
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-throughput drug target discovery using a fully automated proteomics sample preparation platform†
Qiong
Wu‡
 a,
Jiangnan
Zheng‡
ac,
Xintong
Sui‡
a,
Changying
Fu‡
a,
Xiaozhen
Cui
a,
Bin
Liao
a,
Hongchao
Ji
a,
Yang
Luo
a,
An
He
a,
Xue
Lu
a,
Xinyue
Xue
a,
Chris Soon Heng
Tan
*abc and
Ruijun
Tian
*abc
a,
Jiangnan
Zheng‡
ac,
Xintong
Sui‡
a,
Changying
Fu‡
a,
Xiaozhen
Cui
a,
Bin
Liao
a,
Hongchao
Ji
a,
Yang
Luo
a,
An
He
a,
Xue
Lu
a,
Xinyue
Xue
a,
Chris Soon Heng
Tan
*abc and
Ruijun
Tian
*abc
aDepartment of Chemistry, School of Science, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: tianrj@sustech.edu.cn
bResearch Center for Chemical Biology and Omics Analysis, School of Science, Southern University of Science and Technology, 1088 Xueyuan Road, Shenzhen 518055, China. E-mail: chris.tan.sh@gmail.com
cSouthern University of Science and Technology, Guangming Advanced Research Institute, Shenzhen 518055, China
First published on 12th January 2024
Abstract
Drug development is plagued by inefficiency and high costs due to issues such as inadequate drug efficacy and unexpected toxicity. Mass spectrometry (MS)-based proteomics, particularly isobaric quantitative proteomics, offers a solution to unveil resistance mechanisms and unforeseen side effects related to off-targeting pathways. Thermal proteome profiling (TPP) has gained popularity for drug target identification at the proteome scale. However, it involves experiments with multiple temperature points, resulting in numerous samples and considerable variability in large-scale TPP analysis. We propose a high-throughput drug target discovery workflow that integrates single-temperature TPP, a fully automated proteomics sample preparation platform (autoSISPROT), and data independent acquisition (DIA) quantification. The autoSISPROT platform enables the simultaneous processing of 96 samples in less than 2.5 hours, achieving protein digestion, desalting, and optional TMT labeling (requires an additional 1 hour) with 96-channel all-in-tip operations. The results demonstrated excellent sample preparation performance with >94% digestion efficiency, >98% TMT labeling efficiency, and >0.9 intra- and inter-batch Pearson correlation coefficients. By automatically processing 87 samples, we identified both known targets and potential off-targets of 20 kinase inhibitors, affording over a 10-fold improvement in throughput compared to classical TPP. This fully automated workflow offers a high-throughput solution for proteomics sample preparation and drug target/off-target identification.
Introduction
Drug development is a highly inefficient and costly process, which is largely attributed to the inadequate drug efficacy and unexpected toxicity found in the late stages of drug development.1 Considering that current drugs mainly target proteins, proteomics investigations hold promise for revealing potential resistance mechanisms and unexpected side effects resulting from off-targeting pathways.2 Mass spectrometry (MS)-based proteomics, especially isobaric quantitative proteomics, has been widely applied with significant success in the chemical biology field for comprehensive profiling of drug targets at the proteome scale.3–5 For the study of complex biological systems, a large number of samples with multiple conditions and replicates are required to achieve enough statistical power. However, sample preparation for large-scale quantitative proteomic analysis remains a challenge.6 As proteomics workflows typically involve multi-step sample preparation, manually processing hundreds of samples is not only time-consuming but could also introduce variations that affect the overall technical reproducibility. Consequently, automation of sample preparation is increasingly attractive as a solution for enhancing reproducibility through the standardization of sample preparation that reduces both time and costs.The cellular thermal shift assay (CETSA) coupled to MS, also known as thermal proteome profiling (TPP), has emerged as a popular method for identifying drug targets and off-targets based on ligand-induced changes in protein thermal stability.3,7–12 Classical TPP typically involves experiments with ten temperature points, each with two replicates per condition, to estimate the shift in thermal melting temperature (Tm). This results in 40 samples that need to be prepared and labeled with tandem mass tags (TMTs), followed by off-line fractionation steps. To reduce the number of samples and improve analysis throughput, new formats of thermal shift assays, such as proteome integral solubility alteration (PISA),13 isothermal shift assay (iTSA),14 and matrix thermal shift assay (mTSA),15 have been developed. However, the reduction of temperature points could decrease the sensitivity of thermal shift assay.16,17 Like TPP, both PISA and iTSA assays rely on TMT quantification, which necessitates offline fractionation steps and a substantial amount of expensive TMT reagents.18 Recently, label-free data independent acquisition (DIA) quantification was employed in iTSA to further increase throughput.15 Overall, for large-scale drug target identification using TPP, there is an urgent need for an automated and high-throughput sample preparation method.19
Over the past decade, various research groups have harnessed liquid handling systems to develop automated and high-throughput methods for proteomics sample preparation. One crucial step to generate MS-friendly samples involves the removal of detergents that are required to fully lyse cell/tissue samples. However, many approaches, such as the in-StageTip (iST) method, lack the ability to remove detergents and are primarily used for analyzing body fluids.20,21 To address the challenge of handling lysates containing detergents, solvent-induced protein precipitation has been employed for detergent cleanup in sample preparation workflows.22,23 Nonetheless, most of these methods require offline centrifugation or other manual interventions,20,22 and typically involve extended digestion times (often overnight) and multiple sample transfer steps.24 Furthermore, they do not seamlessly integrate TMT labeling. In contrast, building upon the simple and integrated spintip-based proteomics technology (SISPROT),25,26 we have achieved fully automated processing of cell lysates into fractionated peptides within 2–3 hours.27 Despite these advances, a fully automated sample preparation workflow for high-throughput quantitative proteomics is still lacking.
Here, we propose a high-throughput drug target discovery workflow that integrates single-temperature TPP (a.k.a. iTSA), a fully automated proteomics sample preparation platform (autoSISPROT), and DIA quantification (Fig. 1A). The autoSISPROT workflow was developed by combining the all-in-tip sample preparation capabilities of SISPROT with the programmable liquid handling of Agilent AssayMAP Bravo,28 enabling the simultaneous processing of 96 samples in 2.5 hours in a fully automated manner. We thoroughly assessed the performance of autoSISPROT, including both intra- and inter-batch reproducibility, by processing a total of three 96-well plates on three different days. Additionally, by combining with TPP, autoSISPROT can automatically process and TMT-label 40 samples and accurately identify the known target of the well-characterized model drug, methotrexate (MTX). Furthermore, we conducted a comprehensive assessment of two quantitative proteomic methods, namely TMT and DIA, for drug target identification by utilizing a pan-kinase inhibitor, staurosporine. Finally, to enhance the analysis throughput of TPP, we combined autoSISPROT with DIA-based TPP to identify the known targets and potential off-targets of a panel of 20 kinase inhibitors in a fully automated manner.
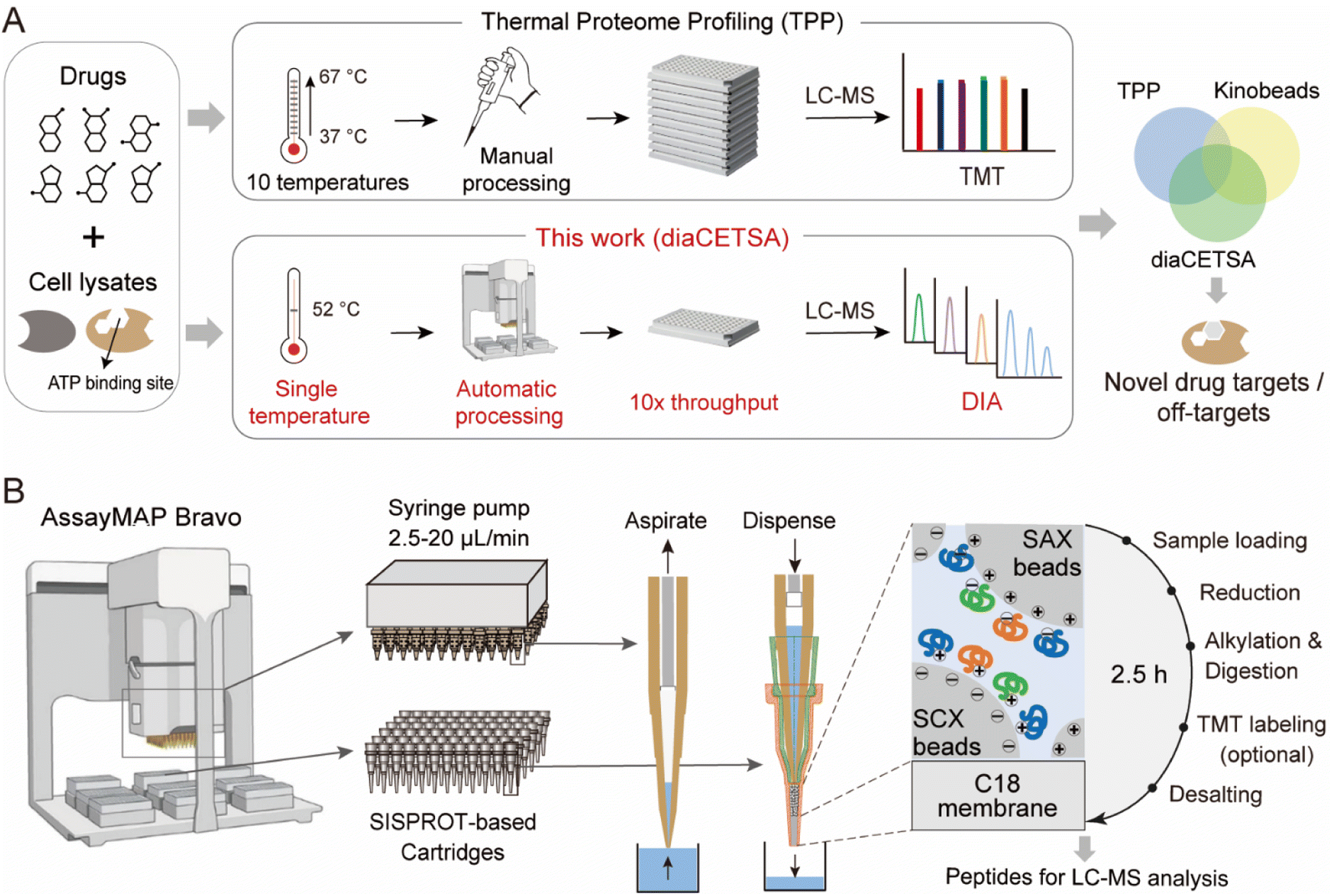 | ||
