
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSwitchover from singlet oxygen to superoxide radical through a photoinduced two-step sequential energy transfer process†
Shengsheng
Yu‡
a,
Rong-Xin
Zhu‡
a,
Kai-Kai
Niu
a,
Ning
Han
 b,
Hui
Liu
a and
Ling-Bao
Xing
*a
b,
Hui
Liu
a and
Ling-Bao
Xing
*a
aSchool of Chemistry and Chemical Engineering, Shandong University of Technology, Zibo, Shandong 255000, P. R. China. E-mail: lbxing@sdut.edu.cn
bDepartment of Materials Engineering, KU Leuven, Leuven 3001, Belgium
First published on 5th January 2024
Abstract
The competitive nature of type II photosensitizers in the transfer of excitation energy for the generation of singlet oxygen (1O2) presents significant challenges in the design of type I photosensitizers to produce the superoxide anion radical (O2˙−). In this study, we present an efficient method for the direct transformation of type II photosensitizers into type I photosensitizers through the implementation of an artificial light-harvesting system (ALHSs) involving a two-step sequential energy transfer process. The designed supramolecular complex (DNPY-SBE-β-CD) not only has the ability to generate 1O2 as type II photosensitizers, but also demonstrates remarkable fluorescence properties in aqueous solution, which renders it an efficient energy donor for the development of type I photosensitizers ALHSs, thereby enabling the efficient generation of O2˙−. Meanwhile, to ascertain the capability and practicality of this method, two organic reactions were conducted, namely the photooxidation reaction of thioanisole and oxidative hydroxylation of arylboronic acids, both of which display a high level of efficiency and exhibit significant catalytic performance. This work provides an efficient method for turning type II photosensitizers into type I photosensitizers by a two-step sequential energy transfer procedure.
Introduction
Photosynthesis is a crucial natural process that enables plants to harness energy from the sun and transform it into biochemical energy, which serves as the foundation for the survival of plants, animals, and microorganisms, making it a vital activity in nature. Moreover, photosynthesis plays a significant role in the material cycling process, ensuring the continuous flow of essential resources.1–3 Scientists are dedicated to the advancement of limitless solar energy. Within biological systems, green plants utilize pigment–protein complexes that consist of numerous closely arranged chlorophyll molecules to capture solar energy and convert it into chemical energy.4–6 Motivated by the wonders of nature, more and more effort has been dedicated to developing photosensitizers that can effectively transform solar energy into chemical energy, emulating the process of photosynthesis in the natural world.7–10 Considerable efforts have been undertaken to facilitate the development of innovative and efficient photosensitizers. However, previous studies have shown that the majority of photosensitizers possess hydrophobic conjugated backbones, resulting in low solubility in aqueous environments and limiting their potential applications.11,12 The development of photosensitizers has entered a new stage with the emergence of supramolecular strategies,13–17 which can be used to modulate the activity of photosensitizers and regulate their photophysical and photochemical properties through non-covalent interactions, providing a novel approach to sensitize oxygen to produce reactive oxygen species (ROS).18–20 Due to their unique advantages, supramolecular photosensitizers have gained significant attention and have emerged as a promising research area, showing broad application prospects in photocatalysis,21–24 photovoltaics,25–28 photodynamic therapy (PDT),29–32 triplet–triplet annihilation-based molecular photon upconversion,33–36 and other fields.At present, the majority of photosensitizers documented in the literature are classified as type II photosensitizers, which produce singlet oxygen (1O2) through direct excitation energy transfer. However, the number of reported cases involving type I photosensitizers that are deemed suitable for practical use is limited. Kida and coworkers reported a noteworthy finding about the functional properties of rhodamine 19 derivatives as type I photosensitizers, which exhibited a significant enhancement in the generation of ROS and a successful PDT effect in mice with tumours, demonstrating the promise of supramolecular assemblies in the advancement of highly efficient type I photosensitizers.37 Very recently, Yang and coworkers employed a supramolecular strategy to facilitate electron transfer via host–guest interaction, resulting in the efficient conversion of a conventional type II photosensitizer into a type I photosensitizer.38 However, the development of type I photosensitizers remains a challenge owing to the competition between energy transfer and electron transfer processes, and also because the excited states of most photosensitizers can be deactivated via the energy transfer pathway.39 Therefore, it is of great significance to convert type II photosensitizers to type I photosensitizers by adjusting the energy transfer and electron transfer of excited states using a simple method.
In the present work, a supramolecular complex was constructed through the co-assembly of a positively charged derivative of 9,10-di(yridine-4-yl)anthracene (DNPY) and a negatively charged sulfobutylether-β-cyclodextrin (SBE-β-CD) via electrostatic interactions. The self-assembling properties of DNPY-SBE-β-CD were investigated using various techniques, including UV-vis absorption spectra, fluorescence emission spectra, zeta potential, 1H-NMR, dynamic light scattering (DLS), and transmission electron microscopy (TEM). The results suggest that DNPY-SBE-β-CD can self-assemble to form spherical structures in aqueous solution and can be used as a type II photosensitizer with high efficiency to produce 1O2, which has been employed to perform photooxidation reactions of thioanisole and its derivatives in water, achieving an impressive yield of 92%. Moreover, the remarkable fluorescence characteristics of DNPY-SBE-β-CD can be effectively utilized as an optimal energy donor for the development of artificial light-harvesting systems (ALHSs). Therefore, a highly effective ALHS has been developed by utilizing DNPY-SBE-β-CD as an energy donor, and RhB and SR101 as energy acceptors. The ALHS incorporates a two-step sequential energy transfer process, starting from DNPY-SBE-β-CD and proceeding to RhB and SR101, which successfully mimics the natural photosynthesis system. It is particularly interesting that compared with the 1O2 produced by the type II photosensitizer DNPY-SBE-β-CD, the construction of the ALHS (DNPY-SBE-β-CD+RhB+SR101) can realize the effective conversion of a type II photosensitizer to a type I photosensitizer, and realize the highly efficient generation of O2˙−, which can be utilized to perform oxidative hydroxylation of arylboronic acids, resulting in a remarkable yield of up to 95%. In this way, the transformation of a type II photosensitizer into a type I photosensitizer was successfully accomplished by constructing an ALHS with a two-step sequential energy transfer process, and applied to different types of photooxidation reactions (Scheme 1).
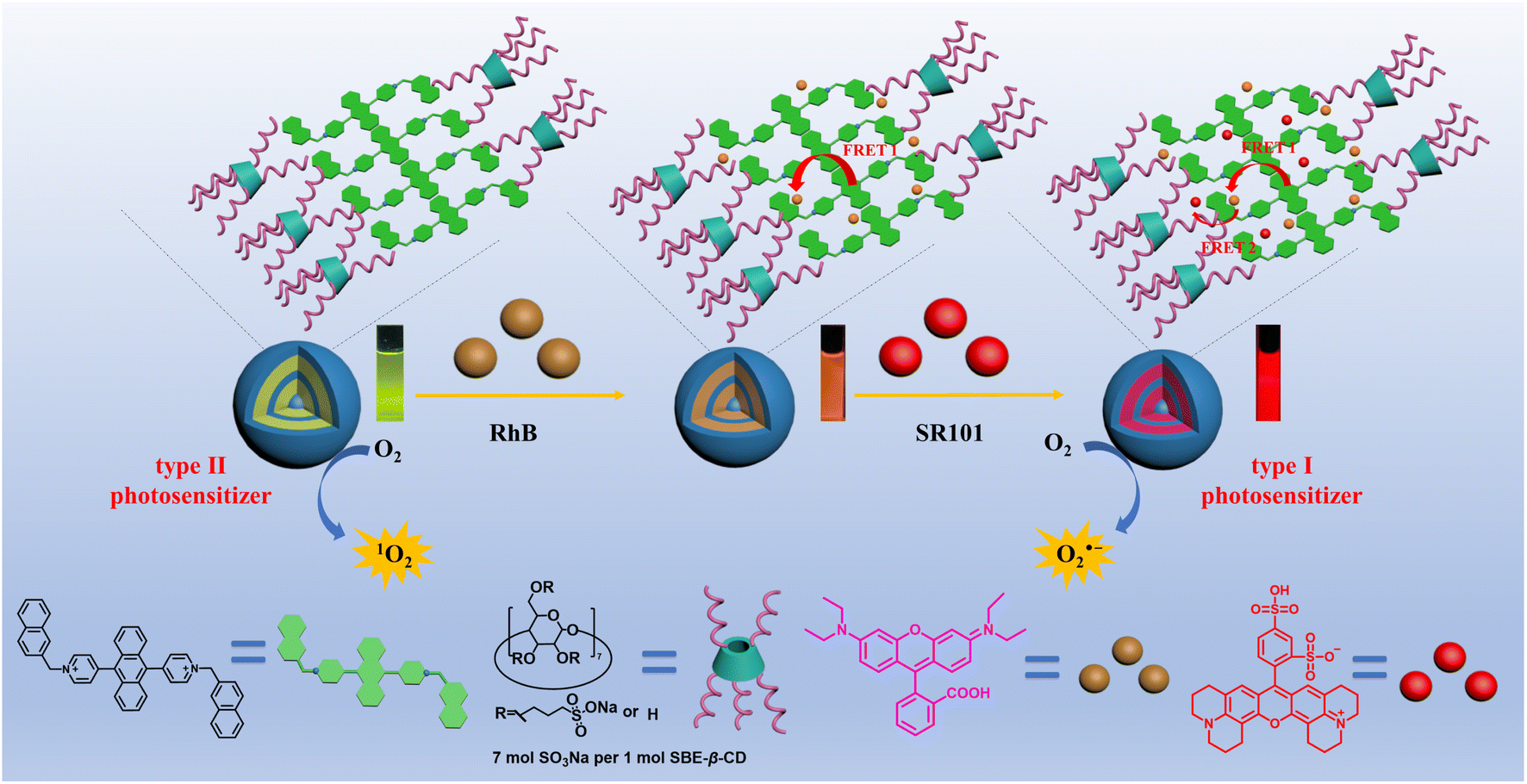 | ||
| Scheme 1 Schematic of the transformation of a type II photosensitizer into a type I photosensitizer by constructing an ALHS with a two-step sequential energy transfer process. | ||
Experimental section
Synthesis of DNPY
A mixture of DPA (0.33 g, 1 mmol), 2-(bromomethyl)naphthalene (0.66 g, 3 mmol), and DMF (20 mL) was stirred at 70 °C for 6 h. The mixture was cooled down to room temperature and then added dropwise to acetonitrile (200 mL), precipitating completely. The precipitate was collected by filtration and the precipitate was dried overnight under vacuum to give DNPY as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.52 (d, J = 6.8 Hz, 4H), 8.38 (d, J = 6.8 Hz, 4H), 8.34 (s, 2H), 8.12 (d, J = 8.5 Hz, 2H), 8.04 (d, J = 15.2 Hz, 4H), 7.87 (d, J = 8.5, 2H), 7.64 (d, J = 6.7 Hz, 8H), 7.59 (d, J = 10.2 Hz, 4H), 6.20 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 156.00, 145.82, 133.57, 133.31, 133.26, 131.82, 131.25, 129.66, 129.60, 128.66, 128.38, 128.27, 127.81, 127.68, 127.42, 126.94, 126.31, 63.80.Results and discussion
DNPY was successfully synthesized by the reaction of DPA with 2-(bromomethyl)naphthalene, giving a yellow solid with a yield of 75% (Scheme S1†). The structure of DNPY was confirmed by 1H NMR (Fig. S3†) and 13C NMR (Fig. S4†). UV-vis absorption, fluorescence emission, and 1H NMR spectroscopy were employed to investigate the formation of the supramolecular complex of DNPY and SBE-β-CD in aqueous solution. As depicted in Fig. S5,† the absorption of DNPY at wavelengths of 250 nm and 409 nm exhibits a gradual decline upon the introduction of SBE-β-CD. However, a noticeable increase in the fluorescence intensity of DNPY at 550 nm is observed, which can be attributed to the electrostatic interactions between DNPY and SBE-β-CD, as well as the formation of a supramolecular complex known as DNPY-SBE-β-CD. Upon the addition of 0.20 equiv. of SBE-β-CD, the fluorescence emission intensity of DNPY achieved its maximum value, accompanied by a transition in the fluorescence colour from yellow to green (Fig. 1). In addition, the quantum yield and fluorescence lifetime of DNPY and DNPY-SBE-β-CD were also measured. After the addition of 0.20 equiv. of SBE-β-CD, the quantum yield of DNPY increased from 10.2% to 14.3%, and the fluorescence lifetime increased from 2.30 ns to 7.04 ns (Fig. S6†). However, subsequent addition of SBE-β-CD resulted in a decrease in the fluorescence emission intensity without an obvious change in the fluorescence colour (Fig. S7†), but the quantum yield was reduced to 7.4%. This phenomenon could be explained by the enhancement of π–π interactions between anthracene groups by electrostatic interaction, leading to fluorescence quenching.40 Therefore, the supramolecular complex of DNPY-SBE-β-CD that demonstrated the highest fluorescence intensity with a DNPY![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SBE-β-CD ratio of 1:0.20 was selected as the subject for investigation. 1H NMR experiments were conducted to further investigate the electrostatic interactions between DNPY and SBE-β-CD, in which the 1H NMR signals of the DNPY protons revealed a significant change in chemical shift upon the addition of 0.20 equiv. of SBE-β-CD (Fig. S8†). The above results demonstrated that DNPY can form a supramolecular complex with SBE-β-CD through electrostatic interactions.
:SBE-β-CD ratio of 1:0.20 was selected as the subject for investigation. 1H NMR experiments were conducted to further investigate the electrostatic interactions between DNPY and SBE-β-CD, in which the 1H NMR signals of the DNPY protons revealed a significant change in chemical shift upon the addition of 0.20 equiv. of SBE-β-CD (Fig. S8†). The above results demonstrated that DNPY can form a supramolecular complex with SBE-β-CD through electrostatic interactions.
 | ||
| Fig. 1 Fluorescence emission spectra of DNPY with gradual addition of SBE-β-CD in aqueous solution (inset: photographs of DNPY and DNPY-SBE-β-CD). [DNPY] = 1.0 × 10−5 mol L−1. | ||
The self-assembly characteristics of DNPY and DNPY-SBE-β-CD were further investigated using DLS, zeta potential, and TEM. The results revealed that the average size of the DNPY aggregates was about 343 nm (Fig. 2a). Upon the addition of SBE-β-CD to the DNPY solution, the average size noticeably increased to approximately 531 nm (Fig. 2b), which can be attributed to the formation of a supramolecular complex through electrostatic interactions. Meanwhile, in the presence of a laser lamp, DNPY and DNPY-SBE-β-CD both exhibited an obvious Tyndall effect, suggesting the formation of aggregates. TEM experiments were further performed to investigate the morphology of DNPY and DNPY-SBE-β-CD. As shown in Fig. 2c, the TEM results revealed that DNPY self-assembled into irregular structures, with aggregate size distributed within the range of 250–450 nm. However, after the addition of 0.20 equiv. SBE-β-CD, spherical structures were observed with average aggregate size of 535 nm (Fig. 2d). This modification might be attributed to the electrostatic interactions between DNPY and SBE-β-CD. The TEM results were consistent with the DLS findings, indicating the formation of a supramolecular complex between DNPY and SBE-β-CD. In addition, zeta potential studies were conducted in an aqueous medium, which revealed that the relative zeta potential of DNPY was +6.50 mV (Fig. S9a†). Nevertheless, when 0.20 equiv. of SBE-β-CD was introduced, the zeta potential of DNPY-SBE-β-CD exhibited a negative value of −2.41 mV (Fig. S9b†). The obtained results provide additional evidence for the development of the supramolecular complex between DNPY and SBE-β-CD through electrostatic interactions.
 | ||
| Fig. 2 DLS plots and TEM images for (a), (c) DNPY and (b), (d) DNPY-SBE-β-CD. [DNPY] = 1.0 × 10−5 mol L−1, [SBE-β-CD] = 2.0 × 10−6 mol L−1. | ||
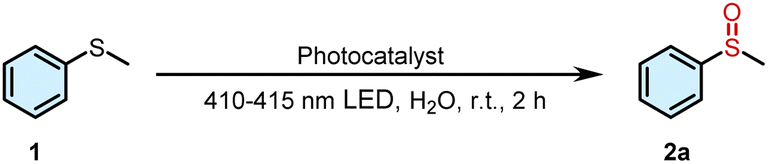
In order to investigate the ability of the supramolecular complex to generate ROS, 9,10-anthracenediylbis (methylene)dimalonic acid (ABDA) and N,N,N′,N′-tetramethyl phenylenediamine (TMPD) were employed as the specific indicators for 1O2 and O2˙−, respectively. ABDA can selectively react with 1O2, resulting in a corresponding decrease in its absorbance. When the aqueous solution of DNPY and DNPY-SBE-β-CD containing ABDA was irradiated, a decrease in the ABDA absorption peak was observed (Fig. S10b and S11b†). The 1O2 quantum yield was measured using Rose Bengal (RB) as the reference photosensitizer, and the 1O2 quantum yield of DNPY-SBE-β-CD (99.4%) was significantly stronger than that of DNPY (59.7%) (Fig. S1, S10, and S11†). In addition, TMPD can selectively react with O2˙−, resulting in a corresponding increase in its absorbance. When UV light was used to irradiate the aqueous solution of DNPY and DNPY-SBE-β-CD containing TMPD, the absorption peak of TMPD was slightly increased (Fig. S12†). Moreover, we measured the O2˙− generation efficiencies of DNPY and DNPY-SBE-β-CD. The amount of O2˙− was quantitatively analyzed by nitroblue tetrazolium (NBT) transformation. NBT, which can react with O2˙− and displays a maximum absorbance at 260 nm, was selected to determine the amounts of O2˙− generated. The NBT conversion percentages of DNPY and DNPY-SBE-β-CD were 6.3% and 9.2%, respectively (Fig. S2 and S13†), which suggests that DNPY and DNPY-SBE-β-CD produce O2˙− with low efficiency. These results indicate that the main ROS produced by DNPY-SBE-β-CD is 1O2, which can promote the photooxidation reaction of thioanisole.41–43 Therefore, we employed DNPY-SBE-β-CD as a photosensitizer to enhance the photooxidation reaction of thioanisole in water. As shown in Table 1, when 0.5 mol% DNPY-SBE-β-CD was added, the yield reached 92% after 2 h of UV irradiation (Fig. S14†). In contrast, the yield obtained with DNPY was 56%. This clearly indicates that DNPY-SBE-β-CD possesses the highest catalytic efficiency for the photooxidation of thioanisole in aqueous solution.
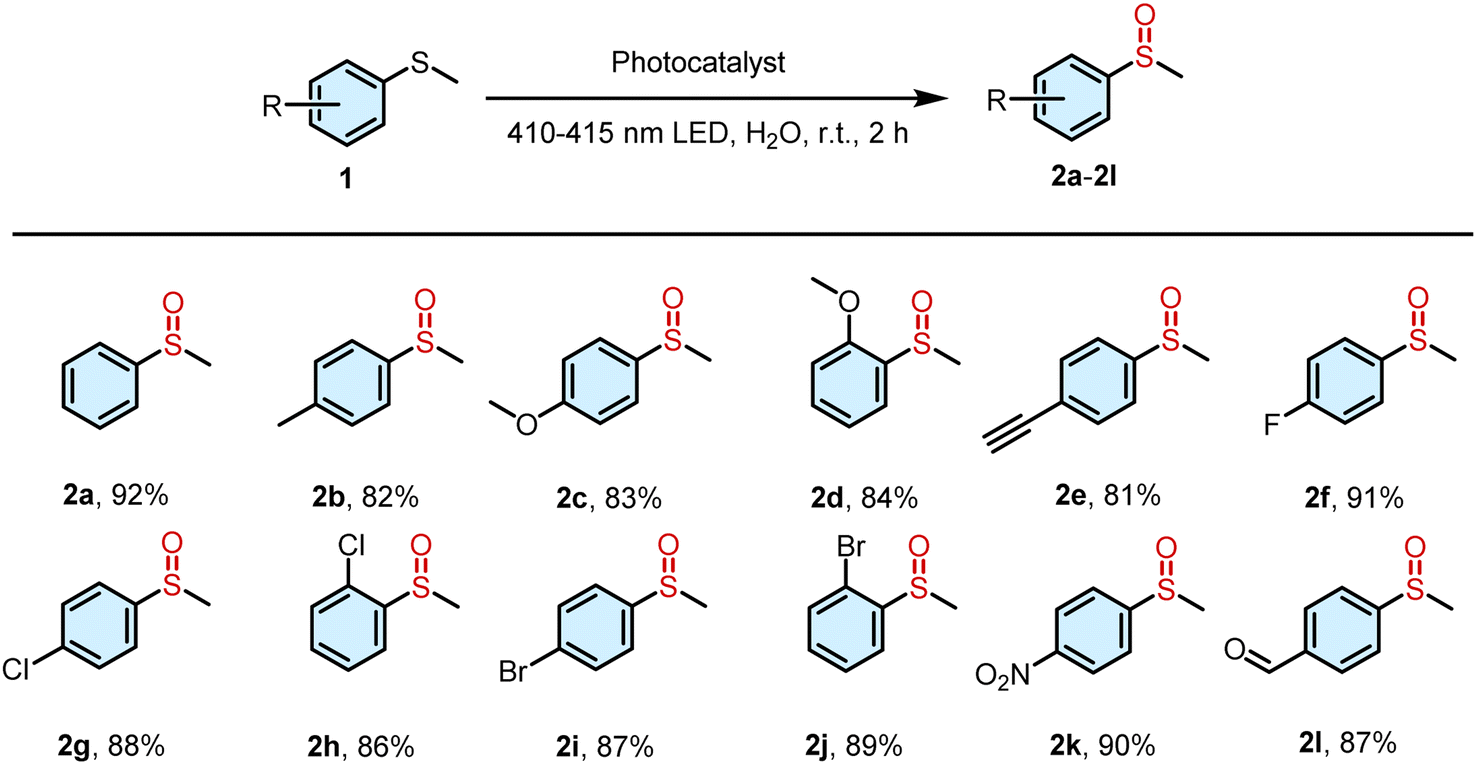
To examine the universality of DNPY-SBE-β-CD as a photocatalyst for enhancing the photooxidation of thioanisole derivatives in aqueous solution, a range of substrates were used to conduct the photooxidation reaction (Fig. S15–S25†). As shown in Table 2, the thioanisole derivatives with electron-donating groups (–CH3 and –OCH3) displayed notable yields (82% for 2b, 83% for 2c, and 84% for 2d). Similarly, the thioanisole derivatives containing electron-withdrawing groups (–F, –Cl, –Br, etc.) also exhibited successful reactions with high yields (81% for 2e, 91% for 2f, 88% for 2g, 86% for 2h, 87% for 2i, 89% for 2j, 90% for 2k, and 87% for 2l). These results demonstrate the significant potential of DNPY-SBE-β-CD for facilitating thioanisole photooxidation reactions under mild conditions.
To explore the mechanism and the active species of the photooxidation reaction of thioanisole and its derivatives, four free radical scavengers, 1,4-benzoquinone (BQ), triethylamine (TEA), potassium iodide (KI), and sodium azide (NaN3), were added to the reaction system to scavenge superoxide anion radicals (O2˙−), hydroxyl radicals (˙OH), holes (h+), and singlet oxygen (1O2), respectively. As shown in Fig. 3a, the introduction of NaN3 resulted in a notable decrease in the yield of the photooxidation reaction, reaching only 15% of the yield. However, the addition of BQ, TEA, and KI under identical reaction conditions did not significantly affect the reaction yields, suggesting that 1O2 serves as the primary active species in the photooxidation reaction of thioanisole. Therefore, DNPY-SBE-β-CD is a typical type II photosensitizer formed by DNPY and SBE-β-CD via electrostatic interactions.
 | ||
| Fig. 3 (a) Control experiments for the thioanisole photooxidation reaction in the presence of different scavengers: NaN3, BQ, TEA, and KI. (b) The proposed mechanism for the photooxidation reaction of thioanisole. | ||
A reaction mechanism for the photooxidation reaction of thioanisole has been proposed based on the experimental results and previous literature (Fig. 3b).44,45 Under light irradiation, DNPY-SBE-β-CD is excited from the ground state [DNPY-SBE-β-CD] to the excited state [DNPY-SBE-β-CD]*, subsequently, the energy of [DNPY-SBE-β-CD]* is transferred to oxygen, leading to the formation of 1O2 and [DNPY-SBE-β-CD]. Then, the substrate interacts with 1O2 to generate the intermediate substance I. Finally, I further reacts with the thioanisole, followed by the loss of H2O to produce the final product. Meanwhile, the thioanisole is oxidized to [thioanisole]˙+, which further reacts with intermediate substance I to form another molecule of target product.41–45
Due to the impressive fluorescence characteristics in the aqueous solution, DNPY-SBE-β-CD shows great promise as an optimal energy donor for the construction of ALHSs. In the fluorescence resonance energy transfer (FRET) process, the presence of a fluorescent acceptor with matching energy is crucial. Hence, for the fabrication of ALHS with DNPY-SBE-β-CD, RhB was chosen as the energy acceptor, because there is a favorable overlap between the absorption band of RhB and the emission band of DNPY-SBE-β-CD (Fig. S26†), in which the absorption band of RhB is at 500–600 nm and the emission band of DNPY-SBE-β-CD is at 525–575 nm. As shown in Fig. 4a, the emission peak of DNPY-SBE-β-CD at 545 nm exhibited a notable decrease upon the gradual addition of RhB, while a distinct fluorescence emission peak appeared at 590 nm, which corresponds to the fluorescence emission of RhB. In addition, the CIE 1931 chromaticity coordinates (Fig. S27†) clearly showed the transition of the fluorescence emission colour from green to orange in the presence of RhB. In addition, the quantum yields and fluorescence lifetimes of DNPY-SBE-β-CD and DNPY-SBE-β-CD+RhB were also measured. The quantum yield of DNPY-SBE-β-CD was 14.3%. With the addition of RhB, the quantum yield increased to 16.1%, however, the fluorescence lifetime decreased from 7.04 ns to 4.04 ns (Fig. 4b). These results indicate that an energy transfer process takes place from DNPY-SBE-β-CD to RhB, with an energy transfer efficiency (ФET) of 76% (Fig. S28a†) and an antenna effect (AE) of 15.7 (Fig. S28b†), when the donor (DNPY-SBE-β-CD) to acceptor (RhB) ratio was 100:10. The results mentioned above suggest that a high-efficiency ALHS was successfully formed between DNPY-SBE-β-CD and RhB in aqueous solution.
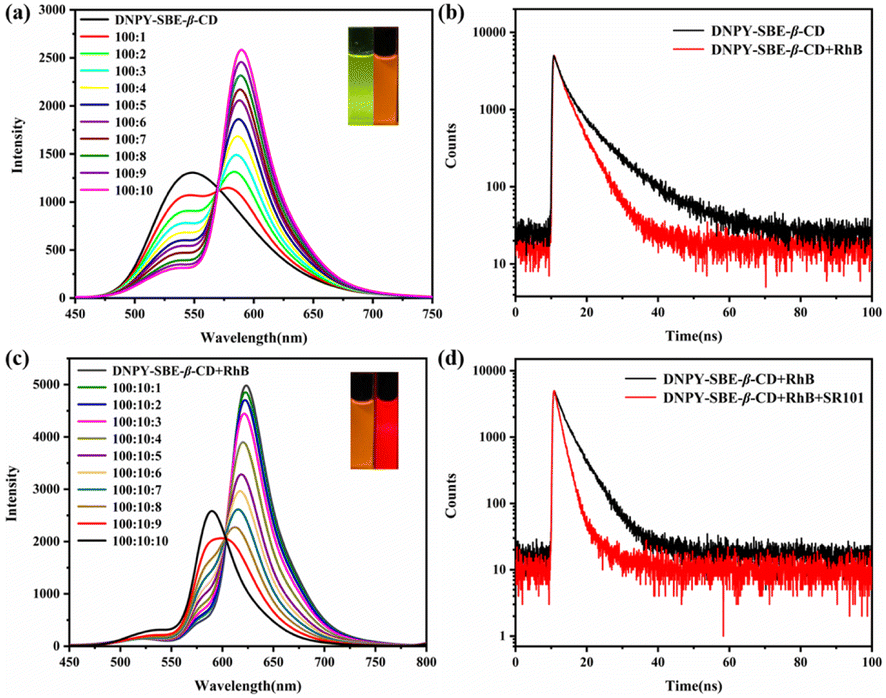 | ||
| Fig. 4 (a) Fluorescence emission spectra of DNPY-SBE-β-CD with gradual addition of RhB (inset: fluorescence colour of DNPY-SBE-β-CD before and after addition of RhB). (b) Time-resolved fluorescence decay curves of DNPY-SBE-β-CD and DNPY-SBE-β-CD+RhB. (c) Fluorescence emission spectra of DNPY-SBE-β-CD+RhB with gradual addition of SR101 (inset: fluorescence colour of DNPY-SBE-β-CD+RhB before and after addition of SR101). (d) Time-resolved fluorescence decay curves of DNPY-SBE-β-CD+RhB and DNPY-SBE-β-CD+RhB+SR101. | ||
In order to further simulate the natural photosynthetic system with multi-step sequential energy transfer,10,46–53 we selected SR101 dye as the second energy acceptor, which exhibits excellent overlap between the absorption band of SR101 and the emission band of DNPY-SBE-β-CD+RhB (Fig. S29†). As shown in Fig. 4c, it can be observed that the fluorescence intensity of DNPY-SBE-β-CD+RhB at 590 nm decreased after the addition of SR101, while the emission of SR101 at 625 nm increased gradually. Meanwhile, as shown in the CIE 1931 chromaticity coordinates, the fluorescence colour changed from orange to red (Fig. S27†). Furthermore, the fluorescence quantum yield of DNPY-SBE-β-CD+RhB increased from 16.1% to 20.1% after the addition of SR101, while the fluorescence lifetime decreased from 4.04 ns to 2.02 ns (Fig. 4d). When the molar ratio of DNPY-SBE-β-CD+RhB (donor) and SR101 (acceptor) was 100:10:10, ФET and AE were calculated to be 81% (Fig. S30a†) and 7.4 (Fig. S30b†), respectively. These results clearly demonstrate the successful construction of an efficient ALHS with a two-step sequential energy transfer process.
In addition, there was a significant overlap between the absorption bands of SR101 and the emission bands of DNPY-SBE-β-CD (Fig. S31†). We attempted to construct an ALHS using SR101 as an energy acceptor and DNPY-SBE-β-CD as an energy donor. According to the data presented in Fig. S32,† there was a significant decrease in the fluorescence emission at 545 nm for DNPY-SBE-β-CD upon the gradual addition of SR101, and a new emission peak appeared at 625 nm, which corresponds to the presence of SR101. At the same time, the fluorescence emission colour of DNPY-SBE-β-CD changed from green to red (Fig. S33†). Moreover, the fluorescence quantum yield of DNPY-SBE-β-CD increased from 14.3% to 16.9%, and the fluorescence lifetime decreased from 7.04 ns to 4.58 ns, after the addition of SR101 (Fig. S34†). These results indicate that the FRET process took place from DNPY-SBE-β-CD (donor) to SR101 (acceptor). The ФET and AE were calculated to be 84% and 7.5, respectively (Fig. S35†). In addition, we also studied the effects of the addition of energy acceptor molecules on the assembly structure of DNPY-SBE-β-CD by DLS and TEM. As shown in Fig. S36,† the structure and size of the assemblies of DNPY-SBE-β-CD did not change significantly after the addition of dye molecules. These results show that the addition of dyes has no obvious effect on the assembly structure.
In order to study the effect of the energy transfer processes on the ROS production, the ability of the ALHS to generate ROS was investigated. ABDA and TMPD were employed as specific indicators for 1O2 and O2˙−, respectively. When the aqueous solutions of DNPY, DNPY-SBE-β-CD, DNPY-SBE-β-CD+RhB, DNPY-SBE-β-CD+SR101, and DNPY-SBE-β-CD+RhB+SR101 containing ABDA were irradiated, a decrease in the absorption peak of ABDA was observed (Fig. S10b, S11b, S37b, S38b and S39b†). The ABDA absorption decay and irradiation time curve proves that they can all produce 1O2, and DNPY-SBE-β-CD+RhB+SR101 exhibits the weakest 1O2 productivity detected by ABDA (Fig. 5a). Furthermore, the 1O2 quantum yield was measured using RB as the reference photosensitizer. The 1O2 quantum yields of DNPY, DNPY-SBE-β-CD, DNPY-SBE-β-CD+RhB, DNPY-SBE-β-CD+SR101, and DNPY-SBE-β-CD+RhB+SR101 were measured as 59.7%, 99.4%, 6.9%, 4.2%, and 5.4%, respectively (Table S1, Fig. S1, S10–S11 and S37–S39†). In contrast, when UV light was used to irradiate the aqueous solutions of DNPY, DNPY-SBE-β-CD, DNPY-SBE-β-CD+RhB, DNPY-SBE-β-CD+SR101, and DNPY-SBE-β-CD+RhB+SR101 containing TMPD, the absorption peak of TMPD increased in all samples, and DNPY-SBE-β-CD+RhB+SR101 led to the strongest increase (Fig. 5b). We also measured the O2˙− generation efficiencies by NBT, which can react with O2˙− and displays a maximum absorbance at 260 nm. The amount of O2˙− was quantitatively inspected by NBT transformation. The O2˙− generation efficiencies of DNPY, DNPY-SBE-β-CD, DNPY-SBE-β-CD+RhB, DNPY-SBE-β-CD+SR101, and DNPY-SBE-β-CD+RhB+SR101 were measured as 6.3%, 9.2%, 19.7%, 24.9%, and 44.1%, respectively (Table S2, Fig. S2, S13, and S40†). Compared with DNPY-SBE-β-CD, the 1O2 quantum yield of DNPY-SBE-β-CD+RhB was reduced 14.4-fold, while the O2˙− generation efficiency was increased 2.1-fold. More interestingly, compared with DNPY-SBE-β-CD, the 1O2 quantum yield of DNPY-SBE-β-CD+RhB+SR101 was reduced 18.4-fold, while the O2˙− generation efficiency was increased 4.8-fold. This distinctly proved that the type II photosensitizer (DNPY-SBE-β-CD) is effectively converted to the type I photosensitizer (DNPY-SBE-β-CD+RhB+SR101) through a two-step sequential energy transfer process.
 | ||
| Fig. 5 (a) Plots of ΔAbs (A0–A) for ABDA at 375 nm upon light irradiation (410–415 nm, 10 W) for different times in the presence of DNPY, DNPY-SBE-β-CD, DNPY-SBE-β-CD+RhB, DNPY-SBE-β-CD+SR101, and DNPY-SBE-β-CD+RhB+SR101 (control: ABDA without any additive). (b) UV-vis absorption spectra for cationic radicals of TMPD generated by indicated samples under the same conditions (control: TMPD without any additive). [DNPY] = 1.0 × 10−5 mol L−1, [SBE-β-CD] = 2.0 × 10−6 mol L−1, [RhB] = 1.0 × 10−6 mol L−1, [SR101] = 1.0 × 10−6 mol L−1. | ||
Subsequently, the ALHS (DNPY-SBE-β-CD+RhB+SR101) with a two-step sequential energy transfer process was employed to provide a reaction platform for the oxidative hydroxylation of arylboronic acid. As shown in Table 3, the addition of 0.5 mol% DNPY-SBE-β-CD+RhB+SR101 (100:20:10:10) resulted in a yield of 93% after being exposed to light irradiation for 12 h (Fig. S41†). In contrast, under the same conditions, the yields of other systems including DNY, DNPY-SBE-β-CD, DNPY-SBE-β-CD+RhB, and DNPY-SBE-β-CD+SR101 were found to be 36%, 40%, 53%, and 57%, respectively. In addition, we also indicated the influence of the base on the catalytic efficiency in the oxidative hydroxylation reaction. The experimental results are shown in Table S3.† When the base of the oxidative hydroxylation reaction was DIPEA, the reaction yield was 93% (entry 1). On reducing the amount of base, the reaction yield was reduced to 75% (entry 2). When the base was changed to triethylamine or trimethylamine, the reaction yields were 88% and 72%, respectively (entry 3 and 4). The above results indicate that the novel ALHS has the potential to serve as an effective catalyst for the oxidative hydroxylation of arylboronic acids. In order to determine the effectiveness of DNPY-SBE-β-CD+RhB+SR101 as a catalyst for oxidative hydroxylation of arylboronic acids, we conducted investigations on various arylboronic acid derivatives. As shown in Table 4, the yields of the arylboronic acid derivatives were 88% (4b), 80% (4c), 92% (4d), 90% (4e), 87% (4f), 91% (4g), 95% (4h), 85% (4i), 85% (4j), 89% (4k), 86% (4l), 85% (4m), 82% (4n), and 84% (4o), respectively (Fig. S42–S55†), indicating the universality of its catalytic activity.
|
|
|||
|---|---|---|---|
| Entry | Conditions | Light irradiation | Yield [%] |
| a Reaction conditions: 4-pyridylboronic acid (0.1 mmol), DIPEA (0.4 mmol), DNPY-SBE-β-CD+RhB+SR101 aqueous solution (0.5 mmol%, 3 mL), 410–415 nm LED, room temperature, 12 h. b Isolated yields. c Without LED. | |||
| 1 | None | Yes | 10 |
| 2 | DIPEA | Yes | 20 |
| 3 | DNPY | Yes | 36 |
| 4 | DNPY-SBE-β-CD | Yes | 40 |
| 5 | DNPY-SBE-β-CD+RhB | Yes | 53 |
| 6 | DNPY-SBE-β-CD+SR101 | Yes | 57 |
| 7 | DNPY-SBE-β-CD+RhB+SR101 | Yes | 93 |
| 8c | DNPY-SBE-β-CD+RhB+SR101 | No | No reaction |


In order to investigate the mechanism and the active species of the oxidative hydroxylation of arylboronic acid derivatives, BQ, TEA, KI, and NaN3 were employed to scavenge superoxide anion radicals (O2˙−), hydroxyl radicals (˙OH), holes (h+), and singlet oxygen (1O2), respectively. As shown in Fig. 6a, the yield of the oxidative hydroxylation of arylboronic acids decreased significantly to only 18% after the addition of BQ. However, the addition of NaN3, TEA, and KI had no significant impact on the reaction yields under the same reaction conditions, indicating that O2˙− is the main active species in the oxidative hydroxylation of arylboronic acids. Therefore, the ALHS with a two-step sequential energy transfer process is a typical type I photosensitizer.
 | ||
| Fig. 6 (a) Control experiments for the oxidative hydroxylation of arylboronic acids in the presence of different scavengers: NaN3, BQ, TEA, and KI. (b) The proposed mechanism for the oxidative hydroxylation of arylboronic acids. | ||
Based on the results of the above experiments and previous literature,54,55 a reasonable mechanism for the photocatalytic reaction of arylboronic acids was proposed (Fig. 6b). Under illumination with light, the excited state [DNPY-SBE-β-CD]* transfers the energy to the acceptor dye to form the excited state [RhB]*, followed by energy transfer from [RhB]* to [SR101] to form [SR101]*. The [SR101]* undergoes an electron transfer process with DIPEA to form [SR101] ˙− and DIPEA˙+, followed by oxygen bursting [SR101] ˙− and formation of O2˙−, which reacts with arylboronic acid to form the intermediate radical anion (II). II grabs a hydrogen atom from DIPEA˙+ to form intermediate III. Finally, intermediate III is rearranged to form intermediate IV, which is subsequently hydrolysed to give the final aryl phenol product.
Conclusion
In summary, we propose a novel approach for the effective conversion of type II photosensitizers to type I photosensitizers using a supramolecular strategy that employs a two-step sequential energy transfer mechanism. The supramolecular complex DNPY-SBE-β-CD exhibits remarkable self-assembly and fluorescence properties in aqueous solution, and it is expected to be used as a type II photosensitizer to generate 1O2, facilitating the photooxidation reaction of thioanisole and its derivatives with the highest yield of 92%. Furthermore, an efficient ALHS with a two-step sequential energy transfer process was constructed to simulate the natural photosynthesis system by using DNPY-SBE-β-CD as the energy donor and two dyes, RhB and SR101, as energy acceptors, which can be used as a supramolecular type I photosensitizer to generate O2˙− to promote the oxidative hydroxylation of arylboronic acid derivatives with the highest yield of 95%. This study not only presents an innovative methodology for constructing a sequential energy transfer ALHS using supramolecular co-assembly facilitated by electrostatic interactions, but also offers an appealing strategy for regulating the types of photosensitizers, and has been successfully applied for efficient photooxidation reactions.Data availability
All supporting data is provided in the ESI.†Author contributions
Shengsheng Yu: conceptualization, writing-review & editing, funding acquisition, resources. Rongxin Zhu: investigation, methodology, formal analysis, writing-original draft. Kai–Kai Niu: writing-review & editing, methodology, data curation. Ning Han: methodology, data curation. Hui Liu: data curation, resources. Ling-Bao Xing: conceptualization, writing-review & editing, supervision, funding acquisition, resources.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (52205210) and the Natural Science Foundation of Shandong Province (ZR2020MB018, ZR2021QB049, and ZR2022QE033).Notes and references
- G. McDermott, S. M. Prince, A. A. Freer, A. M. Hawthornthwaite-Lawless, M. Z. Papiz, R. J. Cogdell and N. W. Isaacs, Nature, 1995, 374, 517–521 CrossRef CAS.
- A. Pannwitz, D. M. Klein, S. Rodríguez-Jiménez, C. Casadevall, H. Song, E. Reisner, L. Hammarström and S. Bonnet, Chem. Soc. Rev., 2021, 50, 4833–4855 RSC.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- Y. Sun, N. C. Giebink, H. Kanno, B. Ma, M. E. Thompson and S. R. Forrest, Nature, 2006, 440, 908–912 CrossRef CAS PubMed.
- A. M. van Oijen, M. Ketelaars, J. Kohler, T. J. Aartsma and J. Schmidt, Science, 1999, 285, 400–402 CrossRef CAS PubMed.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, ACS Appl. Mater. Interfaces, 2012, 4, 2288–2294 CrossRef CAS PubMed.
- G. Sun, W. Qian, J. Jiao, T. Han, Y. Shi, X.-Y. Hu and L. Wang, J. Mater. Chem. A, 2020, 8, 9590–9596 RSC.
- R.-Z. Zhang, H. Liu, C.-L. Xin, N. Han, C.-Q. Ma, S. Yu, Y.-B. Wang and L.-B. Xing, J. Colloid Interface Sci., 2023, 651, 894–901 CrossRef CAS PubMed.
- Y. Wang, R. Zhu, Y. Hang, R. Wang, R. Dong, S. Yu and L.-B. Xing, Polym. Chem., 2023, 14, 248–252 RSC.
- P.-P. Jia, L. Xu, Y.-X. Hu, W.-J. Li, X.-Q. Wang, Q.-H. Ling, X. Shi, G.-Q. Yin, X. Li, H. Sun, Y. Jiang and H.-B. Yang, J. Am. Chem. Soc., 2020, 143, 399–408 CrossRef PubMed.
- P. Sun, G. Wang, H. Hou, P. Yuan, W. Deng, C. Wang, X. Lu, Q. Fan and W. Huang, Polym. Chem., 2017, 8, 5836–5844 RSC.
- Y. Yuan and B. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 14903–14910 CrossRef CAS PubMed.
- M. Yan, X.-B. Liu, Z.-Z. Gao, Y.-P. Wu, J.-L. Hou, H. Wang, D.-W. Zhang, Y. Liu and Z.-T. Li, Org. Chem. Front., 2019, 6, 1698–1704 RSC.
- X. Li, S. Lee and J. Yoon, Chem. Soc. Rev., 2018, 47, 1174–1188 RSC.
- K. Liu, Y. Liu, Y. Yao, H. Yuan, S. Wang, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2013, 52, 8285–8289 CrossRef CAS PubMed.
- K.-X. Teng, L.-Y. Niu, Y.-F. Kang and Q.-Z. Yang, Chem. Sci., 2020, 11, 9703–9711 RSC.
- B. Yuan, H. Wu, H. Wang, B. Tang, J. F. Xu and X. Zhang, Angew. Chem., Int. Ed., 2021, 60, 706–710 CrossRef CAS PubMed.
- R. S. Proctor, A. C. Colgan and R. J. Phipps, Nat. Chem., 2020, 12, 990–1004 CrossRef PubMed.
- Z. Liu, X. Dai, Y. Sun and Y. Liu, Aggregate, 2020, 1, 31–44 CrossRef.
- L. Xia, J. Tian, T. Yue, H. Cao, J. Chu, H. Cai and W. Zhang, Adv. Healthcare Mater., 2022, 11, 2102015 CrossRef CAS PubMed.
- R. Ham, C. J. Nielsen, S. Pullen and J. N. Reek, Chem. Rev., 2023, 123, 5225–5261 CrossRef CAS.
- C. Stoffelen and J. Huskens, Small, 2016, 12, 96–119 CrossRef CAS.
- Y. Kuramochi and O. Ishitani, Inorg. Chem., 2016, 55, 5702–5709 CrossRef CAS.
- Z. Li, Y. Han, Z. Gao and F. Wang, ACS Catal., 2017, 7, 4676–4681 CrossRef CAS.
- Y. Zhong, S. Liu, J. Wang, W. Zhang, T. Tian, J. Sun and F. Bai, APL Mater., 2020, 8, 120706 CrossRef CAS.
- F. D'Souza, A. S. Sandanayaka and O. Ito, J. Phys. Chem. Lett., 2010, 1, 2586–2593 CrossRef.
- F. D'Souza and O. Ito, Chem. Soc. Rev., 2012, 41, 86–96 RSC.
- O. Dumele, J. Chen, J. V. Passarelli and S. I. Stupp, Adv. Mater., 2020, 32, 1907247 CrossRef CAS PubMed.
- C. Li, Y. Gao, R. Huang, L. Fang, Y. Sun, Y. Yang, S. Gou and J. Zhao, ACS Mater. Lett., 2022, 4, 657–664 CrossRef CAS.
- K.-X. Teng, L.-Y. Niu and Q.-Z. Yang, J. Am. Chem. Soc., 2023, 145, 4081–4087 CrossRef CAS PubMed.
- L. Tu, C. Li, X. Xiong, J. Hyeon Kim, Q. Li, L. Mei, J. Li, S. Liu, J. Seung Kim and Y. Sun, Angew. Chem., Int. Ed., 2023, 62, e202301560 CrossRef CAS PubMed.
- B. Huang, P. Wang, Y. Ouyang, R. Pang, S. Liu, C. Hong, S. Ma, Y. Gao, J. Tian and W. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 41038–41046 CrossRef CAS PubMed.
- V. Gray, K. Moth-Poulsen, B. Albinsson and M. Abrahamsson, Coord. Chem. Rev., 2018, 362, 54–71 CrossRef CAS.
- H. Chen, I. Roy, M. S. Myong, J. S. Seale, K. Cai, Y. Jiao, W. Liu, B. Song, L. Zhang, X. Zhao, Y. Feng, F. Liu, R. M. Young, M. R. Wasielewski and J. Fraser Stoddart, J. Am. Chem. Soc., 2023, 145, 10061–10070 CrossRef CAS PubMed.
- R. Pérez-Ruiz, Top. Curr. Chem., 2022, 380, 23 CrossRef PubMed.
- K. Tanaka, W. Ohashi, K. Inafuku, S. Shiotsu and Y. Chujo, Dyes Pigm., 2020, 172, 107821 CrossRef CAS.
- H. Shigemitsu, K. Sato, S. Hagio, Y. Tani, T. Mori, K. Ohkubo, Y. Osakada, M. Fujitsuka and T. Kida, ACS Appl. Nano Mater., 2022, 5, 14954–14960 CrossRef CAS.
- K.-X. Teng, L.-Y. Niu and Q.-Z. Yang, Chem. Sci., 2022, 13, 5951–5956 RSC.
- W. Wu, X. Shao, J. Zhao and M. Wu, Adv. Sci., 2017, 4, 1700113 CrossRef PubMed.
- Y. Kim, S. Shin, T. Kim, D. Lee, C. Seok and M. Lee, Angew. Chem., Int. Ed., 2013, 52, 6426–6429 CrossRef CAS PubMed.
- D.-Y. Zheng, E.-X. Chen, C.-R. Ye and X.-C. Huang, J. Mater. Chem. A, 2019, 7, 22084–22091 RSC.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Commun., 2014, 50, 8177–8180 RSC.
- H. Hao, F. Zhang, X. Dong and X. Lang, Appl. Catal., B, 2021, 299, 120691 CrossRef CAS.
- B. Wu, Y. Liu, Y. Zhang, L. Fan, Q.-Y. Li, Z. Yu, X. Zhao, Y.-C. Zheng and X.-J. Wang, J. Mater. Chem. A, 2022, 10, 12489–12496 RSC.
- C.-J. Wu, X.-Y. Li, T.-R. Li, M.-Z. Shao, L.-J. Niu, X.-F. Lu, J.-L. Kan, Y. Geng and Y.-B. Dong, J. Am. Chem. Soc., 2022, 144, 18750–18755 CrossRef CAS PubMed.
- S. B. Koushik Acharyya, S. Lu, Y. Sun, P. S. Mukherjee and P. J. Stang, Angew. Chem., Int. Ed., 2022, 61, e202200715 CrossRef PubMed.
- D. Zhang, W. Yu, S. Li, Y. Xia, X. Li, Y. Li and T. Yi, J. Am. Chem. Soc., 2021, 143, 1313–1317 CrossRef CAS PubMed.
- M. Hao, G. Sun, M. Zuo, Z. Xu, Y. Chen, X. Y. Hu and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 10095–10100 CrossRef CAS PubMed.
- L. Ji, Y. Sang, G. Ouyang, D. Yang, P. Duan, Y. Jiang and M. Liu, Angew. Chem., Int. Ed., 2019, 58, 844–848 CrossRef CAS PubMed.
- J. J. Li, H. Y. Zhang, X. Y. Dai, Z. X. Liu and Y. Liu, Chem. Commun., 2020, 56, 5949–5952 RSC.
- Q. Song, S. Goia, J. Yang, S. C. L. Hall, M. Staniforth, V. G. Stavros and S. Perrier, J. Am. Chem. Soc., 2020, 143, 382–389 CrossRef PubMed.
- Y. Wang, N. Han, X. L. Li, R. Z. Wang and L. B. Xing, ACS Appl. Mater. Interfaces, 2022, 14, 45734–45741 CrossRef CAS PubMed.
- S. Kuila and S. J. George, Angew. Chem., Int. Ed., 2020, 59, 9393–9397 CrossRef CAS PubMed.
- L. Sun, Y. Yuan, R. Li, W. Zhan, X.-J. Wang, Y. Zhao and X. Han, J. Mater. Chem. A, 2019, 7, 25423–25432 RSC.
- B. Tang, W. Xu, J. F. Xu and X. Zhang, Angew. Chem., Int. Ed., 2021, 60, 9384–9388 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05820d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |