
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrecise activation of C–C bonds for recycling and upcycling of plastics
Hongshun
Ran
ab,
Shuo
Zhang
ab,
Wenyi
Ni
ab and
Yaxuan
Jing
 *ab
*ab
aState Key Laboratory of Pollution Control and Resource Reuse, School of the Environment, Nanjing University, Nanjing 210023, China
bInstitute for the Environment and Health, Nanjing University Suzhou Campus, Suzhou 215163, China. E-mail: yaxuan.jing@nju.edu.cn
First published on 8th December 2023
Abstract
The rapid accumulation of plastic waste has led to a severe environmental crisis and a noticeable imbalance between manufacturing and recycling. Fortunately, chemical upgradation of plastic waste holds substantial promise for addressing these challenges posed by white pollution. During plastic upcycling and recycling, the key challenge is to activate and cleave the inert C–C bonds in plastic waste. Therefore, this perspective delves deeper into the upcycling and recycling of polyolefins from the angle of C–C activation–cleavage. We illustrate the importance of C–C bond activation in polyolefin depolymerization and integrate molecular-level catalysis, active site modulation, reaction networks and mechanisms to achieve precise activation–cleavage of C–C bonds. Notably, we draw potential inspiration from the accumulated wisdom of related fields, such as C–C bond activation in lignin chemistry, alkane dehydrogenation chemistry, C–Cl bond activation in CVOC removal, and C–H bond activation, to influence the landscape of plastic degradation through cross-disciplinary perspectives. Consequently, this perspective offers better insights into existing catalytic technologies and unveils new prospects for future advancements in recycling and upcycling of plastic.
 Hongshun Ran | Hongshun Ran received his Master degree from Jiangsu University in China. Currently, he is pursuing further studies in the group of Prof. Yaxuan Jing at Nanjing University. His research focuses on the upcycling of waste plastics. |
 Shuo Zhang | Shuo Zhang received her B.Sc. degree from Yangtze University in China. She is currently pursuing her Master degree under the supervision of Prof. Yaxuan Jing at Nanjing University. Her research focuses on the upcycling of waste plastics. |
 Wenyi Ni | Wenyi Ni received her B.Sc. degree from Hohai University in China. She is currently pursuing her Master degree under the supervision of Prof. Yaxuan Jing at Nanjing University. Her research focuses on catalytic conversion of biomass. |
 Yaxuan Jing | Yaxuan Jing received his B.Sc. and Ph.D. degrees at East China University of Science and Technology (supervisor: Prof. Yanqin Wang). He studied at National University of Singapore as a joint |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ph.D.
Ph.D.1. Introduction
Plastic, as a manufactured chemical substance, has become a highly competitive material in recent decades due to its favorable attributes, such as cost-effectiveness, lightweight nature, and chemical stability.1–3 It has significantly contributed to the advancement of human society.4,5 Unfortunately, the majority of plastics possess a relatively short lifespan, generally less than a month, and their chemical stability makes the possibility of natural degradation in the short term almost negligible.1,2,6 The rapid accumulation of plastic waste, which is challenging to handle, has resulted in severe environmental pollution, ultimately posing a threat to both Earth's ecosystems and human health.5,7–10 Meanwhile, plastic waste represents one of the most significant potential carbon resources in the modern era.3,11 Approximately 59% of all plastics ever manufactured, equivalent to around 8600 million metric tons, are directly discarded, ultimately ending up in landfill sites, or infiltrating natural ecosystems, such as rivers and oceans.8,12 Only approximately 17% of these plastics are fortunate enough to be reclaimed and used as an energy source.12 Consequently, the imperative lies in achieving environmentally sustainable plastic waste treatment through methods such as physical, chemical, or biological recycling/ upgradation. Among various methods, chemical upgradation has emerged as a promising technology for converting plastic waste into high-value products via chemical processes, including high-quality fuels and valuable chemicals, since that can precisely activate a partial C–C bond under milder environments.6,13,14 Polyolefins, such as polyethylene (PE), polypropylene (PP), polystyrene (PS) and polyvinyl chloride (PVC), have emerged as the predominant plastics on earth, constituting about 74% of the world's total plastic volume.2,6,8,15–18 In recent years, there has been significant interest in systematically and efficiently converting waste polyolefins into high-value products through chemical upgradation. Noteworthily, the most significant structural feature of polyolefins is that they are constructed from monomers by interunit C–C linkages. Consequently, the key scientific question in polyolefin degradation is how to efficiently and precisely activate and cleave their C–C bonds.12,19–22 Although numerous effective strategies have emerged for activating and cleaving C–C bonds, this endeavor encounters inherent challenges due to the chemical inertness of C–C bonds.12The reported strategies for activation–cleavage of C–C bonds in polyolefins mainly include non-catalytic pyrolysis, catalytic cracking, hydrogenolysis, and dehydrogenation-mediated processes.16,23,24 These methods have successfully converted polyolefins into fuels, chemicals, and olefin monomers through the effective activation–cleavage of the C–C bond. Although non-catalytic pyrolysis has emerged as a significant industrial strategy for converting waste polyolefins into chemicals, heat, power, and other materials, its “non-selective” nature imposes constraints on the variety and the economic value of products.6,16 Achieving high-quality fuels and chemicals necessitates a substantial enhancement of the activation strategy for C–C bonds in polyolefins. Advanced catalysts enabling efficient activation–cleavage of C–C bonds in polyolefins would be more advantageous for the chemical upcycling of waste polyolefins.19,25 However, most of the investigations in this area are still in their early stages and necessitate a profound comprehension of the correlation between catalysts and the precise activation–cleavage of C–C bonds. Specifically, elucidating the structure–activity relationship between the properties of the active sites on the catalysts and their capacity for activating C–C bonds, while also considering the impact of various other parameters, will substantially enhance the fundamental understanding of the chemical upgradation of polyolefins. Therefore, comprehensively discussing the reported catalytic systems is necessary to facilitate an in-depth investigation for polyolefin depolymerization based on the lens of C–C bond activation.
Here, we attempt to delve deeper into the depolymerization mechanism of polyolefins from the angle of C–C bond activation–cleavage at the active sites and discuss the various techniques in the degradation of polyolefins and C–C bond activation chemistry (Fig. 1). Three types of C–C bonds involving Caliph–Caliph, Caliph–CCl and Caliph–Carom in polyolefins and their precise activation–cleavage are elucidated. In addition, we draw upon accumulated wisdom from related fields to inspire C–C bond activation in plastics. This includes insights from C–C bond activation in lignin chemistry, alkane dehydrogenation chemistry, C–H bond activation, and C–Cl bond activation in Cl-containing volatile organic compound (CVOC) removal. It is essential to note that this perspective does not aim to provide a comprehensive compilation of publications. Instead, it seeks to offer a well-articulated description of the mechanism underlying C–C bond activation in polyolefins, express insights into improving existing catalytic systems and contribute new perspectives to catalyst design. Readers may refer to the excellent reviews to get a comprehensive understanding of plastic conversion.13,14,26–30
 | ||
| Fig. 1 Schematic illustration of the topics covered in this perspective. | ||
2. Caliph–Caliph activation
C–C bonds, including Caliph–Caliph, Caliph–CCl and Caliph–Carom, are widely present in various types of plastics. Aliphatic carbon chains commonly exist in thermoplastics like polyethylene (PE; global production of 125 Mt per year), polypropylene (PP; global production of 69 Mt per year), and polyvinyl chloride (PVC; global production of 39 Mt per year).17 To distinguish between C–C in PE/PP with C–C in PVC, we use Caliph–Caliph and Caliph–CCl, respectively, to represent them. Noteworthily, Caliph–CCl refers to C–C in PVC, not C–Cl. Although the C–C bonds in PVC also belong to the Caliph–Caliph bonds, its inherent Cl species make the activation of Caliph–CCl in PVC completely different from the activation of Caliph–Caliph in PE/PP. Consequently, we have discussed the precise activation of C–C bonds in waste plastics from three perspectives including Caliph–Caliph, Caliph–CCl and Caliph–Carom bonds. First, the activation of Caliph–Caliph bonds in PE/PP (Fig. 2a) over three catalytic sites including acid sites, metal sites and dual sites in metal–acid bifunctional catalysts is discussed in this section (Fig. 2b–d). After a thorough understanding of the catalytic mechanisms of Caliph–Caliph bond activation–cleavage, we will discuss the research advances of each catalyst type in this piece. This perspective primarily covers the emerging catalytic systems capable of precisely activating Caliph–Caliph bonds, including catalytic cracking (protolytic cracking and β-scission on acid sites), hydrogenolysis (activating Caliph–H and Caliph–Caliph bonds on metal sites), and hydrocracking (synergistic catalysis at dual sites).16 Zeolite catalysts including ZSM-5, ZSM-12, SBA-15, SBA-16, β-zeolites, and so on, play a prominent role in the activation–cleavage of Caliph–Caliph bonds via the catalytic cracking. Metal-loaded catalysts, such as Pt/SiO2, Pt/WO3/ZrO2, Pt/Al2O3, Ru/C, Ru/CeO2, and Ru/WZr, are primarily employed for the activation–cleavage of Caliph–Caliph bonds in hydrogenolysis. On the other hand, bifunctional catalysts can synergistically catalyze hydrocracking, where metal sites activate Caliph–H bonds and acid sites are responsible for the activation of Caliph–Caliph bonds. Bifunctional catalysts commonly comprise noble metal species (i.e., Pt and Pd) and a Brønsted acid component (usually zeolite). Additionally, a series of typical catalysts are displayed in Table 1. In short, waste plastic upcycling via the activation of Caliph–Caliph bonds on three types of catalysts including acid sites, metal sites, and dual sites has aroused extensive research interest. In addition, several other emerging catalytic systems are summarized here, including cross-alkane metathesis31,32 and tandem catalytic strategies.15,33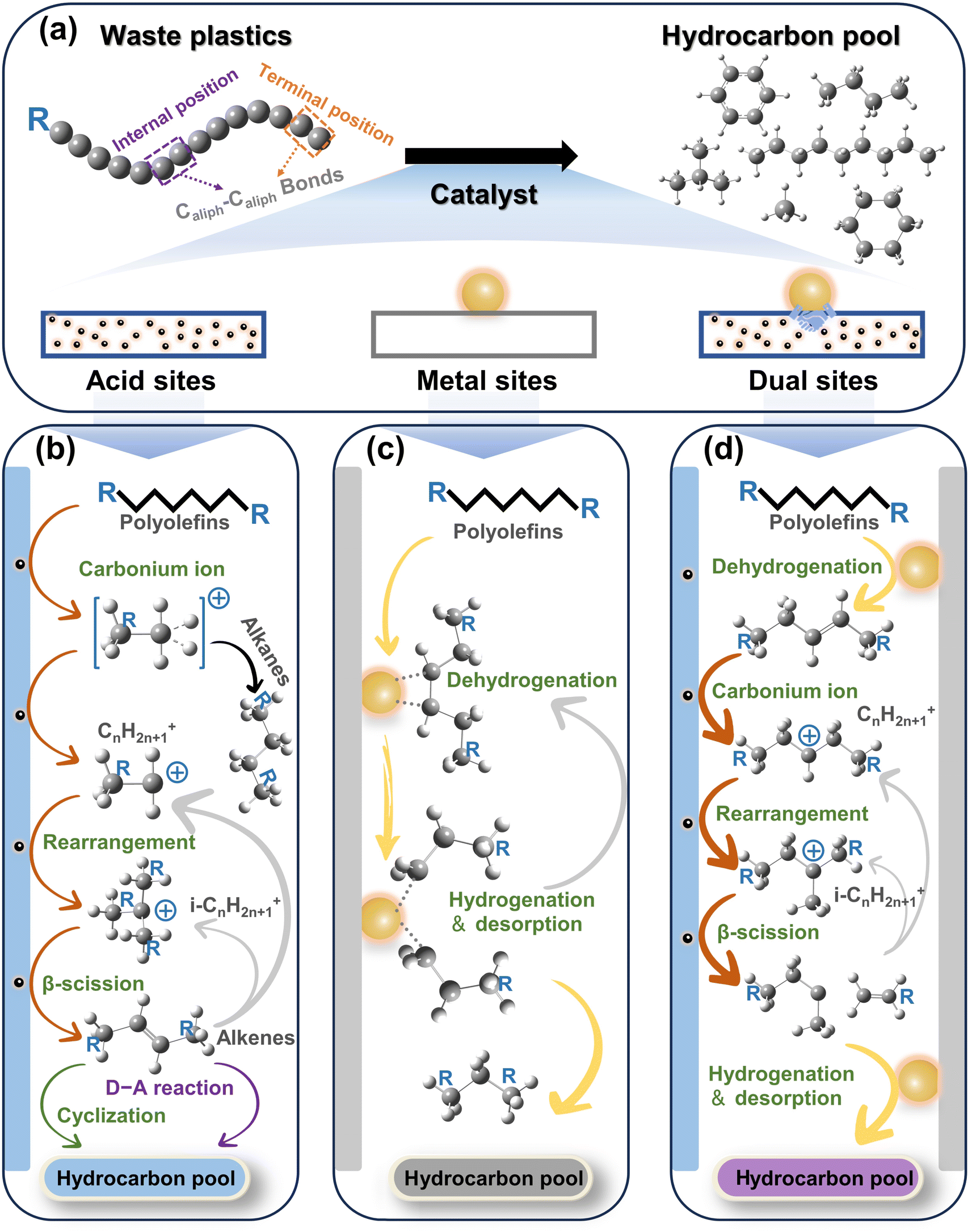 | ||
| Fig. 2 (a) Schematic illustration of the upcycling of polyolefins via Caliph–Caliph bond activation. (b–d) Schematic diagrams of the upgradation of polyolefins when Caliph–Caliph bonds are activated over (b) the acid sites, (c) the metal sites, and (d) the dual sites. | ||
| Active sites | Feedstocks | Catalysts | Reaction conditions | Yields | Ref. |
|---|---|---|---|---|---|
| a G: (gaseous product), L: (liquid product), S: (solid product). | |||||
| Acid sites | HDPE | HZSM-5 (Si/Al2O3 = 80) | 500 °C, N2 | G: 60% (C2–C4) | 54 |
| L: 32% (C5–C11) | |||||
| S: n | |||||
| HDPE | H-ZSM-5 (11.5) | 300 °C, 20 bar H2 | G: >90% | 61 | |
| L: n | |||||
| S: n | |||||
| LDPE | PI-ZSM-5 | 380 °C, 35 mL min−1 N2 | G: 78% (C2–C5) | 63 | |
| L: 17% (C6–C11) | |||||
| S: n | |||||
| LDPE | L-ZSM-5 | 380 °C, 35 mL min−1 N2 | G: 82% (C2–C5) | 63 | |
| L: 18% (C6–C11) | |||||
| S: n | |||||
| LDPE | H-ZSM-5 | 340 °C, 35 mL min−1 N2 | G: 41% (C2–C5) | 63 | |
| L: 29% (C6–C11) | |||||
| S: n | |||||
| HDPE | BEA | 380 °C, 30 mL min−1 N2 | G: n | 73 | |
| L: ∼80% | |||||
| S: n | |||||
| Metal sites | LDPE | Ru/ZrO2 (Ru 5 wt%) | 200 °C, 2 MPa H2 | G: 11% (C1–C4) | 84 |
| L: 89% (C5–C45) | |||||
| S: n | |||||
| HDPE | Ru/ZrO2 (Ru 5 wt%) | 240 °C, 6 MPa H2 | G: 13% (C1–C4) | 84 | |
| L: 86% (C5–C45) | |||||
| S: n | |||||
| PP | Ni-based alloy catalyst | 300 °C, 3 MPa H2 | G: 89% (C1–C4) | 93 | |
| L: n | |||||
| S: n | |||||
| PE | mSiO2/Pt-1.7/SiO2 | 300 °C, 0.89 MPa H2 | G: 66% (C1–C4) | 76 | |
| L: 34% (C10–C37) | |||||
| S: n | |||||
| PE | mSiO2/Pt-2.9/SiO2 | 300 °C, 0.89 MPa H2 | G: 15% | 76 | |
| L: 71% (C10–C37) | |||||
| S: 14% | |||||
| PE | mSiO2/Pt-5.0/SiO2 | 300 °C, 0.89 MPa H2 | G: 12% | 76 | |
| L: 62% (C10–C37) | |||||
| S: 26% | |||||
| LDPE | Ru SAC | 250 °C, 2.0 MPa H2 | G: 3.0% | 100 | |
| L: 95% | |||||
| S: n | |||||
| LDPE | Ru/TiO2-H600 | 240 °C, 2.0 MPa H2 | G: 2.9% | 95 | |
| L: 87% | |||||
| S: n | |||||
| HDPE | mSiO2/Pt/SiO2 (3.5 nm) | 300 °C, 1.7 MPa H2 | G: 22% | 91 | |
| L: 77% | |||||
| S: 1.0% | |||||
| Dual sites | PE | Ru/FAU | 200 °C, 30 bar H2 | G: n | 20 |
| L: 67% | |||||
| S: n | |||||
| LDPE | Pt/WO3/ZrO2 + HY | 250 °C, 30 bar H2 | G: 9.0% (C1–C4) | 104 | |
| L: 83% (C5–C22) | |||||
| S: 6.0% | |||||
| HDPE | Ru/HZSM-5 (300) | 280 °C, 2.0 MPa He | G: 7.4% | 107 | |
| L: 58% | |||||
| S: 30% | |||||
| LDPE | Pt/F–Al2O3 | 280 °C, without H2 | G: 7.0% | 112 | |
| L: 71% | |||||
| S: 12% | |||||
| LDPE | Pt/Cl–Al2O3 | 280 °C, without H2 | G: 4.0% | 112 | |
| L: 72% | |||||
| S: 16% | |||||
The Caliph–Caliph and Caliph–H cleavage mechanisms in polyolefins, as large polymer molecules, remain incompletely elucidated. In contrast, the cleavage of C–C and C–H bonds in small molecules, sharing similar chemical bonds with polyolefins, has undergone a more comprehensive investigation. When discussing the activation–cleavage of C–C bonds in polyolefins, small molecules (e.g., short-chain alkanes) can serve as models, thereby favoring the understanding of bond-breaking mechanisms in polyolefins. The macromolecular structure of polymers often poses challenges for in situ characterization. Since both polyolefins and short-chain alkanes consist of C–C and C–H bonds, employing small molecules as models would facilitate the investigation of these mechanisms in polyolefins, particularly the catalytic nature of C–C and C–H bonds at the molecular level. While this approach may not fully elucidate the activation–cleavage processes in real plastics, it still offers valuable insights. The cleavage of C–C bonds in hydrocarbons often involves the disruption of C–H bond as an initial step. Moreover, the C–H bond exhibits a higher energy barrier, resulting in chemical inertness. Given the chemical similarities between the two fields, many studies on polyolefin degradation draw inspiration from the bond-breaking mechanisms of small-molecule model compounds.34 For example, Pt is recognized as the most active metal in ethane dehydrogenation due to its superior ability to activate C–H bonds.35 This attribution also makes Pt a potential active site for polyolefin degradation. Furthermore, the principle that metals with distinct adsorption capacities for substrate and product will exhibit enhanced product selectivity can be extended to the catalyst design for polyolefin degradation. For example, modifying the size and coordination environment of the metal sites has the potential to reduce the adsorption capabilities of intermediates over the active sites, thereby enhancing product selectivity. While initial steps in small molecules and polyolefin systems share similarities, and the chemical–physical characteristics of cleaving C–C bonds and C–H bonds in small molecules can inspire the depolymerization of polyolefins, there are still significant differences between the two domains. These differences include the accessibility of C–C bonds in plastics with macromolecular structures, mass-transfer capacity during reactions, and the impact of branching on C–C bonds. These factors are essential to consider in polyolefin depolymerization. The likelihood of internal and terminal C–C bonds contacting the active site is comparable in small molecules, while terminal C–C bonds predominate significantly in polymers, particularly those exhibiting a high degree of branching, potentially influencing product distribution. Furthermore, the mass transfer ability differs between small molecules and polymers, especially gaseous small molecules, which requires consideration when assessing the impact of the mechanism on reaction rates. We speculate that when polyolefins are degraded to a certain molecular weight level, they will inevitably follow the C–C bond cleavage mechanism of small molecules. However, whether this mechanism is applicable when plastics are in the form of polymer macromolecules remains a subject requiring in-depth investigation. For instance, in the realm of biomass hydrogenolysis, polymers undergo an initial solvolysis to form a stabilized intermediate before advancing to the hydrogenolysis step. This provides a novel reference point for comprehensively exploring the degradation mechanism of plastics.36 Readers are encouraged to consult related extensive reviews for a comprehensive understanding of bond-breaking mechanisms in small-molecule organic chemistry.37–39
2.1 Acid sites
Primary carbenium ions have higher energy content compared to isocarbenium ions, rendering n-carbon ions (CnH2n+1+) unsuitable for direct β-scissions.47 Furthermore, the alkenes generated by β-scissions might not desorb immediately, or they could re-adsorb onto the Brønsted acid sites after desorption, resulting in the formation of new carbenium ions. Since polyolefins consist of long-chain hydrocarbon molecules, all long-chain carbenium ion intermediates formed from the cleavage of alkanes or olefins will undergo continuous skeletal rearrangement and β-scissions, thereby yielding hydrocarbon products with sufficiently low molecular weight. Furthermore, the branched chains within polyolefin molecules could undergo end-chain scission, resulting in the direct formation of low molecular weight products.50–52
The cleavage of the Caliph–Caliph bond at different positions, whether it's the internal position or the terminal position, results in the formation of various products. In the case of PE, breaking the terminal Caliph–Caliph bond will produce a low molecular weight gaseous product (mainly methane). Conversely, cleaving the Caliph–Caliph bond at the internal position yields liquid products with a slightly higher molecular weight. As a result, the separation of these products is relatively straightforward. In contrast, the products generated from the cleavage of PP are more complex, and the liquid products require further characterization via NMR and/or GC.40 The cleavage of the terminal Caliph1–Caliph3 bond results in the formation of methane, a similar effect can also be observed in the degradation of PE. In contrast, the cleavage of the internal Caliph2–Caliph3 bond yields a relatively complex mixture of short-PP oligomers, when compared with PE. In addition to the structural differences in intermediates and production, the reaction mechanism of catalytic cracking is considered a complex process due to the involvement of a wide variety of secondary reactions, including aromatization, cycloaddition, dehydrogenation, isomerization, oligomerization, etc.53 As a result, it leads to the production of more valuable products, such as alkenes and aromatics. Aromatic hydrocarbons, which belong to an important class of bulk chemicals, hold significant research significance regarding their formation mechanisms.