| Fig. 1 A fully automated and integrated 96-channel proteomics sample preparation platform (autoSISPROT) for high-throughput drug target discovery. (A) The workflows of high-throughput drug targets and off-targets identification by classical thermal proteome profiling (TPP) and our diaCETSA method that combines single temperature CETSA, autoSISPROT and DIA-based protein quantification. (B) The workflow of autoSISPROT. Protein samples in a 96-well plate are processed using the AssayMAP Bravo workstation, which is equipped with 96-well syringes and SISPROT-based cartridges. All the necessary sample preparation steps, including sample loading, protein reduction, alkylation, digestion, TMT labeling, and desalting, are executed by the programed upward aspiration and downward dispensing of the required buffers through the packed cartridges. The autoSISPROT protocol enables the automatic processing of up to 96 protein samples simultaneously, resulting in peptide solutions within 2.5 hours. | ||
Results and discussion
Development of a fully automated 96-channel proteomic sample preparation platform (autoSISPROT)
SISPROT allows for full integration of sample loading, protein reduction, alkylation, digestion, TMT labeling, and desalting, all within a single spintip packed sequentially with a C18 membrane and mixed SCX/SAX beads.25,26,29 We hypothesized that this all-in-tip sample preparation could be automated on AssayMAP Bravo, a microchromatography platform with a 96-channel liquid handling head. This platform can precisely control the flow rates of upward aspiration and downward dispensing within the range of 2.5–20 μL min−1, which allows us to program each sample preparation step on a SISPROT-based cartridge with a desired flow rate and processing time (Fig. 1B). Accordingly, we systematically optimized and significantly reduced the number of buffers used in each step of sample preparation which allows for fully automated operation on the AssayMAP Bravo system with 7 working decks (Fig. S1A†). To ensure an ultra-low dead volume in autoSISPROT, we designed and fabricated the SISPROT-based cartridges that were assembled with the top and bottom tips with specialized shapes (Fig. S1B†). The bottom component was packed with a C18 membrane and mixed SCX/SAX beads in tandem. In the cartridge, proteins are initially trapped on the mixed SCX/SAX beads, followed by reduction, alkylation, and digestion on the beads. Subsequently, peptide desalting is accomplished using the C18 membrane. In summary, autoSISPROT enables the automated processing of up to 96 samples in parallel within 2.5 hours, eliminating the need for manual intervention once the start button is clicked.Comparison of TMT and DIA based quantification for TPP
Current implementations of TPP typically rely on TMT quantification, and the analysis throughput is limited by the number of TMT channels. In comparison, label free quantification (LFQ) with DIA is unlimited in sample numbers and suitable for large-scale drug target identification. Although the use of DIA in TPP analysis has been documented at the time of this writing,30,31 no studies have systematically evaluated the performance of TMT and DIA methods for single temperature TPP analysis. Here, we conducted a comparison between the TMT and DIA methods, referred to as tmtCETSA and diaCETSA, respectively. We used staurosporine, a pan-kinase inhibitor, as the model drug since its targets have been extensively studied.3,14,30 K562 cell lysates were treated with either staurosporine or a vehicle control and subjected to thermal treatment at 52 °C. Afterwards, equal aliquots of the soluble lysates were individually processed using the diaCETSA and tmtCETSA methods (Fig. 2A). To ensure a fair comparison, the number of samples, MS instrument, and MS acquisition time were kept the same for both methods. In comparison to classical TPP, tmtCETSA and diaCETSA exhibited a 4-fold improvement in throughput by reducing the number of samples from 40 to 10. Additionally, diaCETSA was able to reduce time and costs, particularly for TMT labeling and peptide fractionation, compared to tmtCETSA. Within our study, tmtCETSA resulted in 20–30% more proteins compared to diaCETSA, primarily due to the benefit of fractionation (Fig. 2B), which was consistent with previous work.32 Both methods achieved a high Pearson correlation coefficient (>0.98) and low median coefficient of variation (CV; <10%) at the protein level (Fig. 2C and S2A†), demonstrating good quantitative reproducibility and precision for both methods. Furthermore, 120 and 106 proteins were identified as staurosporine targets using tmtCETSA and diaCETSA, respectively (Fig. 2D). The diaCETSA method identified slightly fewer kinase targets (53 kinases) compared to tmtCETSA (67 kinases) (Fig. 2E and Table S1†), which could be attributed to the lower proteomic depth of diaCETSA. Subsequently, we benchmarked our dataset against classical TPP. Overall, 24 kinase targets were found to be shared among these three methods (Fig. 2E). Interestingly, we found that certain non-kinase proteins were identified as targets by both methods. STRING analysis33 of the significant targets revealed that approximately 40% of non-kinase proteins interact with kinases, suggesting that indirect interactions with staurosporine could be identified by thermal stability analysis (Fig. 2F). For instance, non-kinases PDCD10 and PXN were identified as interactors with several kinases in tmtCETSA. Similarly, non-kinase REHB interacted with different kinases, including non-kinases, in diaCETSA (Fig. 2G). Furthermore, we mapped the kinase targets identified by both methods onto the kinome tree (Fig. S2B†). Enrichment analysis of gene ontology (GO) molecular functions of the significant proteins revealed their involvement in various kinase activities, suggesting that these proteins may play functional roles in different kinase-related processes (Fig. S2C†). Notably, we observed that several kinases, such as PRKCB, PRKCD, PRKCI, and SLK, exhibited destabilization upon staurosporine treatment, as identified by both tmtCETSA and diaCETSA.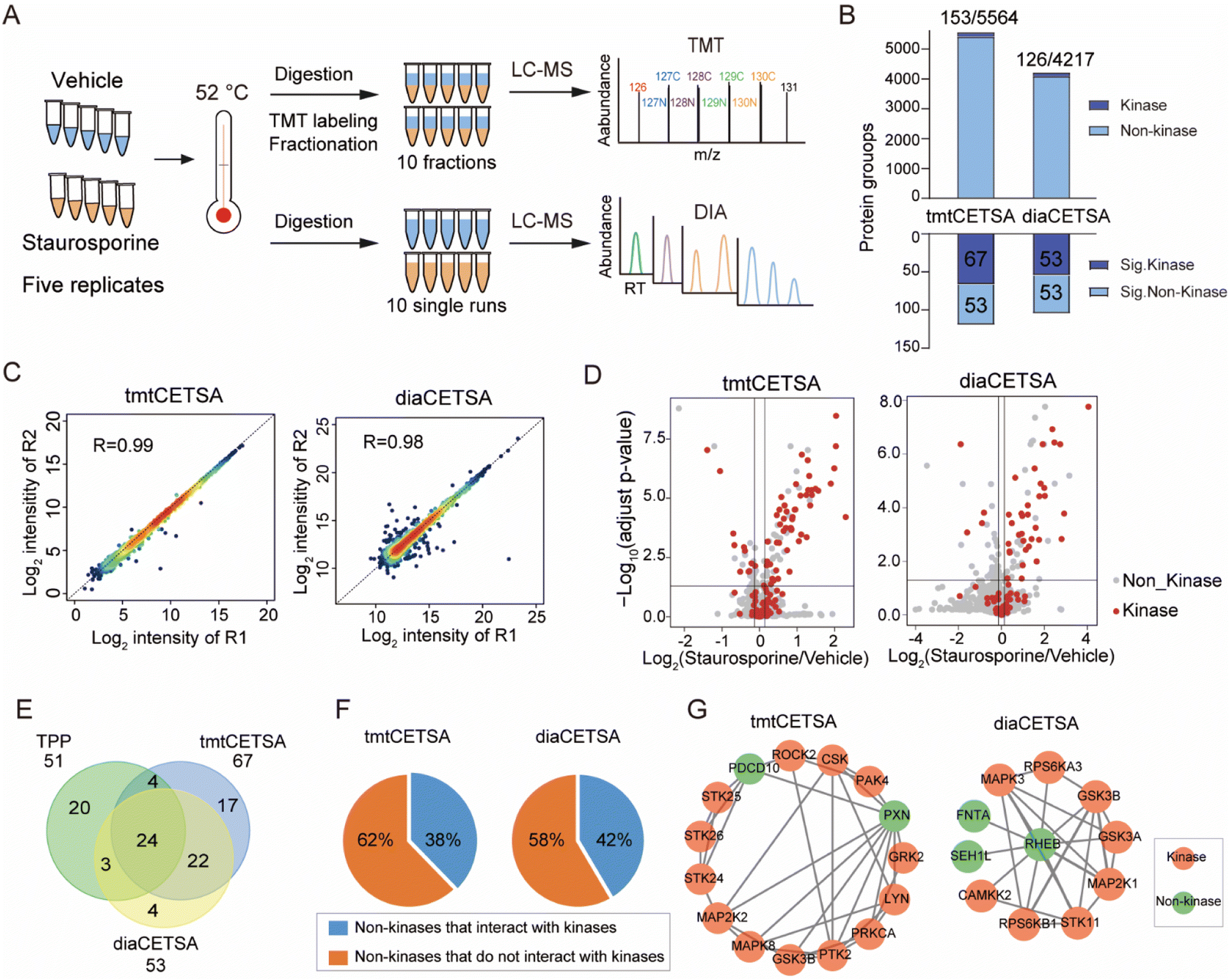 | ||
| Fig. 2 Comparison of the TMT and DIA based CETSA (tmtCETSA and diaCETSA). (A) The workflows of tmtCETSA and diaCETSA. (B) Bar charts showing the number of total proteins, kinases, significant kinases, and significant non-kinases by using tmtCETSA and diaCETSA. (C) Pearson correlation coefficient of protein intensities between two replicates by using tmtCETSA and diaCETSA. (D) Volcano plot visualization of kinase targets from K562 cell lysates, performed at 52 °C using 20 μM staurosporine by using tmtCETSA and diaCETSA. Adjusted p-value = 0.05 is indicated by a solid horizontal line. (E) Venn diagram displaying the kinase targets identified by classical TPP, tmtCETSA and diaCETSA methods. (F) Pie chart displaying the ratio of significant non-kinases that interact with kinases. (G) Interaction map of significant non-kinases PDCD10, PXN (tmtCETSA, right panel) and REHB (diaCETSA, left panel) interacting with kinases, respectively. | ||
We further optimized the diaCETSA method to enhance the depth of proteome analysis. Given the significant impact of the spectral library's quality on DIA data analysis, we initially compared two spectral libraries constructed from K562 cell lysates using different sample preparation approaches. Spectral library 1 was generated using a conventional proteomics workflow, involving protein precipitation and in-solution digestion.34 On the other hand, samples for constructing spectral library 2 underwent heat treatment at 52 °C and were processed using autoSISPROT. Despite Library 1 having a larger capacity (protein number: 9589 versus 6000), more proteins were identified when searching with Library 2 (Fig. S3A†). This demonstrated that the project-specific library does not need to be as extensive as possible but should be built in a manner consistent with the analyzed DIA samples. Additionally, we compared the quantitative performance of DIA mode on two different mass spectrometers, i.e., an Orbitrap Exploris 480 and timsTOF Pro, and found that the Orbitrap Exploris 480 exhibited slightly better protein quantification and identified more drug targets (Fig. S3B–E and Table S2†). These optimized conditions were employed for the subsequent diaCETSA analysis.
Performance of autoSISPROT
We benchmarked the sample preparation performance of the autoSISPROT platform against manual SISPROT for sample preparation of HEK 293T cell lysates in three technical replicates. We found no significant differences in terms of the number of protein groups between autoSISPROT and manual SISPROT, although autoSISPROT identified more peptides than manual SISPROT (Fig. 3A). The median CV for autoSISPROT and manual SISPROT was 5.3% and 7.9%, respectively, indicating that autoSISPROT achieved better quantitative precision (Fig. 3B). The comparison of Pearson correlation coefficients also demonstrated better quantitative reproducibility for autoSISPROT (Fig. 3C), suggesting that automated operation resulted in smaller variations. In autoSISPROT samples, 99.7% of cysteine-containing peptides were alkylated, demonstrating a nearly complete reduction and alkylation rection (Fig. S4A†). Meanwhile, over 94% of the identified peptides had fewer than two missed cleavage sites, indicating high trypsin digestion efficiency (Fig. S4A†). Additionally, the samples processed by autoSISPROT were subjected to DIA analysis, and the results also showed good reproducibility in identification and quantification (Fig. S4B–D†).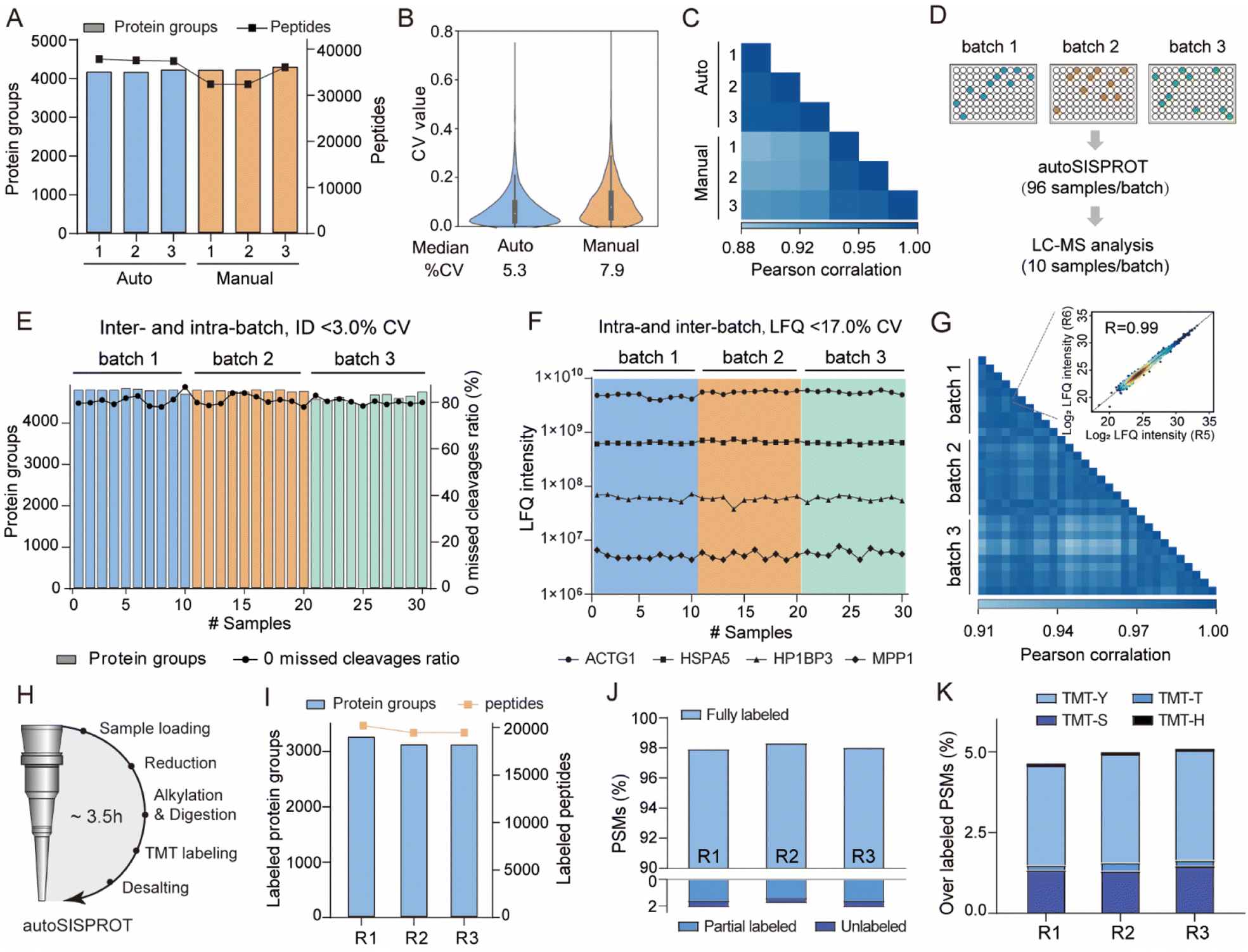 | ||
| Fig. 3 Performance of autoSISPROT. (A) The number of identified protein groups and peptides using autoSISPROT and manual SISPROT under three technical replicates. (B) Violin plots showing the distributions of CVs of protein LFQ intensities between autoSISPROT and manual SISPROT under three technical replicates. (C) Correlation of LFQ intensities of quantified proteins under three technical replicates. (D) Schematic representation of the experimental design. 96-well plates with 10 μg of HEK 293T cell lysates are processed in three batches on three different days. From each batch, ten randomly selected samples are subjected to LC-MS/MS analysis. (E) Protein groups and percentage of PSMs with zero missed cleavages across the three batches. (F) LFQ intensities of four proteins representing the different dynamic ranges are plotted across the three batches. (G) Pearson correlation coefficient of protein LFQ intensities for inter-batch comparison, and the displayed data are filtered for 75% data completeness. The inset graph shows a high correlation (>0.99) between replicate 5 and replicate 6 of batch 1. (H) The workflow for autoSISPROT that integrated TMT labeling. (I) Protein groups and peptide identifications across analytical duplicates. (J) TMT labeling efficiency was evaluated using the proportions of fully labeled, partial, and unlabeled PSMs. (K) Overlabeling efficiency was evaluated using the proportions of serine, threonine, tyrosine and histidine labeled PSMs. | ||
To evaluate the reproducibility of autoSISPROT, we processed three 96-well plates with 10 μg of HEK 293T cell lysates per well in three batches on different days, resulting in a total of 288 individual samples (Fig. 3D). For each batch, we randomly selected ten samples to evaluate intra- and inter-batch reproducibility of autoSISPROT. Across the three batches, we identified an average of 4745 proteins with 80% of them showing zero missed trypsin cleavage sites (digestion was performed at room temperature for 1 hour), and the CVs for both proteins and zero missed trypsin cleavage sites were less than 3% (Fig. 3E and Table S3†). The intensity distributions of quantified peptides were highly consistent (Fig. S4E†), indicating minimal differences in quantification between intra- and inter-batch analyses. The median CVs for batch 1, batch 2, and batch 3 were 10.8, 12.8, and 12.1%, respectively (Fig. S4F†), demonstrating highly consistent protein quantification within each batch. The median CVs of intra-batch were comparable to those obtained by autoSP3, which processed 96-wells containing 10 μg of HeLa cell lysates per well.23 Furthermore, we selected four proteins with different LFQ intensity ranges from 106 to 1010, and the CVs for these proteins were below 17% across the three batches (Fig. 3F). To evaluate inter-batch reproducibility, we calculated Pearson correlation coefficients for proteins with a minimum data completeness of 75% across 30 samples (corresponding to 3403 proteins). Inter-batch comparisons showed highly quantitative reproducibility, with Pearson correlation coefficients exceeding 0.91, indicating no significant differences among the three batches by fully automated sample preparation (Fig. 3G). As expected, higher Pearson correlation coefficients (>0.95) were achieved for intra-batch comparisons. In summary, these results indicate that autoSISPROT exhibits high intra- and inter-batch reproducibility in sample preparation.
Furthermore, we evaluated the TMT labeling performance of autoSISPROT (Fig. 3H). With autoSISPROT, the TMT-labeled protein groups and peptides identified from three technical replicates were highly consistent (Fig. 3I). Using a cost-effective on-column TMT labeling approach,35 autoSISPROT achieved high TMT labeling efficiencies for both peptide N-terminus and lysine residues. Approximately 98% of peptide-spectrum matches (PSMs) were consistently identified as fully labeled peptides, while the percentage of partially labeled and unlabeled PSMs in the three technical replicates was less than 2% (Fig. 3J), which aligns with previous studies.36 Importantly, the fraction of overlabeled PSMs (i.e., off-target TMT labeling on serine, threonine, tyrosine, and histidine) was controlled below 5% (Fig. 3K). These results collectively demonstrate the effective sample preparation performance of autoSISPROT for both label-free and TMT-based quantitative proteomics.
Automatic high-throughput identification of drug targets and off-targets
To evaluate whether autoSISPROT could achieve high-throughput drug target identification, we applied autoSISPROT coupled with diaCETSA to identify targets of 24 drugs including 20 kinase inhibitors (KIs) (Fig. 4A, Tables S4 and S5†). In this study, different drug treatment conditions can share a common vehicle control, which further improves the analysis throughput. These 24 drugs were classified into two groups based on whether they had been previously studied using TPP methods and were processed on different days. Compared to the classical TPP, our method provided a more than 10-fold improvement in throughput by reducing the number of samples from 960 to 87. The sample preparation for classical TPP on this scale would typically take around 48 days, including proteomics digestion, TMT labeling, and off-line fractionation, which usually takes 2 days. In contrast, autoSISPROT processes samples of this scale in less than 2.5 hours, representing a 460-fold improvement in handling time. The number of identified protein groups and enzymatic cleavage efficiency were highly consistent across all sets of samples (Fig. S5A–D†). In addition, median CVs of LFQ intensities were <25% for all 24 drugs (Fig. S5E and F†), demonstrating good processing precision spanning the entire sample preparation procedure from thermal treatment to MS data acquisition. Among the 20 kinase inhibitors, we successfully identified the reported targets for 12 kinase inhibitors (Fig. 4A). The annotated kinome tree shows that these identified kinase targets mostly belong to protein kinase families of GCMC, CAMK, and AGC (Fig. 4B). Furthermore, our method can effectively identify the known targets and yielded complementary results with previously published methods for palbociclib and dinaciclib (Fig. 4C).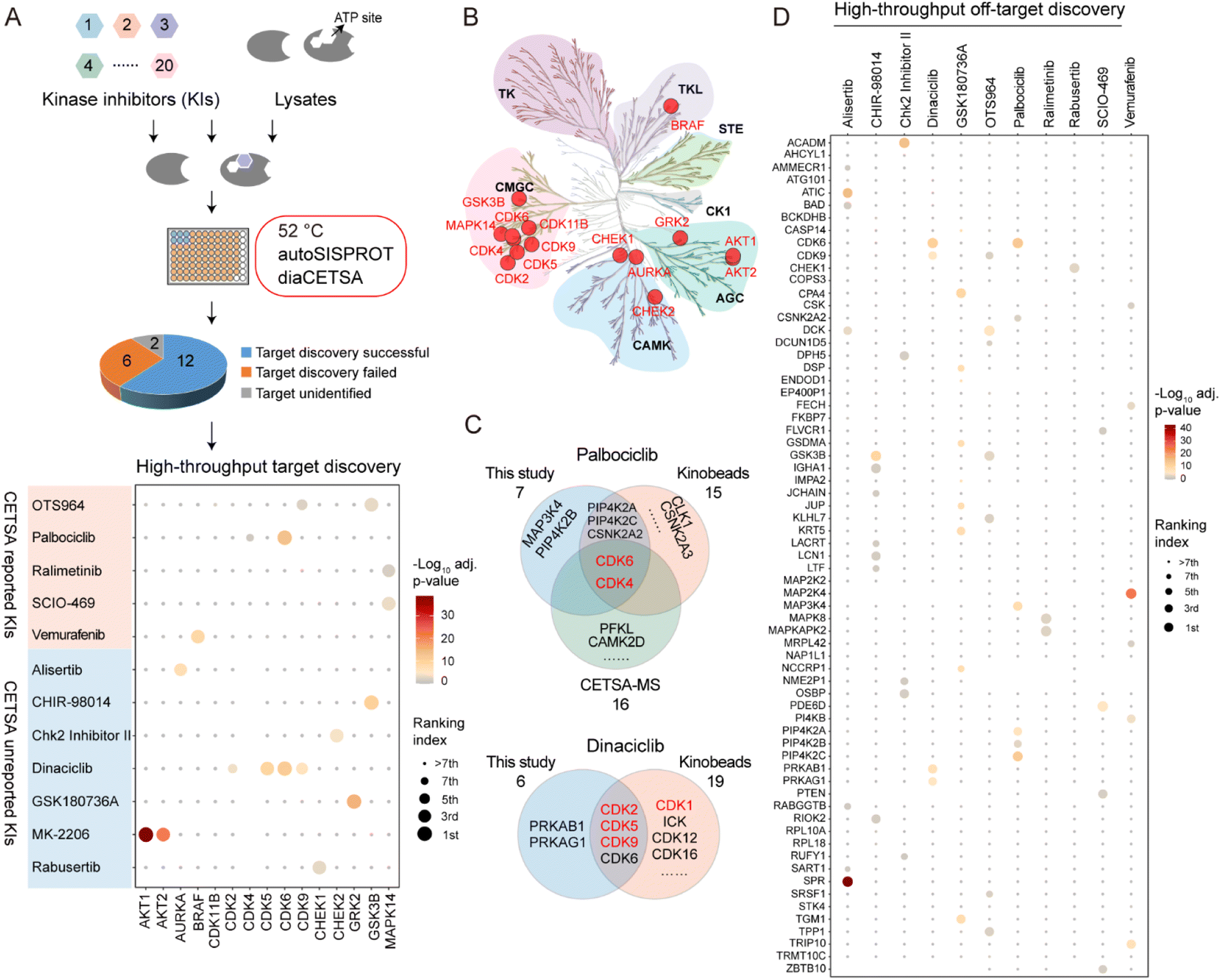 | ||
| Fig. 4 High-throughput drug target identification for kinase inhibitors by combining autoSISPROT and diaCETSA. (A) Workflow for high-throughput identification of targets of kinase inhibitors. Dot plot visualization of target identification of OTS964, palbociclib, ralimetinib, SCIO-469, vemurafenib, alisertib, CHIR-98014, Chk2 Inhibitor II, dinaciclib, GSK180736A, MK-2206, and rabusertib. (B) Kinome tree displaying all identified kinase targets. (C) Venn diagram displaying the common and complementary targets of palbociclib and dinaciclib identified by kinobeads, classical TPP, and our method. The proteins marked in red are the known targets. (D) High-throughput off-target discovery by our method. All identified off-targets of alisertib, CHIR-98014, Chk2 Inhibitor II, dinaciclib, palbociclib, ralimetinib, rabusertib, SCIO-469, and vemurafenib, as well as the top 10 significant off-targets of GSK180736A and OTS964, are shown. | ||
We successfully identified the reported targets for all of the five CETSA-studied KIs, namely OTS964, palbociclib, ralimetinib, SCIO-469, and vemurafenib and seven CETSA-unstudied KIs (Fig. 4A and S6A–L†). Notably, all of the identified kinase targets ranked within the top seven hits, except for OTS964, indicating the high accuracy of our method. Moreover, we accurately identified the targets of four CETSA-studied non-kinase inhibitors: methotrexate, olaparib, panobinostat, and raltitrexed (Fig. S7†). This further demonstrates the robustness of the autoSISPROT and diaCETSA methods. In addition, we conducted target identification for HDAC inhibitors, including fimepinostat, SAHA, trichostatin A (TSA), and the studied panobinostat, to further demonstrate the applicability of our method. As expected, HDAC1 and HDAC2 were successfully identified as targets for all four HDAC inhibitors (Fig. S8†).
Regarding the CETSA-unstudied KIs, target identification could fail due to several reasons. In the case of two KIs, KN-62 and saracatinib, the abundance of their known targets (CaMK2 and SRC) in K562 cells could be too low to determine. For the other six KIs (bafetinib, bosutinib, BS-181, tideglusib, SGC-GAK-1, and roscovitine), their known targets did not exhibit significant differences between the drug and vehicle treatment conditions (Fig. S6M–R†). To determine whether this failure was due to the lack of a thermal stabilization effect, we conducted classical TPP analysis using ten temperature points to identify the drug targets of SGC-GAK-1 (Fig. 5). As a quality control, MTX was chosen to evaluate this autoSISPROT-based workflow (Fig. S9†). K562 cell lysates were treated with either a drug or a vehicle control, with two independent replicates for each condition, followed by thermal treatment at ten different temperature points (Fig. 5A). All samples showed consistent protein identification and high TMT labeling efficiency (Fig. S9A and B†). The boxplot of samples from ten different temperature points exhibited a typical sigmoidal trend (Fig. S9C–J†). A good correlation of Tm assessed with two independent replicates was achieved (Fig. 5B and E), illustrating the high reproducibility of autoSISPROT. The melting curves revealed significant changes in DHFR's thermal stability between the MTX and vehicle treatment conditions (Fig. 5C). As expected, the known target of MTX, dihydrofolate reductase (DHFR), was identified. The shifts of melting point (ΔTm) of DHFR for two independent replicates were very similar (20.4 °C and 18.4 °C, respectively). Moreover, DHFR ranked first based on the score provided by ProSAP software,37 which combines the significance of ΔTm and the goodness of fit. The inset graph also showed that DHFR had the most significant changes in ΔTm (Fig. 5d and Table S6†). However, the known target of SGC-GAK-1, cyclin-G-associated kinase (GAK), did not show significant changes between the drug and vehicle treatment conditions (Fig. 5F–G). Therefore, the failure to identify the target of SGC-GAK-1 is likely attributed to the absence of thermal stabilization effect of SGC-GAK-1 on GAK in cell lysates.
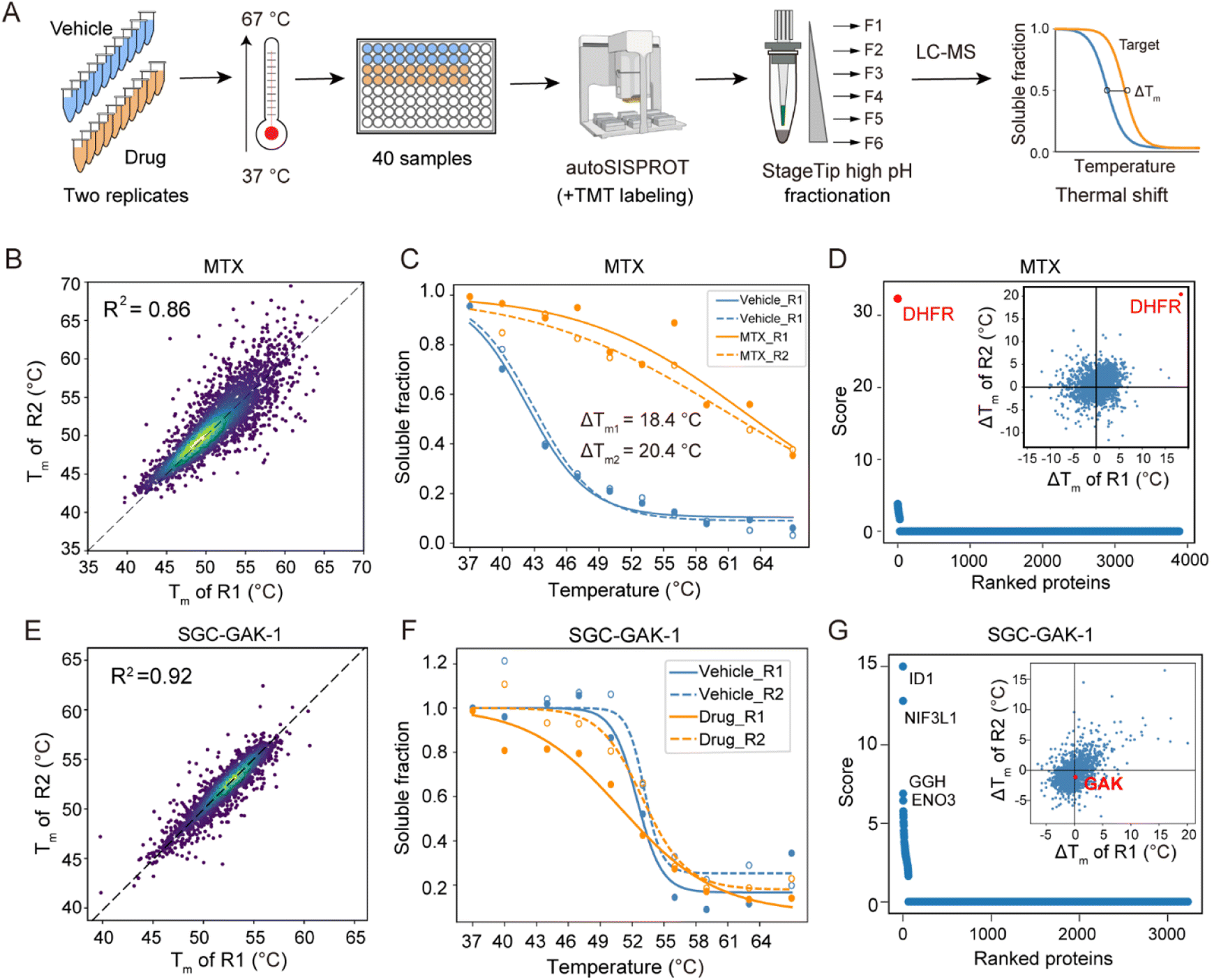 | ||
| Fig. 5 TPP analysis of the drug targets of SGC-GAK-1. (A) The workflow of TMT-based TPP with ten temperature points using autoSISPROT. (B and E) Scatter plot showing the correlation of Tm between two independent replicates for (B) MTX and (E) SGC-GAK-1. (C and F) Melting curves of DHFR in the presence (orange symbols) and absence (blue symbols) of (C) MTX and (F) SGC-GAK-1. Data are representative of two independent experiments. (D and G) Results of drug target identification for (D) MTX and (G) SGC-GAK-1. Proteins are ranked based on their scores generated using ProSAP software. The inset graph shows the scatter plot of ΔTm shifts calculated from the two independent replicates. | ||
Based on the principle of TPP, our method also enabled the high-throughput identification of potential drug off-targets (Fig. 4D). For instance, we also identified the reported off-targets of dinaciclib (CDK6), palbociclib (PIP4K2A, PIP4K2C, and CSNK2A2)38 and vemurafenib (FECH and MAP2K4), along with their known targets. In addition, our results reveal several new potential off-targets for kinase inhibitors (e.g., OTS964, vemurafenib, alisertib, dinaciclib, etc.).
To verify the performance of the autoSISPROT and diaCETSA combination, we utilized targeted parallel reaction monitoring (PRM)-MS analysis to validate the identified drug off-targets (Fig. 6A). Five kinase inhibitors (palbociclib, ralimetinib, vemurafenib, alisertib, and dinaciclib) were selected to treat K562 cell lysates and were subjected to single-temperature TPP coupled with PRM-MS analysis for the quantification of potential off-targets with high sensitivity and high precision. In addition, we also performed isothermal dose–response (ITDR)-MS analysis to determine the IC50 values of these inhibitors. Meanwhile, all of the known targets of palbociclib (CDK4 and CDK6), ralimetinib (MAPK14), vemurafenib (BRAF), alisertib (AURKA), and dinaciclib (CDK2, CDK5, and CDK9) were selected as positive controls for PRM analysis (Fig. S10†). The PRM data confirmed that all of these targets exhibit significantly higher abundance in the drug treatment groups, thereby demonstrating the success of the single-temperature TPP experiments. We selected 25 potential off-targets for PRM-MS analysis, 17 of which were confirmed to be thermally stabilized by corresponding inhibitors (Fig. 6B–D and S11†), including the reported off-targets of dinaciclib (CDK6), palbociclib (PIP4K2C), and vemurafenib (FECH and MAP2K4). It should be noted that the TPP-based analysis cannot distinguish between direct and indirect off-targets, as the interaction partners of off-target proteins may also be stabilized.39 For example, during the identification of ralimetinib's target, MAPK14 (p38α), its binding partner MAPKAPK2 was also identified as an off-target.
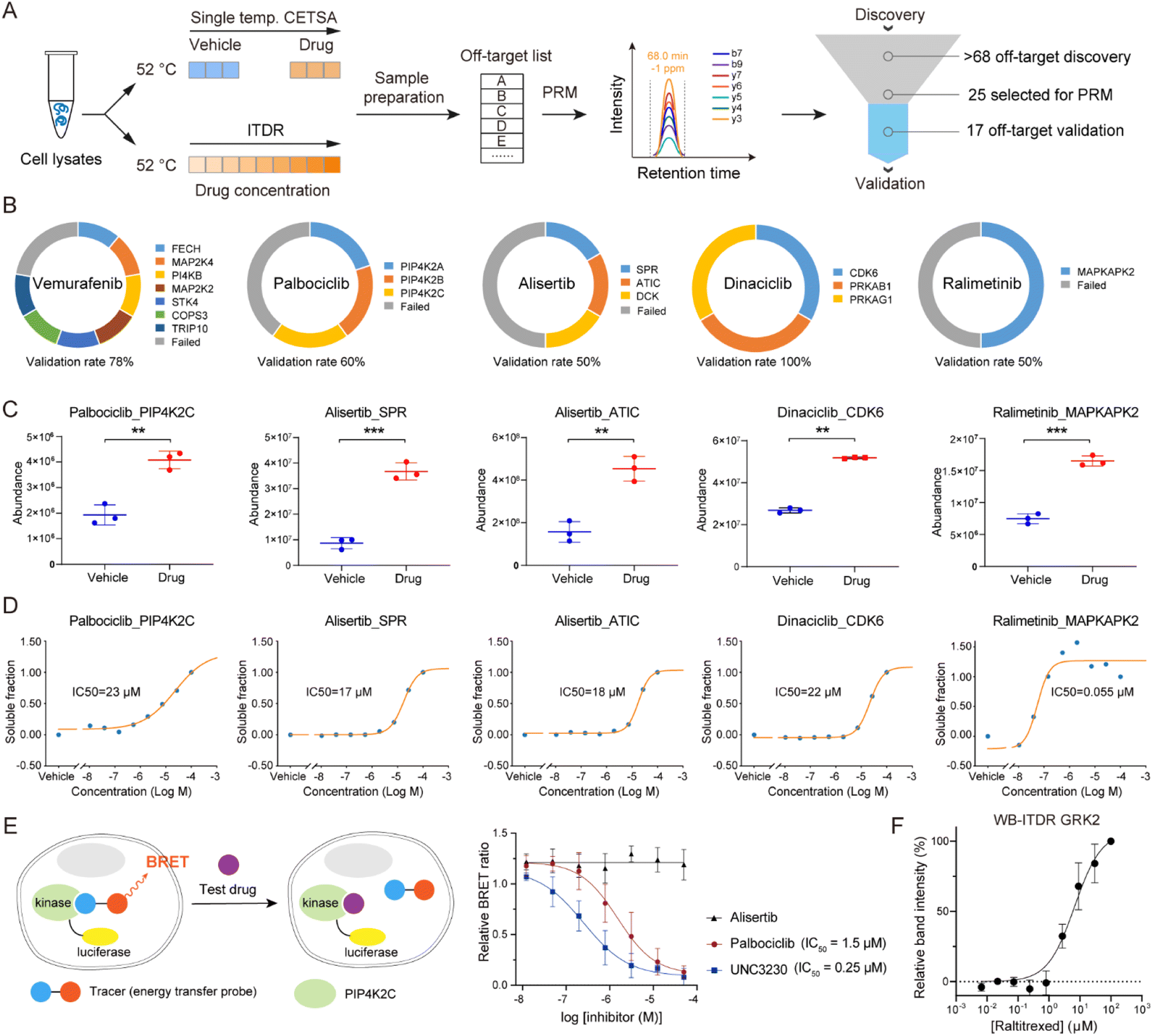 | ||
| Fig. 6 Off-target validation by PRM assay. (A) Workflow for the validation of off-targets via PRM assay that performed in single temperature TPP and ITDR modes. (B) Ring chart displaying the percentages of off-target validation for vemurafenib, palbociclib, alisertib, dinaciclib, and ralimetinib. (C and D) PRM-MS quantification of the selected potential off-targets. (C) K562 cell lysates were treated with a 20 μM drug or vehicle, followed by thermal treatment at 52 °C. (D) ITDR with treatment of eight concentrations (100, 27, 7.3, 2.0, 0.53, 0.14, 0.039, and 0.010 μM) of drug and vehicle, followed by thermal treatment at 52 °C. (E) NanoBRET analysis of palbociclib-PIP4K2C interaction in HEK 293T cells. A known inhibitor (UNC3230) of PIP4K2C was used as the positive control, and alisertib was taken as the negative control. n = 4 biologically independent replicates. (F) Western blot based ITDR for GRK2 at 52 °C. The band intensities were related to the intensities of the DMSO vehicle control samples. GAPDH levels were used to normalize the intensities. Data are reported as mean ± SD of three independent experiments. | ||
To evaluate the engagement of palbociclib with PIP4K2C in living cells, we performed the Nanoluc luciferase-based bioluminescence resonance energy transfer (NanoBRET) assay.40 We observed a palbociclib dose-dependent decrease in NanoBRET signals (Fig. 6E), indicating competitive displacement of the fluorescent tracer. The IC50 of palbociclib against PIP4K2C was determined to be 1.5 μM, which was higher than that of the known PIP4K2C inhibitor UNC3230 (0.25 μM) but still exhibited strong inhibitory potency in living cells. Meanwhile, alisertib was selected as a negative control since it did not show inhibition activity for PIP4K2C in our TPP results. These results validate the intracellular binding of palbociclib with PIP4K2C as determined by diaCETSA. In addition, we conducted classical western blot (WB) based CETSA experiments to validate GRK2 as a putative off-target of raltitrexed (Fig. S7C†). WB-ITDR revealed a weak binding affinity of raltitrexed to GRK2 (IC50 = 6.7 μM), which further demonstrates the utility of our method (Fig. 6F and S12†).
Overall, these results demonstrate that the combination of autoSISPROT and diaCETSA enables the identification of drug targets and off-targets in a fully automated manner and is well-suited for high-throughput drug target identification.
Discussion
The SISPROT technology allows for the seamless integration of multiple steps in protein sample preparation, including preconcentration, reduction, alkylation, digestion, desalting, and high-pH RP-based peptide fractionation, all within a single spintip device. The bead-based sample preparation ensures both high digestion efficiency and easy adaptability for robotic automation. Most of the currently reported automated and high-throughput sample preparation methods rely on in-solution digestion strategies using robotic liquid handling workstations such as Bravo, making it challenging to achieve fully automated and integrated sample preparation. In contrast, autoSISPROT enables fully automated and integrated 96-well sample preparation by performing on-bead digestion in the AssayMAP Bravo system. Through the seamless integration of all sample preparation steps into self-designed cartridges, the end-to-end autoSISPROT method is rapid, completing the processing of 96 samples from protein input to peptide elution in just 2.5 hours. No manual operations are required when performing autoSISPROT, thereby significantly reducing multi-step manual operations, hands-on time, and variability in protein quantification. By integrating the on-column TMT labeling step into the automated protocol, autoSISPROT only takes 3.5 hours to complete the digestion and TMT labeling of 96 samples, providing a streamlined solution for automated and reproducible quantitative proteomics. We demonstrated the excellent performance of autoSISPROT in sample preparation, and importantly, we also showcased its good intra- and inter-batch reproducibility. This was evidenced by CVs for protein identification and quantification below 3.0% and 17%, respectively, as well as Pearson correlation coefficients of more than 0.95 and 0.91 for intra- and inter-batch analyses, respectively.Notably, the cartridge used in autoSISPROT was specifically self-designed, taking reference from a standard 200 μL tip (Fig. S1B†). The cartridge had several key requirements: (1) the inner part of the top tip outlet had a similar geometry to syringes, allowing the cartridges to be picked up by syringes while maintaining air tightness and avoiding leakage. (2) The overfill position of the bottom tips had a similar geometry to the housing of the top tip outlet, enabling them to be assembled in an overfill manner to ensure air tightness. (3) The cartridges needed to be sufficiently stable to withstand the back pressure exerted during packing with SISPROT materials and organic solvents such as ACN. (4) The cartridges should be cost-effective and disposable. Industry-level moldmaking technology and a polypropylene (PP) material were employed for fabricating the cartridges, allowing for mass production of thousands of cartridges at a cost of less than 0.3 US dollars per cartridge.
TPP has been widely used for identifying targets and off-targets of various drugs. Currently, TPP and improved throughput methods such as PISA, iTSA, and mTSA rely on laborious manual sample preparation, making high-throughput drug target identification challenging. Encouragingly, the automated sample preparation platforms developed in this study provide multifunctional options for TPP-based drug target identification. By combining autoSISPROT and TPP technology, drug targets can be identified in an automated manner. Furthermore, autoSISPROT with diaCETSA allowed for the identification of kinase targets for 20 KIs in a fully automated and high-throughput manner. Compared to manual TPP, our automated platform significantly reduced manual operation and hands-on time while improving analysis throughput. By incorporating autoSISPROT and diaCETSA, up to 127 drugs can be analyzed in a single automated operation using 384-well plates, greatly facilitating proteomics-based drug discovery. Collectively, as large-cohort proteomic analysis continues to advance, autoSISPROT will provide a multifunctional and end-to-end solution for automated, robust, and reproducible sample preparation without manual intervention.
Conclusions
We developed autoSISPROT, a workflow that enables simultaneous processing of 96 samples in less than 2.5 hours. Benefiting from its 96-channel all-in-tip operation, protein digestion, peptide desalting, and TMT labeling could be achieved in a fully automated manner. autoSISPROT demonstrates effective sample preparation performance for both label-free and TMT-based quantitative proteomics as well as large-cohort sample preparation. Additionally, we have systematically evaluated the performance of TMT and DIA methods for TPP analysis and highlighted the potential of diaCETSA for target identification. By combining with diaCETSA, autoSISPROT allows for high-throughput and automated identification of the known targets and potential off-targets of 20 kinase inhibitors. The establishment of the autoSISPROT workflow will empower the high-throughput identification of novel drug targets that improves the probability of success for drug development efforts.Experimental
Cell culture and protein extraction
The cancer cell lines HEK 293T and K562 were purchased from American Type Culture Collection. HEK 293T cells were cultured in Dulbecco's modified Eagle's medium (Corning), supplemented with 10% fetal bovine serum (Gibco), 100 U per mL penicillin (Invitrogen), and 100 μg per mL streptomycin (Invitrogen) in a humidified incubator with 5% CO2 at 37 °C. HEK 293T cells were harvested at ∼80% confluence by washing three times with phosphate buffered saline (PBS) buffer and lysed in the lysis buffer containing 20 mM HEPES, pH 7.4, 150 mM NaCl, 600 mM guanidine HCl, 1% n-dodecyl β-D-maltoside (DDM), and protease inhibitors (1 mM PMSF, 1 μg per mL leupeptin, 1 μg per mL pepstatin, and 1 μg per mL aprotinin). The obtained cell lysates were sonicated and centrifuged at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g for ∼30 min at 4 °C. The protein concentration was measured using the BCA assay (Thermo Fisher Scientific, Germany), and the final protein concentration was adjusted to 5 mg mL−1.
000×g for ∼30 min at 4 °C. The protein concentration was measured using the BCA assay (Thermo Fisher Scientific, Germany), and the final protein concentration was adjusted to 5 mg mL−1.
K562 cells were cultured in RPMI 1640 medium (Gibco). Cells were harvested by washing three times with PBS buffer. For CETSA experiments, the K562 cell pellet was resuspended in a lysis buffer containing a final concentration of 50 mM HEPES (pH 7.4), 5 mM β-glycerophosphate, 0.1 mM activated Na3VO4, 10 mM MgCl2, 1 mM tris(β-chloroethyl) phosphate (TCEP, Sigma), and EDTA-free protease inhibitor (Roche) and lysed by three rounds of flash-freeze–thaw cycles (alternating exposure of the samples to liquid nitrogen and 37 °C in a water bath). Mechanical shearing was carried out by passing the thawed suspension through a syringe with a narrow needle several times. The resulting cell lysates were pelleted by centrifugation at 18000×g for approximately 30 min at 4 °C, and the supernatant was collected. The final protein concentration was adjusted to 5 mg mL−1 as determined by the BCA assay. For building the DDA spectral library, the K562 cell pellet was lysed in a lysis buffer containing 8 M urea, 50 mM ammonium bicarbonate (ABC), and a protease inhibitor mixture. The subsequent steps for protein extraction were the same as those for the HEK 293T cell lysate.
Thermal treatment of K562 cell lysates
The procedure for the ten temperature points-based TPP experiments followed a reported protocol.3 Briefly, K562 cell lysates were divided into ten identical aliquots (100 μg per aliquot, 20 μL) and treated with either 20 μM drug in 1% DMSO or with DMSO alone. With two replicates per condition, a total of 40 treated samples were incubated at room temperature for 5 min before heat treatment. Subsequently, all samples were transferred to a PCR machine and heated for 3 min at ten different temperature points (37, 40, 44, 47, 50, 53, 56, 59, 64, and 67 °C), followed by a 5-minute incubation at 4 °C. After heat treatment, the protein aggregates were removed by centrifugation at 18000×g for approximately 30 min at 4 °C, and the supernatant was collected.
For ten temperature points based TPP experiments, thermal treatment was performed similarly to the procedure reported in previous work.3 Briefly, K562 cell lysates were split into ten identical aliquots (100 μg per aliquot, 20 μL) and treated with either 20 μM drugs in 1% DMSO, or with DMSO alone. With two replicates per condition, 40 treated samples were incubated at room temperature for 5 min prior to heat treatment. After incubation, all samples were transferred to a PCR machine and heated for 3 min at ten different temperature points (37, 40, 44, 47, 50, 53, 56, 59, 64, and 67 °C), followed by a 5-min incubation at 4 °C. After heat treatment, the protein aggregates were removed by centrifugation at 18000×g for 30 min at 4 °C, and the supernatant was collected.
For the single temperature point-based TPP experiments, K562 cell lysates were divided into two identical aliquots (100 μg per aliquot, 20 μL), treated with 20 μM drug in 1% DMSO, or with DMSO alone. In the tmtCETSA versus diaCETSA comparison experiment, five replicates per condition were chosen. For high-throughput diaCETSA experiments, six replicates were chosen for the vehicle condition and three replicates for the drug condition, with multiple drug conditions sharing the common vehicle conditions. The treated samples were incubated at room temperature for 5 min and then transferred to a PCR machine for 3 min of heating at 52 °C, followed by a 5-minute incubation at 4 °C. The remaining steps for thermal treatment were the same as described above.
Fabrication and screening of the SISPROT-based cartridges
The preparation of SISPROT-based cartridges strictly followed ten steps. In brief, the SISPROT-based cartridges were constructed by assembling the top and bottom tips. The bottom tips were packed sequentially with three plugs of the C18 membrane (3 M Empore) and mixed-mode ion exchange beads (SCX:SAX = 1:1, Applied Biosystems). The quantity of the C18 membrane and mixed-mode ion exchange beads was adjusted based on the protein amount.
Sample preparation for building the DDA spectral library
For the construction of the DDA spectral library 1, K562 cell lysates underwent in-solution trypsin digestion following a previous study.34 Prior to the digestion reaction, proteins were purified using methanol-chloroform precipitation.41 The resulting peptides were desalted using a Sep-Pak Vac 1cc tC18 cartridge (Waters), dried using SpeedVac, and stored at −20 °C until LC-MS/MS analysis. For building DDA spectral library 2, sample preparation was processed by the autoSISPROT method, as described below.Sample preparation by using autoSISPROT
The autoSISPROT method was carried out using an AssayMAP Bravo equipped with 96-well ultra-low dead volume syringes and the homemade disposable SISPROT-based cartridges.The autoSISPROT workflow involved eleven key steps, which were (1) activation of SISPROT-based cartridges with activation buffer (deck 3), (2) equilibrium of SISPROT-based cartridges with equilibrium buffer (deck 5), (3) loading acidified cell lysates (pH 2.0–3.0, deck 7) into SISPROT-based cartridges, (4) washing SISPROT-based cartridges with activation buffer, (5) washing SISPROT-based cartridges with equilibrium buffer, (6) reduction of disulfide bonds with reduction buffer (deck 6) for 30 min at room temperature, (7) washing with pH change buffer (deck 9) to adjust pH, (8) loading alkylation and digestion buffer (deck 4) for 60 min at room temperature (in darkness), (9) transferring digested peptides from the mixed-mode ion exchange beads onto the C18 membrane with transfer buffer (deck 8), (10) desalting peptides with desalting buffer, and (11) eluting peptides with elution buffer. The eluted peptides were collected into 96-well plates. Finally, the samples were dried using a SpeedVac and stored at −20 °C before LC-MS/MS analysis. During the autoSISPROT operation, no manual operations were required once all buffers were transferred to the corresponding 96-well plates.
For TMT-based quantitative proteomics analysis, the buffer in deck 9 is replaced with HEPES buffer. Then two additional steps are implemented: pH adjustment using HEPES buffer and TMT labeling using a labeling buffer containing 0.4 μg μL−1 of TMT reagents in HEPES buffer. Moreover, a pause step is necessary to replace the sample plate with the TMT reagent plate before TMT labeling.
Sample preparation by using manual SISPROT
We conducted manual SISPROT using the same SISPROT-based cartridges, buffers, and HEK 293T cell lysates as in autoSISPROT. However, in the manual method, all sample preparation steps were carried out using centrifugation, following the procedures described in previous studies.25,26In the tmtCETSA versus diaCETSA comparison experiment, we employed the reported mixed-mode SISPROT for sample preparation with slight modifications.26 After digestion, ten samples from the tmtCETSA experiment were labeled with a TMT10-plex kit (Thermo Fisher Scientific, Germany) using the TMT-based FISAP method.35 Subsequently, the eluants from the tmtCETSA experiments were combined into a single sample, dried using the SpeedVac, and stored at −20 °C before fractionation. On the other hand, ten samples from the diaCETSA experiment were directly eluted without labeling, dried using the SpeedVac and stored at −20 °C before LC-MS/MS analysis.
High-pH reversed phase fractionation
To generate a comprehensive DDA spectral library of K562 cells, peptide fractionation was conducted using a 1260 Infinity II HPLC system (Agilent Technologies) with an XBridge peptide BEH C18 column (2.1 mm i.d. × 150 mm) at a flow rate of 200 μL min−1. Buffer A consisted of 2% ACN in 10 mM ABC (pH 8.0), while buffer B comprised 90% ACN in 10 mM ABC (pH 8.0). Peptides were separated using a 70-minute segmented gradient as follows: 1–9% buffer B in 1 minute, 9–35% buffer B in 50 min, and 35–70% buffer B in 4 min, followed by a 15-minute wash with 70% buffer B. Fractions were collected every 30 seconds and combined into 24 fractions.In the tmtCETSA versus diaCETSA comparison experiment, the TMT-labeled peptide mixture was separated using a 40-minute segmented gradient as follows: 1–13% buffer B in 1 minute, 13–52% buffer B in 29 min, and 52–90% buffer B in 4 min, followed by a 6-minute wash with 90% buffer B. Fractions were collected every 30 seconds and combined into 10 fractions. The obtained fractions were dried using the SpeedVac and stored at −20 °C before LC-MS/MS analysis.
For the ten temperature points based TPP experiments, the TMT-labeled peptide mixture underwent fractionation using C18 StageTip-based high-pH reversed-phase fractionation.35 The peptide mixture was fractionated using 10 μL portions of 18 different elution buffers (3%, 5%, 7%, 9%, 11%, 13%, 15%, 17%, 19%, 21%, 23%, 24%, 26%, 28%, 30%, 35%, 40%, and 80% ACN) in 5 mM ABC at pH 10.0. Subsequently, 18 fractions were combined into six fractions, dried using the SpeedVac, and stored at −20 °C.
LC-MS/MS analysis
Nano-flow LC-MS/MS was performed on an Orbitrap Exploris 480 equipped with an UltiMate 3000 (Thermo Fisher Scientific, Germany), or on a timsTOF Pro equipped with a nanoElute (Bruker Daltonics, Germany). Buffer A was 0.1% FA in water for both the Dionex UltiMate 3000 and nanoElute, and buffer B was 0.1% FA in 80% ACN for the Dionex UltiMate 3000 and 0.1% FA in 100% ACN for the nanoElute. Chromatographic separation was performed via the homemade 100 μm i.d. × 20 cm analytical column packed with 1.9 μm/120 Å C18 beads (Dr Maisch GmbH, Germany) at a flow rate of 500 nL min−1 (Dionex UltiMate 3000) or 300 nL min−1 (nanoElute). When using the Dionex UltiMate 3000 system to separate unlabeled peptides, an 85-minute segmented gradient was applied as follows: 4–8% buffer B in 2 min, 8–28% buffer B in 53 min, 28–36% buffer B in 10 min, and 36–100% buffer B in 9 min, followed by an 11-minute equilibration with 1% buffer B. For separating TMT-labeled HEK 293 T cell peptides, a 75-minute segmented gradient was used: 4–9% buffer B in 2 min, 9–35% buffer B in 51 min, 35–50% buffer B in 10 min, and 50–100% buffer B in 3 min, followed by a 3-minute wash with 100% buffer B and a 6-minute equilibration with 1% buffer B. For the analysis of unlabeled K562 cell peptides, peptide samples were spiked with iRT peptides from Biognosys (Switzerland) for retention time calibration. A 135-minute segmented gradient was used: 4–8% buffer B in 2 min, 8–28% buffer B in 105 min, 28–40% buffer B in 15 min, and 40–99% buffer B in 1 minute, followed by a 5-minute wash with 99% buffer B and a 7-minute equilibration with 1% buffer B. For analyzing TMT-labeled K562 cell peptides, a 135-minute segmented gradient was employed: 4–9% buffer B in 2 min, 9–35% buffer B in 103 min, 35–50% buffer B in 15 min, and 50–100% buffer B in 3 min, followed by a 5-minute wash with 100% buffer B and a 7-minute equilibration with 1% buffer B. When using the nanoElute system to separate unlabeled K562 cell peptides, an 80-minute segmented gradient was applied: 2–24% buffer B in 50 min, 24–36% buffer B in 10 min, and 36–80% buffer B in 10 min, followed by a 10-minute wash with 100% buffer B. For PRM assay, an 85-minute segmented gradient was applied: 4–8% buffer B in 2 min, 8–28% buffer B in 60 min, 28–36% buffer B in 10 min, and 36–100% buffer B in 3 min, followed by a 10-minute equilibration with 1% buffer B.The Orbitrap Exploris 480 instrument was operated in positive ion mode with the following settings: an electrospray voltage of 2.0 kV, a funnel RF lens value of 40, and an ion transfer tube temperature of 320 °C. MS1 scans were performed in the Orbitrap analyzer, covering an m/z range of 350 to 1200, with a resolution of 60000. The automatic gain control (AGC) target value was set to 3 × 106, and the maximum injection time (MIT) was in auto mode. The MS/MS spectra were acquired using DDA mode, with one MS scan followed by 40 MS/MS scans. Precursors were isolated using the quadrupole using a 1.4 Da window, followed by higher-energy collisional dissociation (HCD) fragmentation using a normalized collision energy (NCE) of 30%. Fragment ions were scanned at a resolution of 7500. The AGC target and MIT for MS2 scans were set to standard mode and 15 ms, respectively. The scanned peptides were dynamically excluded for 30 s. Monoisotopic precursor selection was enabled, and peptide charge states from +2 to +6 were selected for fragmentation.
For TMT-labeled peptides, MS1 scans were performed in the Orbitrap analyzer, covering an m/z range of 350 to 1400, with an MIT of 45 ms for MS1 scans. The instrument was set to run in top speed mode with 2 s cycles for the survey and MS/MS scans. HCD fragmentation was performed with a NCE of 36%. Fragment ions were scanned at a resolution of 30000, with an AGC target of 1 × 105 and an MIT of 54 ms. TurboTMT was operated in TMT reagent mode, with a precursor fit threshold of 65% and a fit window of 0.7 Da.
For DIA mode, each MS1 scan was followed by 60 variable DIA windows with 1.0 Da overlap, and the remaining parameter settings were the same as described above. In the tmtCETSA versus diaCETSA comparison samples, the FAIMS Pro device from Thermo Fisher Scientific (Germany) was used. The FAIMS device parameters included an inner electrode temperature of 100 °C, an outer electrode temperature of 100 °C, a carrier gas flow rate of 0 L min−1, an asymmetric waveform with a dispersion voltage of −5000 V, and an entrance plate voltage of 250 V. The selected CV (−45 and −65 V) was applied throughout the LC-MS/MS run for static CV conditions.
For PRM acquisition, target precursors were isolated through a window of 1 Da. The MS/MS spectra were scanned with a resolution of 30000, an AGC target of 1 × 106, and an MIT of 100 ms. The PRM scans were triggered by an unscheduled mode, where targeting precursor ions were repeatedly acquired in the entire elution windows. The PRM inclusion list of alisertib, dinaciclib, palbociclib, ralimetinib, and vemurafenib contained 33, 39, 34, 22, and 41 precursor ions, respectively.
A Bruker timsTOF Pro was operated in positive ion mode using a captive nano-electrospray source at 1500 V. The MS operated in DDA mode for ion mobility-enhanced spectral library generation. The accumulation and ramp time for mass spectra were both set to 100 ms, and the recorded mass spectra ranged from m/z 300 to 1500. Ion mobility was scanned from 0.75 to 1.40 V s cm−2. The overall acquisition cycle consisted of one full TIMS-MS scan and 10 parallel accumulation-serial fragmentation (PASEF) MS/MS scans. During PASEF MS/MS scanning, the collision energy was linearly ramped as a function of mobility from 59 eV at 1/K0 = 1.40 V s cm−2 to 20 eV at 1/K0 = 0.75 V s cm−2. In DIA mode, 32 × 25 Da isolation windows were defined from m/z 400 to 1200. To adapt the MS1 cycle time in diaPASEF, the repetitions were set to 2 in the 16-scan diaPASEF scheme.
Data analysis
The raw data were searched with MaxQuant (version 1.6.2.3) against a human protein database (release 2020_03, 74811 entries). Unless otherwise noted, the same database was used and the default parameters were employed. Trypsin was set as the enzyme with up to two missed cleavages. Carbamidomethylation (+57.021 Da) of cysteine was set as a fixed modification, and protein N-terminal acetylation (+42.011 Da) and oxidation of methionine residues (+15.995 Da) were considered variable modifications. The false discovery rate (FDR) was set to 1% at the site, peptide-spectrum match (PSM), and protein levels. For TMT-labeled samples, Proteome Discoverer™ software (version 2.4.1.15) was used for the search. Precursor mass tolerance was set to 10 ppm, and fragment ions were set to 0.02 Da. TMT tags on lysine residues and the peptide N-terminus (+229.163 Da) were defined as static modifications, while the other fixed and variable modifications remained consistent with those mentioned above. PSMs were validated using the Percolator algorithm, peptides were validated using the Peptide Validator algorithm, and proteins were validated using the Protein FDR Validator algorithm. Proteins were quantified by summing reporter ion counts across all matching PSMs. DIA raw data were searched with Spectronaut (version 15.7) with default parameters.
The output results from MaxQuant were used to generate density plots, heat maps, boxplots, and dot plots using R (version 3.4.0). The output results from Proteome Discoverer or Spectronaut were used to generate violin plots, melting curves, scatter plots of Tm and ΔTm shifts using Python (version 3.9). All the volcano plots were created using the ProSAP37 software and the p-value was calculated to assess the statistical significance of Tm after a Benjamini–Hochberg correction. Molecular function annotation was based on the GO knowledgebase (https://geneontology.org/). Protein–protein relationships were analyzed using STRING (version 11.5; https://string-db.org/). Kinome trees were built using the kinmapbeta tool (https://www.kinhub.org/kinmap/).42
All PRM raw files were processed using Skyline (version 20.2.0.286) to generate XIC and perform peak integration. Data that met the following four criteria: mass difference within ±20 ppm and dot-product (dotp) score ≥0.7 were accepted for further analysis. For single temperature CETSA-RPM, GraphPad Prism (version 8.0.2) was used to perform an unpaired t test and to calculate adjusted p-values for the analysis of significance between vehicle and drug treatment conditions. For the ITDR-PRM data, sigmoidal curve fitting and IC50 calculations were performed using Python (version 3.9).
NanoBRET target engagement assay
The NanoBRET target engagement assay was performed according to the manufacturer's instructions. Briefly, HEK293 cells were transfected with a C-terminally tagged PIP4K2C NanoLuc fusion vector (Promega, #NV1191). After 24 h, cells were counted and diluted to 2 × 105 cells per mL in Opti-MEM containing 4% (v/v) FBS. A K-8 kinase tracer (Promega, #N2620) was added to cells at a final concentration of 0.5 μM before 40 μL per well were added to a 384-well plate containing pre-plated compounds in triplicate. After 2 h of incubation 20 μL per well of substrate and extracellular NanoLuc inhibitor mix (1:166, 1:500 in Opti-MEM) were added. Luminescence signals from the donor (460 nm) and acceptor (610 nm) were measured on a PheraSTAR FSX plate reader after 10 min of incubation at room temperature. Data were analyzed by calculating the ratio of acceptor to donor signal, subtracting the background (transfected cells w/o the tracer), and normalizing to DMSO. IC50 values were calculated by nonlinear regression (dose–response curve fitting) analysis via GraphPad Prism (version 9.1.0).
Western blot analysis
The protein soluble fractions were lysed in 2× Laemmli buffer and heated at 95 °C for 5 min. Then proteins were separated on 10% SDS-PAGE gel and transferred onto PVDF membranes. The membranes were blocked with 5% BSA or non-fat dried milk (NFDM) in TBS with 0.1% Tween20 (TBST) at RT for 2 h, followed by incubation at 4 °C overnight with primary antibodies: anti-GRK2 (Abcam, ab227825, 1:1000) and anti-GAPDH (Beyotime, AF0006, 1:1000). After washing with TBST, membranes were incubated with HRP-conjugated goat anti-mouse IgG (Beyotime, A0216, 1:1000) or goat anti-rabbit IgG (Beyotime, A0208, 1:1000) at RT for 1 h. Chemiluminescence intensities were detected with a Clarity Western ECL Substrate (Bio-Rad) through an Odyssey infrared scanner (LICOR Bioscience) and quantified using ImageJ software.
Data availability
The mass spectrometric raw data have been deposited on ProteomeXchange via the PRIDE partner repository43 with the dataset identifier PXD044006. All other data are provided in the ESI/Data files.†Author contributions
Q. W., J. Z., X. S., R. T. and C. T. designed the experiments. Q. W. and X. S. performed the sample preparation and proteomics experiments. J. Z. C. F. and X. X. conducted biophysical, cell biology, and molecular biology experiments. X. C. and X. L. provided assistance in the preparation of SISPROT-based cartridges. Q. W., H. J., Y. L., A. H., B. L. and J. Z. analyzed the data. R. T. conceived and supervised the project. Q. W., J. Z., C. T. and R. T. wrote the paper.Conflicts of interest
R. T. is a founder of BayOmics, Inc. The other authors declare no competing interests.Acknowledgements
This work was supported by grants from the China State Key Basic Research Program Grants (2021YFA1301601, 2021YFA1302603, 2021YFA1301602, 2020YFE0202200, and 2022YFC3401104), the National Natural Science Foundation of China (22125403, 22074060, 22150610470, 92253304, 22304036, 2201218, and 2210404), the Shenzhen Innovation of Science and Technology Commission (JCYJ20200109140814408, JCYJ20210324120210029, JCYJ20200109141212325, and JSGGZD20220822095200001), and Natural Science Foundation of Guangdong province, China (2019B151502050).Notes and references
- J. W. Scannell, J. Bosley, J. A. Hickman, G. R. Dawson, H. Truebel, G. S. Ferreira, D. Richards and J. M. Treherne, Nat. Rev. Drug Discovery, 2022, 21, 915–931 CrossRef CAS PubMed.
- F. Meissner, J. Geddes-McAlister, M. Mann and M. Bantscheff, Nat. Rev. Drug Discovery, 2022, 21, 637–654 CrossRef CAS PubMed.
- M. M. Savitski, F. B. Reinhard, H. Franken, T. Werner, M. F. Savitski, D. Eberhard, D. Martinez Molina, R. Jafari, R. B. Dovega, S. Klaeger, B. Kuster, P. Nordlund, M. Bantscheff and G. Drewes, Science, 2014, 346, 1255784 CrossRef PubMed.
- J. Zecha, F. P. Bayer, S. Wiechmann, J. Woortman, N. Berner, J. Müller, A. Schneider, K. Kramer, M. Abril-Gil, T. Hopf, L. Reichart, L. Chen, F. M. Hansen, S. Lechner, P. Samaras, S. Eckert, L. Lautenbacher, M. Reinecke, F. Hamood, P. Prokofeva, L. Vornholz, C. Falcomatà, M. Dorsch, A. Schröder, A. Venhuizen, S. Wilhelm, G. Médard, G. Stoehr, J. Ruland, B. M. Grüner, D. Saur, M. Buchner, B. Ruprecht, H. Hahne, M. The, M. Wilhelm and B. Kuster, Science, 2023, 380, 93–101 CrossRef CAS PubMed.
- D. C. Mitchell, M. Kuljanin, J. Li, J. G. Van Vranken, N. Bulloch, D. K. Schweppe, E. L. Huttlin and S. P. Gygi, Nat. Biotechnol., 2023, 41, 845–857 CrossRef CAS PubMed.
- Q. Xiao, F. Zhang, L. Xu, L. Yue, O. L. Kon, Y. Zhu and T. Guo, Adv. Drug Delivery Rev., 2021, 176, 113844 CrossRef CAS PubMed.
- K. V. Huber, K. M. Olek, A. C. Muller, C. S. Tan, K. L. Bennett, J. Colinge and G. Superti-Furga, Nat. Methods, 2015, 12, 1055–1057 CrossRef CAS PubMed.
- I. Becher, T. Werner, C. Doce, E. A. Zaal, I. Togel, C. A. Khan, A. Rueger, M. Muelbaier, E. Salzer, C. R. Berkers, P. F. Fitzpatrick, M. Bantscheff and M. M. Savitski, Nat. Chem. Biol., 2016, 12, 908–910 CrossRef CAS PubMed.
- J. M. Dziekan, G. Wirjanata, L. Dai, K. D. Go, H. Yu, Y. T. Lim, L. Chen, L. C. Wang, B. Puspita, N. Prabhu, R. M. Sobota, P. Nordlund and Z. Bozdech, Nat. Protoc., 2020, 15, 1881–1921 CrossRef CAS PubMed.
- J. Perrin, T. Werner, N. Kurzawa, A. Rutkowska, D. D. Childs, M. Kalxdorf, D. Poeckel, E. Stonehouse, K. Strohmer, B. Heller, D. W. Thomson, J. Krause, I. Becher, H. C. Eberl, J. Vappiani, D. C. Sevin, C. E. Rau, H. Franken, W. Huber, M. Faelth-Savitski, M. M. Savitski, M. Bantscheff and G. Bergamini, Nat. Biotechnol., 2020, 38, 303–308 CrossRef CAS PubMed.
- N. Prabhu, L. Dai and P. Nordlund, Curr. Opin. Chem. Biol., 2020, 54, 54–62 CrossRef CAS PubMed.
- D. M. Molina, R. Jafari, M. Ignatushchenko, T. Seki, E. A. Larsson, C. Dan, L. Sreekumar, Y. Cao and P. Nordlund, Science, 2013, 341, 84–87 CrossRef CAS PubMed.
- M. Gaetani, P. Sabatier, A. A. Saei, C. M. Beusch, Z. Yang, S. L. Lundström and R. A. Zubarev, J. Proteome Res., 2019, 18, 4027–4037 CrossRef CAS PubMed.
- K. A. Ball, K. J. Webb, S. J. Coleman, K. A. Cozzolino, J. Jacobsen, K. R. Jones, M. H. B. Stowell and W. M. Old, Commun. Biol., 2020, 3, 75 CrossRef CAS PubMed.
- C. Ruan, Y. Wang, X. Zhang, J. Lyu, N. Zhang, Y. Ma, C. Shi, G. Qu and M. Ye, Anal. Chem., 2022, 94, 6482–6490 CrossRef CAS PubMed.
- J. Li, J. G. Van Vranken, J. A. Paulo, E. L. Huttlin and S. P. Gygi, J. Proteome Res., 2020, 19, 2159–2166 CrossRef CAS PubMed.
- N. Zinn, T. Werner, C. Doce, T. Mathieson, C. Boecker, G. Sweetman, C. Fufezan and M. Bantscheff, J. Proteome Res., 2021, 20, 1792–1801 CrossRef CAS PubMed.
- X. Lu, B. Liao, S. Sun, Y. Mao, Q. Wu, R. Tian and C. S. H. Tan, Anal. Chem., 2023, 95(37), 13844–13854 CrossRef CAS PubMed.
- H. Ji, X. Lu, S. Zhao, Q. Wang, B. Liao, L. G. Bauer, K. V. M. Huber, R. Luo, R. Tian and C. S. H. Tan, Cell Chem. Biol., 2023, 30(11), 1478–1487 CrossRef CAS PubMed.
- P. E. Geyer, N. A. Kulak, G. Pichler, L. M. Holdt, D. Teupser and M. Mann, Cell. Syst., 2016, 2, 185–195 CrossRef CAS PubMed.
- C. B. Messner, V. Demichev, D. Wendisch, L. Michalick, M. White, A. Freiwald, K. Textoris-Taube, S. I. Vernardis, A. S. Egger, M. Kreidl, D. Ludwig, C. Kilian, F. Agostini, A. Zelezniak, C. Thibeault, M. Pfeiffer, S. Hippenstiel, A. Hocke, C. von Kalle, A. Campbell, C. Hayward, D. J. Porteous, R. E. Marioni, C. Langenberg, K. S. Lilley, W. M. Kuebler, M. Mulleder, C. Drosten, N. Suttorp, M. Witzenrath, F. Kurth, L. E. Sander and M. Ralser, Cell. Syst., 2020, 11, 11–24 CrossRef CAS PubMed.
- A. P. Burns, Y. Q. Zhang, T. Xu, Z. Wei, Q. Yao, Y. Fang, V. Cebotaru, M. Xia, M. D. Hall, R. Huang, A. Simeonov, C. A. LeClair and D. Tao, Anal. Chem., 2021, 93, 8423–8431 CrossRef CAS PubMed.
- T. Müller, M. Kalxdorf, R. Longuespée, D. N. Kazdal, A. Stenzinger and J. Krijgsveld, Mol. Syst. Biol., 2020, 16, e9111 CrossRef PubMed.
- X. Ye, J. Tang, Y. Mao, X. Lu, Y. Yang, W. Chen, X. Zhang, R. Xu and R. Tian, Trends Anal. Chem., 2019, 120, 115667 CrossRef CAS.
- W. Chen, S. Wang, S. Adhikari, Z. Deng, L. Wang, L. Chen, M. Ke, P. Yang and R. Tian, Anal. Chem., 2016, 88, 4864–4871 CrossRef CAS PubMed.
- L. Xue, L. Lin, W. Zhou, W. Chen, J. Tang, X. Sun, P. Huang and R. Tian, J. Chromatogr. A, 2018, 1564, 76–84 CrossRef CAS PubMed.
- X. Lu, Z. Wang, Y. Gao, W. Chen, L. Wang, P. Huang, W. Gao, M. Ke, A. He and R. Tian, Anal. Chem., 2020, 92, 8893–8900 CrossRef CAS PubMed.
- S. Fulton, S. Murphy, J. Reich, Z. Van Den Heuvel, R. Sakowski, R. Smith and S. Agee, J. Lab. Autom., 2011, 16, 457–467 CrossRef CAS PubMed.
- W. Chen, S. Adhikari, L. Chen, L. Lin, H. Li, S. Luo, P. Yang and R. Tian, J. Chromatogr. A, 2017, 1498, 207–214 CrossRef CAS PubMed.
- C. Ruan, Y. Wang, X. Zhang, J. Lyu, N. Zhang, Y. Ma, C. Shi, G. Qu and M. Ye, Anal. Chem., 2022, 94, 6482–6490 CrossRef CAS PubMed.
- A. L. George, F. R. Sidgwick, J. E. Watt, M. P. Martin, M. Trost, J. L. Marín-Rubio and M. E. Dueñas, J. Proteome Res., 2023, 22, 2629–2640 CrossRef CAS PubMed.
- J. Muntel, J. Kirkpatrick, R. Bruderer, T. Huang, O. Vitek, A. Ori and L. Reiter, J. Proteome Res., 2019, 18, 1340–1351 CrossRef CAS PubMed.
- C. von Mering, M. Huynen, D. Jaeggi, S. Schmidt, P. Bork and B. Snel, Nucleic Acids Res., 2003, 31, 258–261 CrossRef CAS PubMed.
- P. Huang, C. Liu, W. Gao, B. Chu, Z. Cai and R. Tian, J. Mass Spectrom., 2021, 56, e4653 CrossRef CAS PubMed.
- Y. Mao, P. Chen, M. Ke, X. Chen, S. Ji, W. Chen and R. Tian, Anal. Chem., 2021, 93, 3026–3034 CrossRef CAS PubMed.
- J. Zecha, S. Satpathy, T. Kanashova, S. C. Avanessian, M. H. Kane, K. R. Clauser, P. Mertins, S. A. Carr and B. Kuster, Mol. Cell. Proteomics, 2019, 18, 1468–1478 CrossRef CAS PubMed.
- H. Ji, X. Lu, Z. Zheng, S. Sun and C. S. H. Tan, Briefings Bioinf., 2022, 23, bbac057 CrossRef PubMed.
- A. L. Chernobrovkin, C. Cázares-Körner, T. Friman, I. M. Caballero, D. Amadio and D. Martinez Molina, SLAS Discovery, 2021, 26, 534–546 CrossRef CAS PubMed.
- C. S. H. Tan, K. D. Go, X. Bisteau, L. Dai, C. H. Yong, N. Prabhu, M. B. Ozturk, Y. T. Lim, L. Sreekumar, J. Lengqvist, V. Tergaonkar, P. Kaldis, R. M. Sobota and P. Nordlund, Science, 2018, 359, 1170–1177 CrossRef CAS PubMed.
- M. B. Robers, M. L. Dart, C. C. Woodroofe, C. A. Zimprich, T. A. Kirkland, T. Machleidt, K. R. Kupcho, S. Levin, J. R. Hartnett, K. Zimmerman, A. L. Niles, R. F. Ohana, D. L. Daniels, M. Slater, M. G. Wood, M. Cong, Y. Q. Cheng and K. V. Wood, Nat. Commun., 2015, 6, 10091 CrossRef CAS PubMed.
- L. Jiang, L. He and M. Fountoulakis, J. Chromatogr. A, 2004, 1023, 317–320 CrossRef CAS PubMed.
- S. Eid, S. Turk, A. Volkamer, F. Rippmann and S. Fulle, BMC Bioinf., 2017, 18, 16 CrossRef PubMed.
- Y. Perez-Riverol, J. Bai, C. Bandla, D. García-Seisdedos, S. Hewapathirana, S. Kamatchinathan, D. J. Kundu, A. Prakash, A. Frericks-Zipper, M. Eisenacher, M. Walzer, S. Wang, A. Brazma and J. A. Vizcaíno, Nucleic Acids Res., 2022, 50, D543–d552 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05937e |
| ‡ These authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2024 |