
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLong-range electrostatic effects from intramolecular Lewis acid binding influence the redox properties of cobalt–porphyrin complexes†
Jose L.
Alvarez-Hernandez‡
 a,
Xiaowei
Zhang‡
b,
Kai
Cui
c,
Anthony P.
Deziel
a,
Sharon
Hammes-Schiffer
*c,
Nilay
Hazari
*a,
Nicole
Piekut
a and
Mingjiang
Zhong
*b
a,
Xiaowei
Zhang‡
b,
Kai
Cui
c,
Anthony P.
Deziel
a,
Sharon
Hammes-Schiffer
*c,
Nilay
Hazari
*a,
Nicole
Piekut
a and
Mingjiang
Zhong
*b
aDepartment of Chemistry, Yale University, New Haven CT 06520, USA. E-mail: nilay.hazari@yale.edu
bDepartment of Chemical and Environmental Engineering, Yale University, New Haven CT 06520, USA. E-mail: mingjiang.zhong@yale.edu
cDepartment of Chemistry, Princeton University, Princeton, NJ 08544, USA. E-mail: shs566@princeton.edu
First published on 9th April 2024
Abstract
A CoII–porphyrin complex (1) with an appended aza-crown ether for Lewis acid (LA) binding was synthesized and characterized. NMR spectroscopy and electrochemistry show that cationic group I and II LAs (i.e., Li+, Na+, K+, Ca2+, Sr2+, and Ba2+) bind to the aza-crown ether group of 1. The binding constant for Li+ is comparable to that observed for a free aza-crown ether. LA binding causes an anodic shift in the CoII/CoI couple of between 10 and 40 mV and also impacts the CoIII/CoII couple. The magnitude of the anodic shift of the CoII/CoI couple varies linearly with the strength of the LA as determined by the pKa of the corresponding metal–aqua complex, with dications giving larger shifts than monocations. The extent of the anodic shift of the CoII/CoI couple also increases as the ionic strength of the solution decreases. This is consistent with electric field effects being responsible for the changes in the redox properties of 1 upon LA binding and provides a novel method to tune the reduction potential. Density functional theory calculations indicate that the bound LA is 5.6 to 6.8 Å away from the CoII ion, demonstrating that long-range electrostatic effects, which do not involve changes to the primary coordination sphere, are responsible for the variations in redox chemistry. Compound 1 was investigated as a CO2 reduction electrocatalyst and shows high activity but rapid decomposition.
Introduction
Metalloenzymes often use secondary coordination sphere effects, such as hydrogen bonding, intramolecular proton shuttles, or oriented electric fields, to achieve high activity and selectivity for a specific transformation.1 This has inspired the design of many transition metal catalysts containing ancillary ligands that stabilize the transition state in the turnover limiting step through secondary coordination sphere effects.2 The vast majority of these systems incorporate functional groups that promote hydrogen bonding or can act as intramolecular proton shuttles.3 In contrast, electric field effects have not been investigated as extensively in synthetic systems.4 This is in part because it is challenging to orient a freely diffusing molecular catalyst so that it experiences a uniform electric field.One attractive strategy for understanding the impact of electric fields on transition metal catalysts in solution involves using an internal electrostatic field.4c,d,f,i–l For example, the binding of a Lewis acidic cation, such as an alkali or alkaline earth metal ion to a prepositioned site on an ancillary ligand, can alter the redox properties of a metal center due to electrostatic effects.5 Crown or aza-crown ethers are attractive as binding sites for cationic Lewis acids (LAs) because they have high and tunable binding constants for alkali or alkaline earth metal cations.6 Consequently, several ligands have been designed which feature pendant crown or aza-crown ether moieties, A–E (Fig. 1).4l,5a,5c,d,5h,7 In many of these systems, one or two of the atoms in the crown (A or B), bind directly to the transition metal center or effect the donor properties of the ligand (C or D), making it challenging to disambiguate changes in the primary coordination sphere from electrostatic effects caused by binding of the LA cation. Further, in A–C, the LA is often close (∼3 Å) to the transition metal, which can reduce the ability of the crown or aza-crown ether to bind the LA cation. We propose that there is value in developing systems where electrostatic effects can be tuned without significantly perturbing the electronic properties of the ligand directly bound to the transition metal, as this allows for independent control of primary and secondary coordination sphere effects and a high LA binding constant.
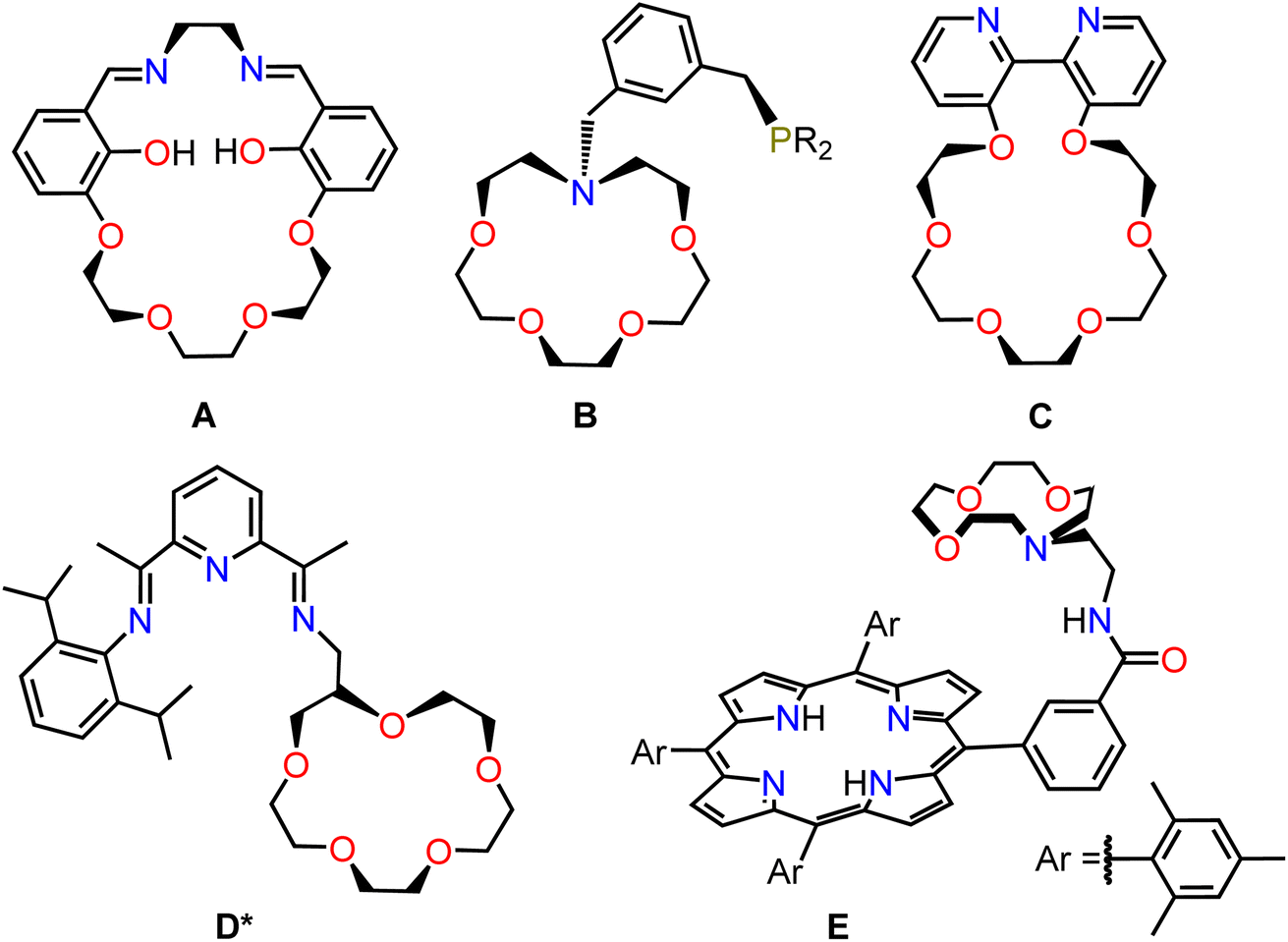 | ||
| Fig. 1 Selected ligands featuring appended crown or aza-crown ether groups reported in the literature. *Ligand D was not isolated independently but was prepared in situ with an Fe coordinated to the pyridinediimine ligand. | ||
Porphyrin-ligated transition metal complexes are used as electro-, photo, and thermal catalysts for a wide variety of societally important transformations, including CO2 reduction,8 O2 reduction,8c,9 H2 evolution,9a,10 polymerization,11 C–H functionalization,12 epoxidation,13 and alcohol oxidation.14 As such, porphyrins are a privileged class of ligand and represent attractive targets for the introduction of crown ether or aza-crown ethers to explore electrostatic effects. There are reports of symmetric porphyrins containing two or four pendant crown ether groups,15 however, this results in complexes that can bind a variable numbers of Lewis acidic cations, which complicates the interpretation of any changes in reactivity caused by LAs. Recently, Zhong et al. prepared a rare example of an asymmetric porphyrin ligand, E,16 containing a single aza-crown ether site for LA binding, which does not directly connect to atoms in the primary coordination sphere (Fig. 1).15f,17 We propose that this porphyrin ligand is optimal for studying internal electrostatic field effects caused by LA binding because the aza-crown ether group should be able to bind a variety of different cationic LAs and is remote from the primary coordination sphere.
In this work, we prepared a CoII complex (1, Fig. 2) supported by ligand E. We show that 1 can bind a variety of LAs and that the binding constant for Li+ is similar to that observed for a free aza-crown ether. Further, for alkali and alkaline earth metal cations, binding to the aza-crown ether causes predictable changes to the redox properties of the Co center. Remarkably, these changes occur despite the LA being 5.6 to 6.8 Å away from the Co center, demonstrating that ‘long-range’ electrostatic effects can be used to tune the redox properties of the transition metal center. Finally, we demonstrate that changes in the redox properties of the Co center caused by LA binding depend on the ionic strength of the solution, which provides a handle for modulating the redox properties of Co without changing the LA or ancillary ligand structure. Overall, this work provides new insight into how to modulate induced electric fields caused by LA binding to an ancillary ligand to change the properties of a transition metal complex.