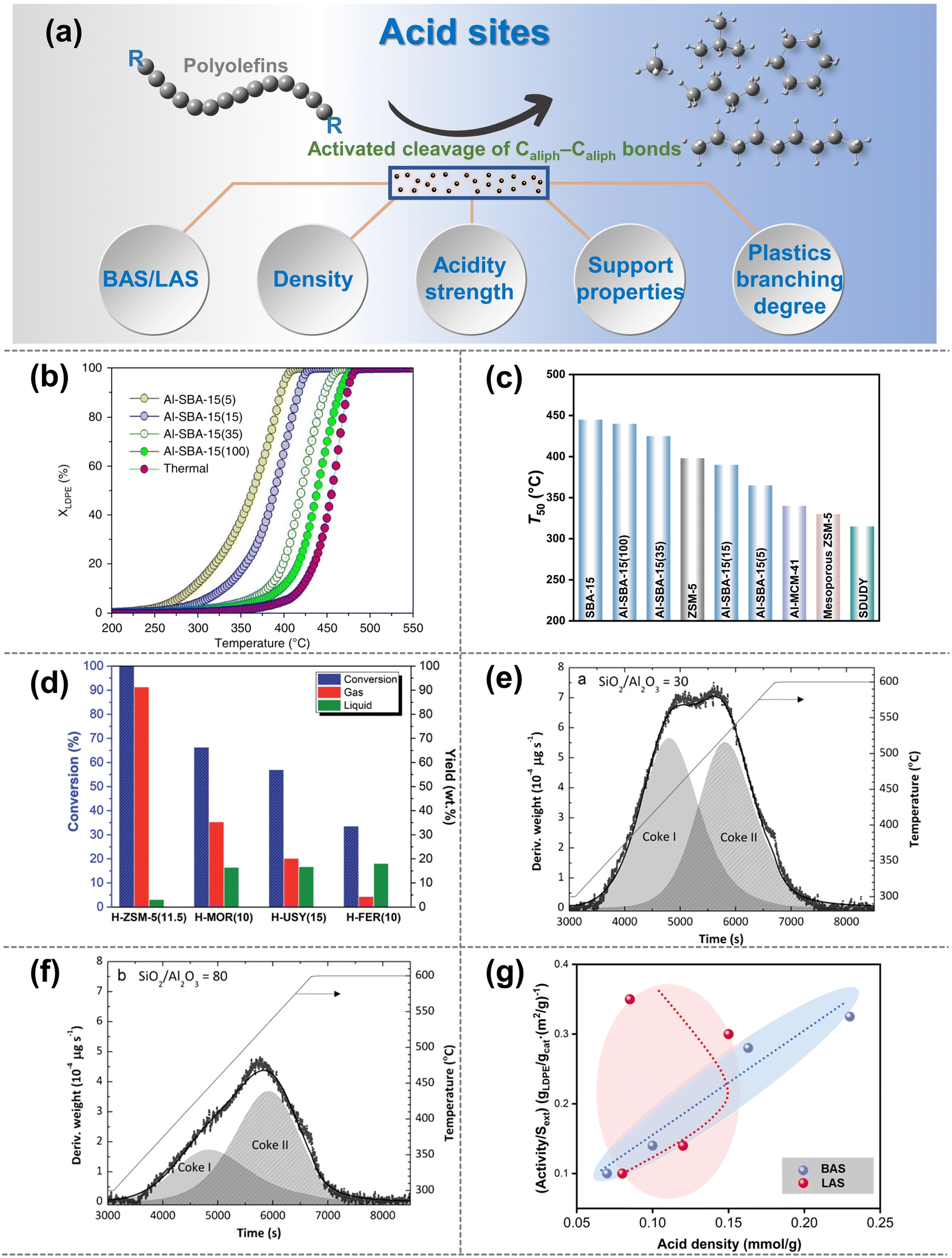 | ||
| Fig. 3 (a) Schematic diagrams of the upgradation of polyolefins when Caliph–Caliph bonds are activated by the acid sites. (b and c) Effect of the density of BAS on the catalytic cracking efficiency and temperature on the catalysts: (b) cracking conversion curves for the Al-SBA-15 series, XLDPE is the conversion of LDPE; (c) temperature at 50% conversion (T50) for the aluminum-based SBA-15 series compared to the reference materials. Data collected and reproduced with permission from ref. 59. Copyright 2019 Nature. (d) Conversion and yield of gas and liquid products as a function of catalyst type with different acidity strengths: H-ZSM-5 (11.5), H-MOR (10), H-USY (15) and H-FER (10), data collected from ref. 61 and copyright 2022 Royal Society of Chemistry. (e and f) Effect of the SiO2/Al2O3 ratio of HZSM-5 zeolite on the coke deposited on the catalysts: (e) HZSM-5 zeolite catalyst with a SiO2/Al2O3 ratio of 30; (f) SiO2/Al2O3 ratio of 80. Data collected from ref. 84. Copyright 2012 Elsevier. (g) Relationship between the activity of LDPE cracking and BAS density or LAS density (reproduced with permission from ref. 67). Copyright 2016 Royal Society of Chemistry. | ||
The initial activation and cleavage of Caliph–Caliph bonds primarily occur at the BAS.16 The physicochemical properties of the BAS will have a significant impact on the activation and cleavage of Caliph–Caliph bonds. Additionally, LAS also plays an important role in the activation of Caliph–Caliph bonds. It is well known that zeolite catalysts are mainly used for the activation of Caliph–Caliph bonds over the acid sites (e.g., ZSM-5, ZSM-12, SBA-15, SBA-16, β-zeolites, etc.).16 First, the density of BAS/LAS is of critical significance for the activation efficiency of Caliph–Caliph bonds. A higher density of BAS/LAS corresponds to a greater number of active sites available for the direct activation of the Caliph–Caliph bonds. This increase in the active site density significantly enhances the efficiency of bond activation, thereby reducing the required reaction temperature. Zhang et al. observed that an increased density of BAS on the Al-SBA-15 achieved a higher activation efficiency and a decreasing trend of the degradation temperature, owing to a greater number of sites capable of directly activating the Caliph–Caliph bonds (Fig. 3b and c).59 Furthermore, similar trends were obtained in the activation cracking of Caliph–Caliph bonds, employing LAS-rich β-zeolites and ZSM-12, respectively.60 Briefly, increasing both LAS and BAS concentrations can boost the activation of Caliph–Caliph bonds, respectively. However, the synergistic effects between the two are still unclear and further exploration is strongly encouraged.
In addition to the density effect of BAS/LAS sites, the acidity strength is also a pivotal factor influencing the activation of Caliph–Caliph bonds and product distribution. Section 2.1.1 has described that the β-scission is a key step in the activation of Caliph–Caliph bonds. In general, active sites with stronger acidity exhibit a greater propensity for inducing β-scissions.16 Consequently, the activation efficiency of Caliph–Caliph bonds and the product distribution can be regulated by adjusting the acidity strength of catalysts. Costa et al. found that among structurally similar catalysts, those with higher BAS strength can enhance the cleavage of the Caliph–Caliph bonds, resulting in a higher percentage of light alkane products (Fig. 3d).61 Furthermore, a similar regularity was demonstrated by Piovano et al. for the catalytic degradation of linear low-density polyethylene (LDPE) by metal-based (Zr, Al) catalysts (Al-SBA-15, Al-SBA-16, Zr-SBA-15, and Zr-SBA-16) with different LAS strengths.62 Obviously, higher acidity strengths in both BAS and LAS significantly favor the cleavage of Caliph–Caliph bonds. Although a high concentration of activation sites and high acidity favor the conversion of polyolefins, excessive acidity can lead to severe coking. For example, Elordi et al. found that the amount of coke deposited on the catalyst surface could be effectively reduced in ZSM-5-catalytic HDPE degradation when the acidity of the catalysts was lowered (Fig. 3e and f).54
Based on the significance of BAS and LAS, the contribution of the BAS and LAS to the activation of Caliph–Caliph bonds will be further distinguished in this section, respectively. In general, the significance of BAS for activating the Caliph–Caliph bonds of polyolefins is recognized. Escola et al. found that the cracking activity of LDPE over ZSM-5 zeolite with different BAS and LAS densities exhibited an almost linear relationship with the BAS density but was stochastically disconnected from the LAS density (Fig. 3g).63 This is attributed to the lower activation energy barrier of Caliph–Caliph bonds on the BAS compared to that of the LAS.64 Although the LAS may not directly affect the activation of Caliph–Caliph bonds, its contribution to the catalytic cracking of polyolefins is still not negligible. Recently, the significance of LAS has been demonstrated again by Kokuryo et al., who found that β-zeolites with a higher density of LAS exhibited superior catalytic performance compared to conventional β-zeolites in the activated cracking of Caliph–Caliph bonds in LDPE.60 The outstanding improvement in catalytic performance may be attributed to the synergistic interaction between the BAS and the adjacent LAS.65–67 Liu et al. further confirmed that the synergistic interaction between BAS and LAS can effectively enhance the acidity of BAS in the catalytic system, increasing the efficiency of Caliph–Caliph bond activation.55 Another explanation is that LAS can enhance paraffin dehydrogenation and the formation of olefinic intermediates, thereby promoting the formation of carbenium and the subsequent fluidized catalytic cracking.53 Briefly, both BAS and LAS play a significant role in the activation of Caliph–Caliph bonds. Notably, the role of LAS in activating Caliph–Caliph bonds needs further exploration compared with the well-studied BAS. The significance of both BAS and LAS for cleaving Caliph–Caliph bonds is substantial. However, these significances are manifested through distinct mechanisms. While the presence of both BAS and LAS does not inherently imply superiority, nor is it advantageous to have an abundance of either, it is imperative to thoroughly explore the intricate relationship between them at the mechanism level. For example, the inherent correlation between BAS and LAS in relation to the activation performance of Caliph–Caliph bonds could be thoroughly investigated by regulating the varying ratios.
Particularly, the pore morphology of catalysts and the branching degree of the plastic are pivotal in facilitating the diffusion and adsorption of reactants and intermediates. While the pore structure does not directly impact the activation process of Caliph–Caliph bonds, it does affect the accessibility of acid sites to polyolefin reactants and intermediates, thereby influencing the activation efficiency and selectivity of Caliph–Caliph bonds.68 Thus, this section discusses the impact of catalyst pore types and plastics' branched chains on the activation of Caliph–Caliph bonds. In the case of micropores, the unique shape-selectivity enable small-molecular intermediates to undergo a series of secondary reactions, ultimately leading to the formation of target products (such as light alkanes). However, they restrict the direct access of large polyolefin molecules to the active site channels, particularly highly branched polymers. As a result, small-sized micropores exhibit shape-selective and confinement effects on the small-molecular reactants, intermediates, and products.69 In contrast to micropores, mesopores effectively alleviate the diffusion limitation of reactants and intermediates on the catalyst's surface, accordingly enhancing the efficiency of Caliph–Caliph activation. However, mesopores tend to exhibit reduced selectivity for the desired products compared to micropores. Furthermore, the diverse branching degree of plastics leads to distinct degradation and diffusion processes in catalysts with identical pore types; meanwhile, this potential connection is intimately linked to the pore morphology.70,71
It has been confirmed that the microporosity of the catalyst exerts a spatial confinement effect on the activation of Caliph–Caliph bonds, which regulates the chemical structure of the products. Manos et al. confirmed that larger microporous structures in Y- and β-zeolites facilitate rapid bimolecular hydrogen transfer reactions, resulting in HDPE conversion with alkanes as the main product.72 Furthermore, during the catalytic cracking of polyolefins over porous catalysts (e.g., ZSM-5, Y-zeolites, USY, β-zeolites, MOR, etc.), liquid products are predominantly generated over these catalysts with larger pore sizes rather than microporous ones. For instance, Park et al. reported that β- and Y-zeolite exhibit higher selectivity for liquid products compared to ZSM-5 and MWW zeolites, which have relatively small micropores. This is attributed to the weak acidity of the catalysts, as well as the presence of relatively large three-dimensional pores that promote the diffusion of liquid hydrocarbon products and prevent deep cracking.73 Apart from micropores, mesopores also play a critical role in effectively cleaving Caliph–Caliph bonds. However, mesopores tend to exhibit reduced selectivity for reactants, intermediates, or products compared to micropores.70 The preparation of catalysts with a rich mesoporous structure can significantly alleviate diffusion limitations and improve the cleavage efficiency of the Caliph–Caliph bond. Bonilla et al. discovered that introducing mesopores enhanced the accessibility of active sites, optimizing the diffusion pathway and greatly improving the efficiency of Caliph–Caliph bond activation.74 The acidity on mesoporous aluminosilicates is lower compared to conventional microporous acid catalysts, but the active centers within the mesopores offer improved accessibility and result in excellent conversion towards gasoline products. In addition to catalyst morphology, the potential connection of the plastics' branching level with catalyst morphology, such as micropores, should also be emphasized. The branching level of plastics influences the diffusion and cleavage efficiency of polymers over acid sites, especially within micropores. Crossley et al. found that in LLDPE with higher branching levels, Caliph–Caliph cleavage predominantly occurs at acid sites.70 LLDPE exhibits better reactivity than HDPE due to the facile protonation of tertiary carbon groups, while increased branching leads to the formation of more stable carbenium ion through β-scission events. From another perspective, HDPE, characterized by a lower branching level and a superior self-diffusion coefficient, accesses acid sites within catalyst micropores more effectively, where the activation–cleavage of Caliph–Caliph bonds occurs. Briefly, LLDPE shows heightened reactivity over surface-located acid sites, underscoring the importance of the branching degree of plastics in the activation–cleavage of Caliph–Caliph bonds. Alternatively, Caliph–Caliph bonds of HDPE are more efficiently cleaved when acid sites are confined to the micropores.70 Deriving from the aforementioned findings, a rational design of active site locations (either in the micropores or on the catalyst's surface) coupled with the shape-selectivity of the micropores could enable a stepwise cleavage of Caliph–Caliph bonds in mixed plastics with different branching levels. This approach is anticipated to enhance product selectivity and degradation efficiency. Extending this strategy to address real-world mixed plastic challenges holds promising prospects. Furthermore, when dealing with highly branched substrates (e.g., PP), exploring methods for selectively cleaving Caliph–Caliph bonds on the main chain, rather than the branched chains, appears as a favorable approach for obtaining multi-carbon alkanes instead of methane.
This section methodically discusses the effects of acid site density, acidic strength, pore morphology, and the branching degree of plastics on the activation–cleavage of Caliph–Caliph bonds. Abundant acid sites and favorable acidic strength generally facilitate the cleavage of Caliph–Caliph bonds, reducing the energy barrier required. However, excessive acid sites and acidic strength may result in increased carbon deposition, highlighting the significance of adjusting the concentration and strength of acid sites during catalyst design. In terms of activation mechanisms, both BAS and LAS play crucial roles in Caliph–Caliph bond activation. The Caliph–Caliph bond activation mechanisms over LAS, however, remain somewhat unclear, necessitating further research. Furthermore, by integrating various pores and the branching degree of plastics in the depolymerization of plastics, it is expected that tandem microporous and mesoporous catalysts could lead to the stepwise degradation of mixed plastics with varying branching degrees. The cleavage of Caliph–Caliph bonds over acid sites typically necessitates harsh conditions, during which the unsaturated intermediates tend to adsorb on the catalyst. This adherence often leads to secondary polymerization and may exacerbate carbon deposition. Hence, designing appropriate acid sites, in conjunction with a variety of pore types, is anticipated to tackle this issue. This strategy aims to lower the energy barrier for Caliph–Caliph bond cleavage and to diminish interactions between intermediates and catalysts. In addition to the direct role of acid site catalysts in activating Caliph–Caliph bonds, the adjustment of metal sites by supports can indirectly influence the activation process. Moreover, acid sites continue to play an essential role in the dual-site strategy. This aspect will be discussed in Section 2.3.
2.2 Metal sites
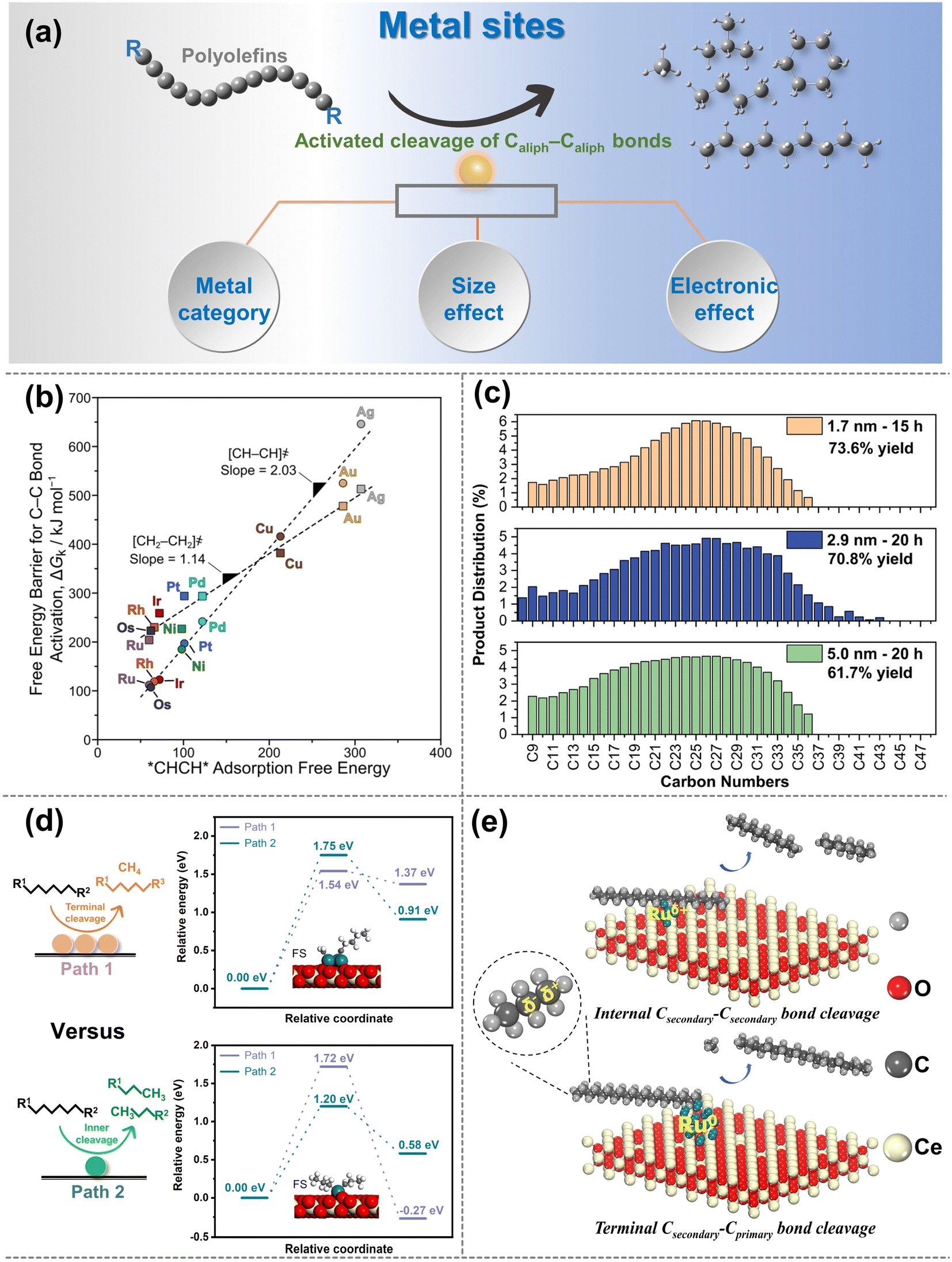 | ||
| Fig. 4 (a) Schematic diagrams of the upgradation of polyolefins when Caliph–Caliph bonds are activated over the metal sites. (b) Free-energy barriers ΔGk for Caliph–Caliph bond activation in *CHCH* (circles) and *CH2CH2* (squares) as a function of *CHCH* adsorption free energies over various metal sites (593 K, 1 bar H2). Data collected from ref. 97. Copyright 2019 American Chemical Society. (c) Carbon number distributions of hydrogenolysis of polyolefins over size-controlled Pt nanoparticles, data collected from ref. 76 and copyright 2022 American Chemical Society. (d) Summarized cleavage modes of polyolefin on conventional SAC and nanocluster catalyst, reproduced with permission from ref. 100 and copyright 2023 Science Partner Journals. (e) Proposed reaction mechanism of polyethylene hydrogenolysis over different Ru chemical states of Ru/CeO2 catalysts. Data collected with permission from ref. 22. Copyright 2023 Wiley. | ||
Noble metals like Ru and Pt are particularly attractive for investigating the activation of Caliph–Caliph bonds in polyolefins, with Ru being recognized as the optimal candidate for activating Caliph–Caliph bonds.76,84,87,91 Metal sites such as Ru, Pt, etc. tend to preferentially activate the Caliph–Caliph bonds at the internal position, whereas Ni, Co, etc. commonly favor the Caliph–Caliph bonds at the terminal position.76,84,94,96 Tamura et al. confirmed the uniqueness of Ru in the activation of Caliph–Caliph bonds and found that Ru-based catalysts (Ru/ZrO2, Ru/CeO2, Ru/SiO2, and Ru/γ-Al2O3) all exhibited outstanding performance in the activation of Caliph–Caliph bonds in LDPE.84 The interaction between the support and the metal sites can adjust the electronic state of the active center to affect the cleavage of Caliph–Caliph bonds, which will be discussed in detail in the subsequent section. Nonetheless, the inherent activation properties of metal sites are undeniably crucial. Previous theoretical calculations have indicated that the Gibbs free energy barrier for the activation–cleavage of Caliph–Caliph bonds on Ru is significantly lower than on other metals (Rh, Pt, Ni, Pd, Cu, Ag, Au) (Fig. 4b) in ethane hydrogenolysis.97 Although further confirmation is needed to generalize this principle to polyolefin degradation, a subtle correlation between these two processes exists. Similarly, the activation–cleavage of Caliph–Caliph bonds on metal sites is also the key step in polyolefin conversion. Consequently, previous reports also identified that the lower energy barrier on Ru-based catalysts enables superior properties in activating the Caliph–Caliph bonds of polyolefins.16,19,26 In addition to noble metals, non-noble metal (Co, Ni) catalysts have also been applied in polyolefin conversion and demonstrate remarkable catalytic performance.67,93,94 For example, Si et al. discovered that Ni-based catalysts have excellent capabilities for the activation–cleavage of terminal Caliph–Caliph bonds at the branched chain in polyolefins, leading to superior selectivity for methane.93 This pattern may be attributed to either the excessively strong adsorption of active sites for the intermediate, leading to deep cleavage or terminal cleavage of Caliph–Caliph bonds. Strategies such as coating the metal site or adjusting the coordination environment are anticipated to facilitate the cleavage of non-terminal Caliph–Caliph bonds more effectively. Basically, high-value fuel range alkanes predominantly occur at Ru and Pt metal sites, while Ni and Co metal sites favor the production of light alkanes due to their preferential activation for Caliph–Caliph bonds at terminal positions. However, Ru/Pt metal sites may also continuously activate Caliph–Caliph bonds leading to methane production, which is closely linked to the size effects of Ru- and Pt-particles, and will be discussed in detail subsequently.98 It is noted that precious noble metals are highly sensitive to the presence of heteroatoms (N, S, Cl, etc.) originating from commercial additives and contaminants, making them susceptible to toxic deactivation. Therefore, extensive research efforts should be dedicated to studying non-noble metal-based catalysts for polyolefin conversion. The following strategies may prove valuable for conducting in-depth investigations. Utilizing the strong interactions between metals and supports can alter the electronic state of metal sites, promoting the activation of internal Caliph–Caliph bonds and leading to products with higher economic value distribution. Moreover, the activation properties of transition metals, except for Co and Ni, for the Caliph–Caliph bonds are worth comprehensive and in-depth exploration in future studies.
The size effect of metal sites can significantly affect activation performance and activation positions of Caliph–Caliph bonds. Specifically, the evolution of the surface population (corners, edges and faces) plays an important role in the Caliph–Caliph bond activation at metal sites with different sizes.99 Reactions in which the reaction rate is significantly modulated by the size effect of metal sites are termed structure-sensitive catalytic reactions and this effect is more pronounced on Pt metal sites.76 Compared with the large-sized particles, the smaller Pt metal site particles uniformly dispersed on the catalyst surface have attracted particular attention due to the outstanding ability of Caliph–Caliph bonds. Incompletely coordinated edge metal sites over the Pt particles are highly active to Caliph–Caliph bond activation compared to metal facets. Smaller-size Pt particles can provide more edge metal sites with outstanding accessibility.28,76 However, a high concentration of coordination-unsaturated smaller-sized Pt particles on the catalyst surface will promote excessive hydrogenolysis of polyolefins, resulting in high yields of low-value light alkanes. As a result, by modulating the structural properties of the metal particles (e.g., size, corners, edges, and faces), high-value liquid products with superior yields and relatively narrow dispersibility can be obtained.28 For instance, Wu and co-workers investigated the rate of the Caliph–Caliph cleavage on different sizes of Pt particles (1.7, 2.9, and 5.0 nm, Fig. 4c) and discovered that the rate constants for the cleavage of the Caliph–Caliph bonds increase progressively larger as the size of the Pt particles decreases.76 This is attributed to the fact that the activation–cleavage of Caliph–Caliph bond on Pt metal sites is a structure-sensitive reaction, and the reaction rate accelerates with the increase in the ratio of metal sites at the edge and corner positions. Similar rules have been observed by Delferro and co-workers.28 Furthermore, the size effect of Ru particles on the activation–cleavage of Caliph–Caliph bonds has also attracted increasing attention.83,84 Multi-point interactions between Ru metal planes and polyolefins can also promote the adsorption of target molecules on planar sites, leading to superior activity, which has rarely been reported on Pt particles. For example, Tamura and co-workers identified a significant correlation between Ru particle sizes and the catalytic performance of Caliph–Caliph activation, and that small- and medium-sized Ru particles exhibited lower selectivity for gas products compared to large Ru particles.84 This could be attributed to the planarity of large-sized Ru metal sites enabling interactions with polyethylene at multiple positions. This multipoint adsorption subsequently facilitates the successive hydrogenolysis of the Caliph–Caliph bonds in adsorbed states of polyethylene, thereby increasing the yield of the gaseous products. While the performance of Ru catalysts is primarily influenced by the particle size, the activation capacity and selective cleavage position of Caliph–Caliph bonds can also be impacted by other factors. Besides the particle size, adsorptive interaction between Ru metal faces and target polyolefin molecules also contributes to the catalytic rate of Caliph–Caliph cleavage, and excessively small Ru particles can decrease this interaction.84 However, the strong adsorption between the polyolefin and Ru sites also leads to the successive hydrogenolysis of Caliph–Caliph bonds, favoring the formation of methane. Consequently, it is still worthwhile to explore methods for optimizing catalytic performance by moderating the size effect in further investigations. Currently, the size effect primarily focuses on Pt and Ru, and the generalizability of this principle to other non-noble metals needs further confirmation. The focal emphasis is not on asserting the superiority of smaller metal-sized particles, instead, the key lies in striking a balance between the high activity associated with small-sized particles and the selectivity of resulting products. This balance point may vary among different metals or may not be applied to non-structure-sensitive reactions. Furthermore, the average spacing between metal sites on the catalyst surface increased due to a more uniform distribution, and smaller sizes expose more active sites. Can reducing the nanoparticles to the single-atom form be more effective in improving their performance for Caliph–Caliph cleavage? Chen et al. discovered that the cluster-like Ru species can catalyze the simultaneous cleavage of two adjacent Caliph–Caliph bonds. In contrast, the Ru single-atom catalyst (Ru-SAC) has independent Ru atoms as active sites and site-selective activation–cleavage modes (Fig. 4d).100 The barriers for activating the internal Caliph–Caliph bonds are relatively favorable on Ru-SAC, and it can prevent the successive breakage of the neighboring Caliph–Caliph bonds due to the absence of a sufficiently close activation center. However, with a monolithic metal site within SAC it could be difficult to perform all the necessary steps involving multiple active centers, such as C–H, C–C and H2 activation, especially in the case of real plastics. This limitation could potentially hinder the catalytic performance. In addressing this challenge of maintaining a low metal loading, the question arises as to whether dual- or poly-atom catalysts can provide a viable solution. Alternatively, a non-selective catalyst that initially generates intermediates from long chains and then converts the intermediates to liquids and/or gaseous products will result in processes with conversion-dependent yields and selectivities. The ideal catalysts could directly cleave long chains into desired short-chain fragments by modulating the size and distribution pattern of the metal sites at the microscopic level, and yield the desired products over a wide conversion range.
The size effect of metal sites on the activation of Caliph–Caliph bonds is to some extent related to the electronic effect. Different types and sizes of metal sites have different interactions with the supports, leading to different electron transfer effects. Next, the chemical states for the activation–cleavage of Caliph–Caliph bonds will be discussed. Metal sites with different valence states may exhibit significantly different catalytic performances and product distributions during the activation of Caliph–Caliph bonds. We previously found that the Ru species supported by different CeO2 crystal structures exhibited differential catalytic properties and site-selective activation ability in the activation of Caliph–Caliph bonds.22 Specifically, the positively charged Ru species preferentially activate the internal Caliph–Caliph bonds, while the metal Ru species prefer to activate the terminal Caliph–Caliph bonds (Fig. 4e). This is because the more positively charged Ru species enables the bonding of the internal C to drive the precise activation of internal Caliph–Caliph bonds due to the electron-donating effect of adjacent alkyl species, accordingly preventing the generation of methane. This pattern substantiates the feasibility of selectively cleaving specified Caliph–Caliph bonds by adjusting the coordination environment of the metal sites, thereby altering their adsorption capacity for intermediates, as highlighted in the preceding section. Moreover, Wang et al. also found that the sites of electron-rich Ru are more active to perform hydrogenation and desorption steps, to inhibit the activation–cleavage of secondary Caliph–Caliph bonds, avoiding the generation of methane.95 Besides, metal–support interactions and support factors (e.g., interfacial effects and geometrical effects) can also change the chemical state of metal sites. This effect could provide valuable insights into the nature behind the excellent performance of Caliph–Caliph bond activation.
Besides the factors directly acting on the metal sites, the physical properties of the pores in the supports can also indirectly affect the accessibility of the metal sites as well as the mass transfer of the intermediates, which in turn affects the activation performance of the Caliph–Caliph bonds and product distribution. Huang and his colleagues obtained a highly designed porous metal site catalyst by anchoring the Pt nanoparticles as metal sites at the closed end of the pores on the surface of mesoporous silica.91 The catalyst enables highly site-selective cleavage of the Caliph–Caliph bonds, affording a narrow product distribution. Consequently, artificial design of the microstructure, such as pore size and length, can further optimize the product distribution. Furthermore, the intricately designed catalyst architecture can effectively facilitate the precise positioning of the target macromolecule in close proximity to metal sites, thereby enhancing the catalytic performance for Caliph–Caliph bond activation.
Here, we systematically discuss the effects of metal site category, size effects, and electronic effects on the activation of Caliph–Caliph bonds. Notably, several scientific issues remaining in size effect and electronic effect need to be investigated, including the selective and synergistic cleavage of Caliph–Caliph bonds using dual-atom catalysts, and selective activation of Caliph–Caliph bonds through the modulation of interfacial and geometrical effects. Meanwhile, the activation mechanisms of the above studies also merit in-depth investigation. It is worth noting that geometric and electronic effects are commonly correlated and both play important roles in the cleavage of Caliph–Caliph bonds. However, current reports tend to investigate the significance of these two factors separately. When only one parameter is chosen as the key focus of the activation of Caliph–Caliph bonds, it becomes difficult to eliminate the influence of the alternative parameter in the experiment. Therefore, future research should aim to study the role of both parameters based on the perspective of a unified view. Furthermore, besides the previously discussed acid sites and metal sites, the synergistic catalysis between acid sites and metal sites (dual sites) is also significant for the activation of the Caliph–Caliph bond, which will be addressed in Section 2.3. In addition to the common homolytic dissociation of H2 over metal catalysts with metal–metal pairs, the heterolytic dissociation on frustrated Lewis acid–base pairs (defective metal oxides, single-atom catalysts, and metal–support interfaces) showed enhanced performance in some catalytic hydrogenation and hydrogenolysis reactions.101 Future focus is encouraged on the exploration of the heterolytic hydrogenolysis of waste plastics over frustrated Lewis acid–base pairs. This approach may potentially allow for the avoidance of using some expensive noble metals. A core question is whether frustrated Lewis acid–base pairs can activate and cleave C–C bonds, even though their capabilities for H2 dissociation have already been identified.
2.3 Dual sites
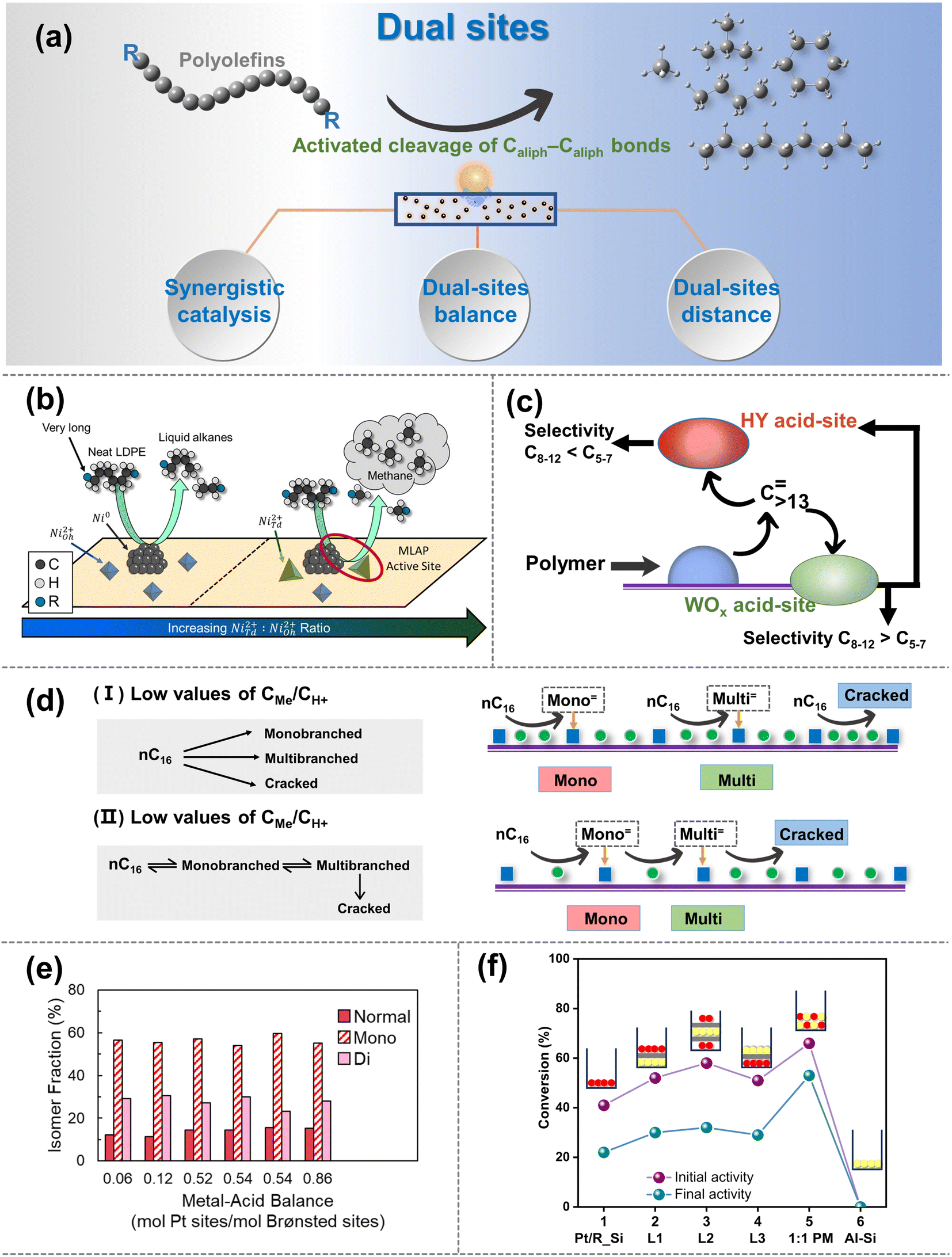 | ||
| Fig. 5 (a) Schematic diagrams of the upgradation of polyolefins when the Caliph–Caliph activation occurs over the dual sites. (b) Schematic picture of the proposed mode by which NiTd2+ promotes methane production, selectivity comparisons are made at 25–45% LDPE deconstruction. Reproduced with permission from ref. 111. Copyright 2023 American Chemical Society. (c) Depiction of main intermediates diffusing over the Pt/WO3/ZrO2 + HY(30) catalyst. Reproduced with permission from ref. 104. Copyright 2021 Science. (d) n-Hexadecane hydroisomerization mechanisms over the Me/S31 catalysts at: (I) low values of CMe/CH+; and (II) high values of CMe/CH+. Reproduced with permission from ref. 117. Copyright 2018 Royal Society of Chemistry. (e) Fraction of normal, monobranched, and dibranched isomers in extractable LDPE hydrocracking over catalysts with different metal–acid balances (MABs). Data collected from ref. 102 and copyright 2021 Elsevier. (f) Conversion obtained for different two-component catalysts based on Pt/R_Si and SIRAL80 (Al–Si). Reproduced with permission from ref. 119 and copyright 2016 Elsevier. | ||
Synergistic catalysis between dual sites can be finely categorized into two types: the synergistic catalysis between metal sites and BAS/LAS within a catalyst, and the synergistic catalysis between multiple catalysts with physically mixed metal sites and acid sites (including BAS and LAS).106 Notably, the former is more prevalent and the latter is primarily composed of catalysts containing metal sites or dual sites, along with acid site-rich zeolites. In synergistic catalysis, acid sites primarily drive the isomerization and/or activation–cleavage of Caliph–Caliph bonds, while metal sites mainly facilitate the hydrogenation/dehydrogenation reactions of polyolefins and intermediates.106 Moreover, in the case of hydrogenolysis, metal sites can directly activate the Caliph–Caliph bond and cleave the Caliph–H bond as discussed in Section 2.2. For example, Vance et al. demonstrated that platinum tungstated zirconia (Pt-WZr) efficiently boosts LDPE hydrocracking. They proposed that Pt sites are responsible for dehydrogenation and re-hydrogenation of LDPE long chains at short times and low temperatures, while the BAS present in Pt–WZr enables the isomerization and the activation–cleavage of the Caliph–Caliph bonds in the intermediates through carbenium ion mechanism.102 Therefore, the activation mechanism over metal sites and the synergistic catalytic mechanism on dual sites coexist within loaded catalyst systems, factors that warrant consideration during the initial catalyst design. A similar synergistic effect was also observed between the Ru with the BAS.109 Interestingly, apart from BAS, LAS can also form synergistic catalysts with metal sites, promoting the cleavage of the Caliph–Caliph bond and enabling position-selective cleavage for product selectivity. For example, Du et al. proposed that the high activity of dual-site catalysts in HDPE upcycling results from the synergistic catalysis between Ru metal sites and LAS of HZSM-5.107 Specifically, Ru metal sites facilitate the dehydrogenation and subsequent hydrogenolysis of long-chain polyolefin molecules, while the LAS in HZSM-5 convert the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of olefinic intermediates to carbenium ions, thereby promoting the cyclization and β-scission.110 Interestingly, Vance's group has proposed an alternative explanation for synergistic catalysis between LAS and metal sites.111 They have observed that the NiTd2+, acting as LAS and closely situated around Ni nanoparticles, can engender a metal Lewis acid pair (MLAP) effect with Ni metal sites (Fig. 5b), which promotes hydrogenation. The MLAP effect can enhance the ability to hydrogenate polyolefins, accelerate the activation–cleavage of the Caliph–Caliph bonds, and increase the extent of terminal cascades, ultimately leading to an increased production of methane. However, a limited number of studies have been reported on the MLAP effect, and further investigation is necessary to extend this pattern to other catalysts with dual sites. The synergistic catalysis between metal sites and BAS/LAS can tune the activity and selectivity to obtain the desired hydrocarbon products. Moreover, synergy is also observed through tandem catalytic systems composed of physically mixed catalysts containing metal sites and acid sites. For instance, Liu and co-workers discovered that catalysts consisting of Pt nanoparticles supported on a mixture of tungsten zirconia (Pt/WO3/ZrO2) and zeolite (HY) exhibit exceptional activity and selectivity in the mild hydrocracking of LDPE due to the synergistic catalysis between Pt/WO3/ZrO2 and HY zeolite.104 It is worth noting that, in addition to the Pt sites and BAS on Pt/WO3/ZrO2, the BAS of HY zeolites also play a crucial role in the cleavage of Caliph–Caliph bonds (Fig. 5c). Specifically, polyolefins initially activate primarily at Pt metal sites, then diffuse to the acid sites of WO3/ZrO2 supports and HY zeolites, where they undergo cleavage of Caliph–Caliph bonds. The intermediate then undergoes subsequent isomerization on WO3/ZrO2 supports and hydrogenated desorption over Pt metal sites. Furthermore, the combination of M/ZSM-5 (M = Ni, Co, Ru) and γ-Al2O3 also shows a similar regularity.105 It is necessary to clarify that researchers commonly prefer to focus the discussion of synergistic catalysis between metal sites and BAS/LAS on a single type of acid site. However, both types of acid sites usually coexist in supports (e.g., zeolite), and the corresponding other one could also have a certain effect. Therefore, subsequent studies are expected to deactivate non-target acidic sites through pretreatment, thereby eliminating interfering factors to obtain more accurate conclusions. Briefly, the synergistic catalysis between acid sites and metal sites can effectively promote the cleavage of Caliph–Caliph bonds, and afford the desired product distribution. Furthermore, precise modulation of the active sites on the dual-site catalysts is expected to expand the boundaries of cleavage rate and selectivity of Caliph–Caliph bonds. Recently, Scott's group further confirmed the pivotal roles of synergistic catalysis between metal and acid sites using dual-site catalysts (Pt/Cl–Al2O3 and Pt /F–Al2O3).112 In this context, Pt metal sites play a crucial role in the dehydrogenation of alkanes and the hydrogenation of olefins, while the robust BAS protonate alkenes to generate carbenium ions that undergo Caliph–Caliph cleavage, isomerization and cyclization. Although the enhanced acidity of BAS facilitates the depolymerization of PE, it is concomitantly associated with the production of undesirable gaseous byproducts and coke. They propose that precise modulation of the moderate acidities of BAS within the supports can facilitate efficient degradation of PE with the highly selective production of valuable surfactant-range alkylaromatics. This suggests significant potential for future research in exploring the precise modulation of active sites in dual-site catalysts. Additionally, synergistic catalysis in dual-site catalysts is closely related to the equilibrium and distance between metal sites and acid sites.
C bonds of olefinic intermediates to carbenium ions, thereby promoting the cyclization and β-scission.110 Interestingly, Vance's group has proposed an alternative explanation for synergistic catalysis between LAS and metal sites.111 They have observed that the NiTd2+, acting as LAS and closely situated around Ni nanoparticles, can engender a metal Lewis acid pair (MLAP) effect with Ni metal sites (Fig. 5b), which promotes hydrogenation. The MLAP effect can enhance the ability to hydrogenate polyolefins, accelerate the activation–cleavage of the Caliph–Caliph bonds, and increase the extent of terminal cascades, ultimately leading to an increased production of methane. However, a limited number of studies have been reported on the MLAP effect, and further investigation is necessary to extend this pattern to other catalysts with dual sites. The synergistic catalysis between metal sites and BAS/LAS can tune the activity and selectivity to obtain the desired hydrocarbon products. Moreover, synergy is also observed through tandem catalytic systems composed of physically mixed catalysts containing metal sites and acid sites. For instance, Liu and co-workers discovered that catalysts consisting of Pt nanoparticles supported on a mixture of tungsten zirconia (Pt/WO3/ZrO2) and zeolite (HY) exhibit exceptional activity and selectivity in the mild hydrocracking of LDPE due to the synergistic catalysis between Pt/WO3/ZrO2 and HY zeolite.104 It is worth noting that, in addition to the Pt sites and BAS on Pt/WO3/ZrO2, the BAS of HY zeolites also play a crucial role in the cleavage of Caliph–Caliph bonds (Fig. 5c). Specifically, polyolefins initially activate primarily at Pt metal sites, then diffuse to the acid sites of WO3/ZrO2 supports and HY zeolites, where they undergo cleavage of Caliph–Caliph bonds. The intermediate then undergoes subsequent isomerization on WO3/ZrO2 supports and hydrogenated desorption over Pt metal sites. Furthermore, the combination of M/ZSM-5 (M = Ni, Co, Ru) and γ-Al2O3 also shows a similar regularity.105 It is necessary to clarify that researchers commonly prefer to focus the discussion of synergistic catalysis between metal sites and BAS/LAS on a single type of acid site. However, both types of acid sites usually coexist in supports (e.g., zeolite), and the corresponding other one could also have a certain effect. Therefore, subsequent studies are expected to deactivate non-target acidic sites through pretreatment, thereby eliminating interfering factors to obtain more accurate conclusions. Briefly, the synergistic catalysis between acid sites and metal sites can effectively promote the cleavage of Caliph–Caliph bonds, and afford the desired product distribution. Furthermore, precise modulation of the active sites on the dual-site catalysts is expected to expand the boundaries of cleavage rate and selectivity of Caliph–Caliph bonds. Recently, Scott's group further confirmed the pivotal roles of synergistic catalysis between metal and acid sites using dual-site catalysts (Pt/Cl–Al2O3 and Pt /F–Al2O3).112 In this context, Pt metal sites play a crucial role in the dehydrogenation of alkanes and the hydrogenation of olefins, while the robust BAS protonate alkenes to generate carbenium ions that undergo Caliph–Caliph cleavage, isomerization and cyclization. Although the enhanced acidity of BAS facilitates the depolymerization of PE, it is concomitantly associated with the production of undesirable gaseous byproducts and coke. They propose that precise modulation of the moderate acidities of BAS within the supports can facilitate efficient degradation of PE with the highly selective production of valuable surfactant-range alkylaromatics. This suggests significant potential for future research in exploring the precise modulation of active sites in dual-site catalysts. Additionally, synergistic catalysis in dual-site catalysts is closely related to the equilibrium and distance between metal sites and acid sites.
The balance between acid sites and metal sites is a crucial parameter for the rational design of dual-site catalysts for polyolefin hydrocracking.16 By combining weakly acidic sites with metal sites possessing strong dehydrogenation properties, the hydroisomerization efficiency of the intermediates can be maximized, albeit with a slight reduction in the activation rates of Caliph–Caliph bonds.113 Conversely, if there are too many acid sites, the intermediates located at the acid sites will undergo excessive cleavage of Caliph–Caliph bonds before the hydrogenation/dehydrogenation reaction, thereby reducing the selectivity of the target product. Moreover, a lack of acid sites will limit the conversion of the intermediates by the polyolefin cleavage.114 Briefly, it is imperative to enhance the catalytic performance by modulating the equilibrium between acid sites and metal sites. The equilibrium between acid sites and metal sites can be adjusted to control the cleavage efficiency and position by changing their quantitative ratio (nMe/nA).107 An ideal dual-site catalyst should achieve a dynamic equilibrium between the hydrogenation/dehydrogenation reaction and the activation–cleavage of Caliph–Caliph bonds. A fundamental requirement for this ideal dynamic catalytic reaction is identified to be that the value of nMe/nA should be sufficiently high.16 The reaction pathway and product distribution over dual-site catalysts with high nMe/nA values are primarily determined by the physicochemical properties of the acid sites. At this stage, the rate-determining step shifts from hydrogenation/dehydrogenation on metal sites to the skeletal rearrangements of olefinic intermediates and/or the activation–cleavage of Caliph–Caliph bonds at acid sites.115 Hence, it is noteworthy that once the nMe/nA ratio reaches the threshold for ideal dual-site catalysts, further increases in nMe/nA do not affect the selectivity of the product based on molecular weight, leading to an increase in the isomeric product.16 Moreover, the ideal dual-site catalyst exhibited a high turnover frequency (TOF) while minimizing excessive cleavage of Caliph–Caliph bonds and coke formation, leading to enhanced isomerization, stability, and activation properties of the intermediate.116 For example, Zhang et al. found that the highest isomeric yield of 72% was obtained on dual-site catalysts with both BAS and two metal site types (Pd and Ni).117 This significant enhancement in isomeric performance is primarily attributed to the equilibrium effect between acid sites and metal sites of the high nMe/nA catalysts (Fig. 5d). Briefly, dual-site catalysts with high nMe/nA ratios tend to exhibit superior performance in the cleavage of Caliph–Caliph bonds, and the modulation of nMe/nA on the dual sites serves as an effective strategy for determining the distribution of isomeric products. Furthermore, in comparison to PE, dual-site catalysts with high nMe/nA ratios primarily exert a more significant impact on the reaction efficiency of PP hydrocracking.20 Rorrer et al. found that dual-site catalysts with high nMe/nA ratios exhibit a more pronounced impact on the cleavage rate of the Caliph–Caliph bonds in PP hydrocracking, while appropriately low nMe/nA ratios in dual-site catalysts may contribute more effectively for PE hydrocracking.20 This can be attributed to the fact that during the degradation of PP, the carbenium ion intermediate is stabilized by methyl branches on the substrate, facilitating the formation of the transition state required for β-scissions of the Caliph–Caliph bonds. In contrast, in the hydrocracking of PE, the additional BAS plays a more beneficial role in the activation–cleavage of Caliph–Caliph bonds. Consequently, for different feedstocks and desired target products, the appropriate equilibrium states between acid sites and metal sites may vary accordingly. Hence, contingent upon distinct substrates and target products, the adjustment of the ideal nMe/nA ratios becomes imperative for attaining the desired results, as opposed to rigidly adhering to a fixed value indefinitely. Overall, the above studies have indicated that the equilibrium states between acid sites and metal sites are closely related to the activation–cleavage of Caliph–Caliph bonds and the isomerization of the intermediate.20,116,117 Interestingly, Vance and coworkers uncovered an entirely different discovery.102 They point out that Pt metal sites are not capable of directly cleaving Caliph–Caliph bonds at low temperatures and short periods, but could dehydrogenate and re-hydrogenate long-chain polyolefins. The subsequent diffusion of intermediates to BAS facilitates skeletal rearrangements and the activation–cleavage of Caliph–Caliph bonds. During this upcycling process, reactants with macromolecular nature can accommodate numerous branching points within a single chain without undergoing β-scission. It will seriously hinder the diffusion of liquid products into acid sites/metal sites to form relatively stable isomeric products. Therefore, the selectivity of branched isohydrocarbons remained independent of the balance between acid sites and metal sites (Fig. 5e). A more comprehensive and in-depth investigation of the equilibrium between acid sites and metal sites, considering their properties and selectivity in cleaving Caliph–Caliph bonds and distribution of isomerization products, is needed to validate the generalization and accuracy of the corresponding rules.
To achieve the appropriate equilibrium between acid sites and metal sites, the distance between dual sites is also crucial as it directly influences the diffusion of olefinic intermediates.16,106 Generally, the distance between dual sites commonly affects the activity and selectivity for cleaving Caliph–Caliph bonds. In terms of activity, the excessive spacing will lead to an over-long path for the diffusion of olefinic intermediates, hindering their hydrogenation over the metal sites and leading to reduced catalytic activity, while the over-tightly distance between acid sites and metal sites may promote the excessive cleavage of Caliph–Caliph bonds. In terms of selectivity, over-long diffusion paths tend to cause excessive cleavage of olefinic intermediates over the acid sites, leading to an increase of low-value alkane products and coke formation. Conversely, over-short diffusion paths promote the selective cleavage of the Caliph–Caliph bond at the terminal site, affording the production of methane. For example, Weisz and colleagues explored the structure–efficiency relationship between the distance of dual sites and the catalytic performance of polyolefin degradation.118 They observed that catalysts with smaller particle sizes (e.g., 5 or 70 μm) yielded higher amounts of isoheptanes compared to catalysts with larger particle sizes (1000 μm). They suggested that the excessive distance leads to an overly long diffusion path for olefinic intermediates, affecting the hydroisomerization on the metal sites and resulting in decreased activity. Meanwhile, overly long diffusion paths easily cause the excessive cracking of olefinic intermediates on acid sites, leading to increased yields of light alkane and coke deposition. Furthermore, Samad et al. also observed that when the distance increased to the millimeter scale, the corresponding catalysts behaved more like monofunctional catalysts, and the activation properties of Caliph–Caliph bonds decreased sharply due to the excessively long diffusion paths of olefinic intermediates (Fig. 5f).119 Briefly, an excessive distance between the acid sites and metal sites has a significant negative impact on the cracking of Caliph–Caliph bonds, as it delays the arrival of olefinic intermediates at the metal sites for the subsequent reaction. On the other hand, how does the over-short distance affect the catalytic performance of Caliph–Caliph bonds? In this aspect, Vance et al. proposed that the close proximity of Ni metal sites and LAS may lead to the MLAP effect (Fig. 5b). This effect favors the hydrogenation of polyolefins and tighter binding of polyolefin chains to the catalyst surface, thereby enhancing the activation–cleavage of polyolefin while simultaneously promoting the selective cleavage of the Caliph–Caliph bond at the terminal site.111 Although the excessively short distance can promote the cleavage of the Caliph–Caliph bond, it may lead to the continuous cleavage of the Caliph–Caliph bond, resulting in an overabundance of light alkanes in the products. Furthermore, it is essential to confirm whether this pattern can be generalized to other dual-site catalysts. Hence, the principle of optimal spacing between dual sites applicable to diverse substrates and target products may be elucidated by structure–activity relationships, and requires further confirmation in future endeavors. Interestingly, from the alternative perspective of the mass transfer properties, Zecevic et al. found that the dual-site catalysts with relatively longer-pitch and better mass transfer capabilities exhibited superior yields of isomeric products.120 They attribute this to the fact that the zeolite's microporous network impedes the diffusive intermediate molecules formed on the metal sites. Additionally, the strong adsorption of intermediate molecules on the acid sites results in slow diffusion and longer residence times. As a result, this prolonged interaction increased the probability of over-cracking and reduced the isomeric product distribution. Conversely, longer-pitch dual-site catalysts with more optimized mass transfer pathways exhibited superior performance. Inspired by the above work, simply requiring only that the acid sites and metal sites be as close as possible to achieve ideal hydrocracking may be imperfect, and an extended concept for the distance between dual sites that is not confined to spatial distances alone.106 Specifically, in addition to the spatial distance between acid sites and metal sites, the mass transfer properties of olefinic intermediates between acid sites and metal sites may also be significant factors that define this distance effect. Excitingly, the manipulation of catalysts by strategically adjusting the distance between their dual sites provides the opportunity to tailor catalyst properties to precisely match the need of the target process.120,121 More generally, we anticipate that strategies for spatially modulating active sites at the nanoscale level will be significantly beneficial for the further development and optimization of emerging multifunctional catalysts.
In summary, we provide a comprehensive analysis of the factors that affect the catalytic performance of Caliph–Caliph bond activation over the dual-site catalysts. In this piece, several key understandings can be obtained. (1) Polyolefins undergo hydrogenation/dehydrogenation reactions on metal sites, while acid sites are mainly responsible for the isomerization and/or the activation–cleavage of Caliph–Caliph bonds.106 (2) Achieving a balance between acid sites and metal sites is widely recognized as a crucial essential factor in the design of ideal dual-site catalysts for efficient and selective polyolefin hydrocracking.16 Dual-site catalysts with higher values of nMe/Na are expected to establish as more favorable dynamic equilibrium between hydrogenation/dehydrogenation reactions and the activation–cleavage of Caliph–Caliph bonds.20 (3) The distance between acid sites and metal sites also significantly affects the activation–cleavage of Caliph–Caliph bonds as it directly influences the diffusion efficiency of olefin intermediates.119 In general, dual-site catalysts with an appropriate distance between acid sites and metal sites tend to exhibit outstanding efficiency and selectivity in the activation–cleavage of Caliph–Caliph bonds.106,120 (4) The hydrogen spillover effect can potentially facilitate synergistic catalysis between dual sites with an extended spacing by promoting the formation of alkyl carbenium ions and accelerating the desorption of carbenium ion intermediates on acid sites.47 Therefore, the development of dual-site catalysts with the hydrogen spillover effect may be a promising strategy for achieving exceptional synergistic catalysis. (5) The mass transfer performance of olefin intermediates between acid sites and metal sites is also significant for understanding distance effects but reports about this aspect are rare and it deserves more comprehensive investigation.
2.4 Other transformation strategies
The previous section discussed the activation–cleavage of the Caliph–Caliph bond on three catalytic sites. In addition to the above catalytic systems, several other emerging technologies are discussed here, including tandem catalytic strategies4,15,33 and cross-alkane metathesis.31,32 The tandem catalytic strategies, which enable selective conversions at relatively lower temperatures and unlock possibilities for decentralized catalytic upgradation, have also garnered extensive research interest. For example, Hartwig's group first conducted a partial dehydrogenation of polyethylene over Ir-tBuPOCOP to break the C–H bond, akin to creaking a “crack” in a tight chain and then performed tandem isomerization and ethenolysis of the desaturated chain to obtain propylene (Fig. 6a).4 This suggests the feasibility of employing tandem catalytic strategies to depolymerize stabilized polyolefins under mild conditions. Furthermore, Zhang and co-workers designed a tandem cracking-alkylation system for polyolefin degradation (Fig. 6b).15 This technique entails the endothermic cleavage of Caliph–Caliph bonds, followed by the exothermic alkylation of isoparaffins, i.e., alkylation of the primary alkenes formed from the Caliph–Caliph cleavage using paraffin, wherein the carbon atoms are retained in the resulting products. By connecting these reactions kinetically and thermodynamically, it becomes possible to achieve complete conversion below 100 °C. The mechanism of tandem cracking–alkylation comprises the following steps: (1) initiation of the carbenium ion chain mechanism involving tert-butyl carbenium ions, (2) skeletal isomerization and cracking of the carbenium ions formed within the polymer strands via β-scission, (3) the production of alkenes resulting from the β-scission cracking with carbenium ions formed from isoalkanes in the alkylation cycle (Fig. 6b).15 The rate of cracking in these coupled processes allows the use of an exceptionally low concentration of isoalkanes in the overall pool of substrates, in contrast to conventional isobutane and n-butene alkylation. Additionally, in the realm of traditional non-catalytic pyrolysis technology with precise control and tandem oxidation catalysis, it is possible to efficiently convert polyolefins into value-added chemicals. For instance, Liu's team has demonstrated that controlled degradation of PE and PP into waxes can be accomplished through temperature gradient pyrolysis, where the deliberate inhibition of over-pyrolysis serves to restrict the yield of small-molecule products.8 Subsequently, combined with the oxidation of manganese stearate and subsequent processing, these waxes are transformed into value-added surfactants. This proposed strategy showcases a new way of upcycling plastics without the need for novel catalysts or complex procedures. It provides better control over the products resulting from plastic depolymerization under mild conditions. Notably, besides its applications in converting single-component plastics, tandem catalytic strategies are anticipated to be applied in the treatment of mixed plastics, mirroring real-life scenarios of plastic waste. For instance, the technology for converting single-component plastic waste can be utilized in a tandem fashion, where the sequence of reactions can be tailored to the distinct physical and chemical properties of each component within the mixed plastics, allowing the efficient extraction and conversion of individual polymers. Despite the requirement for intricate design and meticulous product separation, this tandem catalytic strategy has the potential to address the challenges posed by complex mixed plastics.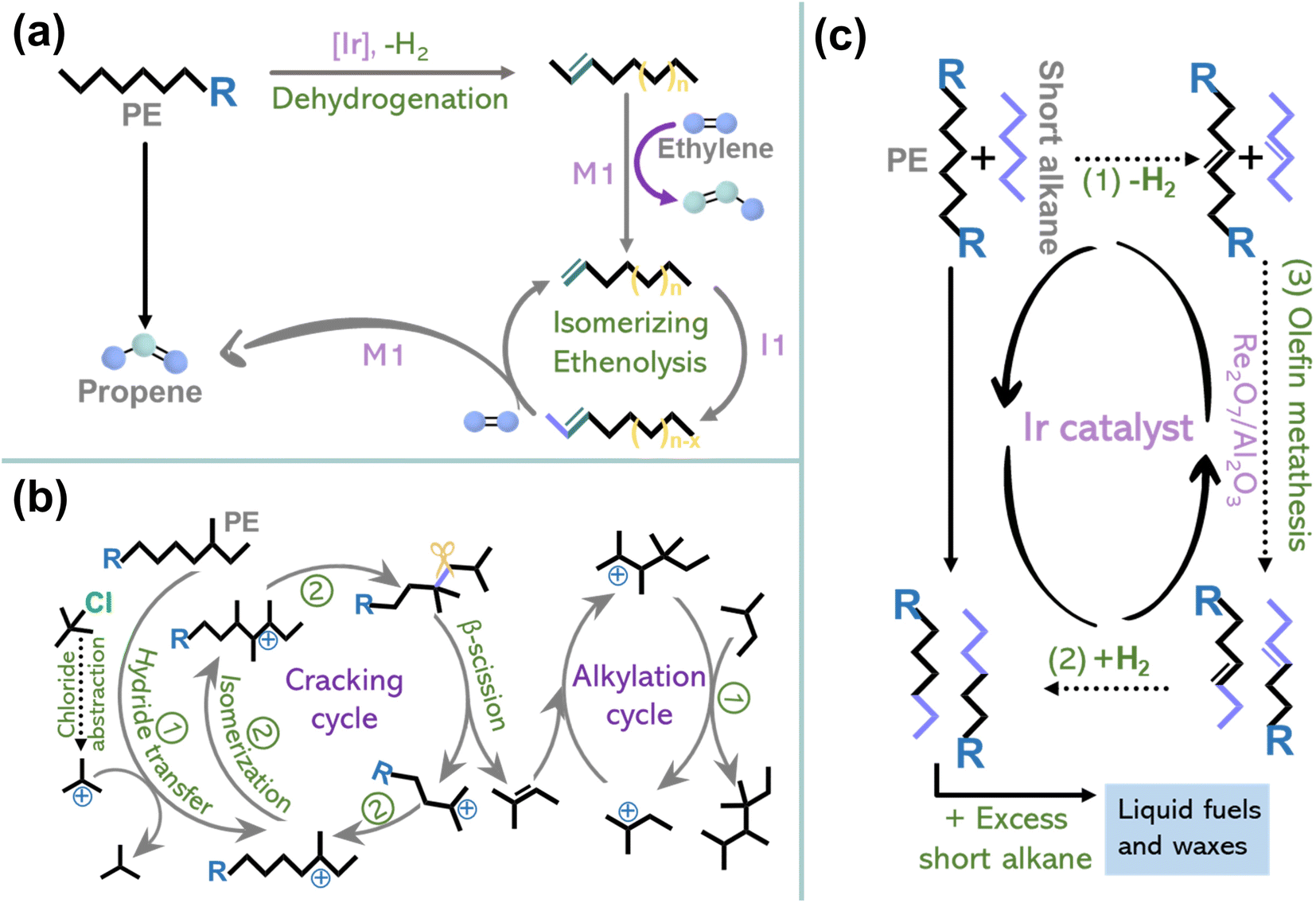 | ||
| Fig. 6 (a) Schematic diagrams of the conversion of PE to propylene by dehydrogenation and tandem isomerization and ethenolysis. Reproduced with permission from ref. 4. Copyright 2022 Science. (b) Schematic diagrams of the reaction mechanism for the tandem cracking–alkylation process of a polyolefin with iC5. Reproduced with permission from ref. 15. Copyright 2023 Science. (c) Schematic diagrams of the degradation of PE through cross-alkane metathesis with light alkanes (for example, n-hexane). Reproduced with permission from ref. 32. Copyright 2016 Science. | ||
In addition to tandem catalytic strategies, cross-alkane metathesis is also an important contributor to polyolefin depolymerization. Recently, Huang and co-workers found that the light alkanes mediated cross-alkane metathesis can selectively convert PE into value-added liquid fuels and waxes (Fig. 6c).32 Metathesis mainly employs the homogeneous Grubbs catalyst and Grubbs–Hoveyda catalyst.122,123 Unlike conventional metathesis, which requires the exchange of Caliph–Caliph bond fragments between unsaturated substrates, cross alkane metathesis operates in the absence of a CC bond in the substrate. Instead, the metathesis mechanism involves a three-step tandem process: (1) dehydrogenation of the paraffin to form an olefin, (2) cross-olefin metathesis, and (3) hydrogenation of the olefin products to convert them back into a paraffin sequence (Fig. 6c).31,32 Metathesis holds great promise for industrial applications as it relies on inexpensive paraffins as feedstock and produces stoichiometric hydrogen during the reaction, no external hydrogen supply is needed. Briefly, this approach couples a noble metal catalyst, responsible for hydrogenation/dehydrogenation, with a metathesis catalyst to achieve synergistic catalysis, facilitating polyolefin degradation and advancing the frontiers of plastic degradation research. More importantly, innovative catalytic systems are still under exploration for converting polyolefins into well-defined and value-added chemicals via precise activation–cleavage of Caliph–Caliph bonds.
In recent years, photocatalysis for the depolymerization of plastics has garnered increasing attention as it can achieve the activation–cleavage of Caliph–Caliph bonds in polymers driven by visible light under mild conditions. Conventionally, photocatalytic degradation of plastics may result in the ready generation of CO2, particularly with inert polyolefins.29 The selective activation and cleavage of Caliph–Caliph bonds through photocatalytic strategies for the transformation of plastics into value-added liquid products remains a significant challenge. To tackle this challenge, Sun and colleagues developed an innovative tandem photocatalytic strategy capable of converting polyolefins into valuable chemicals under simulated natural light. The first step involves the single-unit-cell Nb2O5 photooxidation process to cleave Caliph–Caliph bonds in polyolefins, resulting in the generation of CO2 and H2O. Subsequently, the generated CO2 is further converted in situ into valuable C2 fuels via selective C–C bond coupling driven by photocatalysis.124 The above investigation demonstrates the practical feasibility of photo-catalytically activating and cleaving Caliph–Caliph bonds in polyolefins. However, the yield of C2 fuels is encumbered by the constrained efficiency of current photocatalysts during the initial steps of cleaving Caliph–Caliph bonds. Future advancements are expected to design photocatalysts with dual active sites. These dual-site catalysts would not only efficiently activate and cleave Caliph–Caliph bonds but also favor the following C–C bond coupling, promising a highly efficient conversion of plastics into multiple carbon fuels under natural light. In addition to the direct photocatalytic activation and cleavage of Caliph–Caliph bonds, the pretreatment via incorporating polar elements, such as oxygen and nitrogen, into polyolefins could alter the polarity of the long-chain polymer. This modification can reduce the activation energy barriers for the Caliph–Caliph bonds adjacent to these polar elements during the photocatalytic process, thereby enhancing the efficiency of their activation and cleavage and resulting in a more diversified range of value-added chemicals.125,126
3. Caliph–CCl activation
The challenges associated with plastic upcycling arise not only from the activation of Caliph–Caliph bonds but also from the poisoning and deactivation of active sites caused by the presence of unmanageable chlorine elements in the feedstock, such as polyvinyl chloride (PVC).127–130 For instance, Pidko et al. illustrated through a DFT study that the presence of Cl atoms in polyolefins significantly inhibits the performance of catalysts. The Cl atoms can form stable molecular complexes with the catalyst, thereby increasing the energy barrier for the activation of C–H bonds as the initial step.131 The Cl-inhibition pattern observed by DFT research aligns closely with experimental observations. Specifically, Cl species poison the active sites in catalysts through potent adsorption or electronic interactions. Here, this section provides an overview of several strategies for activating Caliph–CCl bonds and the activation mechanism at each active site forms a holistic view (Fig. 7a) and discusses the current state of research, and the key challenges faced here. As shown in Fig. 7a, we categorize the mainstream strategies for PVC degradation into two groups, namely direct-dechlorination and Cl-transfer strategy: (1) direct-dechlorination involves the chemical removal of chlorine from PVC in the form of HCl, resulting in the generation of polyacetylene and polyolefin-like products. These products can serve as precursors for multifunctional carbon materials or be degraded into small molecules.127 (2) The Cl-transfer strategy involves converting the chlorine element in PVC into Cl-containing organic and inorganic salts.128,129,132,133 The dechlorinated polyethylene (PE)-like polymer can then be further cleaved into value-added short-chain alkanes. The main distinguishing factor between PVC and PP/PE is the presence of polar chlorine atoms (Fig. 7b).127 This property causes the melting temperature of PVC to be very close to its pyrolysis temperature. Additionally, the polarity of the chlorine atoms contributes to PVC's hardness and brittleness, making further conversion relatively challenging. Consequently, the reports on effective PVC degradation are relatively limited, and this area still requires in-depth investigation. It is noteworthy that Caliph–Cl bonds exhibit lower bond energies compared to Caliph–Caliph bonds and Caliph–H bonds (Fig. 7b).127 Therefore, Caliph–Cl bonds could be preferentially cleaved during the chemical degradation of PVC in most cases. | ||
| Fig. 7 (a) Schematic diagrams of the upgradation of polyvinyl chloride when Caliph–CCl bonds are activated over the dual sites. (b) The molecular formula and bond energy of PVC. Reproduced with permission from ref. 127. Copyright 2022 Royal Society of Chemistry. (c) The Schematic picture and the dechlorination effect of [BMIM]Cl on PVC at different temperatures. Reproduced with permission from ref. 134. Copyright 2010 Royal Society of Chemistry. (d) The Schematic picture of the ionic liquid dechlorination/catalytic hydrogenation process. Reproduced with permission from ref. 135. Copyright 2023 Royal Society of Chemistry. (e) Pathways from plastic waste to valuable products. Reproduced with permission from ref. 132 and copyright 2023 Nature. (f) The possible catalytic mechanism of Cl-transfer reaction. Data collected from ref. 141 and copyright 2023 Elsevier. | ||
The general mechanism for direct-dechlorination is as follows: Cl atoms in PVC, being more electronegative than C atoms, can undergo an elimination reaction, resulting in the formation of HCl through interaction with positively charged H atoms. The remaining polyacetylene and PE-like substances can be further converted into alkanes using emerging PE degradation technologies. Ionic liquids, due to their physicochemical properties, have been identified as the primary system for the effective removal of HCl from PVC to form polyacetylene. Zhao and colleagues proposed that both anions and cations in ionic liquids can influence the removal of HCl from PVC and can synergize to achieve a more efficient de-HCl rate (Fig. 7c).134 Given that ionic liquids do not require additional catalysts or organic solvents for the removal of Cl, they offer a low environmental impact and represent a promising strategy for industrial applications. However, the dechlorination products of PVC require further conversion or degradation to yield valuable short-chain alkanes. To streamline reaction steps and reduce separation and energy consumption, additional catalysts such as metal-based catalysts can be introduced into the ionic liquid system. This integration allows for both the de-HCl step and the subsequent conversion of dechlorination products to occur within a one-pot system. This type of metal catalyst can draw inspiration from the work reported by O'Rourke et al.,135 where they proposed that H(CO)Rh(PPh3)3, as a homogeneous Rh catalyst, can hydrogenate polyalkynes into polyolefins (Fig. 7d). This system avoids the isomerization and cyclization reactions that usually occur during conventional heat-treatment processes, providing an innovative improvement in optimizing the PVE dechlorination technology. Unfortunately, it does not address the cleavage of the Caliph–Caliph bond in the hydrogenation products to form more valuable short-chain alkane products. Since polyolefin, the main product of dechlorination, is difficult to solubilize, it can lead to the clogging of industrial reactors, hindering their further conversion to short-chain alkanes.135 Along this line, we believe that by integrating PVC dechlorination technology with the cleavage of the Caliph–Caliph bond in the dechlorinated product, the conversion of PVC into Cl-containing chemicals and high-value short-chain alkanes can be achieved.
In addition to the direct dechlorination into HCl, the Cl-transfer stands out as an efficient strategy to further convert the chlorine in PVC into reusable inorganic salts or organic chemicals through a suitable Cl acceptor. For example, Vlachos et al. developed a catalytic degradation system for Cl-contaminated plastic waste utilizing a two-step strategy.133 First, a magnesium-alumina mixed oxide was used as a chlorine trapping agent to extract chlorine, resulting in the rapid formation of a Cl-contaminated solid inorganic salt (Fig. 7e). Then, the dechlorination plastic waste was converted into a lubricant over the Ru/TiO2.133 This work offers a catalytic path to integrate waste PVC plastic into the supply chain of inorganic salts. Excitingly, we have extended the Cl-transfer strategy into the realm of organochloride preparation132 We employed simple Cl acceptors (tetrahydrofuran, dibutyl ether and butanol) to capture the Cl species to generate value-added organochlorides, leaving an easily manageable PE-like polymer, which can be upcycled into methane over Ru/Al2O3. We identified that the synergistic catalysis of LAS on Al2O3 and Ru metal sites significantly promotes PVC degradation. LAS activates the C–O bond in tetrahydrofuran, while the Ru metal site activates the Caliph–Cl bond and cleaves H2 (Fig. 7f). Although we have successfully integrated waste PVC plastic into the supply chain of organochlorides, the obtained Cl-containing chemicals remain mixtures of various chlorinated organics rather than pure Cl-containing products. The poor selectivity is due to the over-chlorination or condensation of the Cl-containing products generated initially. Therefore, it is highly desirable to optimize hydrogenolysis properties by adjusting the categories, sizes, and electronic effects of the metal sites, in combination with the regulation of acidic supports, to inhibit side reactions during the Cl-transfer process. Moreover, the above study highlights that the key factor of strategies is the design of a suitable Cl acceptor for electrophilic substitution reactions. In the future, additional categories of Cl acceptors can be developed, such as those containing electron-rich conjugated aromatic ring units. Additionally, Ma's group found that the presence of C–O bonds can facilitate PET's capacity as a Cl acceptor.136 This finding highlights the possibility of co-upcycling of PET and PVC, thereby expanding the applicability of the Cl-transfer strategy for PVC upcycling and offering novel insights into the simultaneous treatment of mixed plastic waste.
Briefly, this section focuses on Caliph–CCl bond activation during PVC degradation with significant industrial applications and research value. We believe that the design and optimization of multifunctional catalysts capable of simultaneously performing the Cl-transfer process and the subsequent activation of Caliph–Caliph bonds in remaining polyolefin-like polymers would be a promising research direction. Furthermore, upon the completion of the Cl-transfer process, the yield and product distribution from the degradation of polyolefin-like substances can be optimized by regulating the categories, sizes, and electronic effects of the metal sites.16 In this regard, detailed regulation strategies can be referenced from the discussion on Caliph–Caliph bond activation (Section 2) in the previous chapter, as both remaining polyolefin-like polymers and polyolefins share similar C–C bonds, mainly Caliph–Caliph bonds. Actually, the Caliph–CCl bonds in PVC and the Caliph–Caliph bonds in PE/PP are essentially C–C bonds with a sp3–sp3 hybridization pattern. Consequently, an attempt can be made to draw upon the efficient catalysts reported for the activation–cleavage of the Caliph–Caliph bonds in PE and PP for the activation–cleavage of Caliph–CCl bonds of PVC. What requires consideration is the preferential adsorption of Cl and the poisoning of metal sites by Cl due to the strong bonding interaction between Cl and the active sites. For example, in the case of the extensively reported Ru metal sites, halogens (e.g., Cl (ref. 137) and Br (ref. 138)) typically tend to poison face or bulk sites with relatively high coordination numbers. Therefore, adjusting the coordination number of metal sites can avoid the poison effect of Cl, accordingly allowing the smooth activation–cleavage of the Caliph–CCl bonds in PVC.137,138 This highly selective cleavage mode of the metal site with a partially poisoned state can be extended to other categories of catalysts in future investigation.
4. Caliph–Carom activation
It is widely recognized that the degradation of waste plastic involves not only the activation of Caliph–Caliph and Caliph–CCl bonds but also the significant role played by the activation of Caliph–Carom (sp2–sp3 hybridization) bonds.16,92,127,139,140 Aromatic polymers including polystyrene (PS), polycarbonate (PC), and phenol formaldehyde resin (PF) contain benzene ring groups and are widely used due to their colorlessness, stability, and hardness.139 Therefore, the exploration of recycling or upcycling aromatic plastic waste into aromatic compounds through the activation of Caliph–Carom bonds has sparked widespread research interest. Two main strategies have been developed: one involves the activation by the individual acid sites,141 base sites,142 or metal sites140 of catalysts, and the other involves the activation through dual sites25,92 in metal–acid bifunctional catalysts (Fig. 8a). Here, a series of typical catalysts are displayed in Table 2. A key principle is to regard aromatic plastic waste as an abundant source of aromatics, which can be further converted into aromatic products through the precise activation–cleavage of Caliph–Carom bonds. Briefly, this section will focus on the groundbreaking activation mechanisms of Caliph–Carom bonds over various activation sites while addressing the current research and the significant challenges faced here. | ||
| Fig. 8 (a) Schematic diagrams of the upgradation of aromatic plastic waste when Caliph–Carom bonds are activated over the dual sites. (b) A generic and versatile platform to extract aromatics from PS and upcycle the aromatics to a library of aryl ketones and sulfides. Reproduced with permission from ref. 141. Copyright 2023 Wiley. (c) Recovery of styrene monomer from polystyrene on various catalysts. Reproduced with permission from ref. 142 and copyright 1996 Elsevier. (d) The conversion of PS and the yields of ethylbenzene and styrene using the tandem fixed-bed reactor. Data collected from ref. 140 and copyright 2023 American Chemical Society. (e) Proposed mechanism of catalytic C–O/C–C cleavage over Ru/Nb2O5. Data collected from ref. 146 and copyright 2021 Wiley. (f) Schematic diagrams of the cleavage of C–O and C–C bonds. Data collected from ref. 25 and copyright 2022 American Chemical Society. | ||
| Active sites | Feedstocks | Catalysts | Reaction conditions | Yields | Ref. |
|---|---|---|---|---|---|
| a G: (gaseous product), L: (liquid product), S: (solid product). | |||||
| Acid/base/metal sites | PS | Co–N–Ni | (I: Pyrolysis) 470 °C, 1.5 bar H2, (II: hydrogenation) 200 °C, 1.5 bar H2 | Ethylbenzene: 92% | 140 |
| PS | AlCl3 | 300 °C, N2D-benzene (solvents) | Benzene: 98% | 141 | |
| PS | BaO | 350 °C, N2 | G: n | 142 | |
| L: 93% | |||||
| S: n | |||||
| Dual sites | PC, PS | Ru/M-Nb2O5 | 270 °C, 0.7 MPa H2 | Monocyclic oxygenates: 57% | 25 |
| PS | Fe2N@C | (I: Hydropyrolysis) 480 °C, 0.2 MPa H2, (II: hydrogenation) 280 °C, 0.2 MPa H2 | Ethylbenzene: 81% | 145 | |
| PET, PS, PC, PPO | Ru/Nb2O5 | 320 °C, 0.5 MPa H2 | Aromatic monomers: 79% | 146 | |
The activation–cleavage of Caliph–Carom bonds at acid/base sites involves three main subdivided pathways. The BAS as active centers are discussed first, and these catalysts mainly consist of zeolites such as HMCM-41, ZSM-5, HZSM-5, zeolite-β, and zeolite-Y, etc.139 When the BAS works as an active center to activate the Caliph–Carom bonds of aromatic plastic waste, the aromatic ring on the carbon chain is protonated to form carbonium ion intermediates, including sec-carbonium and tert-carbonium ion intermediates. These intermediates then undergo β-scissions, resulting in the generation of new carbonium ion intermediates and short-chain aromatic products. Intermediates with carbonium ions at the terminal position can yield olefinic products such as styrene or α-methylstyrene, which could undergo further cleavage of the Caliph–Carom bonds at the BAS, resulting in limited yields of aromatic hydrocarbons. Additionally, more stable compounds like benzene or branched-chain saturated aromatics are formed during this process, including toluene, ethylbenzene, and isopropylbenzene. For instance, Marczewski's group confirmed the pivotal role of BAS in the activation–cleavage of Caliph–Carom bonds in aromatic plastics by manipulating the acid–base properties of silica-alumina (SiO2–Al2O3) catalysts.143 However, these reaction pathways remain inconclusive in current reports and warrant further investigation.16 In addition to the BAS, LAS also plays an important role in the activation–cleavage of Caliph–Carom bonds in aromatic plastic waste, but fewer studies have been reported on this aspect. The cleavage mechanism of Caliph–Carom bonds is believed to involve the removal of a hydride anion from the benzylic position in the carbon chain, leading to the formation of a tertiary benzylic carbocation and initiating the production of lower molecular weight products.144 For example, Liu and co-workers employed AlCl3 as a LAS catalyst to activate Caliph–Carom bonds in PS waste, achieving a high selectivity for benzene (97.6%).141 This report proposed a versatile and general “Deg-Up” strategy (Fig. 8b) that expands the existing cleavage strategies of Caliph–Carom bonds and offers inspiration for designing innovative catalytic systems for aromatic plastic upcycling. However, the reported acid-site catalysts do not perform well in terms of styrene selectivity. From this perspective, base site catalysts appear to be highly promising options. For example, a pioneering report has highlighted that the use of base site catalysts (BaO) results in significantly higher styrene yields in comparison to acid-site catalysts (HZSM-5) (Fig. 8c).142 This is attributed to the fact that styrene intermediates undergo further cleavage of Caliph–Carom bonds to form secondary products at the acid site, such as benzene and indanes, whereas no such reactions occur at the base site. Briefly, investigating how to avoid excessive cleavage of Caliph–Carom bonds at the BAS to improve the selectivity for the target product still deserves intensive attention. Furthermore, during the degradation of aromatic plastic waste, the metal sites on the catalyst can induce the hydrogenation reaction, resulting in the efficient hydrogenation of intermediates into ethylbenzene, such as styrene. For example, Li et al. reported an N-bridged Co, Ni dual-atom (Co–N–Ni) catalysis for the efficient hydroconversion of styrene intermediates to the target product, ethylbenzene, following the hydropyrolysis of PS waste.140 The synergistic interaction of Co and Ni atoms within the Co–N–Ni catalyst optimized the adsorption configuration of styrene. Specifically, the CC bond adheres to the Co site while the benzene ring group attaches to the Ni site (Fig. 8d), resulting in a remarkable 95.2 wt% yield, with an impressive 92 wt% yield of ethylbenzene. This report innovatively employs dual-atom metal site catalysts to activate and cleave Caliph–Carom bonds in PS, providing new design ideas for innovative catalysts and catalytic systems for the degradation of aromatic plastic waste.
In addition to the individual acid sites, base sites, and metal sites serving as active centers for the activation–cleavage of Caliph–Carom bonds in aromatic plastic waste, the synergistic effect between acid sites and metal sites also plays a significant role.25,92,145 By strategically regulating the excellent cleavage properties of acid sites for Caliph–Carom bonds and combining them with the efficient hydrogenation ability of metal sites, aromatic hydrocarbons can be produced more efficiently and selectively. For example, we have reported that Ru/Nb2O5 as a multifunctional catalyst can selectively break Caliph–Carom and C–O bonds in mixed aromatic plastics. For the selective Caliph–Carom cleavage, the benzene ring of aromatic plastics is first adsorbed onto the NbOx species. Then, the adsorbed benzene ring is protonated by the BAS to initiate the activation of the Caliph–Carom bonds. Subsequently, the dissociated hydrogen species on Ru clusters attack the weakened Caliph–Carom bonds, leading to its selective cleavage (Fig. 8e).146 Furthermore, we propose an exciting finding that during the activation–cleavage of Caliph–Carom bonds and C–O bonds in aromatic plastic waste, the limited surface of small-sized Ru clusters can inhibit the co-adsorption of H2 and aromatic rings to achieve a high selectivity of aromatics. Noteworthily, the benzene ring on the Ru/Nb2O5 with parallel adsorption patterns undergoes protonation, which seems to be an indirect activation effect for the Caliph–Carom bonds attached to the benzene ring. However, the direct activation of Caliph–Carom bonds is rarely reported and deserves to be investigated. Additionally, a similar synergistic pattern of acid sites and metal sites in the catalytic degradation of PS was also reported by Jiang's group.145 In addition to Caliph–Carom bonds, a certain number of C–O bonds with lower cleavage energy also exist in the aromatic plastic waste. The multifunctional catalysts previously reported for the cleavage of Caliph–Carom bonds in aromatic plastic wastes tend to simultaneously activate Caliph–Carom and C–O bonds, resulting in only hydrocarbons as products. To enhance the industrial value and product diversity of aromatic plastic upcycling, there is a need to design an innovative catalytic system that selectively cleaves the Caliph–Carom bonds without activating C–O bonds, thereby generating value-added organic compounds with O-containing groups. Along this line, we found that a methanol-intoxicated Ru/Nb2O5 catalyst selectively activates the Caliph–Carom bonds while preserving the C–O bonds, resulting in the production of valuable monocyclic phenolic compounds (Fig. 8f).25 This is attributed to the fact that methanol can cover the lower-coordination NbOx species, which is the main site for the C–O bond activation, combined with occupying most of the Ru metal sites with a non-selective pattern, which prevents the C–O cleavage due to its strong dependence on H2 activation. Excitingly, the slight poisoning effect had little effect on the activation–cleavage of Caliph–Carom and still maintains the excellent performance of the Caliph–Carom cleavage. Based on the reverse bond energy cleavage strategies, towards the selective production of Cl-containing molecules from PVC waste, is it possible to selectively poison the active sites of Caliph–Cl cleavage meanwhile preserving the active sites of Caliph–CCl cleavage? This is still a challenge and deserves in-depth investigation.127 Apart from targeting hydrocarbon production, chemical functional groups (e.g., –OH, –COOH, –halogen, etc.) can be introduced into the monomer and the oligomer, converting them into value-added heteroatom-containing chemicals compared to hydrocarbons.3 For example, –COOH groups can be introduced by selectively oxidizing specific types of Caliph–H/Caliph–Carom bonds. Previous reports have indicated that PS can be converted to benzoic acid with an 88% yield, and PP can yield acetic acid with a 63% yield by employing the metal/bromide catalysts in acetic acid or water.147 In recent years, significant advances have been made in the oxidative cleavage of Caliph–Carom bonds of aromatic plastic waste with the assistance of either homogeneous (e.g., p-TsOH·H2O, FeCl3),148,149 or heterogeneous (e.g., g-C3N4)150 photocatalysis. In these instances, aromatic plastics are converted into high-value oxygen-containing chemicals (e.g., benzoic acid, phenethyl alcohol) through the activation of Caliph–Carom bonds and the introduction of oxygen atoms. For example, Stache and co-workers found that photoinduction induces FeCl3 to generate a chlorine radical, which then abstracts an electron-rich hydrogen atom from the PS backbone, forming a benzylic radical. Subsequently, O2 contributes to the formation of O radicals on the PS backbone, followed by the cleavage of Caliph–Carom bonds through β-scission and the formation of intermediates. These intermediates undergo further oxidation by oxygen radicals until benzoic acid is generated as the target product.149 A similar pattern was also observed by Reisner et al., and they proposed that the mechanism of Caliph–Carom photooxidation cleavage via the hydrogen transfer strategy is comparable to small-molecule containing –CH2– groups. However, the absence of a second hydrogen atom in the polymer backbone is crucial for the activation–cleavage of Caliph–Carom bonds.34 The above reports collectively show that Caliph–Carom bonds in PS can be cleaved through photooxidation under mild conditions, leading to the depolymerization of PS into value-added small molecules, such as benzoic acid, rather than CO2. Moreover, homogeneous acid catalysts, such as p-TsOH·H2O, have been recognized to be effective for the activation and cleavage of Caliph–Carom bonds in PS during photooxidation. Notably, McInnes et al. found that the singlet oxygen radical is a crucial intermediate in the activation–cleavage of Caliph–Carom bonds. This singlet oxygen radical initiates hydroperoxidation and subsequent breakage of Caliph–Carom bonds by abstracting a hydrogen atom from the tertiary carbon–hydrogen bond, affording a high yield (>70%) of benzoic acid.148 Notably, the current capacity for treating plastics via photocatalysis remains relatively limited at the laboratory level. Anticipation exists for the development of advanced catalysts to enhance degradation efficiency, thereby advancing the implementation of photocatalytic oxidation within the plastic degradation industry.151 During the photocatalytic oxidation of aromatic plastic wastes, the depth-oxidation of intermediates, such as the benzene ring-opening reaction, is prone to occur, resulting in the production of CO2 as the predominant by-product. Thus, avoiding depth-oxidation remains an immediate challenge requiring urgent attention.29,152 Current research on the activation–cleavage of C–C bonds driven by photocatalysis predominantly focuses on aromatic plastics, with limited attention given to more resistant polyolefins.29 Consequently, there is significant value in further investigating the activation–cleavage of C–C bonds driven by photocatalysis. For instance, exploration of multifunctional photocatalysts featuring diverse active sites, capable of cleaving various bond types such as C–C, C–N, C–O, and C–Cl, holds the potential to enhance the depolymerization efficiency of mixed plastics in real-world. Additionally, the product distribution from the photocatalytic oxidative depolymerization of polyolefins can be anticipated to be comparable to that of hydrogenation, and the ultimate product should be a mixed-acid substance falling within a certain carbon number range. To enhance the selectivity of catalytic degradation for mixed products, there is a need to further refine product selectivity or devise superior separation strategies, especially for mixed-acid substances. For example, both Ru metal clusters and TiO2 carriers can effectively promote the formation of peroxides, thereby catalyzing oxidation reactions.153 Hence, the synergistic cooperation between the two components in Ru/TiO2 catalysts facilitates the highly selective production of carboxylic acid compounds.
In summary, we have systematically discussed the activation–cleavage of Caliph–Carom in aromatic plastic waste over monofunctional catalysts with acid sites, base sites, and metal sites, as well as the multifunctional catalysts with synergistic effect between acid sites and metal sites. The design and development of catalytic systems that can selectively activate Caliph–Carom bonds will lead to higher economic conversion efficiencies and more opportunities for aromatic waste plastic conversion. We believe that a more comprehensive catalysis system can be designed by combining diatomic catalysts with efficient hydroconversion capabilities and supports with appropriately concentrated acid sites for the activation of Caliph–Carom bonds in aromatic plastic waste. By exploring the catalytic mechanisms and designing advanced catalysts, the indirect activation strategy of the Caliph–Carom bonds can be further transformed into selectively direct cleavage patterns for higher aromatic selectivity.
5. Accumulated wisdom in other fields for C–C activation
The preceding discussion reveals that the activation–cleavage of C–C bond in plastics presents several challenges, including the activation of C–H bonds, the selective cleavage of specific C–C bonds, the desorption of intermediates following C–C bond cleavage, and how to eliminate the Cl element during the cleavage of the C–C bonds connected to Cl atoms in PVC. In fact, from a cross-disciplinary angle, these challenges have been well-developed in other areas. Specifically, the selective cleavage of specific C–C bonds in plastics mirrors the well-established cleavage of C–C bonds in lignin, alkane dehydrogenation chemistry could be beneficial for the dehydrogenation step in Caliph–Caliph cleavage within plastics, the cleavage of C–H bonds in plastics can be inspired by the rational design of outstanding catalysts in the realm of C–H bond activation, and the removal of Cl element from VOCs could serve as an inspiration for the removal of Cl element and robust resistance to chlorine toxicity in PVC upcycling. Therefore, it is most probable that the accumulated wisdom in these corresponding fields shares similarities with the depolymerization of plastics and may shed light on the C–C activation in plastics. Here, this section will elaborate and discuss how the accumulated wisdom from other fields, such as C–C bond activation in lignin chemistry, alkane dehydrogenation chemistry, C–H bond activation, and C–Cl bond activation in VOC removal, could inspire the activation of C–C bonds in plastics (Fig. 9). Since these fields share substantial similarities in terms of their polymeric nature, catalytic mechanisms, and key steps, potential catalyst design ideas can be obtained by learning from the emerging technologies in these fields.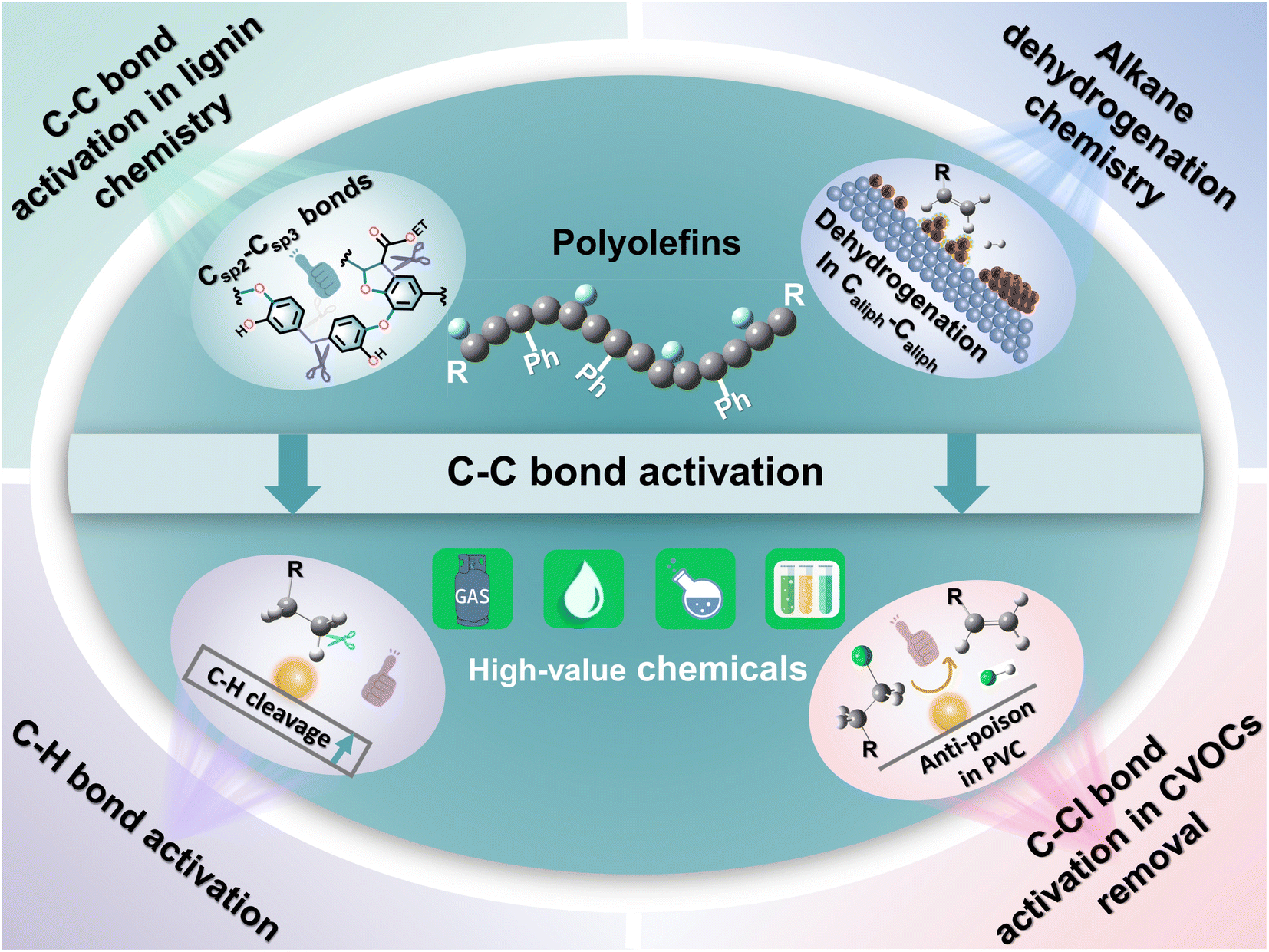 | ||
| Fig. 9 Schematic diagrams of the accumulated wisdom from the fields of C–C bond activation in lignin chemistry, alkane dehydrogenation chemistry, C–Cl bond activation in VOC removal, and C–H bond activation, to inspire the activation of C–C bonds in plastics. | ||
5.1 C–C bond activation in lignin chemistry
Lignin is the only large-volume source of renewable aromatics in nature and its valorization, especially bond activation chemistry has made significant progress.12,154 Previous reports suggest significant similarities in molecular structure, monomeric unit configuration, and the type of bonds connecting the monomeric units between lignin and polyolefins.12,19,154 More interestingly, both feedstocks share very similar C–C bonds, specifically the Csp2–Csp3 bond. Consequently, it is reasonable to believe that the two feedstocks exhibit a high degree of chemical similarity in the activation–cleavage of C–C bonds, and the emerging catalytic strategies in this field share common characteristics. Integrating the fundamental mechanisms and catalyst design principles from breakthrough catalytic conversion strategies in the domains of polyolefin and lignin conversion, from a unified perspective, holds the potential to expand the research boundaries in these fields. Contemporary catalytic systems employed in lignin depolymerization chemistry comprise diverse approaches, such as oxidative cleavage, pyrolysis driven by molecular sieve, and hydrogenolysis. While oxidative cleavage in lignin highly depends on the initiation of oxygen-containing functional groups, pyrolysis driven by molecular sieves can cleave a broad spectrum of C–C bond types, and hydrogenolysis specializes in breaking specific Csp2–Csp3 bonds. Impressively, these catalytic systems have also demonstrated significant effectiveness in the cleavage of C–C bonds in plastics and are extensively used in this field.12,155,156 For example, Nb-based catalysts after suitable modulation have been achieved to efficiently cleave Csp2–Csp3 bonds in polystyrene, polycarbonate, and lignin, further reinforcing the viability of the perspective.19,92 Metathesis, traditionally restricted to feeds with CC bonds, has emerged as an effective strategy for the degradation of aliphatic polyethylene.12 The molecular structure of lignin contains numerous oxygen-containing functional groups, making the specific cleavage strategy of O-linked C–C bonds feasible here. Currently, this unique cleavage strategy of O-linked C–C bonds is primarily employed in biomass, but it also offers prospects for polyolefin degradation, including oxidative cleavage of C–C bonds in polyethylene plastics, and could serve as inspiration for new depolymerization techniques for polar element-containing plastics like PVC. Specifically, the catalyst's ability to selectively cleave Caliph–O and Caliph–Carom bonds in lignin is often influenced by the hydrogenolysis capacity of the metal sites.12,19 For instance, by judiciously adjusting the hydrogenolysis properties of the metal sites, the direct hydrogenolysis reaction can be appropriately inhibited, while simultaneously facilitating intramolecular cyclization. This results in selective generation of indane and its derivatives, instead of monolithic low-value aromatic hydrocarbons. From this perspective, the development of innovative and precise strategies for regulating active sites with targeted activation to cleave specific bonds and selectively generate products is anticipated to apply to polyolefin degradation. Noteworthily, lignin is resistant to decomposition and solubilization due to its abundant hydroxyl groups, which enable the formation of a robust hydrogen bonding network.154 While polyolefins generally lack strong hydrogen bonding networks, depolymerization can still be hindered by the presence of plastic additives.19 Consequently, in the case of lignin depolymerization, the primary focus is identified as disrupting the hydrogen bonding network, whereas when upgrading polyolefins, the emphasis should be on enhancing the resilience of the active sites against potential additive toxicity. It is essential to highlight that lignin contains numerous polar elements whereas polyolefins lack such enrichment. Therefore, to achieve higher product selectivity, the design of the catalytic system should be precisely tailored to address the variations in elemental compositions, thereby maximizing the economic value. Additionally, the solvolysis of polymeric solid materials plays a crucial role in the catalytic valorization of lignin. In this process, water, organic solvents, or their mixtures can serve as solvents. Similarly, solvolysis is anticipated to degrade plastic polymers containing specific elements. For instance, alcoholysis can effectively break C–O and C–N bonds, depolymerizing polyesters into their constituent monomers with high efficiency. Unfortunately, alcoholysis is currently unable to achieve highly efficient cleavage of the inert C–C bonds in plastics. Typically, lignin depolymerization through solvolysis yields lower quantities of value-added monomers compared to solid catalysts, underscoring the necessity and significance of designing efficient solid catalysts. Readers may refer to excellent reviews to get a more comprehensive understanding of lignin conversion.12,155–158
5.2 Alkane dehydrogenation chemistry
