
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical cascade migratory versus ortho-cyclization of 2-alkynylbenzenesulfonamides†
Zhaojiang
Shi‡
 a,
Shicheng
Dong‡
c,
Ting
Liu‡
a,
Wei-Zhen
Wang
a,
Nan
Li
a,
Yaofeng
Yuan
a,
Jun
Zhu
*bc and
Ke-Yin
Ye
*a
a,
Shicheng
Dong‡
c,
Ting
Liu‡
a,
Wei-Zhen
Wang
a,
Nan
Li
a,
Yaofeng
Yuan
a,
Jun
Zhu
*bc and
Ke-Yin
Ye
*a
aKey Laboratory of Molecule Synthesis and Function Discovery (Fujian Province University), College of Chemistry, Fuzhou University, Fuzhou 350108, China. E-mail: kyye@fzu.edu.cn
bSchool of Science and Engineering, The Chinese University of Hong Kong, Shenzhen, Guangdong 518172, China
cState Key Laboratory of Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), Fujian Provincial Key Laboratory of Theoretical and Computational Chemistry, Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: jun.zhu@cuhk.edu.cn
First published on 11th January 2024
Abstract
Efficient control over several possible reaction pathways of free radicals is the chemical basis of their highly selective transformations. Among various competing reaction pathways, sulfonimidyl radicals generated from the electrolysis of 2-alkynylbenzenesulfonamides undergo cascade migratory or ortho-cyclization cyclization selectively. It is found that the incorporation of an extra 2-methyl substituent biases the selective migration of the acyl- over vinyl-linker of the key spirocyclic cation intermediate and thus serves as an enabling handle to achieve the synthetically interesting yet under-investigated cascade migratory cyclization of spirocyclic cations.
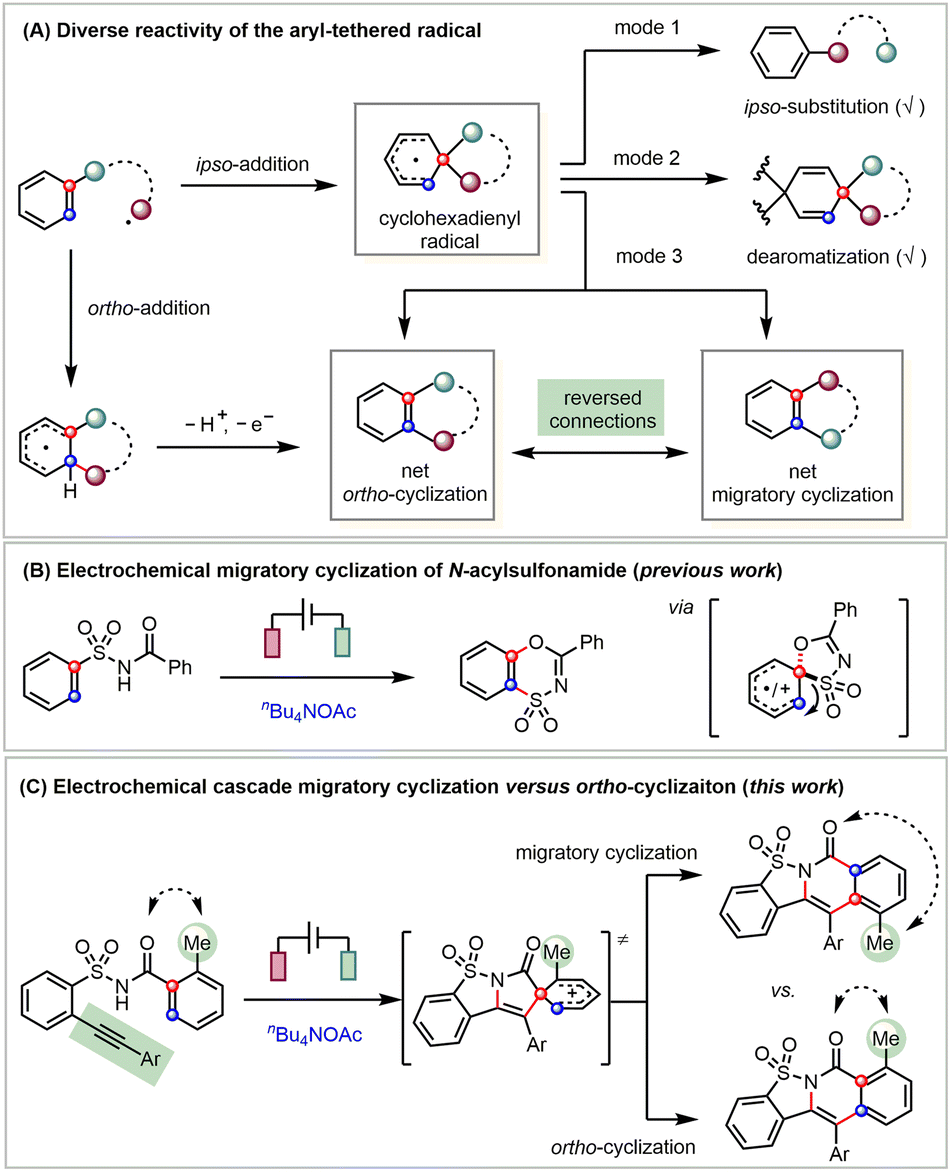
Free radicals are typically highly reactive species yet can still form diverse products through selective transformations.1 Key to the high selectivity of a radical reaction is the efficient control over several competing reaction pathways. For instance, a radical can add to the tethered arene via two pathways (Scheme 1A).2 The straightforward ortho-addition generates a fused cyclohexadienyl radical that further undergoes electron and proton transfer to give the ortho-cyclization product.3 Alternatively, the ipso-addition pathway leads to a spiro cyclohexadienyl radical.4 While there are many studies on ipso-substitution reactivity, known as the Smiles rearrangement (mode 1)5 and dearomatization (mode 2),6 ring expansion of the spirocyclohexadienyl radical is rarely explored (mode 3).7 Apparently, the migratory attitude of the spirocyclic linkers guides the reaction toward two products with reversed connections, i.e., net ortho-cyclization and migratory cyclization. Though both the ortho-addition and ipso-addition can lead to net ortho-cyclization products, there only exist very limited studies on the selective formation of migratory cyclization products.8
 | ||
| Scheme 1 Contents of this study. | ||
Owing to the mild reaction conditions for generating radicals, synthetic electrochemistry9 provides enabling handles for the fine-tuning of the diverse selectivity of radical reactions.10 Even though there are many radical-mediated cascade reactions, very few studies have been carried out on the migratory attitude of different linkers in the in situ generated spirocyclohexadienyl radicals. Understanding the innate migratory attitude of spirocyclic intermediates is not only fundamentally interesting but also potentially serves as an enabling handle for their selective transformations.
Specifically, amidyl radicals are generated either from the homolytic cleavage11a or photocatalysis-facilitated single-electron reduction11b of N–X bonds (X = halogen, O, N, etc.) or photochemical/electrochemical oxidation of N–H bonds.11c Recently, we have reported an electrochemical migratory cyclization of N-acylsulfonamides through an exclusive sulfone migration of the spirocyclohexadienyl intermediate (Scheme 1B).12 This electrochemistry-enabled reactivity is in sharp contrast to an anionic Smiles ipso-rearrangement of the N-acylsulfonamide that only results in its degradation.13
The attachment of an o-alkyne14 shifts the reactivity from the above migratory cyclization to a straightforward aza-cyclization.15 Indeed, we found that linking the aryl acetylene to the ortho-position of the benzenesulfonamide results in the anticipated cascade ortho-cyclization (Scheme 1C).16 More interestingly, the extra incorporation of the 2-methyl substitution favors migratory cyclization. Preliminary mechanistic investigations suggest that this 2-methyl substituent favors the selective migration of the acyl- over vinyl-linker of the key spirocyclic cation intermediate, which helps to achieve the fundamentally interesting yet rarely explored cascade migratory cyclization. Notably, the resultant sultam-fused pyridinones exhibit promising antibacterial activity and the current protocol affords a diverse array of derivatives that should be beneficial to their biological studies.17
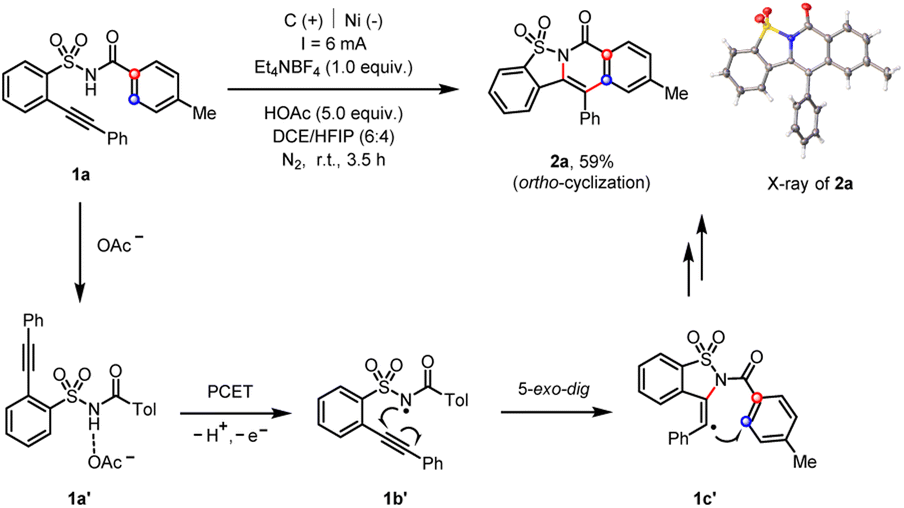
Initially, our extensive optimization of reaction conditions (see ESI†) disclosed that the 2-alkynylbenzenesulfonamide (1a) underwent an electrochemical cascade ortho-cyclization18 to afford the corresponding heterocycle (2a) in 59% yield, whose chemical structure was confirmed by the X-ray diffraction analysis (Scheme 2).19 Among several solvents tested, the combination of dichloroethane (DCE) and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) mixed solvents afforded the cleanest reaction system that facilitated the purification of products. The use of HOAc was crucial to the observed reactivity since HOAc served as the sacrificial oxidant and the resultant acetate acted as a base in the following proton-coupled electron transfer (PCET) process, which was further substantiated both by the cyclic-voltammetry and nuclear-magnetic-resonance studies (see ESI†).11c,20 We proposed that the acid–base complex (1a′) underwent electrochemical oxidation toward the formation of an amidyl radical (1b′). The subsequent 5-exo-dig radical cyclization gave rise to a vinyl radical (1c′), which readily attacked the pendant phenyl moiety to release the ortho-cyclization product (2a).
 | ||
| Scheme 2 Electrochemical cascade ortho-cyclization. | ||
The substrate scope of this electrochemical cascade ortho-cyclization was investigated (Scheme 3). A broad spectrum of substrates with varying electron-donating and electron-withdrawing substituents located at either the arylethynyl (2b–2w), benzoate side (2x–2af), or arylsulfonamide (2ah) moiety were tolerated. The scalability of this transformation was demonstrated by a 4 mmol scale preparation of the desired cascade cyclization heterocycle (2g, 49%). Aromatic iodide (2ac), –CF3 (2ad), and aliphatic halide (2ae) were all tolerated under these electrochemical reaction conditions. Notably, the meta-methyl derivative underwent the cascade ortho-cyclization at the sterically more hindered site in good regioselectivity (2ag, 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 rr). While 1-naphthylene- and 2-thiophene-tethered alkynes (2ai and 2aj) were compatible substrates, the cyclopropyl one (2ak) only afforded the desired product in 28% yield. Unfortunately, aliphatic alkyne and terminal alkene derivatives were not applicable under the current electrolytic conditions (see more unsuccessful substrates in ESI†).
:1 rr). While 1-naphthylene- and 2-thiophene-tethered alkynes (2ai and 2aj) were compatible substrates, the cyclopropyl one (2ak) only afforded the desired product in 28% yield. Unfortunately, aliphatic alkyne and terminal alkene derivatives were not applicable under the current electrolytic conditions (see more unsuccessful substrates in ESI†).
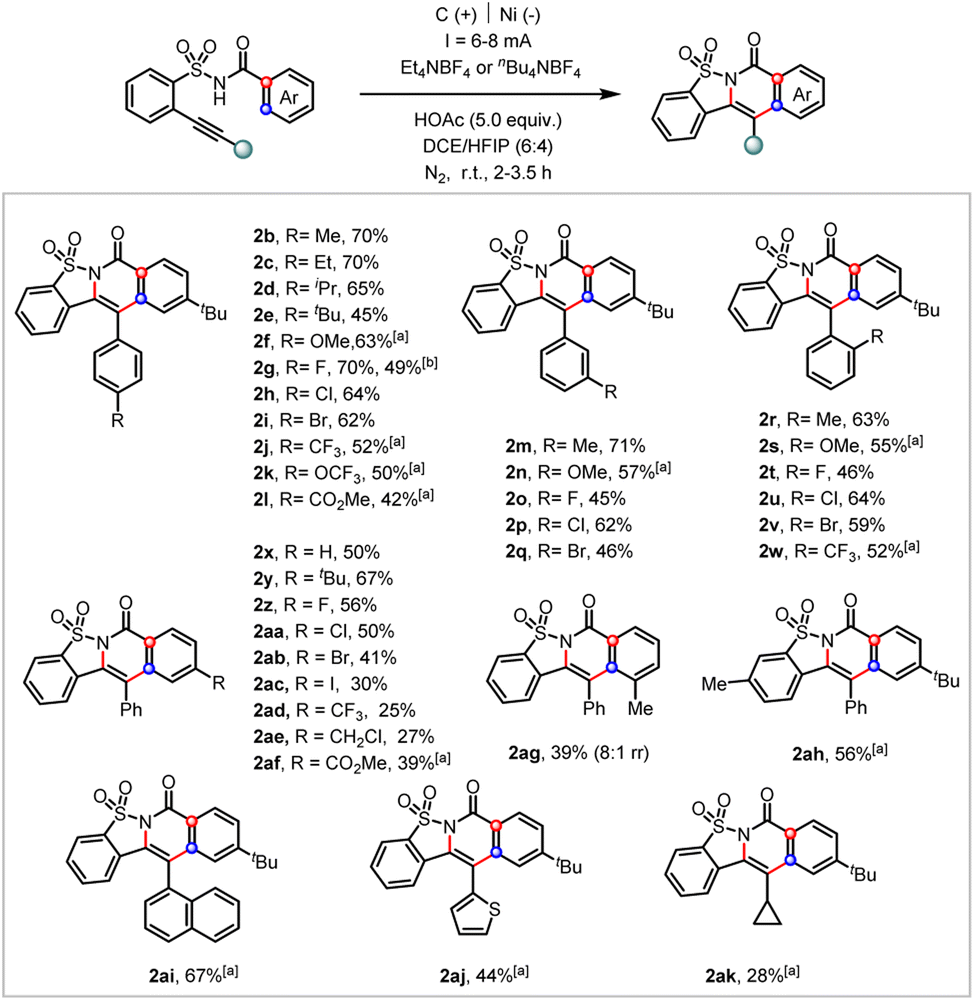 | ||
| Scheme 3 Substrate scope of the electrochemical cascade ortho-cyclization. Reaction conditions: undivided cell, C(+)/Ni(−), substrate (0.2 mmol), Et4NBF4 or nBu4NBF4 (1.0 equiv.), HOAc (5.0 equiv.), CHCl3/HFIP = 6:4, room temperature, I = 6–8 mA, under a N2 atmosphere, 2–3.5 h; a C(+)/Pt(−), nBu4NOAc (1.0 equiv.), Cp2Fe (20 mol%), I = 6–8 mA, 5–7 h, MeCN; b 4 mmol scale reaction. | ||
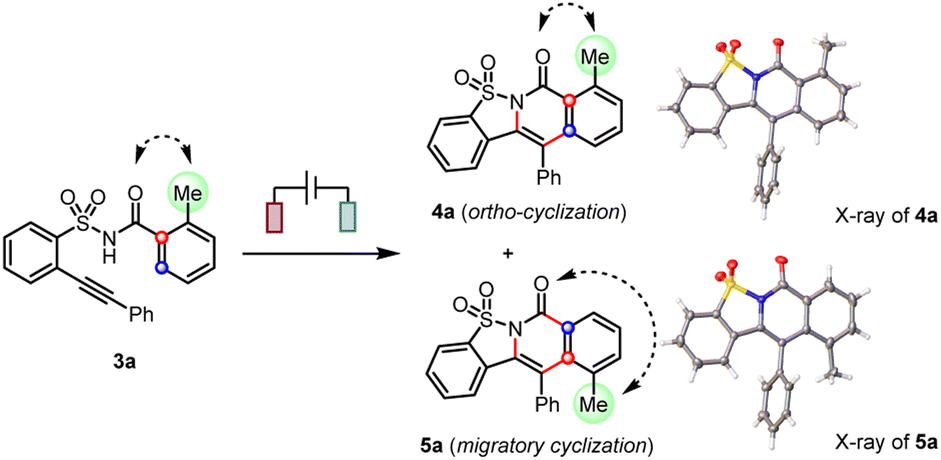
When a methyl group was introduced at the ortho-position (3a), we observed not only the anticipated cascade ortho-cyclization (4a) but also the unexpected migratory cyclization (5a) product (Table 1, entry 1). According to the X-ray diffraction analysis, the migratory product (5a) featured a translocation of the original ortho-methyl to the meta-position. After thorough optimizations (see ESI†), the electrolytic conditions in entry 2 afforded the migratory cyclization (5a) as the major product in the optimal 3.5:1 ratio. While similar results can be obtained without a redox mediator, the incorporation of a catalytic amount of trimethyltriphenylamine as the redox mediator9a,21 in CHCl3/HFIP mixed solvents provided a very clean reaction and thus generally afforded a higher yield.
|
|
|||
|---|---|---|---|
| Entry | Electrolytic conditions | Yielda (4a + 5a, %) | Ratiob (4a:5a) |
| a Isolated yield. b The regioisomeric ratio was determined by 1H NMR analysis of the crude mixture. | |||
| 1 | C(+)|Ni(−) | 54 | 1:1.6 |
| I = 6 mA, Et4NBF4 (1.0 equiv.) | |||
| HOAc (5.0 equiv.), DCE/HFIP (6:4) |
|||
| N2, r.t., 3.5 h | |||
| 2 | C(+)|Pt(−) | 58 | 1:3.5 |
| (4-MePh)3N (20 mol%) | |||
| I = 8 mA, nBu4NOAc (1.0 equiv.) | |||
| HOAc (5.0 equiv.), CHCl3/HFIP (6:4) |
|||
| N2, r.t., 4.5 h | |||
This migratory cyclization reactivity was further examined with a variety of substituted arylethynyl derivatives (Scheme 4). These reactions generally afforded the cascade cyclization products smoothly favoring the migratory product in moderate to good selectivity. It was found that the increase of steric hindrance at the ortho-position of arylethynyls (5e and 5f) or the replacement of the aryl by thiophene (5l) was deleterious to the selectivity. Interestingly, the meta-halo substituent of arylethynyls (5h–5j) afforded the highest preference for the migratory cyclization products (up to 8.6:1) despite moderate yields. Finally, such an electrochemical migratory cyclization was also applicable to the 4-methyl derivative (5m). Unfortunately, attempts to further improve the migratory selectivity via the replacement of the methyl group by other ortho substituents, such as phenyl, ethyl, halide (F or Br), or acetyl were not successful yet (see ESI† for details).
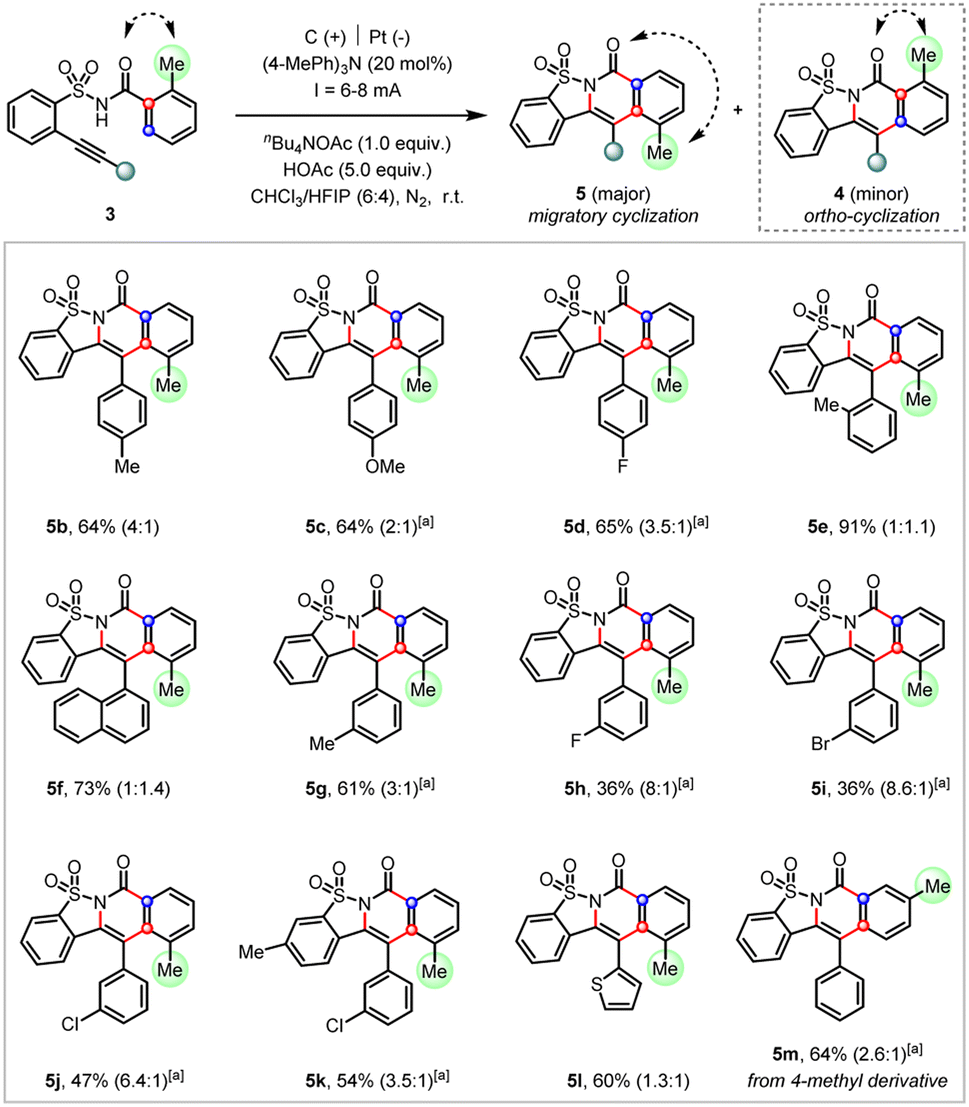 | ||
| Scheme 4 Substrate scope of the electrochemical cascade migratory cyclization. Reaction conditions: undivided cell, C(+)/Pt(−), substrate (0.2 mmol), nBu4NOAc (1.0 equiv.), (4-MePh)3N (20 mol%), HOAc (5.0 equiv.), CHCl3/HFIP = 6:4, room temperature, I = 8 mA, under a N2 atmosphere, 4.5 h. The regioisomeric ratio was determined by 1H NMR analysis of the crude mixture; aI = 6 mA. | ||
While the nitrogen-radical-initiated cascade cyclizations have been heavily investigated, the corresponding migratory cyclization through a spirocyclic intermediate is much less explored. Density-functional-theory calculations (DFT) provided more mechanistic insights into the origin of this selectivity (Scheme 5). We calculated two radical addition pathways for the 2-methyl substituted vinyl radical (A) and found while both pathways were thermodynamically and kinetically favorable, the reaction barrier of ipso-addition is 1.8 kcal mol−1 lower than that of ortho-addition (TS1versusTS1′ is 10.7 versus 8.9 kcal mol−1). By contrast, the energy difference of ipso-addition versus ortho-addition for the analog 4-methyl substituted vinyl radical (A1) is only 0.2 kcal mol−1 (see Fig. S9.3†). Those results suggest the incorporation of a 2-methyl substituent is indeed beneficial to the ipso-addition of the vinyl radical.
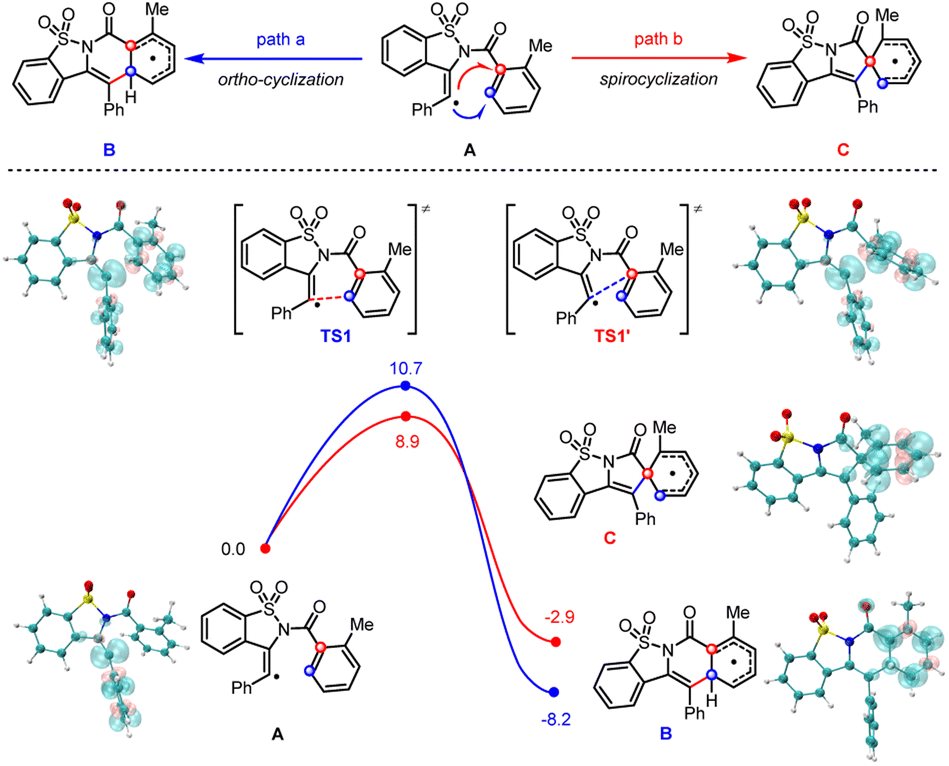 | ||
| Scheme 5 A proposed mechanism and potential energy surface of the addition process for the 2-methyl substituted vinyl radical (A). The Gibbs free energies are given in kcal mol−1. The spin density plots of each species are given (isovalue = 0.005 a.u.). | ||
The spirocyclic radical (C) is known to be readily oxidized into its corresponding spirocyclic cation (C′)22via a radical-polar-crossover process,23 which is supposed to be the key intermediate in migratory cyclization. According to our calculations (Scheme 6), the spirocyclic cation (C′) displayed an elongated Cipso–C1 bond (1.662 Å versus 1.569 Å) and a shortened Cipso–C2 bond (1.536 Å versus 1.556 Å) compared to those of the radical intermediate (C), suggesting that the acyl migration (toward the migratory cyclization) should be more accessible than the vinyl migration (toward net ortho-cyclization) through a spirocyclic cation intermediate.
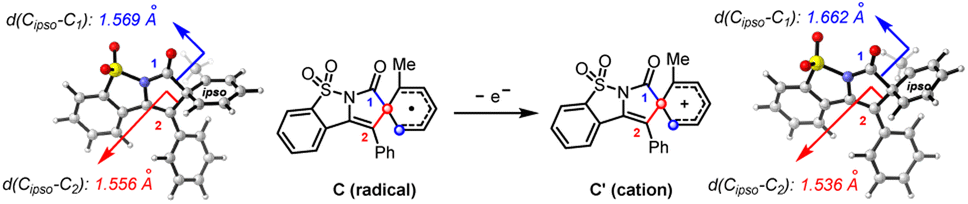 | ||
| Scheme 6 DFT calculations on the spirocyclic radical (C) and cation (C′). | ||
Consistent with the above hypothesis, the cationic acyl migration (path d) was both kinetically and thermodynamically more favorable than the vinyl migration (path c, Scheme 7). However, the small difference between the reaction barriers of these two transition states (7.6 versus 8.1 kcal mol−1) suggested only a moderate preference for migratory cyclization (E) over ortho-cyclization (D) could be achieved, which is also consistent with our experimental observations. By contrast, calculations on the migratory attitudes of the spirocyclic radical intermediate (C) revealed no such preference for acyl migration (see Fig. S9.5†). The proposed migratory mechanism could be further substantiated by a couple of methyl-substituted products, i.e., 4a/5a and 4m/5m, derived from ortho- and para-Me substrates, respectively. Their structures have been unambiguously confirmed by the X-ray diffraction analyses (see ESI†).
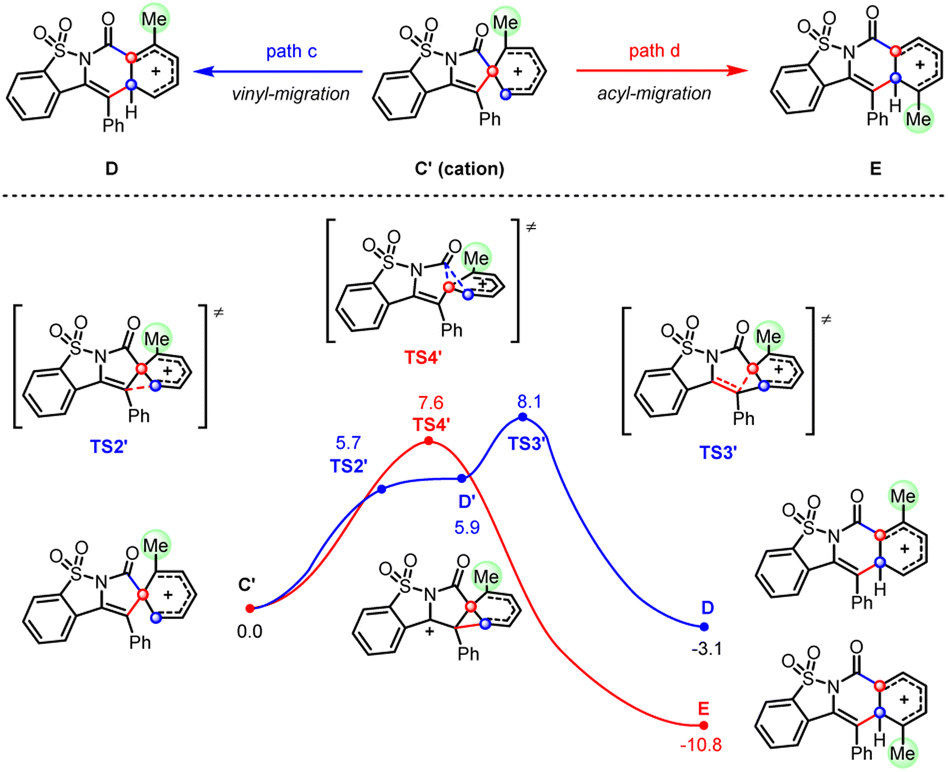 | ||
| Scheme 7 DFT calculations on vinyl- and acyl-migration of spirocation (C′). | ||
Finally, the existence of a spirocyclic cation intermediate was further substantiated by the facile dearomative spirocyclization of a p-methoxyl 2-alkynylbenzenesulfonamide derivative (Scheme 8).6c,24
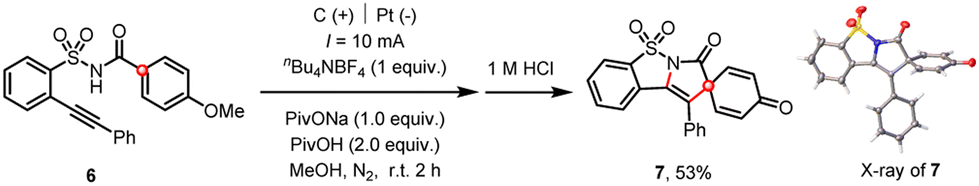 | ||
| Scheme 8 Electrochemical cascade dearomative spirocyclization. | ||
In summary, we have developed an electrochemical cascade ortho-cyclization of 2-alkynylbenzenesulfonamides to afford the corresponding polycyclic heterocycles of promising antibacterial activity. Remarkably, the incorporation of an extra 2-methyl substituent facilitates the formation of a spirocyclic dienyl radical and also favors the cationic migration of the acyl- over vinyl-linker of the key spirocyclic cation, leading to the selective formation of the rarely explored migratory cyclization products. These findings bring new insights into the fine-tuning of the migratory attitude of the linkers of spirocyclic intermediates for selective transformation of radical chemistry.
Data availability
Detailed synthetic procedures and complete characterization data for all new compounds can be found in the ESI.†Author contributions
K. Y. conceived the concept and directed the investigations. Z. S. and T. L. conducted the majority of the experimental work. W. W. and N. L. contributed to the preparation of substrates. J. Z. and S. D. designed and carried out all DFT calculations. K. Y., J. Z., and Y. Y. wrote the manuscript with input from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Zhi-Xiang Yu (Peking University) for the helpful discussions. Financial support from the National Natural Science Foundation of China (No. 22171046) and the Hundred-Talent Project of Fujian (No. 50021113) is gratefully acknowledged.Notes and references
- (a) M. P. Sibi and T. R. Rheault, Enantioselective Radical Reactions, WILEY-VCH Verlag GmbH, Germany, 2001 Search PubMed; (b) J. C. Walton, Analysis of Radicals by EPR, John Wiley & Sons, Ltd, Germany, 2012 Search PubMed.
- (a) D. Liang, W. Yu, N. Nguyen, J. R. Deschamps, G. H. Imler, Y. Li, A. D. MacKerell Jr, C. Jiang and F. Xue, J. Org. Chem., 2017, 82, 3589–3596 CrossRef CAS; (b) G. Revol, T. McCallum, M. Morin, F. Gagosz and L. Barriault, Angew. Chem., Int. Ed., 2013, 52, 13342–13345 CrossRef CAS PubMed.
- (a) N. Fuentes, W. Kong, L. Fernández-Sánchez, E. Merino and C. Nevado, J. Am. Chem. Soc., 2015, 137, 964–973 CrossRef CAS PubMed; (b) E. Brachet, L. Marzo, M. Selkti, B. König and P. Belmont, Chem. Sci., 2016, 7, 5002–5006 RSC; (c) Q.-Q. Zhao, X.-Q. Hu, M.-N. Yang, J.-R. Chen and W.-J. Xiao, Chem. Commun., 2016, 52, 12749–12752 RSC; (d) V. Quint, F. Morlet-Savary, J. F. Lohier, J. Lalevee, A. C. Gaumont and S. Lakhdar, J. Am. Chem. Soc., 2016, 138, 7436–7441 CrossRef CAS PubMed; (e) S. C. Cosgrove, J. M. C. Plane and S. P. Marsden, Chem. Sci., 2018, 9, 6647–6652 RSC. For selective review, see (f) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC; (g) N. Chen and H.-C. Xu, Green Synth. Catal., 2021, 2, 165–178 CrossRef.
- (a) Z.-M. Chen, X.-M. Zhang and Y.-Q. Tu, Chem. Soc. Rev., 2015, 44, 5220–5245 RSC; (b) W. Li, W. Xu, J. Xie, S. Yu and C. Zhu, Chem. Soc. Rev., 2018, 47, 654–667 RSC; (c) J. Xuan and A. Studer, Chem. Soc. Rev., 2017, 46, 4329–4346 RSC; (d) B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505–3521 RSC; (e) A. Bhunia and A. Studer, Chem, 2021, 7, 2060–2100 CrossRef CAS.
- For selective Smiles rearrangement, see (a) W. Kong, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2014, 53, 5078–5082 CrossRef CAS PubMed; (b) T. M. Monos, R. C. McAtee and C. R. J. Stephenson, Science, 2018, 361, 1369–1373 CrossRef CAS; (c) N. Radhoff and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 3561–3565 CrossRef CAS PubMed; (d) W. E. Truce, E. M. Kreider and W. W. Brand, The Smiles and Related Rearrangements of Aromatic Systems, 2011 Search PubMed; (e) X. Wu and C. Zhu, Acc. Chem. Res., 2020, 53, 1620–1636 CrossRef CAS PubMed.
- (a) C. R. Azpilcueta-Nicolas, D. Meng, S. Edelmann and J. P. Lumb, Angew. Chem., Int. Ed., 2023, 62, e202215422 CrossRef CAS PubMed; (b) Z. P. Yang, R. Jiang, Q. F. Wu, L. Huang, C. Zheng and S. L. You, Angew. Chem., Int. Ed., 2018, 57, 16190–16193 CrossRef CAS PubMed; (c) C. Zhang, F. Bu, C. Zeng, D. Wang, L. Lu, H. Zhang and A. Lei, CCS Chem., 2022, 4, 1199–1207 CrossRef CAS; (d) N. Li, Z. Shi, W.-Z. Wang, Y. Yuan and K.-Y. Ye, Chem.–Asian J., 2023, 18, e202300122 CrossRef CAS PubMed; (e) W.-C. Yang, M.-M. Zhang and J.-G. Feng, Adv. Synth. Catal., 2020, 362, 4446–4461 CrossRef CAS; (f) W.-T. Wu, L. Zhang and S.-L. You, Chem. Soc. Rev., 2016, 45, 1570–1580 RSC; (g) W. C. Wertjes, E. H. Southgate and D. Sarlah, Chem. Soc. Rev., 2018, 47, 7996–8017 RSC; (h) J. Bariwal, L. G. Voskressensky and E. V. Van der Eycken, Chem. Soc. Rev., 2018, 47, 3831–3848 RSC.
- (a) D. W. Bayne, A. J. Nicol and G. Tennant, J. Chem. Soc., Chem. Commun., 1975, 782–783 RSC; (b) Y.-R. Chen and W.-L. Duan, J. Am. Chem. Soc., 2013, 135, 16754–16757 CrossRef CAS PubMed; (c) V. K. Soni, H. S. Hwang, Y. K. Moon, S. W. Park, Y. You and E. J. Cho, J. Am. Chem. Soc., 2019, 141, 10538–10545 CrossRef CAS PubMed; (d) C.-H. Luo, P.-L. Wang and C.-C. Chang, J. Org. Chem., 2021, 86, 15033–15044 CrossRef CAS PubMed.
- (a) H. Keum, H. Jung, J. Jeong, D. Kim and S. Chang, Angew. Chem., Int. Ed., 2021, 60, 25235–25240 CrossRef CAS PubMed; (b) L.-R. Song, H. Li, S.-F. Wang, J.-P. Lin, B. Huang and Y.-Q. Long, Chem. Commun., 2022, 58, 8340–8343 RSC.
- (a) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC; (b) T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484–2496 CrossRef CAS; (c) K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed; (d) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC; (e) A. Wiebe, T. Gieshoff, S. Mohle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed; (f) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- (a) L. Ackermann, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed; (b) K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS PubMed; (c) C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed; (d) J. L. Rockl, D. Pollok, R. Franke and S. R. Waldvogel, Acc. Chem. Res., 2020, 53, 45–61 CrossRef PubMed; (e) F. Wang and S. S. Stahl, Acc. Chem. Res., 2020, 53, 561–574 CrossRef CAS PubMed; (f) K. Yamamoto, M. Kuriyama and O. Onomura, Acc. Chem. Res., 2020, 53, 105–120 CrossRef CAS PubMed; (g) Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS PubMed.
- (a) C. Pratley, S. Fenner and J. A. Murphy, Chem. Rev., 2022, 122, 8181–8260 CrossRef CAS PubMed; (b) X.-Y. Yu, Q.-Q. Zhao, J. Chen, W.-J. Xiao and J.-R. Chen, Acc. Chem. Res., 2020, 53, 1066–1083 CrossRef CAS PubMed; (c) P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. McLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Chem. Rev., 2022, 122, 2017–2291 CrossRef CAS PubMed.
- (a) R. Li, D. Yuan, M. Ping, Y. Zhu, S. Ni, M. Li, L. Wen and L.-B. Zhang, Chem. Sci., 2022, 13, 9940–9946 RSC; (b) Z. Shi, Y. Li, N. Li, W.-Z. Wang, H.-K. Lu, H. Yan, Y. Yuan, J. Zhu and K.-Y. Ye, Angew. Chem., Int. Ed., 2022, 61, e202206058 CrossRef CAS PubMed.
- Y. Liang, Y. Simon-Manso, P. Neta, X. Yang and S. E. Stein, J. Am. Soc. Mass Spectrom., 2021, 32, 806–814 CrossRef CAS PubMed.
- (a) Y.-J. Chen, Y.-L. Qu, X. Li and C.-C. Wang, Org. Biomol. Chem., 2020, 18, 8975–8993 RSC; (b) M.-H. Huang, W.-J. Hao, G. Li, S.-J. Tu and B. Jiang, Chem. Commun., 2018, 54, 10791–10811 RSC; (c) Y. Zhang, Z. Cai, S. Warratz, C. Ma and L. Ackermann, Sci. China: Chem., 2022, 66, 703–724 Search PubMed.
- (a) J. Liao, X. Yang, L. Ouyang, Y. Lai, J. Huang and R. Luo, Org. Chem. Front., 2021, 8, 1345–1363 RSC; (b) Y. Liu, C. Zhang, D. Chen and J.-P. Wan, Synthesis, 2023, 55, 2911–2925 CrossRef; (c) Z. Shi, N. Li, W.-Z. Wang, H.-K. Lu, Y. Yuan, Z. Li and K.-Y. Ye, Org. Biomol. Chem., 2022, 20, 4320–4323 RSC.
- (a) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS PubMed; (b) P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 52, 3339–3350 CrossRef CAS PubMed.
- P. C. Zheng, J. Cheng, S. Su, Z. Jin, Y. H. Wang, S. Yang, L. H. Jin, B. A. Song and Y. R. Chi, Chem.–Eur. J., 2015, 21, 9984–9987 CrossRef CAS PubMed.
- (a) Z.-W. Hou, Z. Y. Mao, Y.-Y. Melcamu, X. Lu and H.-C. Xu, Angew. Chem., Int. Ed., 2018, 57, 1636–1639 CrossRef CAS PubMed; (b) Z.-W. Hou, H. Yan, J.-S. Song and H.-C. Xu, Chin. J. Chem., 2018, 36, 909–915 CrossRef CAS; (c) P. Xiong, H.-H. Xu, J. Song and H.-C. Xu, J. Am. Chem. Soc., 2018, 140, 2460–2464 CrossRef CAS PubMed; (d) F. Xu, H. Long, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2019, 58, 9017–9021 CrossRef CAS PubMed.
- S. P. Dunn, M. J. Walters, C. R. Metz, C. F. Beam, W. T. Pennington and M. Krawiec, J. Heterocycl. Chem., 2004, 41, 1005–1009 CrossRef CAS.
- (a) C. Costentin, Chem. Rev., 2008, 108, 2145–2179 CrossRef CAS PubMed; (b) E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef CAS PubMed; (c) X. Hu, G. Zhang, F. Bu, L. Nie and A. Lei, ACS Catal., 2018, 8, 9370–9375 CrossRef CAS; (d) Z.-J. Wu and H.-C. Xu, Angew. Chem., Int. Ed., 2017, 56, 4734–4738 CrossRef CAS PubMed.
- (a) X. Cheng, A. Lei, T.-S. Mei, H.-C. Xu, K. Xu and C. Zeng, CCS Chem., 2022, 4, 1120–1152 CrossRef CAS; (b) C. Ma, P. Fang, Z.-R. Liu, S.-S. Xu, K. Xu, X. Cheng, A. Lei, H.-C. Xu, C. Zeng and T.-S. Mei, Sci. Bull., 2021, 66, 2412–2429 CrossRef CAS PubMed.
- R. J. Wiles and G. A. Molander, Isr. J. Chem., 2020, 60, 281–293 CrossRef CAS PubMed.
- (a) J. A. Murphy, in Radicals in Organic Synthesis, WILEY-VCH Verlag GmbH, Germany, 2001, pp. 298–315 Search PubMed; (b) L. Pitzer, J. L. Schwarz and F. Glorius, Chem. Sci., 2019, 10, 8285–8291 RSC.
- (a) N. Li, Z. Shi, Y. Yuan, Z. Li and K.-Y. Ye, Org. Chem. Front., 2022, 9, 6586–6591 RSC; (b) W. Wei, A. Scheremetjew and L. Ackermann, Chem. Sci., 2022, 13, 2783–2788 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2272008–2272012. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05229j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |