. E. A. Goodall, W. Liang, A. K. Singh, S. Yao, J. Zhang, R. Wentzcovitch, J. Han, J. Liu, W. Jia, D. M. York, W. E, R. Car, L. Zhang and H. Wang, DeePMD-kit v2: A software package for deep potential models, J. Chem. Phys., 2023, 159, 054801, DOI:10.1063/5.0155600/18096887/054801_1_5.0155600.pdf.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in’t Veld, A. Kohlmeyer, S. G. Moore and T. D. Nguyen, et al., LAMMPS-a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, PLUMED 2: New feathers for an old bird, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
- L. Bonati, E. Trizio, A. Rizzi and M. Parrinello, A unified framework for machine learning collective variables for enhanced sampling simulations: mlcolvar, J. Chem. Phys., 2023, 159, 014801, DOI:10.1063/5.0156343/18031428/014801_1_5.0156343.pdf.
- A. Paszke, S. Gross, F. Massa, A. Lerer, J. Bradbury, G. Chanan, T. Killeen, Z. Lin, N. Gimelshein, L. Antiga, A. Desmaison, A. Kopf, E. Yang, Z. DeVito, M. Raison, A. Tejani, S. Chilamkurthy, B. Steiner, L. Fang, J. Bai and S. Chintala, PyTorch: An Imperative Style, High-Performance Deep Learning Library,Advances in Neural Information Processing Systems, ed. H. Wallach, H. Larochelle, A. Beygelzimer, F. d'Alché-Buc, E. Fox and R. Garnett, Curran Associates, Inc., 2019, vol. 32, https://proceedings.neurips.cc/paper_files/paper/2019/file/bdbca288fee7f92f2bfa9f7012727740-Paper.pdf Search PubMed.
- https://github.com/luigibonati/mlcolvar/commit/4387073 .
- A. Stukowski, Visualization and analysis of atomistic simulation data with OVITO-the Open Visualization Tool, Modell. Simul. Mater. Sci. Eng., 2010, 18(1), 015012 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Goedecker, M. Teter and J. Hutter, Separable dual-space gaussian pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS PubMed.
- C. Hartwigsen, S. Goedecker and J. Hutter, Relativistic separable dual-space gaussian pseudopotentials from H to Rn, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 3641–3662 CrossRef CAS.
- D. J. Evans and B. L. Holian, The Nose–Hoover thermostat, J. Chem. Phys., 1985, 83, 4069–4074 CrossRef CAS ), https://pubs.aip.org/aip/jcp/article-pdf/83/8/4069/10987942/<?pdb_no 4069?>4069<?pdb<?db_id PDB?> END?>_1_online.pdf.
- S. Melchionna, G. Ciccotti and B. L. Holian, Hoover NPT dynamics for systems varying in shape and size, Mol. Phys., 1993, 78, 533–544, DOI:10.1080/00268979300100371.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, Canonical sampling through velocity rescaling, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Invernizzi and M. Parrinello, Rethinking metadynamics: from bias potentials to probability distributions, J. Phys. Chem. Lett., 2020, 11, 2731–2736 CrossRef CAS PubMed.
- M. Invernizzi and M. Parrinello, Exploration vs convergence speed in adaptive-bias enhanced sampling, J. Chem. Theory Comput., 2022, 18(6), 3988–3996 CrossRef CAS PubMed.
- M. Bonomi, A. Barducci and M. Parrinello, Reconstructing the equilibrium boltzmann distribution from well-tempered metadynamics, J. Comput. Chem., 2009, 30, 1615–1621, DOI:10.1002/jcc.21305.
- L. Zhang, J. Han, H. Wang, R. Car and W. E, Deep potential molecular dynamics: A scalable model with the accuracy of quantum mechanics, Phys. Rev. Lett., 2018, 120, 143001 CrossRef CAS PubMed.
- D. Zhang, H. Bi, F.-Z. Dai, W. Jiang, L. Zhang, and H. Wang, DPA-1: Pretraining of attention-based deep potential model for molecular simulation, arXiv, 2022, preprint, arXiv:2208.08236, DOI:10.48550/arXiv.2208.08236.
- L. Zhang, J. Han, H. Wang, W. Saidi and R. Car, et al., End-to-end symmetry preserving inter-atomic potential energy model for finite and extended systems, Adv. Neural Inf. Process., 2018, 31, 4436–4446 Search PubMed.
- L. Bonati and M. Parrinello, Silicon liquid structure and crystal nucleation from ab initio deep metadynamics, Phys. Rev. Lett., 2018, 121, 265701 CrossRef CAS PubMed.
- Y. Zhang, H. Wang, W. Chen, J. Zeng, L. Zhang, H. Wang and E. Weinan, DP-GEN: A concurrent learning platform for the generation of reliable deep learning based potential energy models, Comput. Phys. Commun., 2020, 253, 107206 CrossRef CAS.
- https://github.com/EnricoTrizio/sulfur_lambda_transition .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06282a |
| This journal is © The Royal Society of Chemistry 2024 |