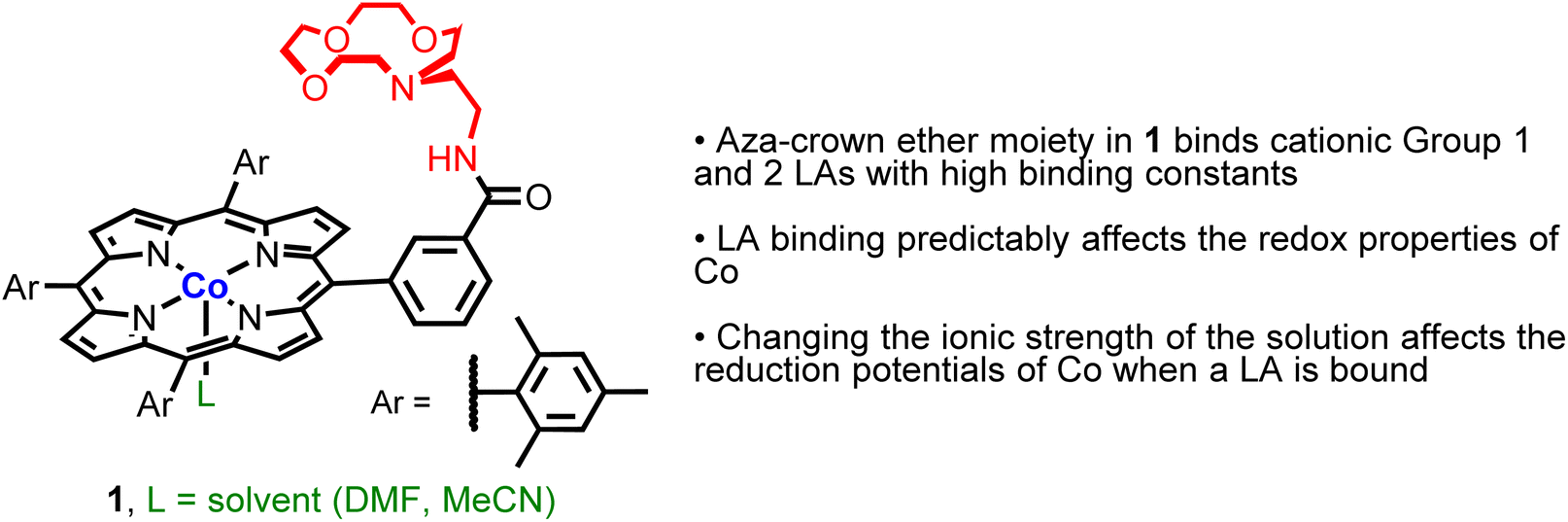 | ||
| Fig. 2 Structure and properties of the new CoII–porphyrin complex 1, which can bind LAs, such as Li+, Na+, K+ Ca2+, Ba2+, and Sr2+, through the pendant aza-crown ether. | ||
Results and discussion
Synthesis and characterization of cobalt–porphyrin complexes
Given that CoII porphyrin complexes are active catalysts for a wide variety of transformations including electrocatalytic CO2 reduction,8 we chose to react ligand E with Co(OAc)2 to form complex 1. The synthesis of 1 followed standard literature procedures,18 but to ensure full metalation of the ligand a five-fold molar excess of Co(OAc)2 was utilized (eqn (1)). Over the course of the reaction, two additional portions of Co(OAc)2 were added to maintain an excess of CoII ions in solution throughout the reaction. To remove excess Co(OAc)2 from 1, our purification procedure included washing the crude reaction mixture with an aqueous solution of sodium ethylenediaminetetraacetate (EDTA), which binds tightly to free CoII ions in solution. As a control compound, we prepared the CoII complex 2, which contains a similar porphyrin ligand to 1, without an intramolecular LA binding site (the full synthesis and characterization data for this new ligand is provided in the SI, see Fig. S1–S3†). Both 1 and 2 were purified by column chromatography and isolated in yields of 86 and 87%, respectively.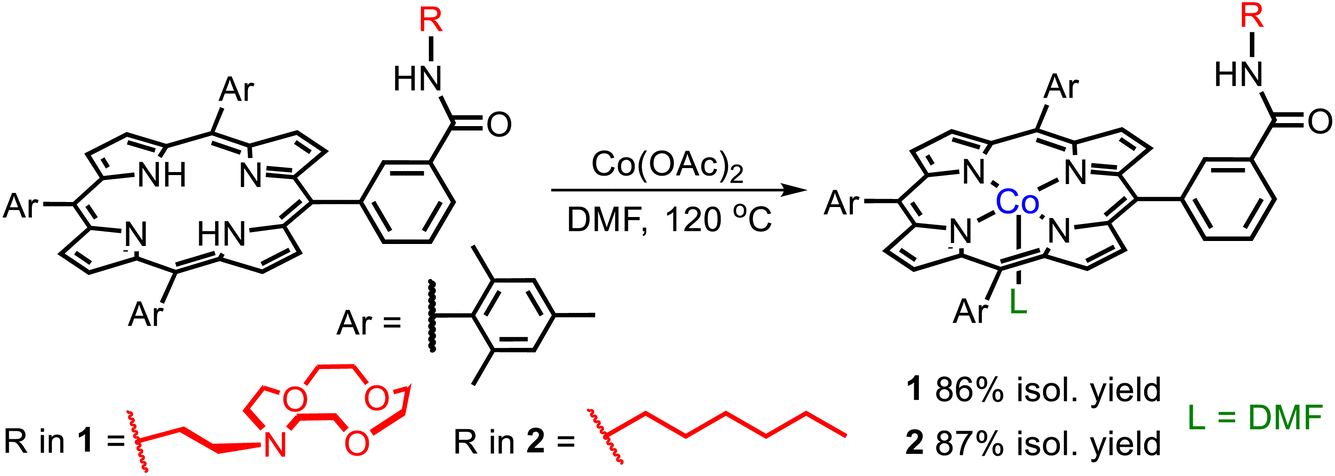 | (1) |
Complexes 1 and 2 were characterized using UV-vis, 1H NMR and EPR spectroscopy and high-resolution mass spectrometry (HRMS) (Fig. S4–S11†). The UV-vis spectrum of 1 is similar to those of other Co porphyrin complexes.19 The spectrum is dominated by a Soret band centered at 413 nm, with a smaller Q band at 530 nm, which has a shoulder at 550 nm. These absorption bands are proposed to originate from porphyrin-centered π–π* electronic transitions.20 The presence of two Q bands serves as an indicator of complete ligand metalation as free deprotonated porphyrins typically exhibit four bands in this spectral region, due to the lower symmetry of the free ligand relative to the metalloporphyrin.20,21 Complex 1 is paramagnetic, and therefore the 1H NMR spectrum of 1 is complicated. In d8-toluene, signals are observed between 0 and 17 ppm with the pyrrolic protons appearing the furthest downfield given their close proximity to the paramagnetic CoII ion. Tentative assignment of the 1H NMR signals is provided in the SI. EPR spectra of Co complexes are often complex, given that 59Co is the only naturally occurring isotope of Co with a nuclear spin of 7/2. The X band EPR spectrum of 1 in toluene at 7 K (Fig. 3a) displays rhombic symmetry with observable hyperfine coupling from 59Co to gx, gy, and gz. The 8 expected peaks can be seen for gx, whereas overlap of gy and gz convolute the splitting. Through simulation of the spectrum, g values (gz = 2.598, gy = 2.375, and gx = 1.955) and the principal values of hyperfine interaction (Az = 330 MHz, Ay = 325 MHz, Ax = 360 MHz) were determined. These values are comparable to previous reports for Co tetraphenyl porphyrin complexes with axial ligands.22 The spectroscopic data for 2 is similar to that for 1 and supports the proposed structure (Fig. S7–S10†). Unfortunately, despite repeated attempts we were unable to grow single crystals of 1 or 2 suitable for analysis using X-ray diffraction, and therefore structural information about 1 was obtained using density functional theory (DFT) calculations (vide infra).
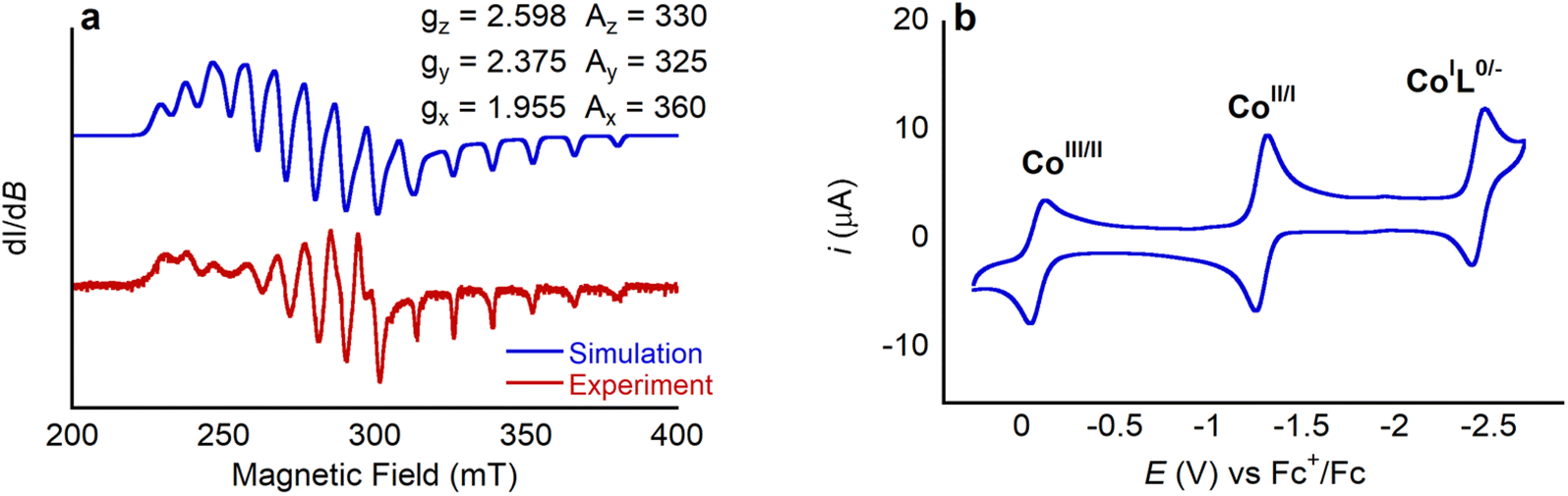 | ||
| Fig. 3 (a) EPR spectrum of 1. (b) Cyclic voltammogram of 1 (0.5 mM) in 0.25 M TBAPF6 in MeCN at 100 mV s−1 under Ar. | ||
Cyclic voltammograms of 1 and 2 were collected in acetonitrile (MeCN) solution under an inert atmosphere. They exhibit three clear redox events, as shown in Fig. 3b for complex 1 (see Fig. S11† for cyclic voltammograms of 2). The first redox wave for 1 at −0.084 V vs. Fc+/Fc (all potentials herein are reported vs. the ferrocenium/ferrocene, Fc+/Fc, internal standard) is assigned as the CoIII/CoII couple based on previous literature reports.23 This couple is quasi-reversible as changing from an octahedral d6 CoIII species to a d7 CoII species leads to axial distortions due to Jahn–Teller effects. The large reorganization energy associated with this distortion decreases the rate of electron transfer from the electrode to 1 in solution, which impacts the reversibility of the couple. The second and third redox couples at −1.288 and −2.459 V are reversible and, based on literature precedent, are assigned as the CoII/CoI couple and a ligand centered reduction.23 Overall, the well-defined redox chemistry observed for 1 makes it ideal for studying the effects of LA binding on its redox properties. Further, the nearly identical cyclic voltammograms observed for 1 and 2 indicates that the incorporation of the pendant LA binding site has negligible impact on the electronic properties of the Co center in 1.
Lewis acid binding to 1
To determine whether complex 1 effectively binds Lewis acidic group I and II cations, we initially performed HRMS experiments. We prepared solutions containing either 1 or 2 in MeCN and added LiOTf (OTf = triflate) or Ca(OTf)2. Triflate was selected as the anion due to its non-coordinating nature and the commercial availability of a wide variety of alkali and alkali earth triflate salts. Whereas solutions of 1 showed peaks at m/z ratios consistent with the binding of Li+ or Ca2+ (Fig. S12–S15†), these peaks were absent in the HRMS of 2 (Fig. S16–S17†). This strongly suggests that 1 can bind LAs but that 2, which lacks the pendant aza-crown ether, cannot. However, this experiment provides no information about the strength of the interaction between 1 and the LA or where binding is taking place.To gain more quantitative information about LA binding, we used 7Li NMR spectroscopy to probe the binding of LiOTf to 1 and 2 in d3-MeCN. We performed the experiments with a 10-fold molar excess of LiOTf relative to 1 (vide infra). Similar behavior was observed with 0.25 M TBAPF6 electrolyte present, added to model electrochemical conditions (Fig. S18–S23†). At room temperature in the presence of 10 equivalents of LiOTf, both 1 and 2 exhibit a single peak at approximately −2.4 ppm (Fig. 4), although the peak for 1 is significantly broader than that of 2. This is consistent with an exchange process involving rapid binding and decoordination of Li+ to the aza-crown ether in 1. At −40 °C, two distinct peaks are observed in the 7Li NMR spectrum for 1 in the presence of LiOTf. We assign the peak at 0.13 ppm to Li+ bound to the aza-crown ether of 1, because similar chemical shifts have been observed for Li+ coordinated to crown ethers in the past,24 and the other to free solvated Li+. Presumably, at −40 °C the exchange between coordinated and solvated Li+ is slow on the NMR timescale. As expected, at −40 °C, only a peak corresponding to solvated Li+ is observed when 2 is cooled in the presence of LiOTf.
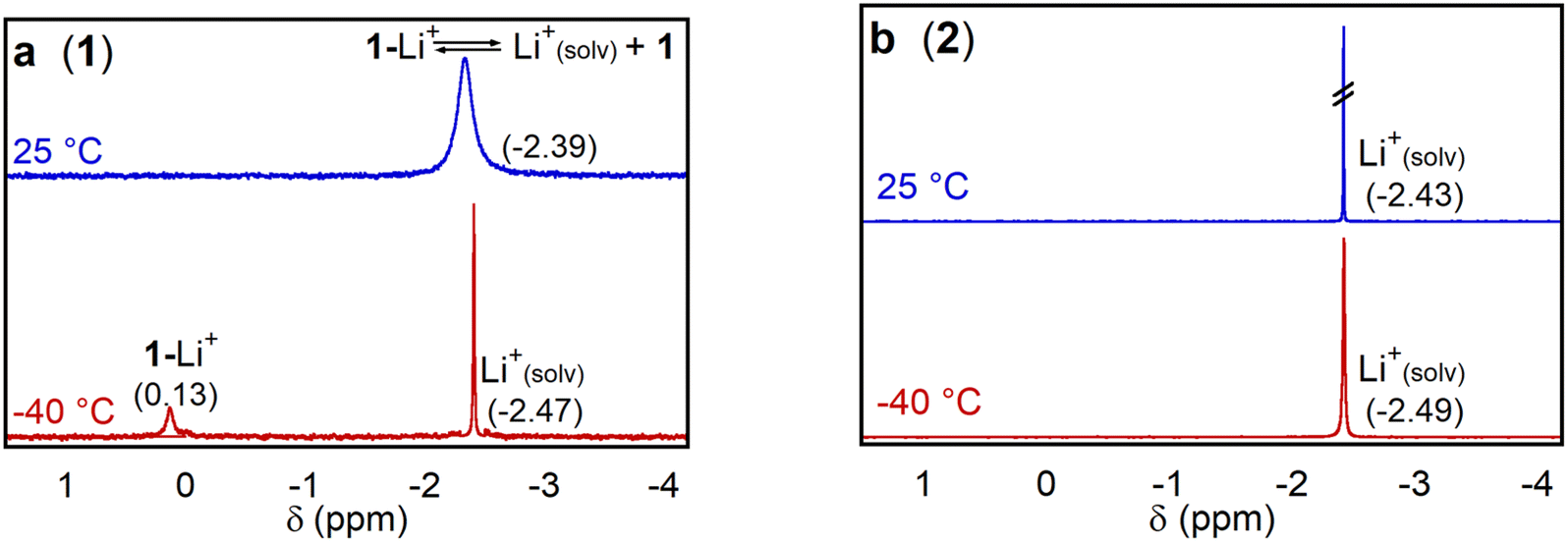 | ||
| Fig. 4 7Li NMR spectra of 15 mM LiOTf with 1.5 mM of (a) 1 and (b) 2 at temperatures of 25 and −40 °C in d3-MeCN. | ||
The observation of a single peak in the 7Li NMR spectra at 25 °C when LiOTf is added to 1 (Fig. 5 and S24†) is indicative of fast exchange on the NMR timescale between coordinated and solvated Li+. It has been demonstrated that, in cases where there is fast exchange between free and complexed metal ions, the observed chemical shift of the resulting single NMR peak can be used to determine the binding constant (Kb).24a,25Kb for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Li+ to 1 adduct was determined from the variation of the 7Li chemical shift with the increasing concentration of LiOTf relative to 1 according to a 1:1 general binding isotherm described by eqn (2),25a
:1 Li+ to 1 adduct was determined from the variation of the 7Li chemical shift with the increasing concentration of LiOTf relative to 1 according to a 1:1 general binding isotherm described by eqn (2),25a
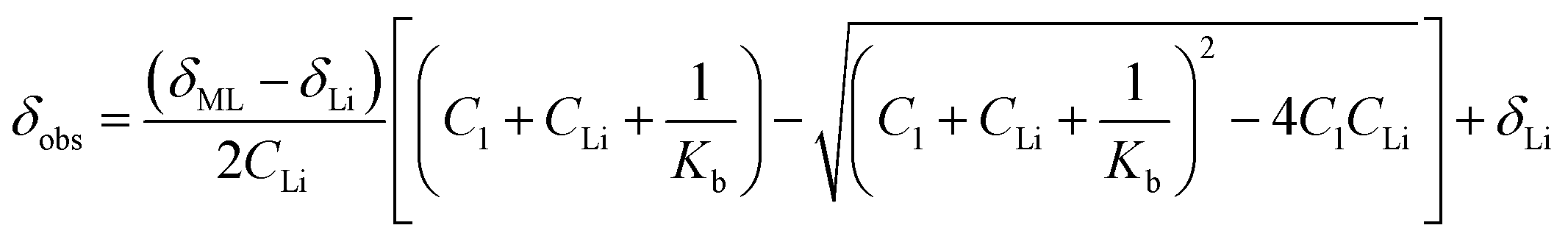 | (2) |
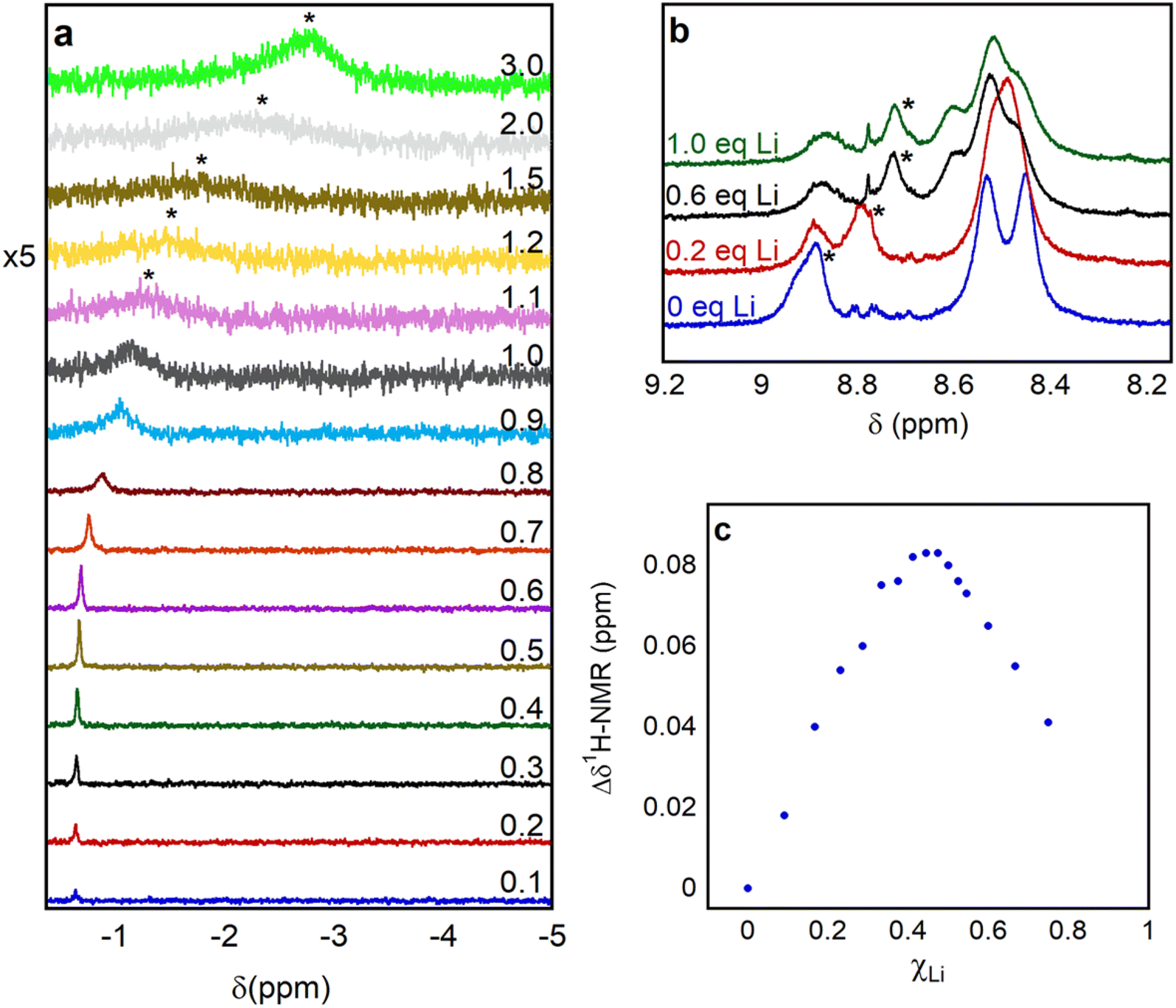 | ||
| Fig. 5 Chemical shift variation in the (a) 7Li NMR and (b) 1H NMR spectra upon adding increasing amounts of LiOTf to a solution containing 1.5 mM of 1 in d3-MeCN. For the 1H NMR spectra, only the peaks that proved more sensitive to added LiOTf are shown. (c) Job plot obtained from the 1H NMR peak initially centered at 8.84 ppm showing a maximum near a Li molar fraction (χLi) 0.5 consistent with formation of a 1:1 adduct between Li and 1. | ||
In principle, changes in the chemical shift of peaks in the 1H NMR spectra of 1 upon the titration of a LA can be used to construct Job plots and estimate Kb in an analogous fashion to 7Li NMR spectroscopy. However, analyzing 1H NMR spectra of 1 in the presence or absence of LAs is complicated by the paramagnetism of 1. Nevertheless, we were able to identify specific peaks in the 1H NMR spectrum of 1 that move predictably upon the addition of Lewis acidic cations (Fig. 5b and S32–S35†). For example, we used the change in the chemical shift of the peak at 8.8 ppm in the presence of LAs to construct Job plots upon addition of a variable number of equivalents of LiOTf, NaOTf, and KOTf to 1 (Fig. 5c, S25, S26, and S34†). The Job plots are consistent with the formation of a 1:1 complex between the LA and 1, although the quality of the data for NaOTf and KOTf is low. Unfortunately, we were unable to use eqn (1) to determine Kb using the change in chemical shift in the 1H NMR spectra upon LA binding as poor fits were observed likely due to the presence of broad and overlapping peaks. When 0.1 equivalents of Ca(OTf)2, which contains a dicationic metal ion, was added to 1, the peak in the 1H NMR spectrum centered at 8.9 ppm broadens significantly more than with the monocationic LAs (Fig. S36†). If more equivalents of Ca(OTf)2 are added, the peak at 8.9 ppm is not observable, presumably because it has broadened into the baseline. This suggests that for 12-O3N aza-crown ether, Kb is higher for Ca2+ than for Li+ and other group I cations, even though an absolute binding constant could not be determined. Consistent with this observation free 12-crown-4 is known to bind more strongly to Ca2+ than to Li+ in acetonitrile.24b
Electrochemical investigation of the binding of group I and II cations to 1
To explore how LA binding impacts the redox properties of 1, we conducted an electrochemical investigation using cyclic voltammetry. Initially, as a control, we added 30 equivalents of TBAOTf to a solution of 1 in MeCN containing 0.25 M TBAPF6 as a supporting electrolyte. The cyclic voltammogram in the presence of TBAOTf was identical to the cyclic voltammogram in its absence (Fig. S37†), indicating that triflate does not interact with 1. Cyclic voltammograms were then recorded in the presence of different amounts of Li+, Na+, K+, Ca2+, Sr2+, and Ba2+ triflate salts (Fig. 6 and S38–S42†). In each case the addition of the LA anodically shifts the position of both the CoIII/CoII and the CoII/CoI couple, as demonstrated in Fig. 6 for one equivalent of Ca(OTf)2. Addition of LA causes the ligand centered reduction at −2.459 V to become irreversible (Fig. S43†) and consequently the CoII/CoI couple also becomes irreversible if the cyclic voltammogram window is extended to include the ligand centered reduction. This observation suggests an electrodegradation pathway in which the LA reacts with the radical-anion species formed upon reduction of the ligand. Hence, we did not extensively explore the impact of LAs on the ligand centered reduction at −2.459 V. Importantly, when LAs are added to solutions of 2, no changes are observed by cyclic voltammetry (Fig. 6). This observation supports the hypotheses that the pendant aza-crown ether can bind LAs and that this binding is crucial to modulating the redox properties of 1.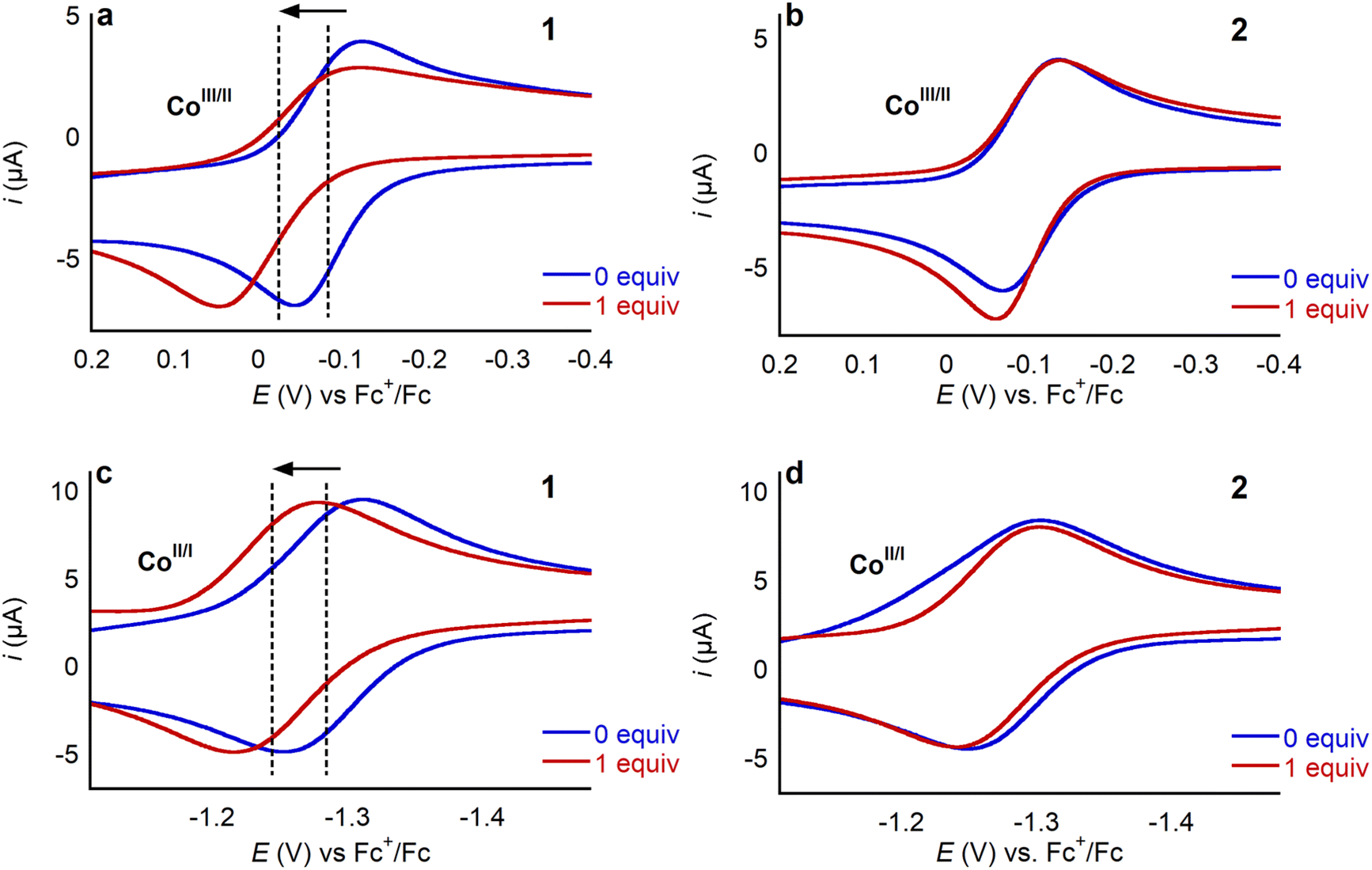 | ||
| Fig. 6 Cyclic voltammograms of the CoIII/CoII (a) and (b) and CoII/CoI (c) and (d) couples of 1 and 2, respectively, in the absence of LA and upon addition of 1 equivalent of Ca(OTf)2, showing that only for 1 does the added LA impact the CV response. Cyclic voltammograms were collected at 100 mV s−1 in 0.25 M TBAPF6 in MeCN under Ar. | ||
The magnitude of the anodic shift of the CoII/CoI couple of 1 increases as the concentration of the LA increases, before stabilizing when a certain number of equivalents of LA are reached (Fig. 7, Tables S2 and S3†). Importantly, the reversibility of the CoII/CoI couple is maintained regardless of the amount of LA salt present. The saturation point of the change in reduction potential is reached with a lower number of equivalents of LA for the dicationic systems compared to the monocationic systems. For example, the changes in the position of the CoII/CoI redox couple stabilize after the addition of 1 equivalent of Ca(OTf)2, whereas 10 equivalents are required for LiOTf. Likely, the CoII/CoI reduction potential stops changing when all the available 1 is bound to the LA. Hence, the different number of equivalents of LA required for the reduction potential to stabilize is related to the different binding affinities of the LA to the aza-crown ether. Given that the aza-crown ether in 1 binds divalent cations more tightly than monocationic LAs (vide supra), it is not surprising that a lower number of equivalents of dicationic LAs is required for the change in reduction potential to stabilize. In the presence of LAs, there is a small peak preceding the CoII/CoI reduction in 1 which may originate from some structural rearrangement or weak adsorption of the complex at the surface of the working electrode. Pre-waves in other transition metal based systems have been broadly interpreted in this way,30 but we do not have experimental evidence to unambiguously assign this small peak. Rinse tests showed no residual features in the cyclic voltammogram, ruling out permanent adsorption of electroactive species to the working electrode.
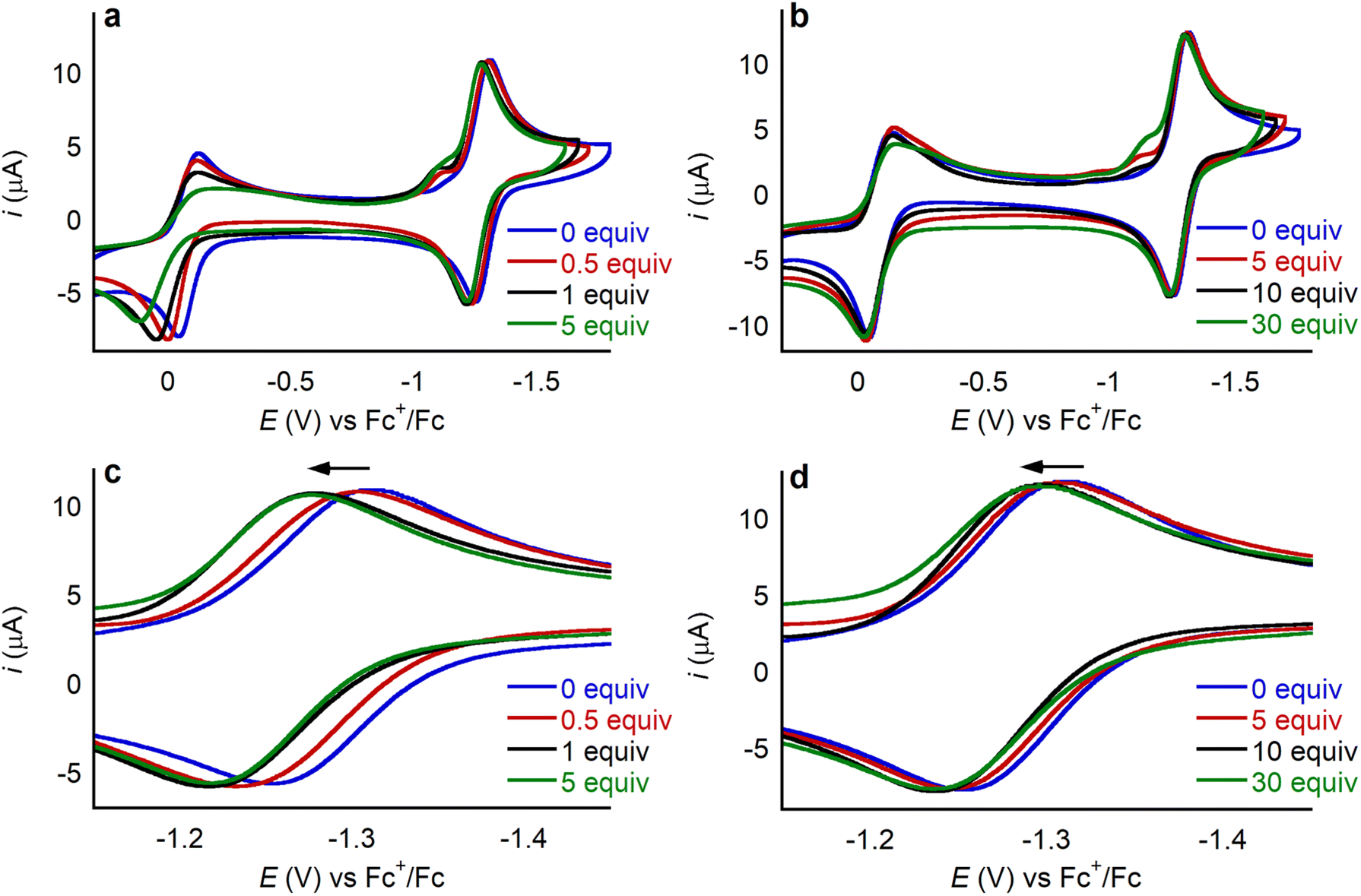 | ||
| Fig. 7 Cyclic voltammograms collected at 100 mV s−1 for 0.5 mM of 1 in 0.25 M TBAPF6 in MeCN under an Ar atmosphere with increasing amounts of (a) Ca(OTf)2 (ΔE1/2 = 34 mV at 1 equiv.) and (b) LiOTf (ΔE1/2 = 15 mV at 10 equiv.). (c) and (d) Zoom in of the CoII/CoI couple in the presence of added Ca(OTf)2 and LiOTf, respectively. | ||
The potential of the CoIII/CoII couple is also affected by the addition of the LA, as evidenced by the anodic shift observed in the presence of Ca(OTf)2 (Fig. 6a). In the case of the CoIII/CoII couple, there is also change in the reversibility. For example, as the amount of Ca(OTf)2 present increases there is an increase in the peak-to-peak separation for the reductive and oxidative voltammetric waves. In the absence of LA, we propose that the CoIII/CoII couple in 1 is only quasi-reversible because of the high reorganization energy associated with axial ligation changes (vide supra). We hypothesize that the solvent reorganization energy, which includes effects from solvent, electrolyte and LA salt, associated with the CoIII/CoII reduction of 1 is higher in the presence of LA. This slows electron transfer, which translates into a larger peak-to-peak separation as well as a decrease in peak current, particularly in the reductive wave.31 The CoIII/CoII couple of 1 does not stop shifting at a given concentration of LA salt as seen for the CoII/CoI couple and instead continues to shift anodically. Further, although the CoII/CoI couple of the control compound 2 does not change upon the addition of LA, there is a small shift in the CoIII/CoII couple, especially for dicationic LAs (Fig. 6b). This shift is much smaller than what is observed for 1 but indicates there is likely some non-specific interaction of the LA with 2. Altogether, for complex 1 the CoIII/CoII couple seems sensitive to bound and free LA, while the CoII/CoI couple is only impacted by the LA ions bound to the aza-crown ether as shown in Fig. 7, hence we have centered our investigation on the CoII/CoI couple of complexes 1 and 2.
The observation that the CoII/CoI couple remains reversible in the presence of LAs allows for the accurate determination of E1/2 in the presence of LAs. We define ΔE1/2 as the maximum difference in the reduction potential of the CoII/CoI couple of 1 in the presence and absence of LA. The maximum difference occurs when enough LA has been added so that the potential of the CoII/CoI couple is no longer shifting and the change in reduction potential has reached its saturation point. We observe that dicationic LAs give bigger changes in ΔE1/2 than monocationic LAs, and the maximum shift that we observe is 38 mV in the case of Sr2+. This change is significantly smaller than changes induced by LA binding in most other systems (for example, Yang and co-workers have observed shifts of up to 270 mV in modified Co Schiff base systems5b), likely because the LA is further away from the transition metal in 1 (vide infra). As observed previously, ΔE1/2 correlates with the Lewis acidity of the bound cations as shown in Fig. 8, when the Lewis acidity of the cation is modelled using the pKa of the aqua complex.5b–d,5g,32 This suggests that electrostatic effects, namely the interaction between the Co center in 1 and the bound LA, are responsible for the differences in reduction potential upon LA binding. Unfortunately, 1 is not stable in the presence of tricationic LAs such as Y3+ and La3+(Fig. S44 and S45†), which would be expected to lead to even bigger changes in ΔE1/2.
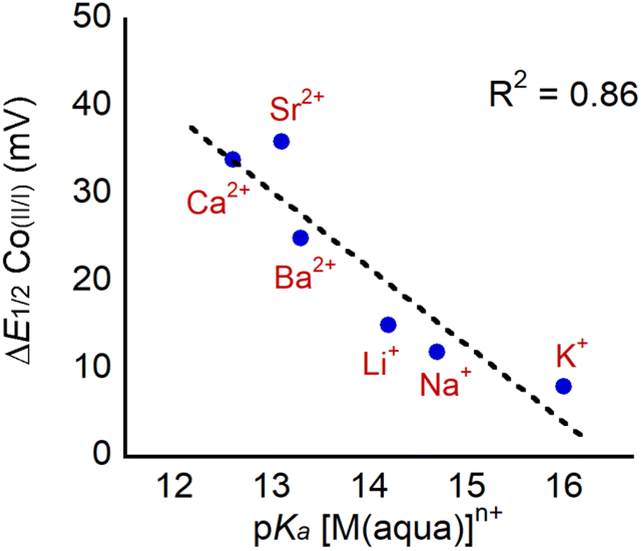 | ||
| Fig. 8 Plot of ΔE1/2 for the CoII/CoI couple in 1 upon LA binding as a function of the Lewis acidity of the cation, which is modelled using the pKa of the corresponding aqua complex. | ||
It is interesting to compare our results to those obtained by Gilbertson and co-workers for an Fe complex containing a pyridinediimine ligand modified with a pendant 15-crown-5 ether (ligand D in Fig. 1) that binds Na+ and Li+ ions.5a In that study, the shift in the quasi-reversible FeII/FeI couple upon Na+ binding (in MeCN containing 0.1 M TBAPF6) is approximately 14 mV, which is similar to the 12 mV shift we observe for 1 bound to Na+. Importantly, the Fe–Na distance in the Fe complex is 6.85 Å in the solid state, while we estimate the Co–LA distance to range between 5.6 and 6.8 Å in our complexes based on DFT calculations (vide infra). When Gilbertson and co-workers shifted from MeCN, which has a high dielectric constant (ε = 37.5), to solvents with lower dielectric constants like dichloromethane (ε = 8.93) or tetrahydrofuran (ε = 7.58) the shift in reduction potential increased to approximately 50 mV for the Na+ bound complex. This solvent dependence is consistent with our interpretation of long-range electrostatic effects being responsible for the changes in the reduction potential of the metal center, as the electric field caused by a point charge is inversely proportional to the dielectric constant of the medium in which the charged particle is immersed.
If electrostatic effects are responsible for the observed changes in reduction potential upon LA binding to 1, it would be expected that the ionic strength (I) of the solution would influence ΔE1/2. This is because ions in solution can order around the 1:LA adduct, and anions can dampen the electrostatic field created by the cationic LA. To modify the ionic strength (I) of the solution, we varied the concentration of the supporting electrolyte (TBAPF6) and then recorded cyclic voltammograms of 1 with one equivalent of Ca(OTf)2 (Fig. 9, S46 and Table S4†). The largest changes in ΔE1/2 for the CoII/CoI couple are observed in solutions with the lowest ionic strength. In fact, a 35 mV difference in ΔE1/2 is observed between the lowest and highest ionic strength. Notably, this is almost as large as the changes observed in ΔE1/2 by varying the identity of the LA (vide supra). Apart from providing evidence for electrostatic effects being responsible for the shifts in reduction potential, this experiment is significant because to our knowledge it is the first example of the use of ionic strength to modulate the redox properties of a complex with an intramolecular LA binding site. Changing the ionic strength of the solution provides a new handle to tune the redox properties of the complex, which in some cases is easier than changing the LA and will give rise to a larger range of reduction potentials. Although lower ionic strengths lead to larger changes in reduction potential, in electrochemical processes it is not recommended to use ionic strengths below 0.1 M. However, Co porphyrin complexes,8–10 such as 1, may be used in conjunction with chemical reductants, in the absence of an electrolyte. In this case, the changes to the reduction potential of 1 that occur with LA binding at low electrolyte concentration will be relevant.
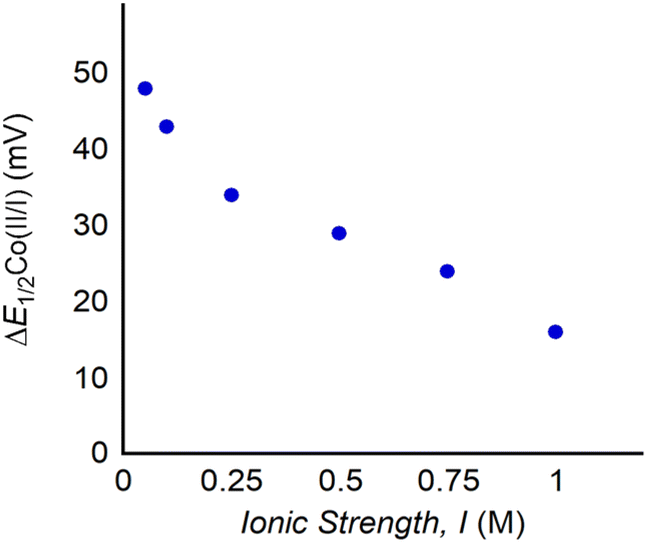 | ||
| Fig. 9 Plot of ΔE1/2 for the CoII/CoI couple of 1 with one equivalent of Ca(OTf)2 at increasing I ranging from 0.05 to 1.0 M TBAPF6. Cyclic voltammograms are provided in Fig. S46.† | ||
Computational studies
DFT calculations were performed to gain information about the structure and electronic properties of 1 both with and without LAs bound. As part of these calculations, we explored the different conformations of the pendant aza-crown ether group that may be present in solution. To reduce the computational cost, the mesityl groups on the porphyrin were replaced by methyl groups. The resulting compound is denoted 1*. Initially, we sampled the possible conformations of one electron reduced 1* (i.e., CoI instead of CoII) with Ca2+ bound to the aza-crown ether group. We selected CoI instead of CoII both because it is diamagnetic, which leads to faster calculations, and because solvent binding does not need to be considered for a square-planar 16-electron complex. Further, given that the cyclic voltammograms of 1 are reversible between CoII and CoI, large conformational changes between complexes in the two oxidation states are likely not occurring. We used the Conformer-Rotamer Ensemble Sampling Tool (CREST)33 to identify 159 conformers of reduced 1* with Ca2+ bound that are local minima at the density functional tight binding (DFTB) level of theory34 in implicit acetonitrile solvent. The Co–Ca2+ distance in these conformers span a wide range, from 5 to 14 Å. The LA-bound aza-crown ether moiety can fold so that is above the porphyrin plane, resulting in a Co–Ca2+ distance close to 5 Å, or the arm connecting the aza-crown ether group to the porphyrin can fully extend, resulting in a much longer Co–Ca2+ distance of ∼14 Å (Fig. S47†). Among these conformers, we selected 12 geometrically distinct structures for further investigation at a higher computational level with the Co center in the paramagnetic CoII oxidation state. The selected structures were optimized at the DFT level of theory using the BP86 functional35 in implicit acetonitrile solvent both with and without an explicit acetonitrile solvent molecule coordinated to Co. The free energy of each conformer was calculated, including zero-point energy and entropic contributions. Additional computational details are provided in the ESI.†The relative stabilities of the selected conformers were characterized by their relative free energies, ΔGrel, where the free energy of the most stable conformer studied was used as the reference at 0 kcal mol−1. The results for 1* with various LAs bound to the aza-crown ether (1*-LA) are shown in Fig. 10a. In these calculations, the axial acetonitrile ligand, expected to be bound in CoII porphyrins,31 was not included to reduce the computational cost. Control calculations on both 1*-Ca2+ and 1*-Li+ indicated that the same trends were observed in the presence and absence of an axial ligand (Fig. S48†). Comparison of the different conformers of 1*-Ca2+ indicates that there is an increase in the relative free energy as a function of the Co–Ca2+ distance. The change in ΔGrel with respect to the Co–Ca2+ distance can be approximately described by a 1/dCo–LA relation (dashed line in Fig. 10a), where dCo–LA is the distance between the Co and Ca2+ centers. The structure of the most stable conformer for 1*-Ca2+ is shown in Fig. 10b. Despite the repulsion between the positively charged CoII and the Ca2+, the LA-bound aza-crown ether prefers to fold onto the porphyrin plane. Presumably, the repulsive interaction between the two cations is attenuated by the aza-crown ether group, and the attractive dispersion interactions between the porphyrin and the aza-crown ether groups are dominant. This leads to a net attraction between the Ca2+-bound aza-crown ether and the Co porphyrin. Conformers similar to the lowest free energy structure of 1*-Ca2+ (shown in Fig. 10b) are found to be the most stable for the other five LAs (Fig. S49†), and the same increase in free energy with increasing distance between the metal centers is observed. For all systems, the free energy difference between the most stable conformer and the others is well beyond the thermal energy at room temperature (0.6 kcal mol−1). We therefore expect that only the most stable conformers exist in solution, with corresponding Co–LA distances ranging from 5.6 to 6.8 Å. The distance is shortest in 1-Li+ and largest in 1-Ba2+, as shown in Fig. 10a, which likely reflects the size of the cations.
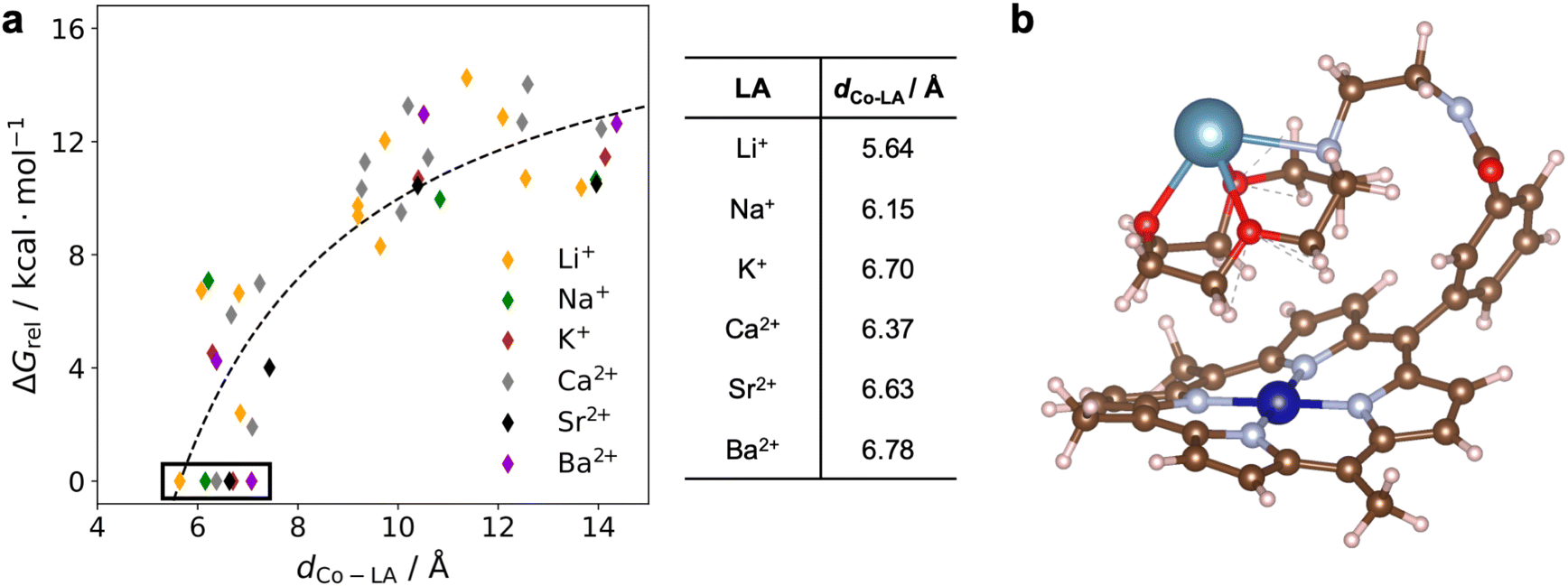 | ||
| Fig. 10 (a) DFT calculated relative free energies of various conformers sampled for complex 1* with different LAs as a function of the Co–LA distance. The axial acetonitrile ligand was not included in these calculations. The most stable conformers are indicated by the black box with their Co–LA distances listed in the table. The black dashed line is used to guide the eye. (b) Optimized geometry of the lowest free energy conformer of 1*-Ca2+. | ||
We also calculated the relative reduction potential of the CoII/CoI couple of 1* for each LA using both the most stable geometry and the geometry with an extended arm (Fig. S47†). The CoII/CoI reduction potential with no LA bound is used as the reference.36 As shown in Fig. 11a, for the most stable conformer, we observe a roughly linear relationship between the relative reduction potential and the LA strength (expressed as the pKa of the corresponding aqua-complex). Apart from the reduction potential of 1*-Ba2+, which is an outlier, the results agree qualitatively with our experimental observations. However, the calculated relative reduction potentials are one order of magnitude larger than the experimental results. We found that this disparity could be partially due to the absence of electrolyte in the computational model (see ESI†). Another source of error in the calculations is that the harmonic approximation was used to compute the zero-point energy and vibrational entropic contributions in the free energy calculations. Since both the porphyrin and the aza-crown ether groups are flexible, the vibrational modes of 1* tend to have strong anharmonic character. Moreover, we replaced the mesityl groups by methyl groups in the calculations. The lower steric bulk of the methyl groups may result in a shorter Co–LA distance than for 1 and therefore may lead to larger reduction potential changes. In contrast to our results with the most stable ‘folded’ geometry, for the geometry with an extended arm, no trend is observed between the CoII/CoI reduction potential and LA strength, as shown in Fig. 11b. The absence of any trend in the extended-arm geometry provides further support that the complexes adopt a folded geometry. Our conclusions also hold without an axial ligand bound to the CoII species (Fig. S49 and S50†).
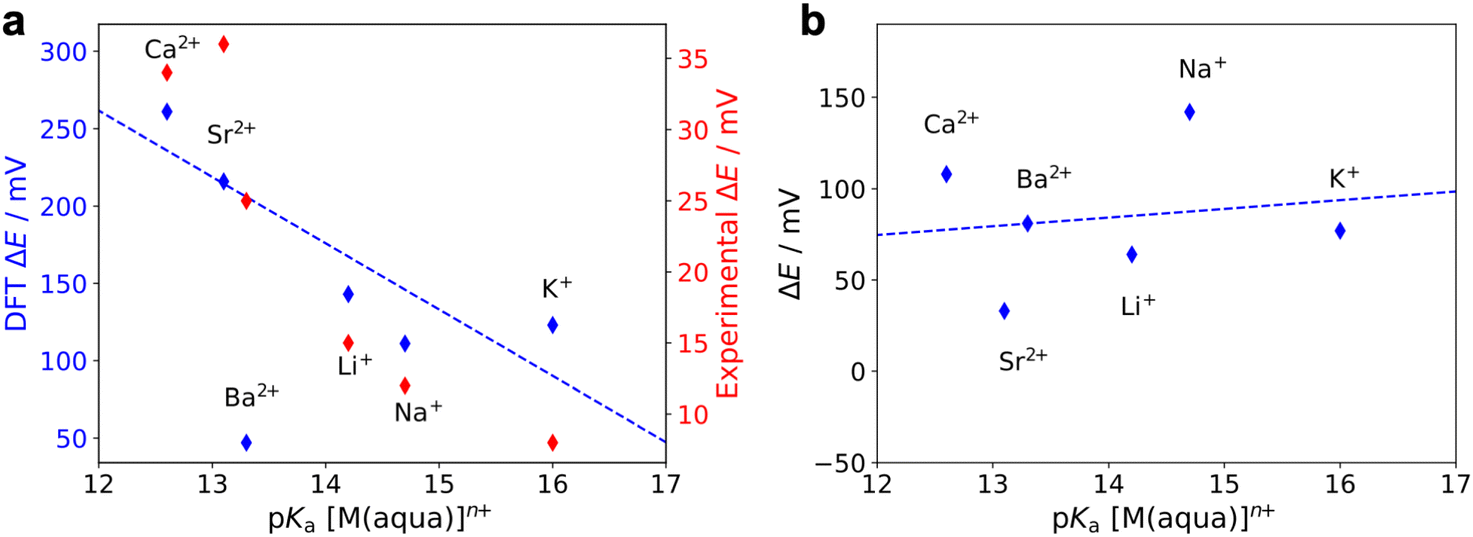 | ||
| Fig. 11 Calculated relative reduction potentials (blue) of the CoII/CoI couple of 1* when LAs are bound to the aza-crown ether as a function of the LA strength. These calculations were performed for (a) the most stable geometry (‘folded’, as shown in Fig. 10b) and (b) the geometry with an extended arm, as in Fig. S42.† ΔE is the reduction potential for the 1*-LA species relative to the reduction potential of 1* with no LA bound in either the folded or extended arm geometry. An axial acetonitrile ligand was explicitly included for the CoII species in these calculations. The data for Ba2+ is excluded in both linear fits. A second y axis in (a) shows the experimental values (red) measured via cyclic voltammetry. | ||
Using the most stable geometry for each 1*-LA system, the electrostatic potential generated by the LA near the Co center, Δϕ, was calculated. This quantity Δϕ is defined as the difference between the total electrostatic potentials of 1*-LA and 1* at the DFT-optimized geometry of 1*-LA. Fig. 12a shows a typical electrostatic potential change caused by the bound Ca2+ ion in the 1*-Ca2+ adduct. A deeper shade of blue indicates a stronger, more positive, potential. The Δϕ generated by different LAs can be quantitatively compared by taking the average within a 1 Å sphere near the Co ion (the ionic radius of CoII is about 0.7 Å)37 as shown in Fig. 12b. Given that Δϕ is determined by subtracting the total electrostatic potentials of two structures that differ only by the presence of the cationic LA, Δϕ reflects the electrostatic effect of a point charge. This explains why the Δϕ values for the divalent cations are approximately double those for the monovalent cations. The differences between LA cations of equal charge arise from the different Co–LA distances determined from geometry optimizations of 1*-LA and the different ionic radii of the LA cations. Altogether, this relatively simple calculation yields Δϕ values that correlate well with the LA strength (Fig. 12b), showing a trend similar to that obtained in our electrochemical experiments (Fig. 8). Our calculations offer proposed geometries for the 1-LA complexes and provide strong support for the hypothesis that the observed changes in the redox properties of 1 upon LA binding are related to electrostatic effects.
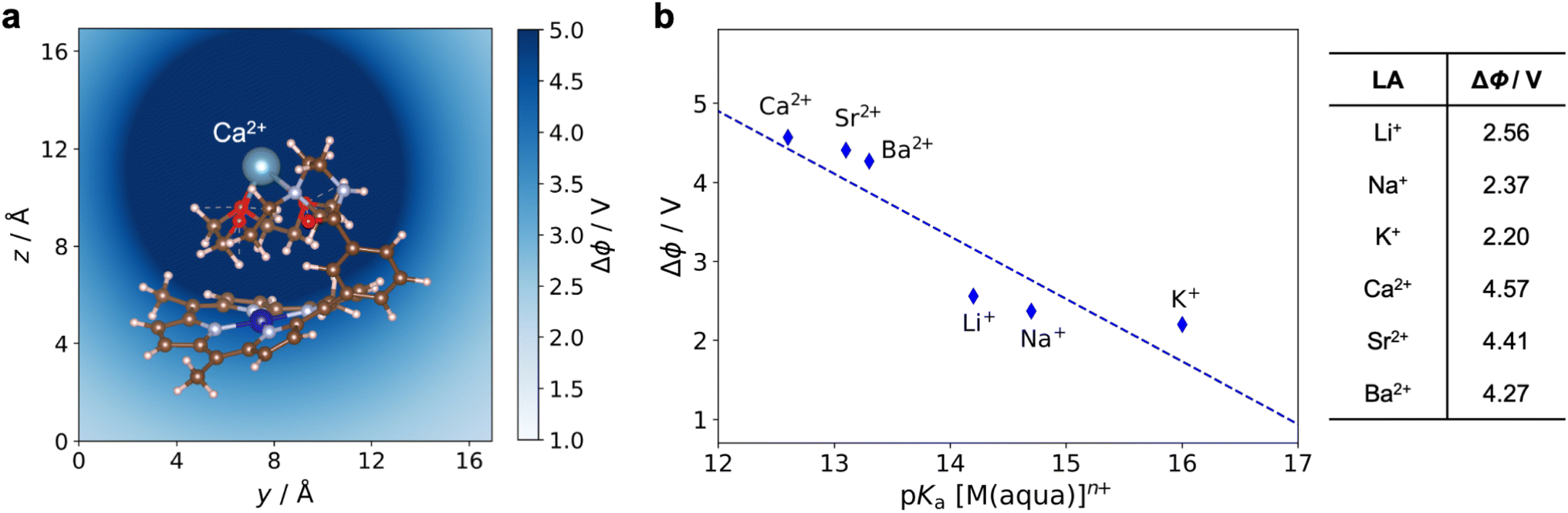 | ||
| Fig. 12 (a) Electrostatic potential (Δϕ) generated by Ca2+ in 1*-Ca2+. (b) Calculated Δϕ generated by the LA at the Co center in 1*-LA as a function of the LA strength. | ||
Electrocatalytic CO2 reduction by 1
CoII porphyrin complexes are well-known electrocatalysts for CO2 reduction, often giving selective conversion to CO.38 Therefore, we explored the ability of 1 to electrocatalytically reduce CO2 with the goal of ultimately assessing whether binding of a cationic LA impacts catalysis. A cyclic voltammogram of 1 under CO2 shows the same CoIII/CoII and CoII/CoI waves described earlier. In addition, there is an irreversible peak approximately 100 mV anodic of the CoII/CoI couple as shown in Fig. 13a, where the new catalytic peak is labeled with an asterisk. This behavior is consistent with a total catalysis regime, specifically the cyclic voltammogram shows the characteristic shape of a KT2 voltammogram as described in Savéant's Kinetic Zone diagram.39 A cyclic voltammogram exhibits a KT2 shape because the rate of catalysis is so fast that even a small fraction of the catalyst in the reduced active form is able to consume all of the substrate, or co-substrate, available in the reaction-diffusion layer quickly. Then, when the potential of the catalysis-initiating redox couple is reached there is no substrate for the catalyst to use and thus the reversible wave is observed. Addition of 10 mM of the Brønsted acid trifluoroethanol (TFE) led to a significant current increase of the catalytic peak, effectively moving the cyclic voltammogram into a KT1 regime (Fig. 13b).39 In this case, the higher concentration of co-substrate (i.e., proton donor, TFE) allows for catalysis to be sustained for longer, so a larger catalytic peak grows that completely obscures the reversible CoII/CoI couple.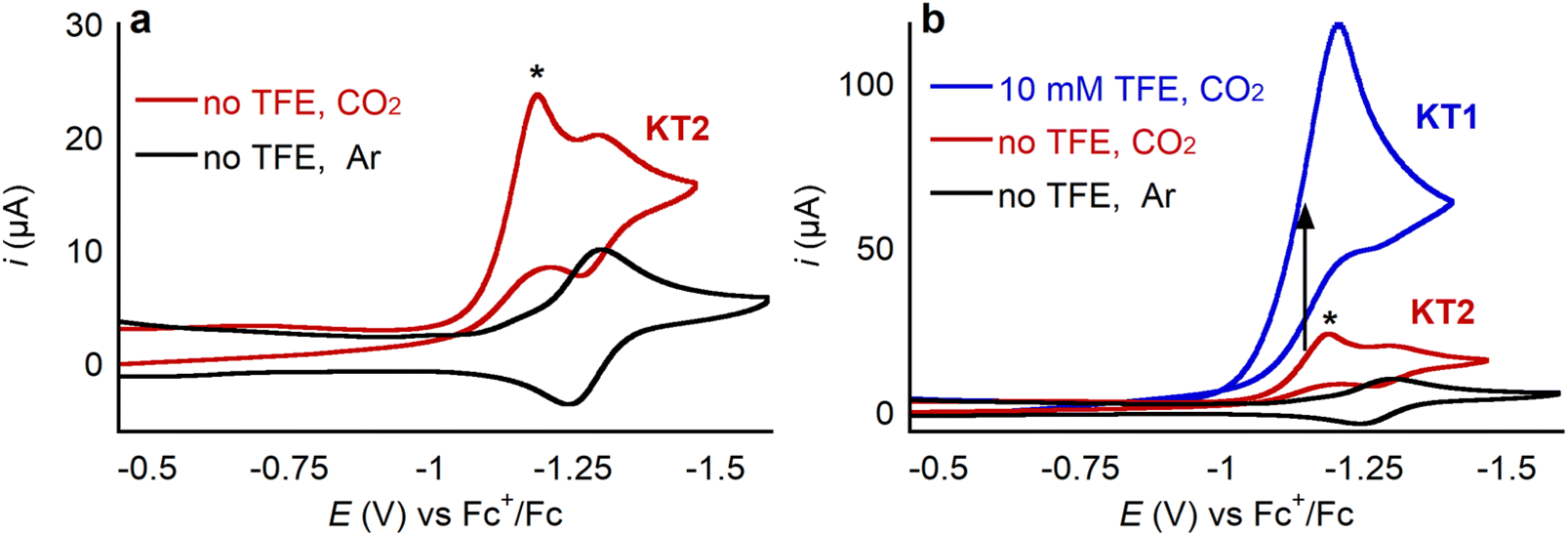 | ||
| Fig. 13 CV of 0.5 mM 1 at 100 mV s−1 in 0.25 M TBAPF6/MeCN under 1 atm of the specified gas, (a) without added proton source (TFE, pKa = 35.4) and (b) with 10 mM of trifluoroethanol. | ||
Despite the cyclic voltammogram of 1 indicating fast initial electrocatalytic CO2 reduction, the complex is unstable under electrocatalytic conditions. When further cyclic voltammograms are collected, the catalytic current diminishes rapidly (Fig. S51†), consistent with loss of catalytic activity, which precluded running controlled potential electrolysis (CPE) experiments to determine the product distribution. Similarly, when the electrocatalytic performance of 1 was evaluated in the presence of LAs, such as Ca(OTf)2, even though there was an initial large increase in current under CO2, rapid catalyst decomposition was observed, preventing detailed analysis. We hypothesize that the nitrogen atom in the aza-crown ether reacts with CO2 leading to a loss of CO2-reduction activity. Consistent with this proposal, the control compound 2 generates significant quantities of CO in electrocatalytic experiments, is stable for over 2 hours of CPE, and displays a stable current (Fig. S52 and S53†). Further, chemical reduction of 1 by KC8 followed by exposure to CO2 leads to formation of a precipitate and a discoloration of the solution. On the other hand, when 2 is treated similarly, it remains in solution with no color change and yields a 1H NMR spectrum consistent with a one-electron reduced CO2-bound species that remains stable for at least 18 hours. Future efforts will be directed toward the synthesis of metalloporphyrin–crown ether complexes that are stable under CO2 to explore the impact of bound LAs in electrocatalytic CO2 reduction.
Conclusions
We have prepared a CoII porphyrin complex, 1, which contains a pendant aza-crown ether group that can bind cationic Group I and II LAs to change the internal electric field. The binding constant for Li+ coordination is similar to free aza-crown ethers, whereas other metal complexes containing pendant aza-crown or crown ether ligands often have significantly lower binding affinities for LAs.5e,27 This is likely because the aza-crown ether group in 1 is remote from the Co center, and none of the atoms associated with the aza-crown ether interact directly with the Co. Strong LA binding is potentially important in electrocatalytic CO2 reduction, where the use of a lower number of equivalents of LA is expected to result in less electrode fouling due to the formation of insoluble carbonates.LA binding causes an anodic shift in both the CoIII/CoII and CoII/CoI redox couples of 1. The magnitude of the CoII/CoI shift varies predictably with the LA strength of the bound cation, increasing linearly with the pKa of the corresponding metal–aqua complex. This result is consistent with previous literature reports,4f,j,5d,g,32b but the magnitude of the shift is smaller than the shifts found for systems in which the LA is positioned closer (∼3 Å) to the metal center.4f,5d,5f,g Our DFT studies imply that the distance between the bound LA and the Co center ranges from 5.6 to 6.8 Å, suggesting that the effects seen in the electrochemical properties of 1 are due to long-range electrostatic interactions. The calculated electrostatic potential generated by the LA near the Co center is also linearly correlated with the pKa of the corresponding metal–aqua complex. Moreover, the magnitude of the shift in the CoII/CoI couple upon LA binding is sensitive to the ionic strength of the solution, with solutions containing higher electrolyte concentration resulting in smaller changes in reduction potential. To our knowledge this is the first time that the ionic strength of the solution has been used as a handle to tune the redox properties of transition metal complexes with intramolecular electric fields. Although the CV response of 1 suggests fast CO2 reduction catalysis, the complex quickly deactivates under CO2, which we attribute to the nitrogen atom in the aza-crown ether group reacting with CO2.
Overall, we have developed a Co porphyrin system in which the redox properties can be tuned via long-range electrostatic effects. Importantly, it is likely that using this system it will be possible to independently modulate the primary coordination sphere and the internal electric field. Future research efforts will be dedicated to the synthesis of structurally related complexes with crown ether groups that are expected to be more stable under the conditions used for electrocatalytic CO2 reduction and will allow catalytic performance to be evaluated as both the primary coordination sphere and magnitude of the internal electric field are varied.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
JLAH: conceptualization, investigation, experimental methodology, writing – original draft. XZ: investigation, synthetic methodology, writing – original draft (synthesis). KC: conceptualization, investigation, computational methodology, writing – original draft (computational studies). APD: investigation. SHS: conceptualization, supervision, writing – review and editing. NH: conceptualization, supervision, writing – review and editing. NP: investigation. MZ: conceptualization, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was solely supported as part of the Center for Hybrid Approaches in Solar Energy to Liquid Fuels (CHASE), an Energy Innovation Hub funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0021173. NH & MZ thank the Yale Planetary Solutions Project for assistance. We thank Prof. Gary Brudvig and Prof. Hailiang Wang for helpful discussions, and Dr Reagan Hooper for assistance with EPR simulations.References
- (a) J. A. Gerlt, M. M. Kreevoy, W. W. Cleland and P. A. Frey, Understanding Enzymic Catalysis: The Importance of Short, Strong Hydrogen Bonds, Chem. Biol., 1997, 4, 259 CrossRef CAS PubMed; (b) G. J. Bartlett, C. T. Porter, N. Borkakoti and J. M. Thornton, Analysis of Catalytic Residues in Enzyme Active Sites, J. Mol. Biol., 2002, 324, 105 CrossRef CAS PubMed; (c) S. D. Fried, S. Bagchi and S. G. Boxer, Extreme Electric Fields Power Catalysis in the Active Site of Ketosteroid Isomerase, Science, 2014, 346, 1510 CrossRef CAS PubMed; (d) S. D. Fried and S. G. Boxer, Measuring Electric Fields and Noncovalent Interactions Using the Vibrational Stark Effect, Acc. Chem. Res., 2015, 48, 998 CrossRef CAS PubMed; (e) P. Hanoian, C. T. Liu, S. Hammes-Schiffer and S. Benkovic, Perspectives on Electrostatics and Conformational Motions in Enzyme Catalysis, Acc. Chem. Res., 2015, 48, 482 CrossRef CAS PubMed; (f) Y. Wu and S. G. Boxer, A Critical Test of the Electrostatic Contribution to Catalysis with Noncanonical Amino Acids in Ketosteroid Isomerase, J. Am. Chem. Soc., 2016, 138, 11890 CrossRef CAS PubMed; (g) S. D. Fried and S. G. Boxer, Electric Fields and Enzyme Catalysis, Annu. Rev. Biochem., 2017, 86, 387 CrossRef CAS PubMed; (h) Y.-W. Lin, Rational Design of Metalloenzymes: From Single to Multiple Active Sites, Coord. Chem. Rev., 2017, 336, 1 CrossRef CAS.
- (a) J. Bonin, C. Costentin, C. Robert, J.-M. Savéant and C. Tard, Hydrogen-Bond Relays in Concerted Proton-Electron Transfers, Acc. Chem. Res., 2011, 45, 372 CrossRef PubMed; (b) S. A. Cook and A. S. Borovik, Molecular Designs for Controlling the Local Environments around Metal Ions, Acc. Chem. Res., 2015, 48, 2407 CrossRef CAS PubMed; (c) K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Non-Covalent Interactions in the Synthesis of Coordination Compounds: Recent Advances, Coord. Chem. Rev., 2017, 345, 54 CrossRef CAS; (d) L. V. Hale and N. K. Szymczak, Hydrogen Transfer Catalysis Beyond the Primary Coordination Sphere, ACS Catal., 2018, 8, 6446 CrossRef CAS.
-
(a) A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. R. DuBois and D. L. DuBois, Hydrogen Oxidation and Production Using Nickel-Based Molecular Catalysts with Positioned Proton Relays, J. Am. Chem. Soc., 2006, 128, 358 CrossRef CAS PubMed;
(b) J. Y. Yang, S. E. Smith, T. Liu, W. G. Dougherty, W. A. Hoffert, W. S. Kassel, M. R. DuBois, D. L. DuBois and R. M. Bullock, Two Pathways for Electrocatalytic Oxidation of Hydrogen by a Nickel Bis(diphosphine) Complex with Pendant Amines in the Second Coordination Sphere, J. Am. Chem. Soc., 2013, 135, 9700 CrossRef CAS PubMed;
(c) C. Costentin, G. Passard, M. Robert and J.-M. Savéant, Pendant Acid−Base Groups in Molecular Catalysts: H-Bond Promoters or Proton Relays? Mechanisms of the Conversion of CO2 to CO by Electrogenerated Iron(0)Porphyrins Bearing Prepositioned Phenol Functionalities, J. Am. Chem. Soc., 2014, 136, 11821 CrossRef CAS PubMed;
(d) S. E. Creutz and J. C. Peters, Exploring Secondary-Sphere Interactions in Fe–NxHy Complexes Relevant to N2 Fixation, Chem. Sci., 2017, 8, 2321 RSC;
(e) S. Roy, B. Sharma, J. Pécaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, Molecular Cobalt Complexes with Pendant Amines for Selective Electrocatalytic Reduction of Carbon Dioxide to Formic Acid, J. Am. Chem. Soc., 2017, 139, 3685 CrossRef CAS PubMed;
(f) H. Sun, Y. Han, H. Lei, M. Chen and R. Cao, Cobalt Corroles with Phosphonic acid Pendants as Catalysts for Oxygen and Hydrogen Evolution From Neutral Aqueous Solution, Chem. Commun., 2017, 53, 6195 RSC;
(g) T. Z. H. Gani and H. J. Kulik, Understanding and Breaking Scaling Relations in Single-Site Catalysis: Methane to Methanol Conversion by FeIV
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, ACS Catal., 2018, 8, 975 CrossRef CAS;
(h) E. M. Nichols, J. S. Derrick, S. K. Nistanaki, P. T. Smith and C. J. Chang, Positional Effects of Second-Sphere Amide Pendants on Electrochemical CO2 Reduction Catalyzed by Iron Porphyrins, Chem. Sci., 2018, 9, 2952 RSC;
(i) D. Dolui, S. Khandelwal, A. Shaik, D. Gaat, V. Thiruvenkatam and A. Dutta, Enzyme-Inspired Synthetic Proton Relays Generate Fast and Acid-Stable Cobalt-Based H2 Production Electrocatalysts, ACS Catal., 2019, 9, 10115 CrossRef CAS;
(j) A. Chapovetsky, M. Welborn, J. M. Luna, R. Haiges, T. F. Miller III and S. C. Marinescu, Pendant Hydrogen-Bond Donors in Cobalt Catalysts Independently Enhance CO2 Reduction, ACS Cent. Sci., 2018, 4, 397 CrossRef CAS PubMed;
(k) N. Queyriaux, D. Sun, J. Fize, J. Pécaut, M. J. Field, M. Chavarot-Kerlidou and V. Artero, Electrocatalytic Hydrogen Evolution with a Cobalt Complex Bearing Pendant Proton Relays: Acid Strength and Applied Potential Govern Mechanism and Stability, J. Am. Chem. Soc., 2020, 142, 274 CrossRef CAS PubMed;
(l) S. Bhunia, A. Rana, S. Hematian, K. D. Karlin and A. Dey, Proton Relay in Iron Porphyrins for Hydrogen Evolution Reaction, Inorg. Chem., 2021, 60, 13876 CrossRef CAS PubMed;
(m) S. Bhunia, A. Ghatak, A. Rana and A. Dey, Amine Groups in the Second Sphere of Iron Porphyrins Allow for Higher and Selective 4e−/4H+ Oxygen Reduction Rates at Lower Overpotentials, J. Am. Chem. Soc., 2023, 145, 3812 CrossRef CAS PubMed;
(n) J. S. Derrick, M. Loipersberger, S. K. Nistanaki, A. V. Rothweiler, M. Head-Gordon, E. M. Nichols and C. J. Chang, Templating Bicarbonate in the Second Coordination Sphere Enhances Electrochemical CO2 Reduction Catalyzed by Iron Porphyrins, J. Am. Chem. Soc., 2022, 144, 11656 CrossRef CAS PubMed.
O, ACS Catal., 2018, 8, 975 CrossRef CAS;
(h) E. M. Nichols, J. S. Derrick, S. K. Nistanaki, P. T. Smith and C. J. Chang, Positional Effects of Second-Sphere Amide Pendants on Electrochemical CO2 Reduction Catalyzed by Iron Porphyrins, Chem. Sci., 2018, 9, 2952 RSC;
(i) D. Dolui, S. Khandelwal, A. Shaik, D. Gaat, V. Thiruvenkatam and A. Dutta, Enzyme-Inspired Synthetic Proton Relays Generate Fast and Acid-Stable Cobalt-Based H2 Production Electrocatalysts, ACS Catal., 2019, 9, 10115 CrossRef CAS;
(j) A. Chapovetsky, M. Welborn, J. M. Luna, R. Haiges, T. F. Miller III and S. C. Marinescu, Pendant Hydrogen-Bond Donors in Cobalt Catalysts Independently Enhance CO2 Reduction, ACS Cent. Sci., 2018, 4, 397 CrossRef CAS PubMed;
(k) N. Queyriaux, D. Sun, J. Fize, J. Pécaut, M. J. Field, M. Chavarot-Kerlidou and V. Artero, Electrocatalytic Hydrogen Evolution with a Cobalt Complex Bearing Pendant Proton Relays: Acid Strength and Applied Potential Govern Mechanism and Stability, J. Am. Chem. Soc., 2020, 142, 274 CrossRef CAS PubMed;
(l) S. Bhunia, A. Rana, S. Hematian, K. D. Karlin and A. Dey, Proton Relay in Iron Porphyrins for Hydrogen Evolution Reaction, Inorg. Chem., 2021, 60, 13876 CrossRef CAS PubMed;
(m) S. Bhunia, A. Ghatak, A. Rana and A. Dey, Amine Groups in the Second Sphere of Iron Porphyrins Allow for Higher and Selective 4e−/4H+ Oxygen Reduction Rates at Lower Overpotentials, J. Am. Chem. Soc., 2023, 145, 3812 CrossRef CAS PubMed;
(n) J. S. Derrick, M. Loipersberger, S. K. Nistanaki, A. V. Rothweiler, M. Head-Gordon, E. M. Nichols and C. J. Chang, Templating Bicarbonate in the Second Coordination Sphere Enhances Electrochemical CO2 Reduction Catalyzed by Iron Porphyrins, J. Am. Chem. Soc., 2022, 144, 11656 CrossRef CAS PubMed. - (a) C. F. Gorin, E. S. Beh and M. W. Kanan, An Electric Field–Induced Change in the Selectivity of a Metal Oxide–Catalyzed Epoxide Rearrangement, J. Am. Chem. Soc., 2012, 134, 186 CrossRef CAS PubMed; (b) C. F. Gorin, E. S. Beh, Q. M. Bui, G. R. Dick and M. W. Kanan, Interfacial Electric Field Effects on a Carbene Reaction Catalyzed by Rh Porphyrins, J. Am. Chem. Soc., 2013, 135, 11257 CrossRef CAS PubMed; (c) I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, Through-Space Charge Interaction Substituent Effects in Molecular Catalysis Leading to the Design of the Most Efficient Catalyst of CO2-to-CO Electrochemical Conversion, J. Am. Chem. Soc., 2016, 138, 16639 CrossRef CAS PubMed; (d) S. Shaik, D. Mandal and R. Ramanan, Oriented Electric Fields as Future Smart Reagents in Chemistry, Nat. Chem., 2016, 8, 1091 CrossRef CAS PubMed; (e) V. M. Lau, W. C. Pfalzgraff, T. E. Markland and M. W. Kanan, Electrostatic Control of Regioselectivity in Au(I)-Catalyzed Hydroarylation, J. Am. Chem. Soc., 2017, 139, 4035 CrossRef CAS PubMed; (f) T. Chantarojsiri, J. W. Ziller and J. Y. Yang, Incorporation of Redox-Inactive Cations Promotes Iron Catalyzed Aerobic C–H Oxidation at Mild Potentials, Chem. Sci., 2018, 9, 2567 RSC; (g) S. Ciampi, N. Darwish, H. M. Aitken, I. Díez-Pérez and M. L. Coote, Harnessing Electrostatic Catalysis in Single Molecule, Electrochemical and Chemical Systems: A Rapidly Growing Experimental Tool Box, Chem. Soc. Rev., 2018, 47, 5146 RSC; (h) C. G. Margarit, N. G. Asimow, M. I. Gonzalez and D. G. Nocera, Double Hangman Iron Porphyrin and the Effect of Electrostatic Nonbonding Interactions on Carbon Dioxide Reduction, J. Phys. Chem. Lett., 2020, 11, 1890 CrossRef CAS PubMed; (i) D. J. Martin and J. M. Mayer, Oriented Electrostatic Effects on O2 and CO2 Reduction by a Polycationic Iron Porphyrin, J. Am. Chem. Soc., 2021, 143, 11423 CrossRef CAS PubMed; (j) N. G. Léonard, R. Dhaoui, T. Chantarojsiri and J. Y. Yang, Electric Fields in Catalysis: From Enzymes to Molecular Catalysts, ACS Catal., 2021, 11, 10923 CrossRef PubMed; (k) A. B. Weberg, S. P. McCollom, L. M. Thierer, M. R. Gau, P. J. Carroll and N. C. Tomson, Using Internal Electrostatic Fields to Manipulate the Valence Manifolds of Copper Complexes, Chem. Sci., 2021, 12, 4395 RSC; (l) A. B. Weberg, R. P. Murphy and N. C. Tomson, Oriented Internal Electrostatic Fields: an Emerging Design Element in Coordination Chemistry and Catalysis, Chem. Sci., 2022, 13, 5432 RSC.
- (a) M. Delgado, J. M. Ziegler, T. Seda, L. N. Zakharov and J. D. Gilbertson, Pyridinediimine Iron Complexes with Pendant Redox-Inactive Metals Located in the Secondary Coordination Sphere, Inorg. Chem., 2016, 55, 555 CrossRef CAS PubMed; (b) A. H. Reath, J. W. Ziller, C. Tsay, A. J. Ryan and J. Y. Yang, Redox Potential and Electronic Structure Effects of Proximal Nonredox Active Cations in Cobalt Schiff Base Complexes, Inorg. Chem., 2017, 56, 3713 CrossRef CAS PubMed; (c) K. Kang, J. Fuller, A. H. Reath, J. W. Ziller, A. N. Alexandrova and J. Y. Yang, Installation of Internal Electric Fields by Non-Redox Active Cations in Transition Metal Complexes, Chem. Sci., 2019, 10, 10135 RSC; (d) A. Kumar, D. Lionetti, V. W. Day and J. D. Blakemore, Redox-Inactive Metal Cations Modulate the Reduction Potential of the Uranyl Ion in Macrocyclic Complexes, J. Am. Chem. Soc., 2020, 142, 3032 CrossRef CAS PubMed; (e) N. S. Idris, J. M. Barlow, S. A. Chabolla, J. W. Ziller and J. Y. Yang, Synthesis and Redox Properties of Heterobimetallic Re(bpyCrown-M)(CO)3Cl Complexes, where M = Na+, K+, Ca2+, and Ba2+, Polyhedron, 2021, 208, 115385 CrossRef CAS; (f) N. G. Léonard, T. Chantarojsiri, J. W. Ziller and J. Y. Yang, Cationic Effects on the Net Hydrogen Atom Bond Dissociation Free Energy of High-Valent Manganese Imido Complexes, J. Am. Chem. Soc., 2022, 144, 1503 CrossRef PubMed; (g) R. R. Golwankar, A. Kumar, V. W. Day and J. D. Blakemore, Revealing the Influence of Diverse Secondary Metal Cations on Redox-Active Palladium Complexes, Chem.–Eur. J., 2022, 28, e202200344 CrossRef CAS PubMed; (h) H. M. Nguyen, H. W. T. Morgan, T. Chantarojsiri, T. A. Kerr, J. Y. Yang, A. N. Alexandrova and N. G. Léonard, Charge and Solvent Effects on the Redox Behavior of Vanadyl Salen–Crown Complexes, J. Phys. Chem. A, 2023, 127, 5324 CrossRef CAS PubMed.
- (a) J. W. Steed, First-and Second-Sphere Coordination Chemistry of Alkali Metal Crown Ether Complexes, Coord. Chem. Rev., 2001, 215, 171 CrossRef CAS; (b) G. W. Gokel, W. M. Leevy and M. E. Weber, Crown Ethers: Sensors for Ions and Molecular Scaffolds for Materials and Biological Models, Chem. Rev., 2004, 104, 2723 CrossRef CAS PubMed; (c) X. Chen, F. Wang, J. Y. Hyun, T. Wei, J. Qiang, X. Ren, I. Shin and J. Yoon, Recent Progress in the Development of Fluorescent, Luminescent and Colorimetric Probes or Detection of Reactive Oxygen and Nitrogen Species, Chem. Soc. Rev., 2016, 45, 2976 RSC.
- S. Acosta-Calle and A. J. M. Miller, Tunable and Switchable Catalysis Enabled by Cation-Controlled Gating with Crown Ether Ligands, Acc. Chem. Res., 2023, 56, 971 CrossRef CAS PubMed.
- (a) G. F. Manbeck and E. Fujita, A Review of Iron and Cobalt Porphyrins, Phthalocyanines and Related Complexes for Electrochemical and Photochemical Reduction of Carbon Dioxide, J. Porphyrins Phthalocyanines, 2015, 19, 45 CrossRef CAS; (b) P. Gotico, Z. Halime and A. Aukauloo, Recent Advances in Metalloporphyrin-Based Catalyst Design Towards Carbon Dioxide Reduction: From Bio-Inspired Second Coordination Sphere Modifications to Hierarchical Architectures, Dalton Trans., 2020, 49, 2381 RSC; (c) Z. Liang, H.-Y. Wang, H. Zheng, W. Zhang and R. Cao, Porphyrin-Based Frameworks for Oxygen Electrocatalysis and Catalytic Reduction of Carbon Dioxide, Chem. Soc. Rev., 2021, 50, 2540 RSC.
- (a) W. Zhang, W. Lai and R. Cao, Energy-Related Small Molecule Activation Reactions: Oxygen Reduction and Hydrogen and Oxygen Evolution Reactions Catalyzed by Porphyrin-and Corrole-Based Systems, Chem. Rev., 2017, 117, 3717 CrossRef CAS PubMed; (b) S. Bhunia, A. Ghatak and A. Dey, Second Sphere Effects on Oxygen Reduction and Peroxide Activation by Mononuclear Iron Porphyrins and Related Systems, Chem. Rev., 2022, 122, 12370 CrossRef CAS PubMed.
- B. B. Beyene and C.-H. Hung, Recent Progress on Metalloporphyrin-Based Hydrogen Evolution Catalysis, Coord. Chem. Rev., 2020, 410, 213234 CrossRef CAS.
- Y. Zhao, M. Yu and X. Fu, Photo-Cleavage of the Cobalt–Carbon Bond: Visible Light-Induced Living Radical Polymerization Mediated by Organo-Cobalt Porphyrins, Chem. Commun., 2013, 49, 5186 RSC.
- (a) H. Lu and X. P. Zhang, Catalytic C–H Functionalization by Metalloporphyrins: Recent Developments and Future Directions, Chem. Soc. Rev., 2011, 40, 1899 RSC; (b) C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Selective Functionalisation of Saturated C–H Bonds with Metalloporphyrin Catalysts, Chem. Soc. Rev., 2011, 40, 1950 RSC.
- (a) E. Rose, B. Andrioletti, S. Zrig and M. Quelquejeu-Ethève, Enantioselective Epoxidation of Olefins with Chiral Metalloporphyrin Catalysts, Chem. Soc. Rev., 2005, 34, 573 RSC; (b) Q.-H. Xia, H.-Q. Ge, C.-P. Ye, Z.-M. Liu and K.-X. Su, Advances in Homogeneous and Heterogeneous Catalytic Asymmetric Epoxidation, Chem. Rev., 2005, 105, 1603 CrossRef CAS PubMed.
- (a) B. Meunier, Metalloporphyrins as Versatile Catalysts for Oxidation Reactions and Oxidative DNA Cleavage, Chem. Rev., 1992, 92, 1411 CrossRef CAS; (b) M. Pagliaro, S. Campestrini and R. Ciriminna, Ru-Based Oxidation Catalysis, Chem. Soc. Rev., 2005, 34, 837 RSC.
- (a) V. Thanabal and V. Krishnan, Porphyrins with Multiple Crown Ether Voids: Novel Systems for Cation Complexation Studies, J. Am. Chem. Soc., 1982, 104, 3643 CrossRef CAS; (b) H. Van Willigen and T. Chandrashekar, ENDOR Study of Copper(II) Crown Porphyrin Dimerization, J. Am. Chem. Soc., 1986, 108, 709 CrossRef CAS; (c) H. Shinmori and A. Osuka, Extended Molecular Assembly of Crown Ether Appended Meso–Meso Coupled Diporphyrin, Tetrahedron Lett., 2000, 41, 8527 CrossRef CAS; (d) H. Shinmori, H. Furuta and A. Osuka, Effective Face-to-Face Dimerization of a Crown Ether Appended N-Confused Porphyrin, Tetrahedron Lett., 2002, 43, 4881 CrossRef CAS; (e) M. Jahan, N. Safari, H. Khosravi, A. Moghimi and B. Notash, Crown Ether-Appended Porphyrins and Metalloporphyrins: Synthesis, Characterization and Metal Ions Interaction, Polyhedron, 2005, 24, 1682 CrossRef CAS; (f) P. Even and B. Boitrel, Crown Porphyrins, Coord. Chem. Rev., 2006, 250, 519 CrossRef CAS; (g) E. A. Mikhalitsyna, V. S. Tyurin, I. A. Zamylatskov, V. N. Khrustalev and I. P. Beletskaya, Synthesis, Characterization and Cation-Induced Dimerization of New Aza-Crown Ether-Appended Metalloporphyrins, Dalton Trans., 2012, 41, 7624 RSC.
- X. Zhang, F. Lin, M. Cao and M. Zhong, Rare Earth–Cobalt Bimetallic Catalysis Mediates Stereocontrolled Living Radical Polymerization of Acrylamides, Nat. Synth., 2023, 2, 855 CrossRef.
- (a) P. Kuś, Tetraphenylporphyrins Monosubstituted with a Crown Ether in One Phenyl Ring. Synthesis and Characterization, Monatsh. Chem., 1997, 128, 911 CrossRef; (b) H. Tsukube, M. Wada, S. Shinoda and H. Tamiaki, Porphyrinatoerbium–Crown Ether Conjugate for Synergistic Binding and Chirality Sensing of Zwitterionic Amino Acids, Chem. Commun., 1999, 1007 RSC; (c) S. Iwata, M. Suzuki, M. Shirakawa and K. Tanaka, Cation and Anion Recognition of Crown Ether-armed Metalloporphyrin, Supramol. Chem., 1999, 11, 135 CrossRef CAS; (d) S. J. Lange, J. W. Sibert, A. G. M. Barrett and B. M. Hoffman, Synthesis and Coordination Chemistry of Unsymmetrical Tetraazaporphyrins Containing Single Oxathia- and Thiacrown Substituents, Tetrahedron, 2000, 56, 7371 CrossRef CAS; (e) Y. Chen and K. Wang, Azacrown[N,S,O]-Modified Porphyrin Sensor for Detection of Ag+, Pb2+, and Cu2+, Photochem. Photobiol. Sci., 2013, 12, 2001 CrossRef CAS PubMed.
- (a) E. B. Fleischer, J. M. Palmer, T. S. Srivastava and A. Chatterjee, Thermodyamic and Kinetic Properties of an Iron-Porphyrin System, J. Am. Chem. Soc., 1971, 93, 3162 CrossRef CAS PubMed; (b) T. Nakazono, A. R. Parent and K. Sakai, Cobalt Porphyrins as Homogeneous Catalysts for Water Oxidation, Chem. Commun., 2013, 49, 6325 RSC.
- F. A. Tezcan, J. R. Winkler and H. B. Gray, Probing Protein Folding with Substitution-Inert Metal Ions. Folding Kinetics of Cobalt (III)-Cytochrome c, J. Am. Chem. Soc., 1999, 121, 11918 CrossRef CAS.
- R. Giovannetti, in Macro to Nano Spectroscopy, ed. J. Uddin, IntechOpen, 2012 Search PubMed.
- J. G. Kleingardner, B. Kandemir and K. L. Bren, Hydrogen Evolution From Neutral Water Under Aerobic Conditions Catalyzed by Cobalt Microperoxidase-11, J. Am. Chem. Soc., 2014, 136, 4 CrossRef CAS PubMed.
- G. Däges and J. Hüttermann, ESR and ENDOR of Pentacoordinated Cobalt(II) Porphyrins, J. Phys. Chem., 1992, 96, 4787 CrossRef.
- (a) M. Isaacs, J. C. Canales, A. Riquelme, M. Lucero, M. J. Aguirre and J. Costamagna, Contribution of the Ligand to the Electroreduction of CO2 Catalyzed by a Cobalt(II) Macrocyclic Complex, J. Coord. Chem., 2003, 56, 1193 CrossRef CAS; (b) V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Splitting Water with Cobalt, Angew. Chem., Int. Ed., 2011, 50, 7238 CrossRef CAS PubMed; (c) B. H. Solis, A. G. Maher, T. Honda, D. C. Powers, D. G. Nocera and S. Hammes-Schiffer, Theoretical Analysis of Cobalt Hangman Porphyrins: Ligand Dearomatization and Mechanistic Implications for Hydrogen Evolution, ACS Catal., 2014, 4, 4516 CrossRef CAS.
- (a) E. Karkhaneei, A. Afkhami and M. Shamsipur, Nuclear Magnetic Resonance Study of Lithium Ion Complexes with Several Crown Ethers in Binary Acetonitrile-Nitromethane Mixtures, J. Coord. Chem., 1996, 39, 33 CrossRef CAS; (b) M. Shamsipur and T. Madrakian, Competitive NMR Study of the Complexation of Some Alkaline Earth and Transition Metal Ions with 12-crown-4, 15-crown-5 and benzo-15-crown-5 in Acetonitrile Solution Using the Lithium-7 Nucleus as a Probe, J. Coord. Chem., 1999, 52, 139 CrossRef.
- (a) P. Thordarson, Determining Association Constants from Titration Experiments in Supramolecular Chemistry, Chem. Soc. Rev., 2010, 40, 1305 RSC; (b) A. Kumar and J. D. Blakemore, On the Use of Aqueous Metal-Aqua pKa Values as a Descriptor of Lewis Acidity, Inorg. Chem., 2021, 60, 1107 CrossRef CAS PubMed; (c) R. R. Golwankar, T. Davis Curry II, C. J. Paranjothi and J. D. Blakemore, Molecular Influences on the Quantification of Lewis Acidity with Phosphine Oxide Probes, Inorg. Chem., 2023, 62, 9765 CrossRef CAS PubMed.
- A. J. Smetana and A. I. Popov, Lithium-7 Nuclear Magnetic Resonance and Calorimetric Study of Lithium Crown Complexes in Various Solvents, J. Solution Chem., 1980, 9, 183 CrossRef CAS.
- J. B. Smith, A. M. Camp, A. H. Farquhar, S. H. Kerr, C.-H. Chen and A. J. M. Miller, Organometallic Elaboration as a Strategy for Tuning the Supramolecular Characteristics of Aza-Crown Ethers, Organometallics, 2019, 38, 4392 CrossRef CAS.
- (a) M. Hammouche, D. Lexa, M. Momenteau and J.-M. Savéant, Chemical Catalysis of Electrochemical Reactions. Homogeneous Catalysis of the Electrochemical Reduction of Carbon Dioxide by lron(“0”) Porphyrins. Role of the Addition of Magnesium Cations, J. Am. Chem. Soc., 1991, 113, 8455 CrossRef CAS; (b) M. D. Sampson and C. P. Kubiak, Manganese Electrocatalysts with Bulky Bipyridine Ligands: Utilizing Lewis Acids To Promote Carbon Dioxide Reduction at Low Overpotentials, J. Am. Chem. Soc., 2016, 138, 1386 CrossRef CAS PubMed.
- A. Zhanaidarova, H. Steger, M. H. Reineke and C. P. Kubiak, Chelated [Zn(cyclam)]2+ Lewis Acid Improves the Reactivity of the Electrochemical Reduction of CO2 by Mn Catalysts with Bulky Bipyridine Ligands, Dalton Trans., 2017, 46, 12413 RSC.
- K. J. Lee, B. D. McCarthy and J. L. Dempsey, On Decomposition, Degradation, and Voltammetric Deviation: The Electrochemist's Field Guide to Identifying Precatalyst Transformation, Chem. Soc. Rev., 2019, 48, 2927 RSC.
- J.-M. Savéant, Elements of Molecular and Biomolecular Electrochemistry, An Electrochemical Approach to Electron Transfer Chemistry, John Wiley & Sons, 2006 Search PubMed.
- (a) E. Y. Tsui and T. Agapie, Reduction Potentials of Heterometallic Manganese–Oxido Cubane Complexes Modulated by Redox-Inactive Metals, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 10084 CrossRef CAS PubMed; (b) E. Y. Tsui, R. Tran, J. Yano and T. Agapie, Redox-Inactive Metals Modulate the Reduction Potential in Heterometallic Manganese–Oxido Clusters, Nat. Chem., 2013, 5, 293 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Automated Exploration of the Low-Energy Chemical Space with Fast Quantum Chemical Methods, Phys. Chem. Chem. Phys., 2020, 22, 7169 RSC.
- C. Bannwarth, S. Ehlert and S. Grimme, GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions, J. Chem. Theory Comput., 2019, 15, 1652 CrossRef CAS PubMed.
- (a) A. D. Becke, Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS PubMed; (b) J. P. Perdew, Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef PubMed.
- B. H. Solis and S. Hammes-Schiffer, Proton-Coupled Electron Transfer in Molecular Electrocatalysis: Theoretical Methods and Design Principles, Inorg. Chem., 2014, 53, 6427 CrossRef CAS PubMed.
- R. D. Shannon, Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- (a) S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. Zho, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Covalent Organic Frameworks Comprising Cobalt Porphyrins for Catalytic CO2 Reduction in Water, Science, 2015, 349, 1208 CrossRef CAS PubMed; (b) X.-M. Hu, M. H. Rønne, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Enhanced Catalytic Activity of Cobalt Porphyrin in CO2 Electroreduction upon Immobilization on Carbon Materials, Angew. Chem., Int. Ed., 2017, 56, 6468 CrossRef CAS PubMed; (c) A. Call, M. Cibian, K. Yamamoto, T. Nakazono, K. Yamauchi and K. Sakai, Highly Efficient and Selective Photocatalytic CO2 Reduction to CO in Water by a Cobalt Porphyrin Molecular Catalyst, ACS Catal., 2019, 9, 4867 CrossRef CAS; (d) X. Zhang, M. Cibian, A. Call, K. Yamauchi and K. Sakai, Photochemical CO2 Reduction Driven by Water-Soluble Copper(I) Photosensitizer with the Catalysis Accelerated by Multi-Electron Chargeable Cobalt Porphyrin, ACS Catal., 2019, 9, 11263 CrossRef CAS; (e) J. L. Alvarez-Hernandez, A. A. Salamatian, J. H. Han and K. L. Bren, Potential- and Buffer-Dependent Selectivity for the Conversion of CO2 to CO by a Cobalt Porphyrin-Peptide Electrocatalyst in Water, ACS Catal., 2022, 12, 14689 CrossRef CAS PubMed.
- (a) J.-M. Savéant and K. B. Su, Homogeneous Redox Catalysis of Electrochemical Reaction. Part VI, Zone Diagram Representation of the Kinetic Regimes, J. Electroanal. Chem., 1984, 171, 341 CrossRef; (b) C. Costentin and J.-M. M. Savéant, Multistep Molecular Catalysis of Electrochemical Reactions: Benchmarking of Homogeneous Catalysts, ChemElectroChem, 2014, 1, 1226 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional information about selected experiments, characterizing data, NMR spectra, cyclic voltammograms, Job plots, and computational details. See DOI: https://doi.org/10.1039/d3sc06177a |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |