
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Disrupting the Hofmeister bias in salt liquid–liquid extraction with an arylethynyl bisurea anion receptor†
Hazel A.
Fargher‡
 a,
Lætitia H.
Delmau
b,
Vyacheslav S.
Bryantsev
c,
Michael M.
Haley
*a,
Darren W.
Johnson
*a and
Bruce A.
Moyer
*c
a,
Lætitia H.
Delmau
b,
Vyacheslav S.
Bryantsev
c,
Michael M.
Haley
*a,
Darren W.
Johnson
*a and
Bruce A.
Moyer
*c
aDepartment of Chemistry and Biochemistry, Materials Science Institute, University of Oregon, Eugene, OR 97403-1253, USA. E-mail: haley@uoregon.edu; dwj@uoregon.edu
bRadioisotope Science and Technology Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831-6384, USA
cChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831-6119, USA. E-mail: moyerba@ornl.gov
First published on 7th March 2024
Abstract
Host-mediated liquid–liquid extraction is a convenient method for the separation of inorganic salts. However, selective extraction of an anion, regardless of its hydrophilicity or lipophilicity as qualitatively described by its place in the Hofmeister series, remains challenging. Herein we report the complete disruption of the Hofmeister-based ordering of anions in host-mediated extraction by a rigidified tweezer-type receptor possessing remarkably strong anion-binding affinity under the conditions examined. Experiments introduce a convenient new method for determination of anion binding using phosphorus inductively coupled plasma mass spectrometry (ICP-MS) to measure extraction of tetra-n-butylphosphonium (TBP+) salts from water into nitrobenzene, specifically examining the disrupting effect of the added arylethynyl bisurea anion receptor. In the absence of the receptor, the salt partitioning follows the expected Hofmeister-type ordering favoring the larger, less hydrated anions; the analysis yields the value −24 kJ mol−1 for the standard Gibbs energy of partitioning of TBP+ cation from water into nitrobenzene at 25 °C. Selectivity is markedly changed by the addition of receptor to the nitrobenzene and is concentration dependent, giving rise to three selectivity regimes. We then used SXLSQI liquid–liquid equilibrium analysis software developed at Oak Ridge National Laboratory to fit host-mediated extraction equilibria for TBP+ salts of Cl−, Br−, I−, and NO3− to the distribution data. While the reverse-Hofmeister 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding of the anions by the receptor effectively cancels the Hofmeister selectivity of the TBPX partitioning into nitrobenzene, formation of unexpected 2:1 receptor:anion complexes favoring Cl− and Br− dominates the selectivity at elevated receptor concentrations, producing the unusual order Br− > Cl− > NO3− > I− in anion distribution wherein a middle member of the series is selected and the most lipophilic anion is disfavored. Density functional theory calculations confirmed the likelihood of forming 2:1 complexes, where Cl− and Br− are encapsulated by two receptors adopting energetically competitive single or double helix structures. The calculations explain the rare non-Hofmeister preference for Br−. This example shows that anion receptors can be used to control the selectivity and efficiency of salt extraction regardless of the position of the anion in the Hofmeister series.
:1 binding of the anions by the receptor effectively cancels the Hofmeister selectivity of the TBPX partitioning into nitrobenzene, formation of unexpected 2:1 receptor:anion complexes favoring Cl− and Br− dominates the selectivity at elevated receptor concentrations, producing the unusual order Br− > Cl− > NO3− > I− in anion distribution wherein a middle member of the series is selected and the most lipophilic anion is disfavored. Density functional theory calculations confirmed the likelihood of forming 2:1 complexes, where Cl− and Br− are encapsulated by two receptors adopting energetically competitive single or double helix structures. The calculations explain the rare non-Hofmeister preference for Br−. This example shows that anion receptors can be used to control the selectivity and efficiency of salt extraction regardless of the position of the anion in the Hofmeister series.
Introduction
Separation of inorganic ions from complex solutions is a major challenge,1 with applications in mining,2 environmental remediation,3,4 resource recovery,5 nuclear-fuel recycle,6,7 and waste treatment,8 to name just a few. Advancing this field, host-mediated liquid–liquid extraction (LLE) has emerged as a powerful approach to impart enhanced selectivity and high binding affinities for desired guest ions.9–12 Research into host-mediated LLE has explored the influence of receptors on cation,13 anion,14 and ion-pair15 extraction. A significant body of this work has focused on the development of cation receptors, whereas complementary studies on anion receptors have lagged in comparison.10 In the absence of an anion receptor, the selectivity of partitioning from water into an organic phase is biased toward anions with increasing size or decreasing charge density, owing to the dominance of hydration energy vs. weaker solvation afforded by typical water-immiscible organic solvents.16,17 Thus, one obtains the hydration-based ordering originally noticed by Hofmeister,18 which persists widely in diverse chemical processes controlled by ion hydration.19 While Hofmeister-type selectivity is extremely useful, it has been particularly challenging to achieve desirable selectivity for small, highly hydrated anions in LLE. Even in the presence of anion receptors, one most often observes attenuated20 or perturbed21,22 Hofmeister ordering, as can be further influenced positively or negatively by ion-pairing. Given that most design strategies for anion receptors rely on hydrogen-bond donor groups, which in LLE must compete with the strong hydrogen-bonding received by the anion in the aqueous phase, extraordinary selectivity and affinity are needed. Accordingly, we recognize that overcoming the Hofmeister bias in LLE requires maximizing strong, complementary, and preorganized donor groups, in line with now-classical principles.23,24 Herein we present a case in which a rigidified tweezer-type receptor neatly fulfills these requirements, using novel methodology to show how LLE selectivity transitions from normal- to disrupted-Hofmeister behavior in the tug of war between the selectivity of anion partitioning and anion binding.We have introduced a family of arylethynyl bisurea receptors and studied their anion binding ability in homogenous organic solutions.25–29 The rigidity of the arylethynyl pivot is thought to impart greater anion binding affinity and selectivity in this tweezer-type receptor, which also features an active C–H donor group to complement its four N–H donors. We have also shown that the arylethynyl urea scaffold can be used in the design of receptors capable of selective extraction of hydrogen sulfate from sulfuric acid.30 Now we hypothesize that our receptors can be used to disrupt the Hofmeister type selectivity in LLE. Within the general framework of ways to deploy host-mediated LLE,10 this idea has previously been illustrated in so-called dual-host31 salt extraction (combination of cation and anion receptors) or synergized anion-exchange21 (combination of anion receptor with anion exchanger) systems. While it has been possible to use anion receptors alone to effect weak salt partitioning with inorganic cations in special cases,21 strong salt extraction is readily achievable with the use of sufficiently lipophilic cations such as long-chain quaternary ammoniums.21,32 To test our hypothesis, we use established arylethynyl bisurea anion receptor R (Fig. 1)33,34 with the lipophilic tetrabutylphosphonium (TBP+) cation to provide a novel handle for measuring salt extraction via phosphorus inductively coupled plasma mass spectrometry (ICP-MS).
 | ||
| Fig. 1 Anion receptor R used in this study. | ||
Methods
Approach
Anion receptor R was chosen for this study due to its high anion-binding affinity in organic solvents, simple expected 1:1 host:guest binding stoichiometry, and negligible water solubility.33,34 The magnitude of the partition ratio of R between the organic and aqueous phases is estimated to be 1.1 × 1010 by the calculated logP function in ChemDraw,35 which is excessively sufficient to ensure that the exceedingly small fraction of R partitioned to the aqueous phase can be neglected. For this study, R was synthesized according to previously reported methods.33 TBP salts (TBPX, where X− = Cl−, Br−, I−, and NO3−) were chosen for extraction experiments so that the change in aqueous P concentrations before and after extraction, measured by ICP-MS, could be used to measure salt partitioning. TBP salts were synthesized by reaction of aqueous TBPOH with the corresponding acid (HCl, HBr, HI, HNO3) and characterized by 31P NMR spectroscopy. Density functional theory (DFT) calculations were conducted to validate the possibility of forming stable 2:1 receptor:anion complexes with Cl− and Br−. Further details on syntheses, materials, instrumentation, equipment, and DFT calculations are described in ESI.†
Liquid–liquid extraction experiments
As detailed in ESI,† aqueous solutions containing variable concentrations of TBPX (0.02–0.03 mM, determined by ICP-MS) were equilibrated with equal volumes of purified nitrobenzene containing variable concentrations of R (2, 1.5, 1, 0.5, 0.1, 0.07, 0.05, and 0 mM). The maximum concentration of 2 mM was constrained by the solubility of R in nitrobenzene (<5 mM). After centrifugation, samples of the aqueous phases were removed and analyzed for P by ICP-MS before ([TBP]0) and after ([TBP]aq) equilibration. The distribution ratio DP, defined as [TBP]org/[TBP]aq, was then determined, where [TBP]org is known by mass balance from the difference [TBP]0 − [TBP]aq.Results and discussion
General equilibrium model
The host-mediated LLE system investigated herein can be represented by a simple model given by the equilibria depicted in Fig. 2. Shown is salt partitioning of TBP+ cation and anion X− from the aqueous to the organic phase, followed by complexation of the anion by R. Polar nitrobenzene provides for complete ion-pair dissociation in the organic phase at the low concentrations used, simplifying both analysis and understanding of binding. For simplicity of illustration consistent with our initial hypothesis, we include here only the anion host–guest complex with 1:1 binding stoichiometry with the understanding that general complex stoichiometries (RmXn−) can be accommodated (as in fact found necessary) in a straightforward, albeit more complicated, manner. The equations defining equilibrium constants are given in ESI.† The overall host-mediated extraction constant (Kex±) is the product of the salt partitioning constant (Kp) and the formation constant (Kf) of anion binding with host R (eqn (1)). Kex± and Kp are determined directly from experiment, from which Kf is derived.| Kex±(TBPRX) = Kp(TBPX) × Kf(RX−) | (1) |
 | ||
| Fig. 2 Equilibria present in host-mediated LLE of TBPX salts. Only a 1:1 RX− complex is shown for simplicity, though other stoichiometries are possible. | ||
Salt partitioning equilibria
Normal Hofmeister-type selectivity was observed in LLE of TBPX salts by nitrobenzene without added R. As shown in Fig. 3, the obtained four plots of salt distribution ratios expressed as logDP,0vs. aqueous salt concentration at equilibrium ([TBPX]aq) follow the order I− > NO3− > Br− > Cl−. Corresponding data for liquid–liquid extractions of 0.0014–12.62 mM TBPX salts are given in Tables S1–S4.† A flat dependence is observed as expected (eqn (S7)†), where a slight activity-coefficient-induced curvature at the higher TBPX concentrations is captured by equilibrium analysis (solid lines in Fig. 3) using the program SXLSQI36,37 (Solvent eXtraction Least SQuares—Ion; see ESI†). The analysis yields corresponding equilibrium constants (logKp) as given in Table 1. Given the known values of the standard Gibbs energies of anion partitioning,  ,38 the previously unknown value of
,38 the previously unknown value of  was found to be −24 ± 1 kJ mol−1, revealing the push–pull dynamics of favorable cation partitioning overcoming unfavorable anion partitioning. Within experimental error, the observed value of
was found to be −24 ± 1 kJ mol−1, revealing the push–pull dynamics of favorable cation partitioning overcoming unfavorable anion partitioning. Within experimental error, the observed value of  turns out to be the same as that for the more commonly used tetra-n-butyl ammonium cation.39
turns out to be the same as that for the more commonly used tetra-n-butyl ammonium cation.39
 | ||
| Fig. 3 Experimental and calculated logDP,0vs. equilibrium log[TBPX]aq for TBPCl, TBPBr, TBPI, and TBPNO3. Solid lines represent the calculated behavior using SXLSQI (vide infra). | ||
Kp for TBPCl, TBPBr, TBPI, and TBPNO3 partitioning from water into nitrobenzene at 25 °C calculated by SXLSQI and determination of the standard Gibbs energy of TBP+ partitioning,  a
a
| Salt | Average logKp |
Equilibrium |
|
|
|---|---|---|---|---|
a Single-ion standard Gibbs energies of partitioning shown here use the TATB (tetraphenylarsonium tetraphenylborate) extrathermodynamic assumption.40 Values of logKp were converted to corresponding Gibbs energies according to  = −2.303RTlogKp. See ESI for full discussion. = −2.303RTlogKp. See ESI for full discussion.
|
||||
| TBPCl | −1.94 ± 0.09 | TBP+(aq) + Cl−(aq) ⇌ TBP+(org) + Cl−(org) | 35 | −24 ± 1 |
| TBPBr | −0.81 ± 0.02 | TBP+(aq) + Br−(aq) ⇌ TBP+(org) + Br−(org) | 29 | −24 ± 1 |
| TBPI | 0.97 ± 0.03 | TBP+(aq) + I−(aq) ⇌ TBP+(org) + I−(org) | 18 | −24 ± 1 |
| TBPNO3 | 0.070 ± 0.007 | TBP+(aq) + NO3−(aq) ⇌ TBP+(org) + NO3−(org) | 24 | −24 ± 1 |
| Average = | −24 ± 1 | |||


Host-mediated extraction disrupts Hofmeister selectivity
DP are plotted against log[R]0 for Cl−, Br−, I−, and NO3− in Fig. 4a–d, respectively; see ESI† for additional plots (Fig. S11†). With decreasing values of the host concentration, logDP asymptotically approaches the baseline values of logDP,0 shown for each salt in Fig. 3 (demarked by solid lines in Fig. 4a and b). With increasing host concentration, however, extraction for all anions becomes more favorable, and at the highest initial R concentration (2 mM), an obviously disrupted Hofmeister bias emerges in terms of overall extraction strength (DP): NO3− (9.54 ± 0.06) ∼ Br− (9.4 ± 0.1) > I− (7.11 ± 0.08) > Cl− (5.80 ± 0.06).
 | ||
| Fig. 4 Experimentally determined logDP plotted as a function of log[R]0 for (a) TBPCl, (b) TBPBr, (c) TBPI, and (d) TBPNO3, where [R]0 is the intitial concentration of R. Solid lines demark DP,0 for each salt. Linear dashed segments denote an approximate slope analysis, which suggests formation of a 1:1 anion complex RX− for nitrate with mixed 1:1 and 2:1 species RmX− for chloride and bromide according to predicted slope 0.5m. Enhancement for TBPI is insufficient for slope analysis. (See ESI†). | ||
:1 and 2:1 RmX− complexes. While the simple model shown in Fig. 2 hypothesized a 1:1 host:anion binding stoichiometry, the dashed-line behavior shown in Fig. 4 suggests that 2:1 binding is also important for chloride and bromide, with special implications for selectivity (see ESI†). More exact modeling of the extraction data using the SXLSQI liquid–liquid equilibrium analysis software36,37 corroborated the qualitative insight from slope analysis and yielded extraction constants for all four anions, including iodide. Equilibria for the host-mediated models considered most valid for each anion are shown together with corresponding calculated logKex± in Table 2. The complete set of data (Tables S1–S8†) was treated, estimating activity coefficients for species in both phases in the calculation using parameters given in Tables S11–S14.† Plots comparing observed and calculated points are shown in Fig. S12–S15.†
Kex±) using SXLSQI for host-mediated extraction of TBPCl, TBPBr, TBPI, and TBPNO3 from water into nitrobenzene at 25 °Ca
| TBPX | Equilibrium | LogKex± |
|---|---|---|
|
a Observed values of DP were taken from Tables S5–S8, yielding the corresponding output values of refined logKex±. In the fitting of the extraction data for each anion, the corresponding logKp values were fixed at the values shown in Table 1 to account for the background extraction of TBPX by nitrobenzene alone.
|
||
| TBPCl | TBP+(aq) + Cl−(aq) + R(org) ⇌ TBP+(org) + RCl−(org) | 3.9 ± 0.2 |
| TBP+(aq) + Cl−(aq) + 2R(org) ⇌ TBP+(org) + R2Cl−(org) | 6.7 ± 0.2 | |
| TBPBr | TBP+(aq) + Br−(aq) + R(org) ⇌ TBP+(org) + RBr−(org) | 4.17 ± 0.06 |
| TBP+(aq) + Br−(aq) + 2R(org) ⇌ TBP+(org) + R2Br−(org) | 7.20 ± 0.07 | |
| TBPI | TBP+(aq) + I−(aq) + R(org) ⇌ TBP+(org) + RI−(org) | 4.34 ± 0.06 |
| TBPNO3 | TBP+(aq) + NO3−(aq) + R(org) ⇌ TBP+(org) + RNO3−(org) | 4.65 ± 0.03 |
Using the equilibrium constants given in Tables 1 and 2 to calculate smoothed distribution profiles illustrates the progression from normal to disrupted Hofmeister ordering (Fig. 5). Dominant partitioning of TBPX yields normal Hofmeister behavior at low concentration of R, giving way to disrupted Hofmeister ordering controlled by host-mediated extraction. At mid concentrations of R, the distribution ratios tend to converge with five crossover points. As the steepness of the chloride and bromide curves increases owing to the onset of 2:1 R2X− complex formation, the distribution ratios diverge, giving rise to the order Br− > Cl− > NO3− > I− at the highest plotted concentrations. Selection of a middle member of the Hofmeister series is quite unusual in LLE systems as is rejection of the largest, most lipophilic anion in the series.
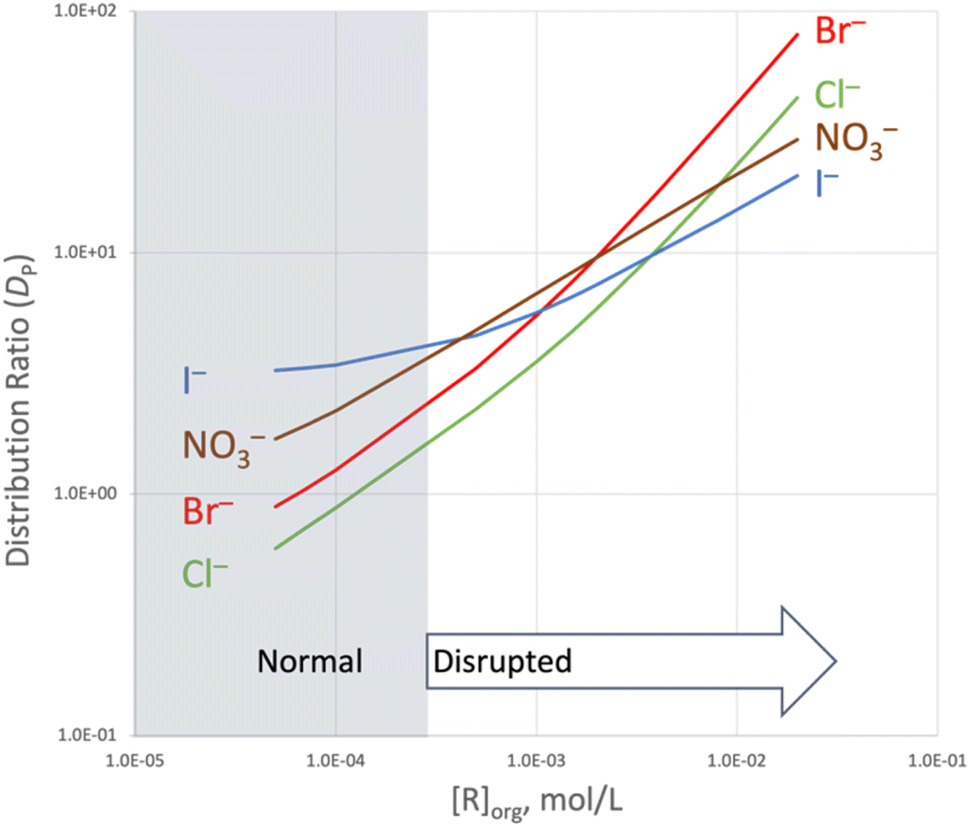 :1 and 2:1 binding in disrupting Hofmeister selectivity. From the logKex± values shown in Table 2 for formation of 1:1 complexes, one may see that the Hofmeister ordering is largely canceled by the anion host. The rendered order NO3− > I− > Br− > Cl− of log Kex for 1:1 complexes is weak, with a spread of only 0.7 log units vs. a spread of 2.0 log units for salt partitioning (Table 1). The propensity of chloride and bromide to form 2:1 complexes proves decisive at higher receptor concentrations, where the corresponding 2:1 logKex± values are three log units higher than the corresponding 1:1 logKex± values. Extrapolating the model to 20 mM receptor (Fig. 5) accentuates the effect of the 2:1 binding on DP.
:1 and 2:1 binding in disrupting Hofmeister selectivity. From the logKex± values shown in Table 2 for formation of 1:1 complexes, one may see that the Hofmeister ordering is largely canceled by the anion host. The rendered order NO3− > I− > Br− > Cl− of log Kex for 1:1 complexes is weak, with a spread of only 0.7 log units vs. a spread of 2.0 log units for salt partitioning (Table 1). The propensity of chloride and bromide to form 2:1 complexes proves decisive at higher receptor concentrations, where the corresponding 2:1 logKex± values are three log units higher than the corresponding 1:1 logKex± values. Extrapolating the model to 20 mM receptor (Fig. 5) accentuates the effect of the 2:1 binding on DP.
Binding constants provide further insight. More familiar to coordination chemists, anion binding constants logKf for each salt with R are obtained here by subtracting logKp from logKex±. We have generally found that binding constants determined by rigorous analysis of LLE data match those obtained by other methods (for example, see ref. 31). These results are shown in Table 3. It may be seen that the 1:1 binding constants follow the reversed-Hofmeister order of extraction Cl− > Br− > NO3− > I−. Flexible anion receptors often follow this trend, where the hydrogen bond strength increases with decreasing anion size or increasing charge density. Although R binds Cl− an order of magnitude more favorably than Br−, the binding of a second R with the RX− complex is statistically at least as large for Br− as it is for Cl−.
Kf) for anion binding by R in water-saturated nitrobenzene as inferred from host-mediated extraction of TBPCl, TBPBr, TBPI, and TBPNO3 from water into nitrobenzene at 25 °Ca
| TBPX | Equilibrium | LogKf |
|---|---|---|
|
a Values of logKf were obtained by subtracting values logKp from logKex± for each salt taken from Tables 1 and 2, respectively.
|
||
| TBPCl | R (org) + Cl−(org) ⇌ RCl−(org) | 5.8 ± 0.2 |
| 2R(org) + Cl−(org) ⇌ R2Cl−(org) | 8.6 ± 0.1 | |
| R (org) + RCl−(org) ⇌ R2Cl−(org) | 2.8 ± 0.2 | |
| TBPBr | R (org) + Br−(org) ⇌ RBr−(org) | 4.98 ± 0.05 |
| 2R(org) + Br−(org) ⇌ R2Br−(org) | 8.02 ± 0.07 | |
| R (org) + RBr−(org) ⇌ R2Br−(org) | 3.03 ± 0.09 | |
| TBPI | R (org) + I−(org) ⇌ RI−(org) | 3.78 ± 0.05 |
| TBPNO3 | R (org) + NO3−(org) ⇌ RNO3−(org) | 4.58 ± 0.03 |
Structural evidence provides clues regarding the observed 1:1 and 2:1 anion binding stoichiometries. Previous DFT calculations for 1:1 complexes reveal that the O-atoms of nitrate are accommodated in an approximately planar array of the available N–H and C–H donors of the arylethynyl host structure.33 In accord with criteria described for nitrate binding by urea hydrogen-bond donors,40 the two urea groups of the host bind along edges of the nitrate anion. The saturation of the nitrate coordination sites by a single R molecule logically explains the tendency of the stoichiometry to remain 1:1. By contrast, an X-ray structure shows a non-planar configuration of the urea donor groups for 1:1 binding of the smaller chloride anion, one urea twisted below and one above the plane of the central benzene ring in an opposing manner. Chloride can structurally accommodate four urea donor groups,40 lending a structural basis for 2:1 binding of R to chloride or bromide. Possibly iodide would exhibit 2:1 binding in this way, but solubility limitations prevented our testing sufficiently high R concentrations. We believe that the lack of ion pairing in nitrobenzene adds another factor favoring 2:1 binding. Anion binding by R and its various modifications was studied previously using chloroform as the solvent,33 where the much lower dielectric constant (ε = 4.81)46 promotes ion pairing. Displacement of the associated cation in the C+[RX−] complex ion pair by a second molecule of R represents an electrostatic energy penalty, weakening its binding. Experimentally, three-fold lower concentrations of R were used in the reported NMR titrations, and high residuals were seen in the early points of the data fits,33 suggesting some formation of 2:1 species had still occurred.
DFT calculations supported the formation of 2:1 complexes with Cl− and Br− implied by the equilibrium analysis and additionally shed light on the unusual selectivity observed for Br−. The structural modification of 1:1 complexes to allow for the inclusion of two ligands in either a ‘sandwich’ or intertwined configuration has yielded two unique assemblies exhibiting competitive energetics. As shown in Fig. 6, two receptors are symmetrically arranged around each anion, resulting in the formation of either a single or a double helicate structure. Thermodynamic calculations (Table S16†) for R(org) + RX−(org) ⇌ R2X−(org) (X = Cl− and Br−) confirmed the stability of the 2:1 complexes, both in the gas phase and with the inclusion of an implicit solvent (nitrobenzene). We find that the formation of the 2:1 complexes with Br− is slightly more favorable than with Cl−, which is uncommon, as the more charge-diffuse Br− ion is expected to form weaker bonds with hydrogen bond donors. Examination of hydrogen bond distances, as detailed in Table S17,† shows that both Cl− and Br− strongly interact with the four urea groups in the two ligands. Yet, the extent to which the hydrogen bonds are elongated compared to the those in the 1:1 complexes is slightly larger for Cl− compared to Br−. This result aligns with the geometric patterns of binding, as the larger Br− ion offers greater spatial accessibility for binding in the tight assembly of two receptors.
 | ||
| Fig. 6 Optimized molecular structures of the complexes with Br− forming the single helix (left) and double helix (right) of R using DFT and implicit solvent model for nitrobenzene. | ||
Finally, we note the remarkably high binding constants determined for all anions with R, which we ascribe to structural rigidity as well as solvent effects. Although a reorganization penalty typically weakens host–guest complexation in flexible hosts, the conjugated system of our receptor imparts significant rigidity compared with many tweezer-type hosts, likely reducing the effect of this penalty. In addition, we have previously suggested that shape-persistent hosts may experience a greater solvation penalty and a greater ground-state destabilization in more polar solvents, compared with flexible hosts.41 The properties of the nitrobenzene diluent used here also favor stronger binding. In Table 4 we compare logKf for 1:1 binding in nitrobenzene (dielectric constant ε = 34.82)42 to binding constants of the same host in H2Osat. CHCl3 (ε ≈ 4.9)43 and 10% DMSO-d6/CD3CN (ε ≈ 42).43 Although solvent-system polarity is often a predictor of binding strength,42,44R binds all anions much more favorably in nitrobenzene compared to the two other solvent systems, irrespective of the solvent system dielectric strength. The hydrogen bond accepting and donating ability of 10% DMSO-d6/CD3CN provides for solvation of the host and the anion guest, competing with anion binding.45 One of the strongest electron-pair donors among organic solvents,44 DMSO in particular would be expected to engage the hydrogen bond donor groups of R, in accord with the lowest binding constants observed in Table 4. As mentioned above, ion pairing in the case of water-saturated CHCl3 by the TBA+ counter cation impedes binding.
Kf for 1:1 binding of the halides and nitrate with R in two different solvent systems compared with nitrobenzene at 25 °C
Conclusions
This work confirms our hypothesis that a previously published arylethynyl bisurea receptor R33,34 with enhanced rigidity would disrupt the Hofmeister-type selectivity in LLE of anions. This host enhances the extraction of four anions as TBP+ salts into nitrobenzene at 25 °C. Normal Hofmeister-type extraction selectivity (I− > NO3− > Br− > Cl−) progresses to an unusual disrupted Hofmeister selectivity (Br− > Cl− > NO3− > I−) favoring a middle member of the series and rejecting the most lipophilic anion (Fig. 5). The distribution ratios for the TBPX salts were used to calculate equilibrium constants for extraction and binding using the SXLSQI equilibrium modeling. Based on the equilibrium constants, Table 5 summarizes the underlying influence of binding on LLE selectivity of the anions, categorizing the selectivity shift as the concentration of receptor increases. Partitioning follows the normal Hofmeister order. Binding at low host concentrations follows a reversed Hofmeister order for 1:1 stoichiometry, which largely cancels the normal Hofmeister order obtained at zero host concentration; however, the disrupted Hofmeister selectivity at the elevated host concentrations turns on unexpected 2:1 binding. DFT calculations corroborate the assembly of 2:1 complexes with Cl− and Br−, indicating the possibility of forming single and double helical structures with R and explaining the selectivity for Br−. We consider the DFT support for the formation of 2:1 complexes via helicate assembly to be a breakthrough in the understanding of our rigidified tweezer molecules. To date, evidence for formation of 2:1 species has been scant, and no structural insight has been hitherto revealed, providing ample fodder for future investigation by, for example, adding electron-withdrawing groups or added structural constraints to the molecular structure to modulate selectivity.
| Regime | Dominant equilibrium | Selectivity |
|---|---|---|
|
a See Table 1 for logKp values.
b See Table 3 for logKf values.
c See Table 2 for logKex± values and Fig. 5 for comparison of distribution ratios DP as a function of [R].
|
||
| Partitioning (no host)a | TBP+(aq) + X−(aq) ⇌ TBP+(org) + X−(org) | I− > NO3− > Br− > Cl− (normal) |
| 1:1 bindingb |
R (org) + X−(org) ⇌ RX−(org) | Cl− > Br− > NO3− > I− (reverse) |
| 2:1 bindingb |
2R(org) + X−(org) ⇌ R2X−(org) | Cl− > Br− ≫ NO3−, I− (disrupted) |
| Host-mediated extractionc | TBP+(aq) + X−(aq) + R(org) ⇌ TBP+(org) + RX−(org) | NO3− > I− > Br− > Cl− (weakly disrupted) |
| Host-mediated extractionc | TBP+(aq) + X−(aq) + 2R(org) ⇌ TBP+(org) + R2X−(org) | Br− > Cl− ≫ NO3−, I− (disrupted) |
A useful by-product of this investigation is the introduction of experimental methodology for the convenient study of anion extraction and binding. The method employs ICP-MS instrumentation routinely available to many experimenters to follow the distribution of the TBP+ cation. The value −24 kJ mol−1 has been determined for its single-ion partitioning from water into nitrobenzene at 25 °C.
In general, development of selective anion receptors could be hugely beneficial in LLE applications, from improving extraction efficiencies,17 to targeting anions of interest,11,12 and even influencing cation selectivity.47 Except where Hofmeister selectivity is desired, advances will hinge on understanding and controlling the selectivity of anion binding. As we and others still appreciate, disrupted Hofmeister ordering in LLE is not often observed and exceedingly difficult to achieve,20–22,31,48–51 challenged especially as applications demand inexpensive materials. Despite voluminous progress on anion receptors and recognition,52–56 the field remains in a dynamic and exciting state of development.
Data availability
Experimental methods, materials, raw data, data treatment, computational methods, and computational results are provided in ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Workforce Development for Teachers and Scientists, Office of Graduate Student Research (SCGSR) program. The SCGSR program is administered by the Oak Ridge Institute for Science and Education (ORISE) for the DOE. ORISE is managed by ORAU under contract number DE-SC0014664. This work was also supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division (B. A. M. and V. S. B.) and the NIH (R01-GM087398 to D. W. J./M. M. H.). This work was also supported by the Bradshaw and Holzapfel Research Professorship in Transformational Science and Mathematics to DWJ. ICP-MS instrumentation was supported by NSF award CHE-2117614 from the Major Research Instrumentation program. We also thank Liam Twight for ICP-MS instrument support. This research used resources of the Compute and Data Environment for Science (CADES) at the Oak Ridge National Laboratory, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC05-00OR22725. Additionally, this study utilized the resources of the National Energy Research Scientific Computing Center (NERSC), a U.S. Department of Energy Office of Science User Facility located at Lawrence Berkeley National Laboratory, operated under Contract No. DE-AC02-05CH11231 using NERSC award BES-ERCAP 0027044. All opinions expressed in this paper are the authors' and do not necessarily reflect the policies and views of DOE, ORAU, or ORISE.References
- R. M. Izatt, S. R. Izatt, R. L. Bruening, N. E. Izatt and B. A. Moyer, Chem. Soc. Rev., 2014, 43, 2451–2475 RSC.
- F. Xie, T. A. Zhang, D. Dreisinger and F. Doyle, Miner. Eng., 2014, 56, 10–28 CrossRef CAS.
- G. Naidu, S. Ryu, R. Thiruvenkatachari, Y. Choi, S. Jeong and S. Vigneswaran, Environ. Pollut., 2019, 247, 1110–1124 CrossRef CAS PubMed.
- B. Moss, Philos. Trans. R. Soc., B, 2008, 363, 659–666 CrossRef CAS PubMed.
- N. F. Attia, M. A. Jawad and A. Al-Saffar, Desalin. Water Treat., 2016, 57, 21201–21210 CrossRef CAS.
- Reprocessing and Recycling of Spent Nuclear Fuel, ed. R. Taylor, Woodhead, Amsterdam, 2015 Search PubMed.
- Advanced Separation Techniques for Nuclear Fuel Reprocessing and Radioactive Waste Treatment, ed. K. L. Nash and G. J. Lumetta, Woodhead, Amsterdam, 2011 Search PubMed.
- Science and Technology for Disposal of Radioactive Tank Wastes, ed. W. W. Schulz and N. J. Lombardo, Plenum Press, New York, 1998 Search PubMed.
- B. G. Cox and H. Schneider, Coordination and Transport Properties of Macrocyclic Compounds in Solution, Elsevier, London, 1992 Search PubMed.
- B. A. Moyer, P. V. Bonnesen, R. Custelcean, L. H. Delmau and B. P. Hay, Kem. Ind., 2005, 54, 65–87 CAS.
- G. I. Vargas-Zúñiga, Q. He and J. L. Sessler, Liquid-Liquid Separations by Supramolecular Systems, in Ion Exchange and Solvent Extraction, ed. B. A. Moyer, CRC Press, Boca Raton, FL, 2019, vol. 23, pp. 45–82 Search PubMed.
- S. Kundu, T. K. Egboluche and M. A. Hossain, Acc. Chem. Res., 2023, 56, 1320–1329 CrossRef CAS PubMed.
- H. K. Frensdorff, J. Am. Chem. Soc., 1971, 93, 4684–4688 CrossRef CAS.
- N. H. Evans and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11716–11754 CrossRef CAS PubMed.
- S. K. Kim and J. L. Sessler, Chem. Soc. Rev., 2010, 39, 3784–3809 RSC.
- B. A. Moyer and P. V. Bonnesen, Physical Factors in Anion Separations, in Supramolecular Chemistry of Anions, ed. A. Bianchi, K. Bowman-James and E. García-España, Wiley-VCH, New York, NY, 1997, pp. 1–44 Search PubMed.
- T. G. Levitskaia, L. Maya, G. J. Van Berkel and B. A. Moyer, Inorg. Chem., 2007, 46, 261–272 CrossRef CAS PubMed.
- F. Hofmeister, Archiv fur Experimentelle Pathologie und Pharmakologie, 1887, 24, 247–260 CrossRef.
- W. Kunz, J. Henle and B. W. Ninham, Curr. Opin. Colloid Interface Sci., 2004, 9, 19–37 CrossRef CAS.
- K. Kavallieratos and B. A. Moyer, Chem. Commun., 2001, 1620–1621 RSC.
- C. J. Borman, P. V. Bonnesen and B. A. Moyer, Anal. Chem., 2012, 84, 8214–8221 CrossRef CAS PubMed.
- T. G. Levitskaia, M. Marquez, J. L. Sessler, J. A. Shriver, T. Vercouter and B. A. Moyer, Chem. Commun., 2003, 2248–2249 RSC.
- D. J. Cram, J. Inclusion Phenom., 1988, 6, 397–413 CrossRef CAS.
- J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH, Weinheim, 1995 Search PubMed.
- C. N. Carroll, B. A. Coombs, S. P. McClintock, C. A. Johnson, O. B. Berryman, D. W. Johnson and M. M. Haley, Chem. Commun., 2011, 47, 5539–5541 RSC.
- J. V. Gavette, N. S. Mills, L. N. Zakharov, C. A. Johnson, D. W. Johnson and M. M. Haley, Angew. Chem., Int. Ed., 2013, 52, 10270–10274 CrossRef CAS PubMed.
- M. D. Hartle, R. J. Hansen, B. W. Tresca, S. S. Prakel, L. N. Zakharov, M. M. Haley, M. D. Pluth and D. W. Johnson, Angew. Chem., Int. Ed., 2016, 55, 11480–11484 CrossRef CAS PubMed.
- H. A. Fargher, N. Lau, L. N. Zakharov, M. M. Haley, D. W. Johnson and M. D. Pluth, Chem. Sci., 2018, 10, 67–72 RSC.
- H. A. Fargher, R. A. Nickels, T. P. de Faria, M. M. Haley, M. D. Pluth and D. W. Johnson, RSC Adv., 2021, 11, 26581–26585 RSC.
- C.-L. Deng, J. P. Bard, J. A. Lohrman, J. E. Barker, L. N. Zakharov, D. W. Johnson and M. M Haley, Angew. Chem., Int. Ed., 2019, 58, 3934–3938 CrossRef CAS PubMed.
- K. Kavallieratos, J. C. Bryan, R. A. Sachleben, G. J. Van Berkel, O. D. Espetia, M. A. Kelly, A. Danby, K. Bowman-James and B. A. Moyer, in Fundamentals and Applications of Anion Separations, ed. B. A. Moyer and R. P. Singh, Springer, Boston, MA, 2004, pp. 125–150 Search PubMed.
- N. J. Williams, V. S. Bryanstev, R. Custelcean, C. A. Seipp and B. A. Moyer, Supramol. Chem., 2016, 28, 176–187 CrossRef CAS.
- B. W. Tresca, R. J. Hansen, C. V. Chau, B. P. Hay, L. N. Zakharov, M. M. Haley and D. W. Johnson, J. Am. Chem. Soc., 2015, 137, 14959–14967 CrossRef CAS PubMed.
- H. A. Fargher, N. Lau, H. C. Richardson, P. H.-Y. Cheong, M. M. Haley, M. D. Pluth and D. W. Johnson, J. Am. Chem. Soc., 2020, 142, 8243–8251 CrossRef CAS PubMed.
- ChemDraw Professional, version 22.2.0.3300 Search PubMed.
- C. F. Baes Jr, SXLSQI: A Program for Modeling Solvent Extraction Systems, ORNL/TM-13604, Oak Ridge National Laboratory, Oak Ridge, TN, 1998 Search PubMed.
- Y. Deng, R. A. Sachleben and B. A. Moyer, J. Chem. Soc., Faraday Trans., 1995, 91, 4215–4222 RSC.
- Y. Marcus, Ion Properties, Marcel Dekker, New York, NY, 1997 Search PubMed.
- J. Rais, Collect. Czech. Chem. Commun., 1971, 36, 3253–3262 CrossRef CAS.
- B. P. Hay, T. K. Firman and B. A. Moyer, J. Am. Chem. Soc., 2005, 127, 1810–1819 CrossRef CAS PubMed.
- T. J. Sherbow, H. A. Fargher, M. M. Haley, M. D. Pluth and D. W. Johnson, J. Org. Chem., 2020, 85, 12367–12373 CrossRef CAS PubMed.
- J. A. Riddick, W. B. Bunger and T. K. Sakano, Organic Solvents: Physical Properties and Methods of Purification, Wiley-Interscience, New York, 4th edn, 1986 Search PubMed.
- A. Jouyban and S. Soltanpour, J. Chem. Eng. Data, 2010, 55, 2951–2963 CrossRef CAS.
- Y. Marcus, M. J. Kamlet and R. W. Taft, J. Phys. Chem., 1988, 92, 3613–3622 CrossRef CAS.
- Y. Liu, A. Sengupta, K. Raghavachari and A. H. Flood, Chem, 2017, 3, 411–427 CAS.
- J. L. Cook, C. A. Hunter, C. M. R. Low, A. Perez-Velasco and J. G. Vinter, Angew. Chem., Int. Ed., 2007, 46, 3706–3709 CrossRef CAS PubMed.
- N. J. Williams, S. Roy, C. O. Reynolds, R. Custelcean, V. S. Bryantsev and B. A. Moyer, Chem. Commun., 2019, 55, 3590–3593 RSC.
- Y. Liu, W. Zhao, C.-H. Chen and A. H. Flood, Science, 2019, 365, 159–161 CrossRef CAS PubMed.
- A. L. Sisson, J. P. Clare and A. P. Davis, Chem. Commun., 2005, 5263–5265 RSC.
- M. Zaleskaya, Ł. Dobrzycki and J. Romański, Int. J. Mol. Sci., 2020, 21, 9465 CrossRef CAS PubMed.
- J. F. Neal, A. Saha, M. M. Zerkle, W. Zhao, M. M. Rogers, A. H. Flood and H. C. Allen, J. Phys. Chem. A, 2020, 124, 10171–10180 CrossRef CAS PubMed.
- The Supramolecular Chemistry of Anions, ed. A. Bianchi, K. Bowman-James and E. García-España, VCH, Weinheim, 1997 Search PubMed.
- Fundamentals and Applications of Anion Separations, ed. B. A. Moyer and R. P. Singh, Kluwer Academic/Plenum, New York, 2004 Search PubMed.
- J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry, The Royal Society of Chemistry, Cambridge, 2006 Search PubMed.
- Anion Coordination Chemistry, ed. K. Bowman-James, A. Bianchi and E. Garcia-Espana, Wiley-VCH, Weinheim, 2011 Search PubMed.
- L. K. Macreadie, A. M. Gilchrist, D. A. McNaughton, W. G. Ryder, M. Fares and P. A. Gale, Chem, 2022, 8, 46–118 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and 31P NMR spectra of TBPX salts, ICP-MS sample preparation and calibration, TBPX concentrations measured by ICP-MS, temperature control studies, and SXLSQI fitting parameters and model curves. See DOI: https://doi.org/10.1039/d3sc05922g |
| ‡ Department of Chemistry, University of Texas at Austin, Austin, TX 78712, USA. |
| This journal is © The Royal Society of Chemistry 2024 |