The conversion of alkanes derived from natural gas and shale gas into target olefins is a crucial reaction process for the production of commodity chemicals.159 The molecular structure of alkanes closely resembles that of polyolefins, characterized by inert C–C and C–H bonds. In particular, as above mentioned, the initial step in the whole polyolefin degradation that occurs on metal sites is dehydrogenation, endowing the potential to learn the accumulated wisdom in alkane dehydrogenation chemistry to inspire the Caliph–Carom bond activation in polyolefins. In the field of alkane dehydrogenation, both oxidative dehydrogenation and direct dehydrogenation are widely used. However, the efficiency of alkane conversion and olefin selectivity in oxidative dehydrogenation is generally less favorable compared to catalytic dehydrogenation. This is attributed to the interference caused by O2 or other oxidizing agents, which interact with the active sites and compete for adsorption with alkanes and intermediates.160 Metals such as Pt, Pd, Ni, and Sn are typically active sites of catalytic dehydrogenation due to their effective C–H bond-breaking capabilities. Similar to alkane dehydrogenation chemistry, in plastic degradation, the interactions and reaction energy barriers among substrates, intermediates, and products with the active sites largely determine the catalytic performance and selectivity of the reaction system.35,160 Previous studies have confirmed that fully exposed cluster catalysts (FECCs) used in alkane dehydrogenation can facilitate dehydrogenation due to their unique geometrical and electronic structures. They efficiently decrease the free energies of the intermediates and products on the FECCs.35 Therefore, employing the active site structure of these FECCs is expected to enhance the reactivity of polyolefin degradation.16,35 This approach aims to achieve highly efficient polyolefin degradation under mild conditions by controlling the geometric and electronic characteristics of the active sites. Moreover, on FECCs, the electron-rich surface shows promise in reducing the bonding interaction between metal sites and olefinic intermediates through electrostatic repulsion.35 This regularity is also anticipated to mitigate carbon deposition resulting from the deep activation of C–C bonds over the active site during polyolefin depolymerization and generate the desired olefin with high selectivity.160 Consequently, the precise control of the interaction between the support and the metals to tune the appropriate bonding interaction is expected to convert polyolefins into short-chain olefin products with high selectivity. Along this line, conducting more systematic investigations was strongly encouraged. In the realm of alkane dehydrogenation, in addition to noble metals like Pd and Pt, several other metals, including Ni and Co, can activate C–H bonds in alkanes, but their selectivity for olefins is relatively poor.160 A similar issue arises in the domain of polyolefin degradation, where the catalytic performance of transition metals like Ni and Co is notably inferior to that of noble metals, such as Pt and Pd.3,161 Recent reports suggest that corresponding metal sulfides of Ni and Co species can enhance olefin selectivity, which is attributed to the more favorable spacing between metal sites within the sulfides.160,162,163 This approach offers novel insights for achieving highly selective activation of C–C bond cleavage in polyolefins by regulating the distance between metals. Noteworthily, both polyolefin degradation and alkane dehydrogenation involve C–H bond activation, but the C–C bond cleavage is also crucial for polyolefin degradation, whereas alkane dehydrogenation demands highly selective C–H bond activation while limiting C–C bond activation properties. This is a significant contradiction between the two fields, and when seeking to apply excellent alkane dehydrogenation catalysts in polyolefin degradation, they cannot be directly replicated. Instead, they must be undergoing precise adjusting based on a profound comprehension of the structure–activity relationship. Enhancing their C–C bond cleaving activity is imperative while ensuring superior dehydrogenation performance. Intriguingly, certain catalysts previously abandoned due to side reactions in alkane dehydrogenation may find a renewed purpose in polyolefin degradation. Furthermore, the alkane dehydrogenation for selectively synthesizing value-added products, such as α-olefins, constitutes a crucial aspect of alkane upgradation. Nevertheless, this endeavor faces significant scientific challenges, including the high activation energy barrier of C–H bonds, necessitating considerable external energy input. Additionally, selectivity is constrained by the uncontrollable cleavage site of C–H bonds, resulting in the undesired formation of non-terminal olefins. The interaction between alkene products and catalysts is stronger compared to that with alkanes, thus limiting catalytic efficiency. The structure–activity relationship in the context of alkane dehydrogenation shows promise for exploration through the development of novel, efficient, and highly selective dehydrogenation catalyst systems. Additionally, the terminal functionalization of olefinic intermediates, including silylation, carbonylation, and amine methylation, can be achieved by utilizing multi-active-site catalysts with synergistic catalysis to enhance the production of terminal olefins. These strategies are also expected to apply to the depolymerization of plastics that share structural similarities with alkanes. Additionally, from a process design standpoint, strategies that can generate hydrogen spontaneously can serve as a hydrogen source for polyolefin degradation. For instance, tandem hydrogenolysis/aromatization stands as an exemplary representation of such strategies.33 This integrated approach to process design will be anticipated to yield increased energy efficiency and greater economic benefits.5.3 C–H bond activation
In organic reaction processes, especially in the conversion of small alkane molecules such as methane into high-value chemicals, direct C–H activation eliminates the need for pre-functionalization. This significantly reduces the number of steps while minimizing the production of undesired by-products, making the strategy of direct C–H bond activation both economical and environmentally friendly.164–166 However, selective activation of inert C–H bonds remains a challenge.164,167 Fortunately, in the realm of C–H bond activation, noble metal catalysts such as Pt, Ru, and Rh continue to demonstrate exceptional catalytic efficiency, paralleling their effectiveness in plastic degradation. Moreover, transition metals including Ni, Mn, Fe, and Co exhibit noteworthy C–H bond activation capabilities, although their application in polymer depolymerization is not as widespread.16,165 Specifically, Ni, Mn, Fe, and Co-based catalysts, characterized metal sites with low coordination, have emerged as a potent approach for C–H activation, facilitating diverse reactions such as alkylations, alkenylations, and arylations.165 The modulation of low coordination in non-noble metal sites presents a promising alternative to traditional noble metal-based catalysts for achieving efficient plastic depolymerization. Similarly, the activation of C–H bonds, particularly as the initial step of C–C bond activation, holds great significance in polyolefin degradation. As an illustration, the activation of C–H bonds holds particular significance in facilitating the introduction of polar elements, such as O, into intermediates for more diversified and valuable products.8 Therefore, it is very promising to apply the innovative catalytic systems in the field of C–H bond activation to polyolefin degradation with appropriate adjustments. For instance, metal sites with lower coordination and defective sites on the catalyst exhibit higher reactivity towards C–H bond activation, especially for non-noble metals.164 Therefore, combining these two factors in the hydrogenolysis of polyolefins is expected to enhance the hydrogenation ability of metal sites. Moreover, the presence of a directing group on the reactants can enhance nearby C–H activation by further optimizing the bonding capability between the target group and the metal sites.167 The directing group effectively lowers the activation energy barrier of C–H bonds, resulting in milder reaction conditions. This strategy shows promise in improving the reaction conditions for the activation–cleavage of C–C bonds in polyolefins, particularly the hydrogenolysis technique that is closely associated with C–H activation. Such as the hydrogenolysis of polyolefins over the metal sites, the introduction of a directing group onto polyolefins serves to augment the accessibility of long carbon chains to metal sites, thus effectively increasing the rate of C–C cleavage. It is important to note that there is typically a significant interplay between C–H bond activation, olefin desorption, and deep C–H bond activation in light-chain alkanes.38 Generally, the deep C–H bond activation results in undesired carbon deposition, and therefore, balancing C–H bond activation with olefin desorption on the catalyst's surface should be given particular attention. This objective is expected to be accomplished through the regulation of active site spacing or the modification of active site accessibility. Nevertheless, significant disparities exist in the mass transfer property requirements for reactions between these two fields. Additionally, although both C–C bond activation in polyolefin degradation and C–H bond activation involve cleaving the C–H bond, the latter requires strict control of cleavage properties for C–C bond activation over the active site. These aforementioned critical considerations must be carefully addressed when incorporating state-of-the-art catalytic systems from the C–H bond activation into the realm of C–C bond cleavage in polyolefins.5.4 C–Cl bond activation in CVOC removal
Volatile organic compounds (VOCs) are organic pollutants with a boiling point below 260 °C at room temperature (20 °C) and atmospheric pressure (101.325 kPa).168,169 Their high vapor pressure, low water solubility, and toxicity make them significant contributors to air pollution. Among them, the degradation and conversion of Cl-containing volatile organic compounds (CVOCs) is the key research direction. In the degradation of CVOCs, the effective development of catalysts with excellent overall performance that can successfully avoid the toxication of the active site by chlorine has attracted increasing attention.170,171 In the domain of CVOC removal, catalytic oxidation technology has achieved widespread application due to its operational simplicity, technological maturity, and effective purification capabilities. Previous reports indicate that a well-designed catalyst for CVOC removal should possess strong redox capacity, an appropriate amount of acid sites, and robust resistance to chlorine toxicity.172 Since PVC contains chlorine, similar to CVOCs, the challenges associated with CVOC removal are equally applicable to PVC degradation. Inspired by the above clues, we considered whether the breakthrough strategy already available for CVOCs could be applied to the activation–cleavage of C–C bonds in PVC. For example, the active sites employed in the catalytic oxidation strategy of CVOC degradation exhibit robust oxidizing capabilities, which allow them to effectively mitigate poisoning by chlorine.169,173 This strategy is rarely reported in PVC degradation and warrants systematic exploration. Specifically, by adjusting the active site, the desired redox capacity and acid site distribution to promote the formation and desorption of HCl during the upgradation of CVOCs can be obtained.171 Additionally, the degradation process of CVOCs involves two distinct stages: the initial cleavage of C–Cl bonds and the subsequent deep oxidation of intermediates. Consequently, enhancing chlorine resistance can be achieved through the deliberate design of catalyst microstructures, physically segregating these two distinct reaction steps. For instance, Wang et al. innovatively developed separated two-stage Ce/TiO2–Cu/CeO2 catalysts.175 In this configuration, the cleavage of C–Cl bonds and the subsequent deep oxidation of CO occurred separately in distinct catalyst regions. Moreover, the advantageous interaction between the CuO component and the chlorine species avoided the poisoning of real active sites. Excitingly, the degradation of PVC similarly involves two sequential stages: the initial cleavage of C–Cl bonds followed by the cleavage of C–C bonds within the PE-like polymers. Hence, this offers a novel approach to addressing the problem of active site inactivation caused by the strong bonding interaction between HCl and the active site during the activation of C–C bonds in PVC. Nonetheless, it is necessary to consider the impact of differing mass transfer effects when establishing a correlation between the cleavage of C–Cl bonds in CVOCs and polyolefins. This distinction arises from the fact that polyolefins typically participate in the reaction as solids or liquids, while CVOCs are predominantly in the gaseous form. Consequently, differences in mass transfer properties between these two reaction processes need dedicated attention in future investigations.6. Future outlook
The nature of C–C bond activation including Caliph–Caliph, Caliph–CCl, and Caliph–Carom, as well as recent developments in catalytic systems for plastic waste conversion, is comparatively discussed here, with special attention given to C–C bond activation chemistry. Significantly, the activation of C–C bonds in plastics exhibits varying degrees of correlation with C–C bond activation in lignin chemistry, alkane dehydrogenation chemistry, C–Cl bond activation in VOC removal, and C–H bond activation. Consequently, there are sufficient opportunities for the upgradation of waste plastics using similar strategies in areas where the above fields are extensively developed. Further investigations into catalyst design, activation sites, and the challenges associated with overcoming catalyst poisoning remain crucial topics. Furthermore, by approaching the current state of polyolefin depolymerization from the vantage point of C–C bond activation, we gain insights into which scientific challenges already possess viable solutions, which remain unexplored or require to be further addressed, and drawing a string of promising opportunities may be harnessed in the depolymerization of plastics. In light of the existing challenges in plastic recycling and upcycling, we outline several future directions in activation–cleavage of the C–C bond in polymers and the conversion of real plastic wastes. These directions aim to deepen the understanding of how C–C bond activation can be applied more rationally in plastic depolymerization and attempt to indicate a more favorable direction for plastic conversion (Fig. 10). These directions are expected to inspire the design of novel catalytic systems characterized by high efficiency and selectivity under mild reaction conditions, which will continue to play a beneficial role in the future chemical upcycling of plastic waste. | ||
| Fig. 10 Schematic diagrams of the outlook directions for the upgradation of waste plastics. | ||
6.1 Activation–cleavage of the C–C bond in polymers
(1) While the principles governing recycling and upcycling of waste plastics are well-established, plastics involve extremely large molecular weights and their exploration poses a challenge, unlike the exploration of small-molecule organic chemistry. This necessitates the development of advanced characterization and detection techniques for the in situ analysis of the detailed activation–cleavage of C–C bonds, which is crucial for elucidating the bond-cleavage mechanisms. Comprehensive characterization is particularly beneficial for exploring the catalytic degradation of waste plastics. Concurrently, advancements in characterization techniques will enhance the understanding of degradation kinetics and product selectivity.(2) The introduction of specific functional groups (such as those containing oxygen, nitrogen, and halogens) into polymers is currently feasible through the activation of C–H bonds. This process weakens the attached C–C bonds, facilitating their cleavage and yielding value-added heteroatom-containing chemicals. An intriguing question arises: is it possible to selectively activate C–H bonds with equal atomic spacing to introduce specific functional groups consistently? Attaining this holds the potential to achieve single-product selectivity in the production of value-added multi-carbon organic compounds. Such a strategy, aimed at selectively weakening C–C bonds with uniform atomic spacing, would not only avoid the generation of economically less valuable C1 molecules but also enhance the efficiency of carbon recycling.
(3) The thermodynamic cleavage of inert C–C bonds currently necessitates significant external energy input, demanding harsh reaction conditions. Such conditions prove counterproductive for the desired industrial-scale degradation of waste plastics. Consequently, reducing the activation–cleavage barrier of inert C–C bonds remains a significant challenge. Tandem catalysis strategies could provide a valid solution, enabling polymer degradation under milder conditions. For instance, the tandem cracking–alkylation process enhances the reactivity of inert C–C bonds through a highly ionic reactive environment, thereby reducing the energy of ionic transition states.15 This approach facilitates the conversion of polyolefins into liquid alkyl products at temperatures below 100 °C. The development of additional tandem catalytic strategies is highly recommended for future endeavors.
(4) Last but not least, the deliberate design and development of advanced catalysts for application in the degradation of plastics bear substantial importance. For example, the catalysts' pore structure can be tailored to correspond with the branching degree of substrates and the structural characteristics of targeted products. The adjustment of metal size and coordination environment can improve the adsorption capacity of substrates, intermediates, and products on active sites. Moreover, the construction of active sites with diverse functions at suitable intervals enables the realization of more potent synergistic catalytic effects. All these strategies significantly contribute to the evolution of advanced catalysts for the activation–cleavage of C–C bonds in polymers. Additionally, from the perspective of practical industrial applications, the stability of catalysts is of great significance. Unfortunately, current works primarily emphasize their performance in the activation–cleavage of C–C bonds, with limited reports addressing the methods to enhance the stability, especially under high-temperature and/or high-pressure reaction conditions. Consequently, the challenge remains in designing catalysts that combine outstanding catalytic performance with enduring stability, and further exploration in this direction is strongly encouraged.
6.2 Conversion of plastic wastes
(1) Although efficient catalysts have been developed for cleaving C–C bonds in plastic waste, solid catalysts in actual plastic conversion all possess the same drawback namely poor diffusion of solid feedstock on solid catalysts and low efficiency due to poor solid–solid contact between the catalyst and the reactants. Thus, the development of hierarchical porous/two-dimension catalysts and the design of advanced solvent systems to improve mass transfer between waste plastics and solid catalysts are regarded as promising strategies for real plastic depolymerization.(2) While it is well-established that most plastic wastes consist of complex mixtures, current reports have predominantly focused on the activation of C–C bonds in single polymer types. In mixed polymer wastes, reactions in one polymer might be influenced by others, and typically, a singular active site isn't adequate for the concurrent activation of C–C bonds across all mixed plastics. For instance, plastics containing heteroatoms often cannot be co-converted with those lacking heteroatoms. This is primarily because the presence of heteroatoms, including various additives, can poison the active sites of catalysts. Such deactivation reduces conversion efficiency, leads to the formation of undesired by-products, and can potentially damage the processing equipment. Consequently, a comprehensive plastic sorting process is essential to pre-sort plastic waste, allowing distinct single-function catalysts to facilitate the depolymerization of varied plastic types. Alternatively, advanced multifunctional catalysts, with versatile active sites could effectively activate C–C bonds in different environments, leading to efficient depolymerization of mixed plastics. Afterward, products undergo secondary categorization based on molecular weights and distinct chemical properties, which may be more cost-effective than classifying mixed plastics in some cases. For instance, Ru/Nb2O5 as a multifunctional catalyst can selectively break Caliph–Carom and C–O bonds in mixed aromatic plastics and achieve a high selectivity of aromatics.146 Additionally, the RuSA-CoAlO catalyst leverages the distinctive electronic structure of the Ru sites to modulate the adsorption energy of intermediates, thereby facilitating the efficient decomposition of mixed plastics.174 All the above suggests that advanced catalysts play a crucial role in advancing the realm of degradation of mixed plastics. Furthermore, from the perspective of conversion technology, polyolefin and polyester plastics can simultaneously undergo upcycling through hydrogenolysis. In terms of product outcomes, aromatic plastics (e.g., PET, PS, PC, PPO) can be simultaneously depolymerized into small aromatic molecules. Additionally, the synergistic effects in the co-conversion of diverse plastics, such as PVC and PET, serve to enhance the overall efficiency of the process.
(3) Until now, most reports on plastic degradation have focused primarily on the efficient and selective cleavage of C–C bonds, while disregarding the influence of additives in real plastics on the catalytic systems. For instance, HDPE commonly contains processing aids to improve plasticity. Moreover, plastics used in external packaging typically require additives, such as ultraviolet (UV) stabilizers or antistatic agents. This is a common trend across most commercial plastics, where the composition of additives is complex, typically including polar elements such as nitrogen, oxygen, and halogens, as well as functional groups like ester, phenolic, and cyanurate. To address this limitation, an exploration of the influence mechanism should be first conducted. Subsequently, a notable challenge is the deactivation of catalysts due to strong interactions between metal sites and polar elements. Several useful approaches are recommended: (a) nitrogen-containing functional groups could poison acidic sites via acid-base interaction. Balancing the acidity strength and catalytic activity should be pursued to resist poisoning and maintain activity; (b) to resist the deactivation of metal sites, it is suggested to wrap a thin crystal layer of carrier around the metal site through the strong metal–support interactions to reduce the adsorption capacity of the metal site for polar elements; (c) modulating the active sites at the atom-scale level will effectively optimize the adsorption configuration of substrates over the catalyst, thereby isolating the active site with the above functional groups, which can be favorable for improving the anti-interference capability.
(4) Microplastics, comprising small synthetic plastic fragments, pose a critical global challenge that requires urgent attention. Microplastics mainly include plastic particles smaller than 1 μm and fibers range from 1 μm to 1 mm in size. The potential negative impacts of microplastics may be associated with the leaching of monomers and additives from polymers, some of which are known to be toxic or carcinogenic, thereby posing significant health risks. While conventional microplastic removal methods, such as adsorption and membrane technologies, have demonstrated some effectiveness, it is crucial to prioritize the chemical transformation of microplastics into innocuous substances (e.g., CO2 and H2O) or their upcycling into value-added chemicals. Inspired by the tandem catalytic strategy in the conversion of waste plastics, the design of advanced oxidative processes, combining functional membranes with adjustable porosity, is anticipated to achieve the capture and in situ catalytic degradation of microplastics. Additionally, the rational design of dual-functional site catalysts based on interfacial engineering can achieve excellent product selectivity by optimizing the adsorption configuration of microplastics and intermediates over the catalyst surface. More significantly, addressing the root cause—waste plastics—requires the implementation of effective production, management, and recycling strategies to prevent their invasion of the ecosystem through natural weathering, leading to the formation of microplastics.
Author contributions
Y. J. conceived the Perspective, supervised the project and revised the manuscript. H. R. conducted the literature search and wrote the manuscript. All authors participated in discussions and revisions.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported financially by the NSFC of China (22102056) and start-up funding for high-level talent at Nanjing University.References
- X. Y. Wang, Y. S. Gao and Y. Tang, Sustainable Developments in Polyolefin Chemistry: Progress, Challenges, and Outlook, Prog. Polym. Sci., 2023, 143, 101713 CrossRef CAS.
- M. Y. Chu, W. L. Tu, S. Q. Yang, C. Y. Zhang, Q. Y. Li, Q. Zhang and J. X. Chen, Sustainable Chemical Upcycling of Waste Polyolefins by Heterogeneous Catalysis, Susmat, 2022, 2, 161–185 CrossRef CAS.
- M. Q. Zhang, M. Wang, B. Sun, C. Q. Hu, D. Q. Xiao and D. Ma, Catalytic Strategies for Upvaluing Plastic Wastes, Chem, 2022, 8, 2912–2923 CAS.
- R. J. Conk, S. Hanna, J. X. Shi, J. Yang, N. R. Ciccia, L. Qi, B. J. Bloomer, S. Heuvel, T. Wills, J. Su, A. T. Bell and J. F. Hartwig, Catalytic Deconstruction of Waste Polyethylene with Ethylene to Form Propylene, Science, 2022, 377, 1561–1566 CrossRef CAS PubMed.
- K. P. Sullivan, A. Z. Werner, K. J. Ramirez, L. D. Ellis, J. R. Bussard, B. A. Black, D. G. Brandner, F. Bratti, B. L. Buss, X. Dong, S. J. Haugen, M. A. Ingraham, M. O. Konev, W. E. Michener, J. Miscall, I. Pardo, S. P. Woodworth, A. M. Guss, Y. Román-Leshkov, S. S. Stahl and G. T. Beckham, Mixed Plastics Waste Valorization through Tandem Chemical Oxidation and Biological Funneling, Science, 2022, 378, 207–211 CrossRef CAS PubMed.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Chemical and Biological Catalysis for Plastics Recycling and Upcycling, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- A. Ahrens, A. Bonde, H. W. Sun, N. K. Wittig, H. C. D. Hammershoj, G. M. F. Batista, A. Sommerfeldt, S. Frolich, H. Birkedal and T. Skrydstrup, Catalytic Disconnection of C-O Bonds in Epoxy Resins and Composites, Nature, 2023, 617, 730–737 CrossRef CAS PubMed.
- Z. Xu, N. E. Munyaneza, Q. K. Zhang, M. Q. Sun, C. Posada, P. Venturo, N. A. Rorrer, J. Miscall, B. G. Sumpter and G. L. Liu, Chemical Upcycling of Polyethylene, Polypropylene, and Mixtures to High-Value Surfactants, Science, 2023, 381, 666–671 CrossRef CAS PubMed.
- V. Nava, S. Chandra, J. Aherne, M. B. Alfonso, A. M. Antao-Geraldes, K. Attermeyer, R. Bao, M. Bartrons, S. A. Berger, M. Biernaczyk, R. Bissen, J. D. Brookes, D. Brown, M. Canedo-Arguelles, M. Canle, C. Capelli, R. Carballeira, J. L. Cereijo, S. Chawchai, S. T. Christensen, K. S. Christoffersen, E. de Eyto, J. Delgado, T. N. Dornan, J. P. Doubek, J. Dusaucy, O. Erina, Z. Ersoy, H. Feuchtmayr, M. L. Frezzotti, S. Galafassi, D. Gateuille, V. Goncalves, H. P. Grossart, D. P. Hamilton, T. D. Harris, K. Kangur, G. B. Kankilic, R. Kessler, C. Kiel, E. M. Krynak, A. Leiva-Presa, F. Lepori, M. G. Matias, S. S. Matsuzaki, Y. McElarney, B. Messyasz, M. Mitchell, M. C. Mlambo, S. N. Motitsoe, S. Nandini, V. Orlandi, C. Owens, D. Ozkundakci, S. Pinnow, A. Pociecha, P. M. Raposeiro, E. I. Room, F. Rotta, N. Salmaso, S. S. S. Sarma, D. Sartirana, F. Scordo, C. Sibomana, D. Siewert, K. Stepanowska, U. N. Tavsanoglu, M. Tereshina, J. Thompson, M. Tolotti, A. Valois, P. Verburg, B. Welsh, B. Wesolek, G. A. Weyhenmeyer, N. C. Wu, E. Zawisza, L. Zink and B. Leoni, Plastic Debris in Lakes and Reservoirs, Nature, 2023, 619, 317–322 CrossRef CAS PubMed.
- H. T. Pinheiro, C. MacDonald, R. G. Santos, R. Ali, A. Bobat, B. J. Cresswell, R. Francini, R. Freitas, G. F. Galbraith, P. Musembi, T. A. Phelps, J. P. Quimbayo, T. Quiros, B. Shepherd, P. V. Stefanoudis, S. Talma, J. B. Teixeira, L. C. Woodall and L. A. Rocha, Plastic Pollution on the World's Coral Reefs, Nature, 2023, 619, 311–316 CrossRef CAS PubMed.
- D. Ma, Transforming End-of-Life Plastics for a Better World, Nat. Sustain., 2023, 6, 1142–1143 CrossRef.
- K. Lee, Y. Jing, Y. Wang and N. Yan, A Unified View on Catalytic Conversion of Biomass and Waste Plastics, Nat. Rev. Chem, 2022, 6, 635–652 CrossRef PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Critical Advances and Future Opportunities in Upcycling Commodity Polymers, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- O. Guselnikova, O. Semyonov, E. Sviridova, R. Gulyaev, A. Gorbunova, D. Kogolev, A. Trelin, Y. Yamauchi, R. Boukherroub and P. Postnikov, “Functional Upcycling” of Polymer Waste Towards the Design of New Materials, Chem. Soc. Rev., 2023, 52, 4755–4832 RSC.
- W. Zhang, S. Kim, L. Wahl, R. Khare, L. Hale, J. Z. Hu, D. M. Camaioni, O. Y. Gutierrez, Y. Liu and J. A. Lercher, Low-Temperature Upcycling of Polyolefins into Liquid Alkanes Via Tandem Cracking-Alkylation, Science, 2023, 379, 807–811 CrossRef CAS PubMed.
- Z. W. Dong, W. J. Chen, K. Q. Xu, Y. Liu, J. Wu and F. Zhang, Understanding the Structure–Activity Relationships in Catalytic Conversion of Polyolefin Plastics by Zeolite-Based Catalysts: A Critical Review, ACS Catal., 2022, 12, 14882–14901 CrossRef CAS.
- A. J. Martín, C. Mondelli, S. D. Jaydev and J. Pérez-Ramírez, Catalytic Processing of Plastic Waste on the Rise, Chem, 2021, 7, 1487–1533 Search PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Production, Use, and Fate of All Plastics Ever Made, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- Y. Guo, Y. Jing, Q. Xia and Y. Wang, Nbox-Based Catalysts for the Activation of C–O and C–C Bonds in the Valorization of Waste Carbon Resources, Acc. Chem. Res., 2022, 55, 1301–1312 CrossRef CAS PubMed.
- J. E. Rorrer, A. M. Ebrahim, Y. Questell-Santiago, J. Zhu, C. Troyano-Valls, A. S. Asundi, A. E. Brenner, S. R. Bare, C. J. Tassone, G. T. Beckham and Y. Román-Leshkov, Role of Bifunctional Ru/Acid Catalysts in the Selective Hydrocracking of Polyethylene and Polypropylene Waste to Liquid Hydrocarbons, ACS Catal., 2022, 12, 13969–13979 CrossRef CAS.
- B. Yan, C. X. Shi, G. T. Beckham, E. Y. X. Chen and Y. Román-Leshkov, Electrochemical Activation of C–C Bonds through Mediated Hydrogen Atom Transfer Reactions, ChemSusChem, 2022, 15, e202102317 CrossRef CAS PubMed.
- S. Lu, Y. Jing, S. Jia, M. Shakouri, Y. Hu, X. Liu, Y. Guo and Y. Wang, Enhanced Production of Liquid Alkanes from Waste Polyethylene Via the Electronic Effect-Favored Csecondary−Csecondary Bond Cleavage, ChemCatChem, 2023, 15, e202201375 CrossRef CAS.
- K. Zheng, Y. Wu, Z. X. Hu, S. M. Wang, X. C. Jiao, J. C. Zhu, Y. F. Sun and Y. Xie, Progress and Perspective for Conversion of Plastic Wastes into Valuable Chemicals, Chem. Soc. Rev., 2023, 52, 8–29 RSC.
- H. Q. Li, J. Y. Wu, Z. Jiang, J. Z. Ma, V. M. Zavala, C. R. Landis, M. Mavrikakis and G. W. Huber, Hydroformylation of Pyrolysis Oils to Aldehydes and Alcohols from Polyolefin Waste, Science, 2023, 381, 660–665 CrossRef CAS PubMed.
- Y. Jing, M. Shakouri, X. Liu, Y. Hu, Y. Guo and Y. Wang, Breaking C–C Bonds and Preserving C–O Bonds in Aromatic Plastics and Lignin Via a Reversing Bond Energy Cleavage Strategy, ACS Catal., 2022, 12, 10690–10699 CrossRef CAS.
- K. Zheng, Y. Wu, Z. Hu, S. Wang, X. Jiao, J. Zhu, Y. Sun and Y. Xie, Progress and Perspective for Conversion of Plastic Wastes into Valuable Chemicals, Chem. Soc. Rev., 2023, 52, 8–29 RSC.
- J. Chen, L. Zhang, L. Wang, M. Kuang, S. Wang and J. Yang, Toward Carbon Neutrality: Selective Conversion of Waste Plastics into Value-Added Chemicals, Matter, 2023, 6, 3322–3347 CrossRef CAS.
- G. Celik, R. M. Kennedy, R. A. Hackler, M. Ferrandon, A. Tennakoon, S. Patnaik, A. M. LaPointe, S. C. Ammal, A. Heyden, F. A. Perras, M. Pruski, S. L. Scott, K. R. Poeppelmeier, A. D. Sadow and M. Delferro, Upcycling Single-Use Polyethylene into High-Quality Liquid Products, ACS Cent. Sci., 2019, 5, 1795–1803 CrossRef CAS PubMed.
- H. Zhou, Y. Wang, Y. Ren, Z. Li, X. Kong, M. Shao and H. Duan, Plastic Waste Valorization by Leveraging Multidisciplinary Catalytic Technologies, ACS Catal., 2022, 12, 9307–9324 CrossRef CAS.
- M. Chu, Y. Liu, X. Lou, Q. Zhang and J. Chen, Rational Design of Chemical Catalysis for Plastic Recycling, ACS Catal., 2022, 12, 4659–4679 CrossRef CAS.
- A. S. Goldman, A. H. Roy, Z. Huang, R. Ahuja, W. Schinski and M. Brookhart, Catalytic Alkane Metathesis by Tandem Alkane Dehydrogenation-Olefin Metathesis, Science, 2006, 312, 257–261 CrossRef CAS PubMed.
- X. Jia, C. Qin, T. Friedberger, Z. Guan and Z. Huang, Efficient and Selective Degradation of Polyethylenes into Liquid Fuels and Waxes under Mild Conditions, Sci. Adv., 2016, 2, e1501591 CrossRef PubMed.
- F. Zhang, M. H. Zeng, R. D. Yappert, J. K. Sun, Y. H. Lee, A. M. LaPointe, B. Peters, M. M. Abu-Omar and S. L. Scott, Polyethylene Upcycling to Long-Chain Alkylaromatics by Tandem Hydrogenolysis/Aromatization, Science, 2020, 370, 437–441 CrossRef CAS PubMed.
- T. Li, A. Vijeta, C. Casadevall, A. S. Gentleman, T. Euser and E. Reisner, Bridging Plastic Recycling and Organic Catalysis: Photocatalytic Deconstruction of Polystyrene Via a C–H Oxidation Pathway, ACS Catal., 2022, 12, 8155–8163 CrossRef CAS PubMed.
- X. Chen, M. Peng, D. Xiao, H. Liu and D. Ma, Fully Exposed Metal Clusters: Fabrication and Application in Alkane Dehydrogenation, ACS Catal., 2022, 12, 12720–12743 CrossRef CAS.
- L. Chen, A. P. van Muyden, X. J. Cui, Z. F. Fei, N. Yan, G. Laurenczy and P. J. Dyson, Lignin First: Confirming the Role of the Metal Catalyst in Reductive Fractionation, JACS Au, 2021, 1, 729–733 CrossRef CAS PubMed.
- S. Najari, S. Saeidi, P. Concepcion, D. D. Dionysiou, S. K. Bhargava, A. F. Lee and K. Wilson, Oxidative Dehydrogenation of Ethane: Catalytic and Mechanistic Aspects and Future Trends, Chem. Soc. Rev., 2021, 50, 4564–4605 RSC.
- Y. Wang, P. Hu, J. Yang, Y.-A. Zhu and D. Chen, C–H Bond Activation in Light Alkanes: A Theoretical Perspective, Chem. Soc. Rev., 2021, 50, 4299–4358 RSC.
- K. Nogi and H. Yorimitsu, Carbon-Carbon Bond Cleavage at Allylic Positions: Retro-Allylation and Deallylation, Chem. Rev., 2021, 121, 345–364 CrossRef CAS.
- Y. Peng, Y. Wang, L. Ke, L. Dai, Q. Wu, K. Cobb, Y. Zeng, R. Zou, Y. Liu and R. Ruan, A Review on Catalytic Pyrolysis of Plastic Wastes to High-Value Products, Energy Convers. Manage., 2022, 254, 115243 CrossRef CAS.
- I. Vollmer, M. J. F. Jenks, R. M. Gonzalez, F. Meirer and B. M. Weckhuysen, Plastic Waste Conversion over a Refinery Waste Catalyst, Angew. Chem., Int. Ed., 2021, 60, 16101–16108 CrossRef CAS PubMed.
- X. Hou, N. Ni, Y. Wang, W. J. Zhu, Y. Qiu, Z. H. Diao, G. Z. Liu and X. W. Zhang, Roles of the Free Radical and Carbenium Ion Mechanisms in Pentane Cracking to Produce Light Olefins, J. Anal. Appl. Pyrolysis, 2019, 138, 270–280 CrossRef CAS.
- M. Boronat and A. Corma, Are Carbenium and Carbonium Ions Reaction Intermediates in Zeolite-Catalyzed Reactions?, Appl. Catal., A, 2008, 336, 2–10 CrossRef CAS.
- F. C. Jentoft and B. C. Gates, Solid-Acid-Catalyzed Alkane Cracking Mechanisms: Evidence from Reactions of Small Probe Molecules, Top. Catal., 1997, 4, 1–13 CrossRef CAS.
- S. Kotrel, H. Knozinger and B. C. Gates, The Haag-Dessau Mechanism of Protolytic Cracking of Alkanes, Microporous Mesoporous Mater., 2000, 35–6, 11–20 CrossRef.
- A. Corma and A. V. Orchilles, Current Views on the Mechanism of Catalytic Cracking, Microporous Mesoporous Mater., 2000, 35–6, 21–30 CrossRef.
- J. Weitkamp, Catalytic Hydrocracking-Mechanisms and Versatility of the Process, ChemCatChem, 2012, 4, 292–306 CrossRef CAS.
- M. N. Mazar, S. Al-Hashimi, M. Cococcioni and A. Bhan, Beta-Scission of Olefins on Acidic Zeolites: A Periodic Pbe-D Study in H-ZSM-5, J. Phys. Chem. C, 2013, 117, 23609–23620 CrossRef CAS.
- Y. V. Kissin, Chemical Mechanisms of Catalytic Cracking over Solid Acidic Catalysts: Alkanes and Alkenes, Catal. Rev.: Sci. Eng., 2001, 43, 85–146 CrossRef CAS.
- S. E. Levine and L. J. Broadbelt, Detailed Mechanistic Modeling of High-Density Polyethylene Pyrolysis: Low Molecular Weight Product Evolution, Polym. Degrad. Stab., 2009, 94, 810–822 CrossRef CAS.
- L. L. Fan, L. Liu, Z. G. Xiao, Z. Y. Su, P. Huang, H. Y. Peng, S. Lv, H. W. Jiang, R. Ruan, P. Chen and W. G. Zhou, Comparative Study of Continuous-Stirred and Batch Microwave Pyrolysis of Linear Low-Density Polyethylene in the Presence/Absence of HZSM-5, Energy, 2021, 228, 120612 CrossRef CAS.
- L. L. Fan, Z. Y. Su, J. B. Wu, Z. G. Xiao, P. Huang, L. Liu, H. W. Jiang, W. G. Zhou, S. Y. Liu and R. Ruan, Integrating Continuous-Stirred Microwave Pyrolysis with Ex-Situ Catalytic Upgrading for Linear Low-Density Polyethylene Conversion: Effects of Parameter Conditions, J. Anal. Appl. Pyrolysis, 2021, 157, 105213 CrossRef CAS.
- V. Mortezaeikia, O. Tavakoli and M. S. Khodaparasti, A Review on Kinetic Study Approach for Pyrolysis of Plastic Wastes Using Thermogravimetric Analysis, J. Anal. Appl. Pyrolysis, 2021, 160, 105340 CrossRef CAS.
- G. Elordi, M. Olazar, M. Artetxe, P. Castaño and J. Bilbao, Effect of the Acidity of the HZSM-5 Zeolite Catalyst on the Cracking of High Density Polyethylene in a Conical Spouted Bed Reactor, Appl. Catal., A, 2012, 415–416, 89–95 CrossRef CAS.
- X. He, Y. Tian, L. Guo, C. Qiao and G. Liu, Fabrication of Extra–Framework Al in ZSM–5 to Enhance Light Olefins Production in Catalytic Cracking of N–Pentane, J. Anal. Appl. Pyrolysis, 2022, 165, 105550 CrossRef CAS.
- C. M. Lok, J. Van Doorn and G. Aranda Almansa, Promoted ZSM-5 Catalysts for the Production of Bio-Aromatics, a Review, Renewable Sustainable Energy Rev., 2019, 113, 109248 CrossRef.
- J. Agullo, N. Kumar, D. Berenguer, D. Kubicka, A. Marcilla, A. Gómez, T. Salmi and D. Y. Murzin, Catalytic Pyrolysis of Low Density Polyethylene over H–B, H–Y, H-Mordenite, and H-Ferrierite Zeolite Catalysts: Influence of Acidity and Structures, Kinet. Catal., 2007, 48, 535–540 CrossRef CAS.
- J. Socci, A. Osatiashtiani, G. Kyriakou and T. Bridgwater, The Catalytic Cracking of Sterically Challenging Plastic Feedstocks over High Acid Density Al-SBA-15 Catalysts, Appl. Catal., A, 2019, 570, 218–227 CrossRef CAS.
- Z. Zhang, K. Gora-Marek, J. S. Watson, J. Tian, M. R. Ryder, K. A. Tarach, L. López-Pérez, J. Martínez-Triguero and I. Melián-Cabrera, Recovering Waste Plastics Using Shape-Selective Nano-Scale Reactors as Catalysts, Nat. Sustain., 2019, 2, 39–42 CrossRef.
- S. Kokuryo, K. Miyake, Y. Uchida, A. Mizusawa, T. Kubo and N. Nishiyama, Defect Engineering to Boost Catalytic Activity of Beta Zeolite on Low-Density Polyethylene Cracking, Mat. Today Sustain, 2022, 17, 100098 CrossRef.
- C. S. Costa, H. D. Thi, K. M. Van Geem, M. R. Ribeiro and J. M. Silva, Assessment of Acidity and the Zeolite Porous Structure on Hydrocracking of HDPE, Sustain. Energy Fuels, 2022, 6, 3611–3625 RSC.
- V. Finelli, V. Gentilin, G. Mossotti, G. Ricchiardi, A. Piovano, V. Crocellà and E. Groppo, The Role of Porosity and Acidity in the Catalytic Upcycling of Polyethylene, Catal. Today, 2023, 419, 114142 CrossRef CAS.
- A. Peral, J. M. Escola, D. P. Serrano, J. Přech, C. Ochoa-Hernández and J. Čejka, Bidimensional ZSM-5 Zeolites Probed as Catalysts for Polyethylene Cracking, Catal. Sci. Technol., 2016, 6, 2754–2765 RSC.
- Q. Li, K. C. Hunter and A. L. L. East, A Theoretical Comparison of Lewis Acid Vs Bronsted Acid Catalysis for n-Hexane → Propane + Propene, J. Phys. Chem. A, 2005, 109, 6223–6231 CrossRef CAS PubMed.
- S. Schallmoser, T. Ikuno, M. F. Wagenhofer, R. Kolvenbach, G. L. Haller, M. Sanchez-Sanchez and J. A. Lercher, Impact of the Local Environment of Brønsted Acid Sites in ZSM-5 on the Catalytic Activity in N-Pentane Cracking, J. Catal., 2014, 316, 93–102 CrossRef CAS.
- S. Li, A. Zheng, Y. Su, H. Zhang, L. Chen, J. Yang, C. Ye and F. Deng, Brønsted/Lewis Acid Synergy in Dealuminated Hy Zeolite: A Combined Solid-State NMR and Theoretical Calculation Study, J. Am. Chem. Soc., 2007, 129, 11161–11171 CrossRef CAS PubMed.
- Z. Yu, S. Li, Q. Wang, A. Zheng, X. Jun, L. Chen and F. Deng, Brønsted/Lewis Acid Synergy in H–ZSM-5 and H–MOR Zeolites Studied by 1h and 27al DQ-MAS Solid-State NMR Spectroscopy, J. Phys. Chem. C, 2011, 115, 22320–22327 CrossRef CAS.
- E. G. Derouane, J.-M. André and A. A. Lucas, A Simple Van Der Waals Model for Molecule-Curved Surface Interactions in Molecular-Sized Microporous Solids, Chem. Phys. Lett., 1987, 137, 336–340 CrossRef CAS.
- M. Hartmann, A. G. Machoke and W. Schwieger, Catalytic Test Reactions for the Evaluation of Hierarchical Zeolites, Chem. Soc. Rev., 2016, 45, 3313–3330 RSC.
- A. C. Jerdy, L. Trevisi, M. Monwar, M. Á. González-Borja, R. Abbott, L. Lobban and S. Crossley, Deconvoluting the Roles of Polyolefin Branching and Unsaturation on Depolymerization Reactions over Acid Catalysts, Appl. Catal., B, 2023, 337, 122986 CrossRef CAS.
- R. A. Hackler, J. V. Lamb, I. L. Peczak, R. M. Kennedy, U. Kanbur, A. M. LaPointe, K. R. Poeppelmeier, A. D. Sadow and M. Delferro, Effect of Macro- and Microstructures on Catalytic Hydrogenolysis of Polyolefins, Macromolecules, 2022, 55, 6801–6810 CrossRef CAS.
- G. Manos, A. Garforth and J. Dwyer, Catalytic Degradation of High-Density Polyethylene on an Ultrastable-Y Zeolite. Nature of Initial Polymer Reactions, Pattern of Formation of Gas and Liquid Products, and Temperature Effects, Ind. Eng. Chem. Res., 2000, 39, 1203–1208 CrossRef CAS.
- J. W. Park, J.-H. Kim and G. Seo, The Effect of Pore Shape on the Catalytic Performance of Zeolites in the Liquid-Phase Degradation of HDPE, Polym. Degrad. Stab., 2002, 76, 495–501 CrossRef CAS.
- A. Bonilla, D. Baudouin and J. Pérez-Ramírez, Desilication of Ferrierite Zeolite for Porosity Generation and Improved Effectiveness in Polyethylene Pyrolysis, J. Catal., 2009, 265, 170–180 CrossRef CAS.
- S. D. Jaydev, A. J. Martin and J. Perez-Ramirez, Direct Conversion of Polypropylene into Liquid Hydrocarbons on Carbon-Supported Platinum Catalysts, ChemSusChem, 2021, 14, 5179–5185 CrossRef CAS PubMed.
- X. Wu, A. Tennakoon, R. Yappert, M. Esveld, M. S. Ferrandon, R. A. Hackler, A. M. LaPointe, A. Heyden, M. Delferro, B. Peters, A. D. Sadow and W. Huang, Size-Controlled Nanoparticles Embedded in a Mesoporous Architecture Leading to Efficient and Selective Hydrogenolysis of Polyolefins, J. Am. Chem. Soc., 2022, 144, 5323–5334 CrossRef CAS PubMed.
- V. Cappello, P. Sun, G. Zang, S. Kumar, R. Hackler, H. E. Delgado, A. Elgowainy, M. Delferro and T. Krause, Conversion of Plastic Waste into High-Value Lubricants: Techno-Economic Analysis and Life Cycle Assessment, Green Chem., 2022, 24, 6306–6318 RSC.
- I. L. Peczak, R. M. Kennedy, R. A. Hackler, R. Wang, Y. Shin, M. Delferro and K. R. Poeppelmeier, Scalable Synthesis of Pt/SrTiO3 Hydrogenolysis Catalysts in Pursuit of Manufacturing-Relevant Waste Plastic Solutions, ACS Appl. Mater. Interfaces, 2021, 13, 58691–58700 CrossRef CAS PubMed.
- C. Jia, S. Xie, W. Zhang, N. N. Intan, J. Sampath, J. Pfaendtner and H. Lin, Deconstruction of High-Density Polyethylene into Liquid Hydrocarbon Fuels and Lubricants by Hydrogenolysis over Ru Catalyst, Chem Catal., 2021, 1, 437–455 CrossRef CAS.
- L. Chen, Y. Zhu, L. C. Meyer, L. V. Hale, T. T. Le, A. Karkamkar, J. A. Lercher, O. Y. Gutiérrez and J. Szanyi, Effect of Reaction Conditions on the Hydrogenolysis of Polypropylene and Polyethylene into Gas and Liquid Alkanes, React. Chem. Eng., 2022, 7, 844–854 RSC.
- J. E. Rorrer, C. Troyano-Valls, G. T. Beckham and Y. Román-Leshkov, Hydrogenolysis of Polypropylene and Mixed Polyolefin Plastic Waste over Ru/C to Produce Liquid Alkanes, ACS Sustain. Chem. Eng., 2021, 9, 11661–11666 CrossRef CAS.
- J. E. Rorrer, G. T. Beckham and Y. Román-Leshkov, Conversion of Polyolefin Waste to Liquid Alkanes with Ru-Based Catalysts under Mild Conditions, JACS Au, 2021, 1, 8–12 CrossRef CAS PubMed.
- L. Chen, L. C. Meyer, L. Kovarik, D. Meira, X. I. Pereira-Hernandez, H. Shi, K. Khivantsev, O. Y. Gutiérrez and J. Szanyi, Disordered, Sub-Nanometer Ru Structures on CeO2 Are Highly Efficient and Selective Catalysts in Polymer Upcycling by Hydrogenolysis, ACS Catal., 2022, 12, 4618–4627 CrossRef CAS.
- M. Tamura, S. Miyaoka, Y. Nakaji, M. Tanji, S. Kumagai, Y. Nakagawa, T. Yoshioka and K. Tomishige, Structure-Activity Relationship in Hydrogenolysis of Polyolefins over Ru/Support Catalysts, Appl. Catal., B, 2022, 318, 121870 CrossRef CAS.
- Y. Nakaji, M. Tamura, S. Miyaoka, S. Kumagai, M. Tanji, Y. Nakagawa, T. Yoshioka and K. Tomishige, Low-Temperature Catalytic Upgrading of Waste Polyolefinic Plastics into Liquid Fuels and Waxes, Appl. Catal., B, 2021, 285, 119805 CrossRef CAS.
- P. A. Kots, T. Xie, B. C. Vance, C. M. Quinn, M. D. de Mello, J. A. Boscoboinik, C. Wang, P. Kumar, E. A. Stach, N. S. Marinkovic, L. Ma, S. N. Ehrlich and D. G. Vlachos, Electronic Modulation of Metal-Support Interactions Improves Polypropylene Hydrogenolysis over Ruthenium Catalysts, Nat. Commun., 2022, 13, 5186 CrossRef CAS PubMed.
- P. A. Kots, S. Liu, B. C. Vance, C. Wang, J. D. Sheehan and D. G. Vlachos, Polypropylene Plastic Waste Conversion to Lubricants over Ru/TiO2 Catalysts, ACS Catal., 2021, 11, 8104–8115 CrossRef CAS.
- C. Wang, K. Yu, B. Sheludko, T. Xie, P. A. Kots, B. C. Vance, P. Kumar, E. A. Stach, W. Zheng and D. G. Vlachos, A General Strategy and a Consolidated Mechanism for Low-Methane Hydrogenolysis of Polyethylene over Ruthenium, Appl. Catal., B, 2022, 319, 121899 CrossRef CAS.
- C. Wang, T. Xie, P. A. Kots, B. C. Vance, K. Yu, P. Kumar, J. Fu, S. Liu, G. Tsilomelekis, E. A. Stach, W. Zheng and D. G. Vlachos, Polyethylene Hydrogenolysis at Mild Conditions over Ruthenium on Tungstated Zirconia, JACS Au, 2021, 1, 1422–1434 CrossRef CAS PubMed.
- J. D. Wei, J. Y. Liu, W. H. Zeng, Z. C. Dong, J. K. Song, S. B. Liu and G. Z. Liu, Catalytic Hydroconversion Processes for Upcycling Plastic Waste to Fuels and Chemicals, Catal. Sci. Technol., 2023, 13, 1258–1280 RSC.
- A. Tennakoon, X. Wu, A. L. Paterson, S. Patnaik, Y. C. Pei, A. M. LaPointe, S. C. Ammal, R. A. Hackler, A. Heyden, I. I. Slowing, G. W. Coates, M. Delferro, B. Peters, W. Y. Huang, A. D. Sadow and F. A. Perras, Catalytic Upcycling of High-Density Polyethylene Via a Processive Mechanism, Nat. Catal., 2020, 3, 893–901 CrossRef CAS.
- Y. X. Jing, Y. Q. Wang, S. Y. Furukawa, J. Xia, C. Y. Sun, M. J. Hulsey, H. F. Wang, Y. Guo, X. H. Liu and N. Yan, Towards the Circular Economy: Converting Aromatic Plastic Waste Back to Arenes over a Ru/Nb2O5 Catalyst, Angew. Chem., Int. Ed., 2021, 60, 5527–5535 CrossRef CAS PubMed.
- X. Si, J. Chen, Z. Wang, Y. Hu, Z. Ren, R. Lu, L. Liu, J. Zhang, L. Pan, R. Cai and F. Lu, Ni-Catalyzed Carbon–Carbon Bonds Cleavage of Mixed Polyolefin Plastics Waste, J. Energy Chem., 2023, 85, 562–569 CrossRef CAS.
- G. Zichittella, A. M. Ebrahim, J. Zhu, A. E. Brenner, G. Drake, G. T. Beckham, S. R. Bare, J. E. Rorrer and Y. Román-Leshkov, Hydrogenolysis of Polyethylene and Polypropylene into Propane over Cobalt-Based Catalysts, JACS Au, 2022, 2, 2259–2268 CrossRef CAS PubMed.
- W. Tu, M. Chu, X. Wang, X. Wang, Y. Li, W. Yang, M. Cao, L. Wang, Y. Li, T.-K. Sham, Y. Cui, Q. Zhang and J. Chen, SMSI-Induced Charge Transfer for Selective Hydrogenolysis of Polyolefins, Appl. Catal., B, 2023, 339, 123122 CrossRef CAS.
- S. S. Borkar, R. Helmer, S. Panicker and M. Shetty, Investigation into the Reaction Pathways and Catalyst Deactivation for Polyethylene Hydrogenolysis over Silica-Supported Cobalt Catalysts, ACS Sustainable Chem. Eng., 2023, 11, 10142–10157 CrossRef CAS.
- A. Almithn and D. Hibbitts, Comparing Rate and Mechanism of Ethane Hydrogenolysis on Transition-Metal Catalysts, J. Phys. Chem. C, 2019, 123, 5421–5432 CrossRef CAS.
- W.-T. Lee, F. D. Bobbink, A. P. van Muyden, K.-H. Lin, C. Corminboeuf, R. R. Zamani and P. J. Dyson, Catalytic Hydrocracking of Synthetic Polymers into Grid-Compatible Gas Streams, Cell Rep. Phys. Sci., 2021, 2, 100332 CrossRef CAS.
- A. Le Valant, F. Drault, C. Maleix, C. Comminges, R. Beauchet, Y. Batonneau, L. Pirault-Roy, C. Especel and F. Epron, Effect of the Metallic Particle Size of Supported Pt Catalysts on Methylcyclopentane Hydrogenolysis: Understanding of the Ring Opening Products Distribution by a Geometric Approach, J. Catal., 2018, 367, 234–243 CrossRef CAS.
- M. Chu, X. Wang, X. Wang, X. Lou, C. Zhang, M. Cao, L. Wang, Y. Li, S. Liu, T.-K. Sham, Q. Zhang and J. Chen, Site-Selective Polyolefin Hydrogenolysis on Atomic Ru for Methanation Suppression and Liquid Fuel Production, Research, 2023, 6, 0032 CrossRef CAS PubMed.
- Y. X. Jing and Y. Q. Wang, Heterolytic Dissociation of H2 and Bond Activation: Spotting New Opportunities from a Unified View, Chem Catal., 2023, 3, 100515 CrossRef CAS.
- B. C. Vance, P. A. Kots, C. Wang, Z. R. Hinton, C. M. Quinn, T. H. Epps, L. T. J. Korley and D. G. Vlachos, Single Pot Catalyst Strategy to Branched Products Via Adhesive Isomerization and Hydrocracking of Polyethylene over Platinum Tungstated Zirconia, Appl. Catal., B, 2021, 299, 120483 CrossRef CAS.
- Z. R. Hinton, P. A. Kots, M. Soukaseum, B. C. Vance, D. G. Vlachos, T. H. Epps and L. T. J. Korley, Antioxidant-Induced Transformations of a Metal-Acid Hydrocracking Catalyst in the Deconstruction of Polyethylene Waste, Green Chem., 2022, 24, 7332–7339 RSC.
- S. Liu, P. A. Kots, B. C. Vance, A. Danielson and D. G. Vlachos, Plastic Waste to Fuels by Hydrocracking at Mild Conditions, Sci. Adv., 2021, 7, eabf8283 CrossRef CAS PubMed.
- W.-T. Lee, A. van Muyden, F. D. Bobbink, M. D. Mensi, J. R. Carullo and P. J. Dyson, Mechanistic Classification and Benchmarking of Polyolefin Depolymerization over Silica-Alumina-Based Catalysts, Nat. Commun., 2022, 13, 4850 CrossRef CAS PubMed.
- J. I. Mirena, J. W. Thybaut, G. B. Marin, J. A. Martens and V. V. Galvita, Impact of the Spatial Distribution of Active Material on Bifunctional Hydrocracking, Ind. Eng. Chem. Res., 2021, 60, 6357–6378 CrossRef CAS.
- J. Du, L. Zeng, T. Yan, C. Wang, M. Wang, L. Luo, W. Wu, Z. Peng, H. Li and J. Zeng, Efficient Solvent- and Hydrogen-Free Upcycling of High-Density Polyethylene into Separable Cyclic Hydrocarbons, Nat. Nanotechnol., 2023, 18, 772–779 CrossRef CAS PubMed.
- M. Guisnet, “Ideal” Bifunctional Catalysis over Pt-Acid Zeolites, Catal. Today, 2013, 218–219, 123–134 CrossRef CAS.
- L. Chen, J. B. Moreira, L. C. Meyer and J. Szanyi, Efficient and Selective Dual-Pathway Polyolefin Hydro-Conversion over Unexpectedly Bifunctional M/TiO2-Anatase Catalysts, Appl. Catal., B, 2023, 335, 122897 CrossRef CAS.
- C. A. A. Monteiro, D. Costa, J. L. Zotin and D. Cardoso, Effect of Metal–Acid Site Balance on Hydroconversion of Decalin over Pt/Beta Zeolite Bifunctional Catalysts, Fuel, 2015, 160, 71–79 CrossRef CAS.
- B. C. Vance, S. Najmi, P. A. Kots, C. Wang, S. Jeon, E. A. Stach, D. N. Zakharov, N. Marinkovic, S. N. Ehrlich, L. Ma and D. G. Vlachos, Structure–Property Relationships for Nickel Aluminate Catalysts in Polyethylene Hydrogenolysis with Low Methane Selectivity, JACS Au, 2023, 3, 2156–2165 CrossRef CAS PubMed.
- J. Sun, Y.-H. Lee, R. D. Yappert, A. M. LaPointe, G. W. Coates, B. Peters, M. M. Abu-Omar and S. L. Scott, Bifunctional Tandem Catalytic Upcycling of Polyethylene to Surfactant-Range Alkylaromatics, Chem, 2023, 9, 2318–2336 CAS.
- H. Deldari, Suitable Catalysts for Hydroisomerization of Long-Chain Normal Paraffins, Appl. Catal., A, 2005, 293, 1–10 CrossRef CAS.
- W. Wang, C.-J. Liu and W. Wu, Bifunctional Catalysts for the Hydroisomerization of N-Alkanes: The Effects of Metal–Acid Balance and Textural Structure, Catal. Sci. Technol., 2019, 9, 4162–4187 RSC.
- N. Batalha, L. Pinard, C. Bouchy, E. Guillon and M. Guisnet, N-Hexadecane Hydroisomerization over Pt-Hbea Catalysts. Quantification and Effect of the Intimacy between Metal and Protonic Sites, J. Catal., 2013, 307, 122–131 CrossRef CAS.
- J. Francis, E. Guillon, N. Bats, C. Pichon, A. Corma and L. J. Simon, Design of Improved Hydrocracking Catalysts by Increasing the Proximity between Acid and Metallic Sites, Appl. Catal., A, 2011, 409–410, 140–147 CrossRef CAS.
- Y. Zhang, W. Wang, X. Jiang, X. Su, O. V. Kikhtyanin and W. Wu, Hydroisomerization of N-Hexadecane over a Pd–Ni2P/SAPO-31 Bifunctional Catalyst: Synergistic Effects of Bimetallic Active Sites, Catal. Sci. Technol., 2018, 8, 817–828 RSC.
- P. B. Weisz, Polyfunctional Heterogeneous Catalysis, Adv. Catal., 1962, 13, 137–190 CAS.
- J. E. Samad, J. Blanchard, C. Sayag, C. Louis and J. R. Regalbuto, The Controlled Synthesis of Metal–Acid Bifunctional Catalysts: The Effect of Metal: Acid Ratio and Metal-Acid Proximity in Pt Silica–Alumina Catalysts for n-Heptane Isomerization, J. Catal., 2016, 342, 203–212 CrossRef CAS.
- J. Zecevic, G. Vanbutsele, K. P. de Jong and J. A. Martens, Nanoscale Intimacy in Bifunctional Catalysts for Selective Conversion of Hydrocarbons, Nature, 2015, 528, 245–248 CrossRef CAS PubMed.
- Y. Yamada, C.-K. Tsung, W. Huang, Z. Huo, S. E. Habas, T. Soejima, C. E. Aliaga, G. A. Somorjai and P. Yang, Nanocrystal Bilayer for Tandem Catalysis, Nat. Chem., 2011, 3, 372–376 CrossRef CAS PubMed.
- A. Gallo, A. Fong, K. C. Szeto, J. Rieb, L. Delevoye, R. M. Gauvin, M. Taoufik, B. Peters and S. L. Scott, Ligand Exchange-Mediated Activation and Stabilization of a Re-Based Olefin Metathesis Catalyst by Chlorinated Alumina, J. Am. Chem. Soc., 2016, 138, 12935–12947 CrossRef CAS PubMed.
- S. Lwin and I. E. Wachs, Olefin Metathesis by Supported Metal Oxide Catalysts, ACS Catal., 2014, 4, 2505–2520 CrossRef CAS.
- X. C. Jiao, K. Zheng, Q. X. Chen, X. D. Li, Y. M. Li, W. W. Shao, J. Q. Xu, J. F. Zhu, Y. Pan, Y. F. Sun and Y. Xie, Photocatalytic Conversion of Waste Plastics into C2 Fuels under Simulated Natural Environment Conditions, Angew. Chem., Int. Ed., 2020, 59, 15497–15501 CrossRef CAS PubMed.
- Y. T. Jiang, H. Y. Zhang, L. F. Hong, J. J. Shao, B. W. Zhang, J. J. Yu and S. Chu, An Integrated Plasma-Photocatalytic System for Upcycling of Polyolefin Plastics, ChemSusChem, 2023, 16 CAS.
- S. J. Zhou, Z. N. Zhang and R. Zeng, Upcycling of Polypropylene Wastes Via Catalytically C–H Modification with Polar Olefins, ChemCatChem, 2023, e202300929 CrossRef.
- X. Jiang, B. Zhu and M. Zhu, An Overview on the Recycling of Waste Poly(Vinyl Chloride), Green Chem., 2023, 25, 6971–7025 RSC.
- M. K. Assefa and M. E. Fieser, Divergent Silylium Catalysis Enables Facile Poly(Vinyl Chloride) Upcycling to Poly(Ethylene-Co-Styrene) Derivatives, J. Mater. Chem. A, 2023, 11, 2128–2132 RSC.
- S. Svadlenak, S. Wojcik, O. Ogunlalu, M. Vu, M. Dor, B. W. Boudouris, D. Wildenschild and K. A. Goulas, Upcycling of Polyvinyl Chloride to Hydrocarbon Waxes Via Dechlorination and Catalytic Hydrogenation, Appl. Catal., B, 2023, 338, 123065 CrossRef CAS.
- B. Feng, Y. Guo, X. Liu and Y. Wang, Transforming PVC Plastic Waste to Benzene Via Hydrothermal Treatment in a Multi-Phase System, Green Chem., 2023, 25, 8505–8509 RSC.
- A. A. Kolganov, A. Sreenithya and E. A. Pidko, Homogeneous Catalysis in Plastic Waste Upcycling: A DFT Study on the Role of Imperfections in Polymer Chains, ACS Catal., 2023, 13, 13310–13318 CrossRef CAS.
- B. Feng, Y. Jing, X. Liu, Y. Guo and Y. Wang, Waste PVC Upcycling: Transferring Unmanageable Cl Species into Value-Added Cl-Containing Chemicals, Appl. Catal., B, 2023, 331, 122671 CrossRef CAS.
- P. A. Kots, B. C. Vance, C. M. Quinn, C. Wang and D. G. Vlachos, A Two-Stage Strategy for Upcycling Chlorine-Contaminated Plastic Waste, Nat. Sustain., 2023, 6, 1258–1267 CrossRef.
- K. Oster, A. Tedstone, A. J. Greer, N. Budgen, A. Garforth and C. Hardacre, Dehydrochlorination of PVC in Multi-Layered Blisterpacks Using Ionic Liquids, Green Chem., 2020, 22, 5132–5142 RSC.
- G. O'Rourke, T. Hennebel, M. Stalpaert, A. Skorynina, A. Bugaev, K. Janssens, L. Van Emelen, V. Lemmens, R. De Oliveira Silva, C. Colemonts, P. Gabriels, D. Sakellariou and D. De Vos, Catalytic Tandem Dehydrochlorination–Hydrogenation of PVC Towards Valorisation of Chlorinated Plastic Waste, Chem. Sci., 2023, 14, 4401–4412 RSC.
- R. Cao, M.-Q. Zhang, Y. Jiao, Y. Li, B. Sun, D. Xiao, M. Wang and D. Ma, Co-Upcycling of Polyvinyl Chloride and Polyesters, Nat. Sustain., 2023, 6, 1685–1692 CrossRef.
- X. Wang, P. Wu, Z. Wang, L. Zhou, Y. Liu, H. Cheng, M. Arai, C. Zhang and F. Zhao, Chlorine-Modified Ru/TiO2 Catalyst for Selective Guaiacol Hydrodeoxygenation, ACS Sustain. Chem. Eng., 2021, 9, 3083–3094 CrossRef CAS.
- D. Wu, Q. Wang, O. V. Safonova, D. V. Peron, W. Zhou, Z. Yan, M. Marinova, A. Y. Khodakov and V. V. Ordomsky, Lignin Compounds to Monoaromatics: Selective Cleavage of C−O Bonds over a Brominated Ruthenium Catalyst, Angew. Chem., Int. Ed., 2021, 60, 12513–12523 CrossRef CAS PubMed.
- C. Marquez, C. Martin, N. Linares and D. De Vos, Catalytic Routes Towards Polystyrene Recycling, Mater. Horiz., 2023, 10, 1625–1640 RSC.
- R. Li, Z. Zhang, X. Liang, J. Shen, J. Wang, W. Sun, D. Wang, J. Jiang and Y. Li, Polystyrene Waste Thermochemical Hydrogenation to Ethylbenzene by a N-Bridged Co, Ni Dual-Atom Catalyst, J. Am. Chem. Soc., 2023, 145, 16218–16227 CrossRef CAS PubMed.
- N. E. Munyaneza, C. Posada, Z. Xu, V. De Altin Popiolek, G. Paddock, C. McKee and G. Liu, A Generic Platform for Upcycling Polystyrene to Aryl Ketones and Organosulfur Compounds, Angew. Chem., Int. Ed., 2023, 62, e202307042 CrossRef CAS PubMed.
- Z. Zhibo, S. Nishio, Y. Morioka, A. Ueno, H. Ohkita, Y. Tochihara, T. Mizushima and N. Kakuta, Thermal and Chemical Recycle of Waste Polymers, Catal. Today, 1996, 29, 303–308 CrossRef CAS.
- M. Marczewski, E. Kamińska, H. Marczewska, M. Godek, G. Rokicki and J. Sokołowski, Catalytic Decomposition of Polystyrene. The Role of Acid and Basic Active Centers, Appl. Catal., B, 2013, 129, 236–246 CrossRef CAS.
- B. Pukánszky, J. P. Kennedy, T. Kelen and F. Tüdős, Cationic Reactions in the Melt, Polym. Bull., 1981, 5, 469–476 CrossRef.
- Z. Zhang, D. Li, J. Wang and J. Jiang, Cascade Upcycling Polystyrene Waste into Ethylbenzene over Fe2N@C, Appl. Catal., B, 2023, 323, 122164 CrossRef CAS.
- Y. Jing, Y. Wang, S. Furukawa, J. Xia, C. Sun, M. J. Hülsey, H. Wang, Y. Guo, X. Liu and N. Yan, Towards the Circular Economy: Converting Aromatic Plastic Waste Back to Arenes over a Ru/Nb2O5 Catalyst, Angew. Chem., Int. Ed., 2021, 60, 5527–5535 CrossRef CAS PubMed.
- W. Partenheimer, Valuable Oxygenates by Aerobic Oxidation of Polymers Using Metal/Bromide Homogeneous Catalysts, Catal. Today, 2003, 81, 117–135 CrossRef CAS.
- Z. Huang, M. Shanmugam, Z. Liu, A. Brookfield, E. L. Bennett, R. Guan, D. E. Vega Herrera, J. A. Lopez-Sanchez, A. G. Slater, E. J. L. McInnes, X. Qi and J. Xiao, Chemical Recycling of Polystyrene to Valuable Chemicals Via Selective Acid-Catalyzed Aerobic Oxidation under Visible Light, J. Am. Chem. Soc., 2022, 144, 6532–6542 CrossRef CAS PubMed.
- S. Oh and E. E. Stache, Chemical Upcycling of Commercial Polystyrene Via Catalyst-Controlled Photooxidation, J. Am. Chem. Soc., 2022, 144, 5745–5749 CrossRef CAS PubMed.
- R. C. Cao, M. Q. Zhang, C. Q. Hu, D. Q. Xiao, M. Wang and D. Ma, Catalytic Oxidation of Polystyrene to Aromatic Oxygenates over a Graphitic Carbon Nitride Catalyst, Nat. Commun., 2022, 13, 4809 CrossRef CAS PubMed.
- J. Ye, Y. P. Chen, C. Gao, C. Wang, A. D. Hu, G. W. Dong, Z. Chen, S. G. Zhou and Y. J. Xiong, Sustainable Conversion of Microplastics to Methane with Ultrahigh Selectivity by a Biotic-Abiotic Hybrid Photocatalytic System, Angew. Chem., Int. Ed., 2022, 61, e202213244 CrossRef CAS PubMed.
- S. Chu, B. W. Zhang, X. Zhao, H. S. Soo, F. Wang, R. Xiao and H. Y. Zhang, Photocatalytic Conversion of Plastic Waste: From Photodegradation to Photosynthesis, Adv. Energy Mater., 2022, 12, 2200435 CrossRef CAS.
- K. L. Wang, R. R. Jia, P. Cheng, L. Y. Shi, X. Wang and L. Huang, Highly Selective Catalytic Oxi-Upcycling of Polyethylene to Aliphatic Dicarboxylic Acid under a Mild Hydrogen-Free Process, Angew. Chem., Int. Ed., 2023, 62, e202301340 CrossRef CAS PubMed.
- C. Zhang, X. Shen, Y. Jin, J. Cheng, C. Cai and F. Wang, Catalytic Strategies and Mechanism Analysis Orbiting the Center of Critical Intermediates in Lignin Depolymerization, Chem. Rev., 2023, 123, 4510–4601 CrossRef CAS PubMed.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chemicals from Lignin: An Interplay of Lignocellulose Fractionation, Depolymerisation, and Upgrading, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- Z. H. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Bright Side of Lignin Depolymerization: Toward New Platform Chemicals, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- C. L. Chio, M. Sain and W. S. Qin, Lignin Utilization: A Review of Lignin Depolymerization from Various Aspects, Renewable Sustainable Energy Rev., 2019, 107, 232–249 CrossRef CAS.
- F. Li, Y. Q. Zhao, L. Xue, F. Y. Ma, S. Y. Dai and S. X. Xie, Microbial Lignin Valorization through Depolymerization to Aromatics Conversion, Trends Biotechnol., 2022, 40, 1469–1487 CrossRef CAS PubMed.
- Y. Dai, X. Gao, Q. Wang, X. Wan, C. Zhou and Y. Yang, Recent Progress in Heterogeneous Metal and Metal Oxide Catalysts for Direct Dehydrogenation of Ethane and Propane, Chem. Soc. Rev., 2021, 50, 5590–5630 RSC.
- C. Li and G. Wang, Dehydrogenation of Light Alkanes to Mono-Olefins, Chem. Soc. Rev., 2021, 50, 4359–4381 RSC.
- P. A. Kots, B. C. Vance and D. G. Vlachos, Polyolefin Plastic Waste Hydroconversion to Fuels, Lubricants, and Waxes: A Comparative Study, React. Chem. Eng., 2021, 7, 41–54 RSC.
- G. Wang, C. Li and H. Shan, Highly Efficient Metal Sulfide Catalysts for Selective Dehydrogenation of Isobutane to Isobutene, ACS Catal., 2014, 4, 1139–1143 CrossRef CAS.
- G. Wang, Z. Meng, J. Liu, C. Li and H. Shan, Promoting Effect of Sulfur Addition on the Catalytic Performance of Ni/MgAl2O4 Catalysts for Isobutane Dehydrogenation, ACS Catal., 2013, 3, 2992–3001 CrossRef CAS.
- P. Schwach, X. Pan and X. Bao, Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, 3d Transition Metals for C–H Activation, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- J. A. Labinger and J. E. Bercaw, Understanding and Exploiting C–H Bond Activation, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- T. Dalton, T. Faber, F. Glorius and C. -H. Activation, Toward Sustainability and Applications, ACS Cent. Sci., 2021, 7, 245–261 CrossRef CAS PubMed.
- A. Cabanes, F. J. Valdés and A. Fullana, A Review on VOCs from Recycled Plastics, Sustainable Mater. Technol., 2020, 25, e00179 CrossRef CAS.
- P. Wu, X. Jin, Y. Qiu and D. Ye, Recent Progress of Thermocatalytic and Photo/Thermocatalytic Oxidation for VOCs Purification over Manganese-Based Oxide Catalysts, Environ. Sci. Technol., 2021, 55, 4268–4286 CrossRef CAS PubMed.
- P. Ebrahimbabaie and J. Pichtel, Biotechnology and Nanotechnology for Remediation of Chlorinated Volatile Organic Compounds: Current Perspectives, Environ. Sci. Pollut. Res., 2021, 28, 7710–7741 CrossRef CAS PubMed.
- C. Pang, R. Han, Y. Su, Y. Zheng, M. Peng and Q. Liu, Effect of the Acid Site in the Catalytic Degradation of Volatile Organic Compounds: A Review, Chem. Eng. J., 2023, 454, 140125 CrossRef CAS.
- Y. Su, K. X. Fu, C. H. Pang, Y. F. Zheng, C. F. Song, N. Ji, D. G. Ma, X. B. Lu, C. X. Liu, R. Han and Q. L. Liu, Recent Advances of Chlorinated Volatile Organic Compounds' Oxidation Catalyzed by Multiple Catalysts: Reasonable Adjustment of Acidity and Redox Properties, Environ. Sci. Technol., 2022, 56, 9854–9871 CrossRef CAS PubMed.
- M. S. Kamal, S. A. Razzak and M. M. Hossain, Catalytic Oxidation of Volatile Organic Compounds (VOCs) – a Review, Atmos. Environ., 2016, 140, 117–134 CrossRef CAS.
- Z. D. Zhang, J. Wang, X. H. Ge, S. L. Wang, A. Li, R. Z. Li, J. Shen, X. Liang, T. Gan, X. D. Han, X. S. Zheng, X. Z. Duan, D. S. Wang, J. C. Jiang and Y. D. Li, Mixed Plastics Wastes Upcycling with High-Stability Single-Atom Ru Catalyst, J. Am. Chem. Soc., 2023, 145, 22836–22844 CrossRef CAS PubMed.
- W. Haiqiang, A two-stage Ce/TiO2–Cu/CeO2 catalyst with separated catalytic functions for deep catalytic combustion of CH2Cl2, Chem. Eng. J., 2016, 290, 147–153 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |