
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sunlight-driven and gram-scale vanillin production via Mn-defected γ-MnO2 catalyst in aqueous environment†
Qingping
Ke
 a,
Yurong
Zhang
a,
Chao
Wan
*ab,
Jun
Tang
a,
Shenglai
Li
c,
Xu
Guo
a,
Minsu
Han
*d,
Takashi
Hamada
e,
Sameh M.
Osman
f,
Yunqing
Kang
*g and
Yusuke
Yamauchi
degh
a,
Yurong
Zhang
a,
Chao
Wan
*ab,
Jun
Tang
a,
Shenglai
Li
c,
Xu
Guo
a,
Minsu
Han
*d,
Takashi
Hamada
e,
Sameh M.
Osman
f,
Yunqing
Kang
*g and
Yusuke
Yamauchi
degh
aSchool of Chemistry and Chemical Engineering, Anhui University of Technology, Ma'anshan 243002, China. E-mail: wanchao@zju.edu.cn
bCollege of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310058, China
cDepartment of Materials Science and Chemical Engineering, Stony Brook University, New York 11794, USA
dAustralian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland, Brisbane, Queensland 4072, Australia. E-mail: minsu.han@uq.edu.au
eDepartment of Materials Process Engineering, Graduate School of Engineering, Nagoya University, Nagoya 464-8603, Japan
fChemistry Department, College of Science, King Saud University, P.O. Box 2455, Riyadh, 11451, Saudi Arabia
gResearch Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: yqkang@toki.waseda.jp
hDepartment of Chemical and Biomolecular Engineering, Yonsei University, Seoul 03722, South Korea
First published on 19th February 2024
Abstract
The production of vanillin from biomass offers a sustainable route for synthesizing daily-use chemicals. However, achieving sunlight-driven vanillin synthesis through H2O activation in an aqueous environment poses challenges due to the high barrier of H2O dissociation. In this study, we have successfully developed an efficient approach for gram-scale vanillin synthesis in an aqueous reaction, employing Mn-defected γ-MnO2 as a photocatalyst at room temperature. Density functional theory calculations reveal that the presence of defective Mn species (Mn3+) significantly enhances the adsorption of vanillyl alcohol and H2O onto the surface of the γ-MnO2 catalyst. Hydroxyl radical (˙OH) species are formed through H2O activation with the assistance of sunlight, playing a pivotal role as oxygen-reactive species in the oxidation of vanillyl alcohol into vanillin. The Mn-defected γ-MnO2 catalyst exhibits exceptional performance, achieving up to 93.4% conversion of vanillyl alcohol and 95.7% selectivity of vanillin under sunlight. Notably, even in a laboratory setting during the daytime, the Mn-defected γ-MnO2 catalyst demonstrates significantly higher catalytic performance compared to the dark environment. This work presents a highly effective and promising strategy for low-cost and environmentally benign vanillin synthesis.
Introduction
Vanillin, a highly popular spice worldwide, is extensively used in food additives, perfumes, and daily-use chemicals.1 Traditionally, it is derived from fossil fuels through the Solvay route,2 which involves the oxidation of vanillyl alcohol to vanillin using methanol as a solvent. In addition to the Solvay route, about 15% of vanillin is directly isolated from depolymerized lignin derivatives through upcycling,3–5 presenting an eco-friendly method that avoids reliance on fossil fuels. However, only approximately 2% of lignin currently undergoes conversion to value-added chemicals, with the remaining 98% being incinerated for energy in the pulp and paper industry. Consequently, a significant challenge in terms of sustainability and environmental protection lies in the valorization of lignin.6–9 Among potential approaches, the synthetic vanillin market, producing around 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons per year, primarily relies on fossil-based routes. Chemical conversions of lignin-derived compounds, such as vanillyl alcohol, offer a sustainable and cost-effective route to replenish the synthetic vanillin market.10 Unfortunately, most lignin-to-vanillin processes operate in organic solvents, requiring high temperature/O2 pressure,6 and consuming artificial energy due to recrystallization and solvent extraction steps.11 In response to these drawbacks, the development of sunlight-driven organic transformations through the construction of novel heterogeneous catalyst materials that are efficient and separable even in aqueous reactions holds promise for the sustainable synthesis of valuable chemicals.12
000 tons per year, primarily relies on fossil-based routes. Chemical conversions of lignin-derived compounds, such as vanillyl alcohol, offer a sustainable and cost-effective route to replenish the synthetic vanillin market.10 Unfortunately, most lignin-to-vanillin processes operate in organic solvents, requiring high temperature/O2 pressure,6 and consuming artificial energy due to recrystallization and solvent extraction steps.11 In response to these drawbacks, the development of sunlight-driven organic transformations through the construction of novel heterogeneous catalyst materials that are efficient and separable even in aqueous reactions holds promise for the sustainable synthesis of valuable chemicals.12
The water-participated route for alcohol oxidation signifies a noteworthy advancement in organic synthesis,13 offering a novel and sustainable approach to vanillin synthesis. However, this oxidation process encounters challenges due to the energy-demanding O–H bond activation of H2O.14 While light-driven H2O dissociation has emerged as an attractive method to activate the O–H bond, the limited success cases of this process mainly rely on noble metal catalysts and artificial light sources, such as a Xenon lamp.15 An effective strategy for manipulating the H2O dissociation pathway, including the energy requirement for O–H bond activation and the chemical nature of the active metal/oxygen species, involves the coordination engineering of metal oxide catalysts. The coordination engineering of bismuth (Bi) and oxygen (O) sites in the BiOBr catalyst has successfully demonstrated the enhancement of ethylbenzene oxidation through sunlight-driven reduction of the adsorption barrier for H2O activation.15 However, investigations into the sunlight-driven synthesis of vanillin in an aqueous environment using metal oxides remain relatively scarce.
MnO2, a transition metal oxide, has garnered significant attention as a heterogeneous catalyst for catalytic oxidation reactions, owing to its inherent advantages, including multivalence (Mn2+, Mn3+, and Mn4+)16 and a variable structure (α-, γ-, ε-, R-, and β-MnO2).17 Among these structures, γ-MnO2, characterized by a disordered structure comprising the intergrowth of β-MnO2 and R-MnO2, stands out as one of the most extensively studied manganese dioxides.18,19 Herein, we report the successful fabrication of a Mn-defected γ-MnO2 catalyst for the water-participated oxidation of vanillyl alcohol to vanillin in an aqueous reaction under sunlight illumination. Our approach offers several notable advantages (Table S1†): (1) the use of H2O as a green reaction medium and oxygen source, promoting environmental sustainability; (2) utilization of sunlight as the energy source for H2O activation, reducing reliance on artificial light sources; (3) conducting the reaction at room temperature, minimizing energy requirements, and enabling milder reaction conditions; (4) the possibility of achieving gram-scale reactions, allowing for large-scale production; (5) a separable, additive-free, and carbon-efficient protocol, enhancing the overall efficiency and sustainability of the process.
Results and discussion
γ-MnO2 catalysts with defects in Mn species were obtained through a hydrothermal process (Fig. 1a, see ESI† for preparation details). The crystal structures of the as-prepared γ-MnO2 catalysts were characterized using powder X-ray diffraction (PXRD). As shown in Fig. 1b and S1,† regardless of the amount of urea, the synthesized γ-MnO2, γ-MnO2(1), and γ-MnO2(10) catalysts (1 and 10 referring to the feeding amount of urea) exhibit peaks at 2θ = 22.4°, 37.0°, 42.3°, and 56.0°, corresponding to the (120), (131), (300), and (160) planes, respectively. These planes are well indexed to the layered γ-MnO2 (JCPDS-14-0644).20,21 For comparison, MnO2 catalysts with different crystal structures, such as α-MnO2, β-MnO2, and ε-MnO2, were synthesized using different raw materials22 and characterized by PXRD (Fig. S2†). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) analyses reveal that the as-prepared γ-MnO2 particles exhibit a spherical shape with a size of 6 μm (Fig. 1c), in which nanorods with a thickness of several tens of nanometers are assembled (Fig. 1d and e). In the selected area electron diffraction (SAED) pattern (Fig. 1f) and high-resolution TEM (HRTEM) image (Fig. 1g) of γ-MnO2 nanorods, crystalline spaces of 0.24 and 0.39 nm corresponding to the (131)23,24 and (120)25,26 planes of γ-MnO2, respectively, are observed, consistent with the XRD results. Energy-dispersive X-ray spectroscopy (EDS) mapping confirms the uniform dispersion of Mn and O throughout the γ-MnO2 particle (Fig. 1h). Notably, no N residue is observed for γ-MnO2 according to X-ray photoelectron spectroscopy (XPS) characterization (Fig. S3a†). Both γ-MnO2(1) and γ-MnO2(10) particles synthesized in the presence of urea exhibit a morphology observed in SEM images (Fig. S4†), a crystal structure observed in TEM images (Fig. S5†), and N2 adsorption–desorption isotherms (Fig. S6†) similar to γ-MnO2 synthesized without urea.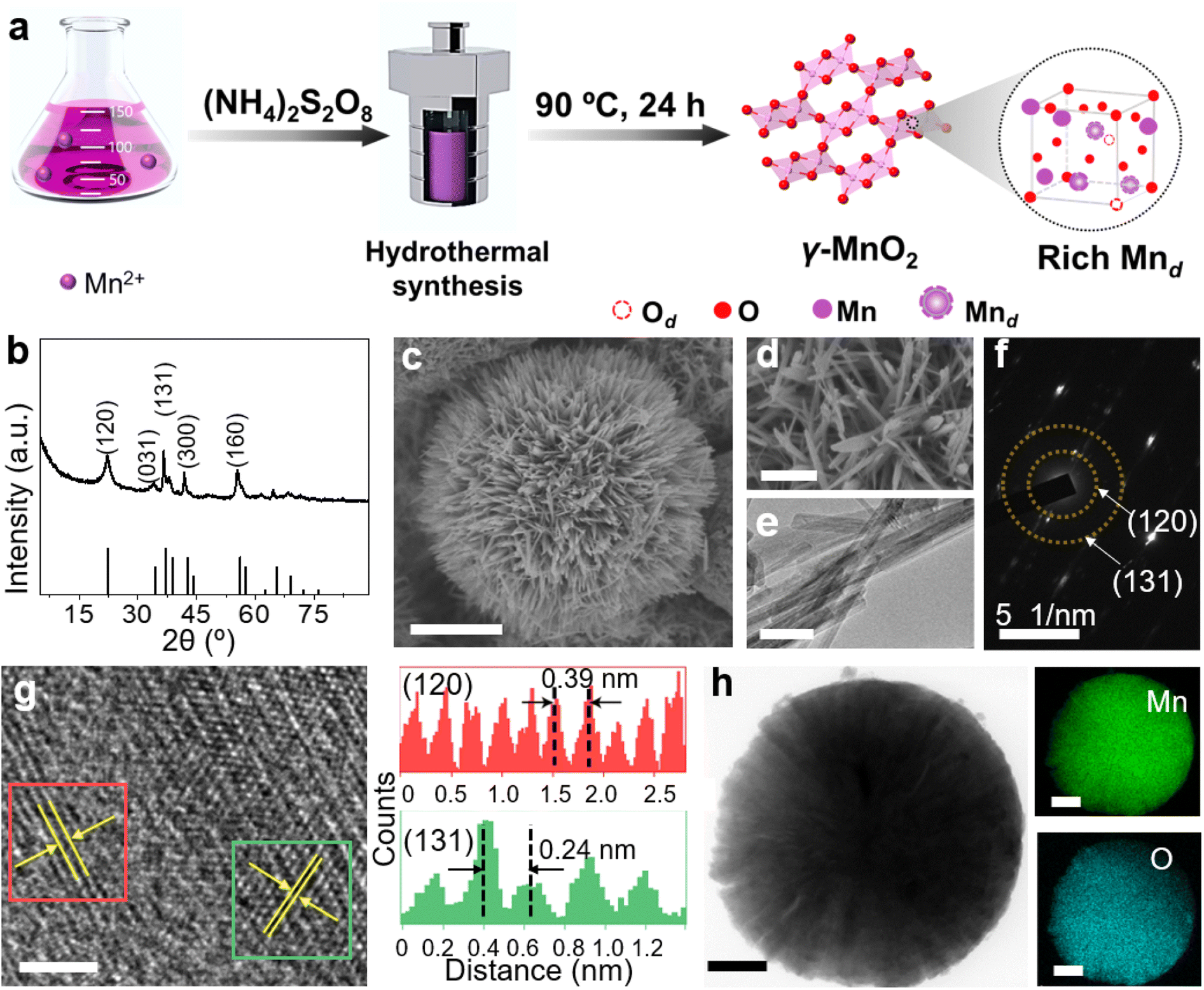 | ||
| Fig. 1 (a) Schematic synthesis process of γ-MnO2 catalysts with Mn defects (Mnd). (b) PXRD pattern, (c and d) SEM images, (e) TEM image, (f) SAED pattern, and (g) HRTEM image (left) and corresponding intensity plot (right) of γ-MnO2 catalyst. (h) EDX elemental maps showing the distributions of Mn and O. Scale bars: (c) 2 μm, (d) 500 nm, (e) 50 nm, (g) 2 nm, (h) 1 μm. | ||
XPS is a regular and powerful tool used to identify the surface elemental species and electronic states of materials. Fig. 2 and S7–S10† show the Mn 3s, Mn 2p, and O 1s XPS patterns of the as-prepared α-MnO2, β-MnO2, ε-MnO2, and γ-MnO2 catalysts. The ΔEs (binding energy between two peaks of Mn 3s multiplet splitting) of the γ-MnO2 catalysts in the range of 4.5–5.1 eV confirm the presence of Mn3+ species (Fig. 2a),27 indicating the coexistence of both Mn3+ and Mn4+ species in these catalysts. The average oxidation states (AOS) of Mn species are calculated using the following formula.28,29
| AOS = 8.956 − 1.126ΔEs |
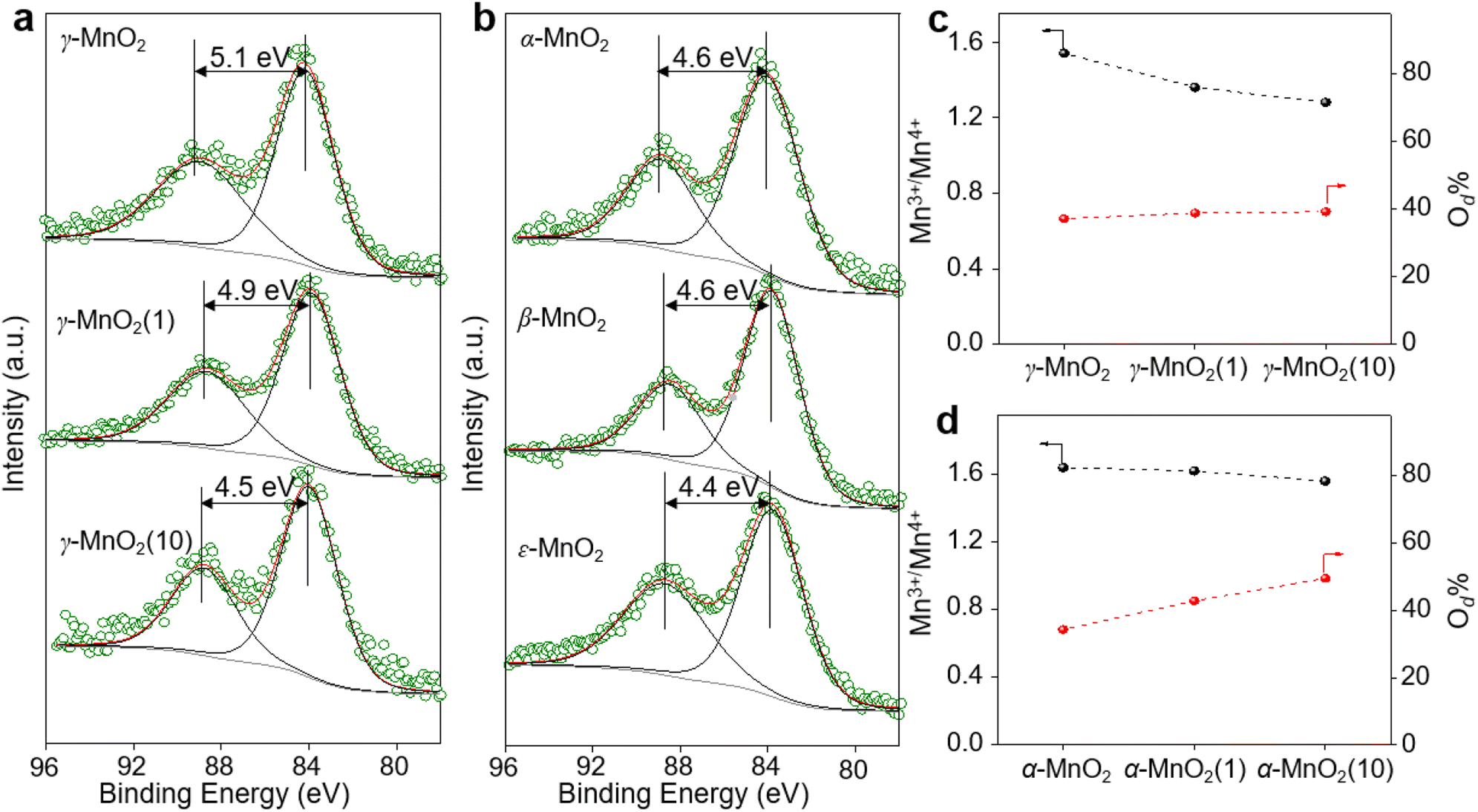 | ||
| Fig. 2 Mn 3s XPS spectra for (a) γ-MnO2 with different amounts of urea added in the synthesis and (b) MnO2 with different crystal structures. The molar ratio of Mn3+/Mn4+ and percentage of oxygen defects (Od) in different (c) γ-MnO2 and (d) α-MnO2. | ||
As shown in Fig. 2a, the calculated AOS values are 3.2, 3.4, and 3.9 for γ-MnO2, γ-MnO2(1), and γ-MnO2(10), respectively, consistent with the Mn3+/Mn4+ ratios (Fig. S7a–c,† Mn3+/Mn4+ = 1.54, 1.36, and 1.28 for γ-MnO2, γ-MnO2(1), and γ-MnO2(10) particles, respectively). It's worth noting that the oxygen defects (Od) remain almost constant in γ-MnO2, γ-MnO2(1), and γ-MnO2(10) (37.2–39.3%), as shown in Fig. S7d–f.† Interestingly, MnO2 with different crystal structures exhibits varying degrees of Mn defects (Mnd) associated with Mn3+ species (Fig. 2b). The calculated AOS values are 3.8, 3.8, and 4.0 for α-MnO2, β-MnO2, and ε-MnO2, respectively, with corresponding ratios of Mn3+/Mn4+ being 1.51, 1.50, and 1.47 (Fig. S8†). The highest ratio of Mn3+/Mn4+ (1.54) in γ-MnO2 among the MnO2 is ascribed to its most abundant Mnd caused by the coordination unsaturation between the lattice oxygen and lattice Mn. Typically, unsaturated metal species and Od species, which are active in various oxidation reactions, coexist in metal oxides.30,31 It is well known that Mnd, caused by coordination unsaturation between lattice oxygen and lattice Mn, can be tuned by inducing Od or adjusting non-metal dopant amounts.32–34 Contrary to the almost constant presence of Od species (Fig. 2c), significantly higher N dopants are observed in γ-MnO2(1) and γ-MnO2(10) compared to γ-MnO2 (Fig. S3†).35 The N anionic (N3−) dopants possess excess negative charges compared to the O anionic (O2−),36 leading to a higher AOS of Mn species due to the charge compensation for higher N dopants. A similar higher AOS of Mn species in MnO2 was previously observed with boron doping.37 For comparison, we synthesized α-MnO2 catalysts with different Od sites. As shown in Fig. 2d and S9,† surface Od sites in α-MnO2 catalysts significantly increase after the addition of urea during the hydrothermal process, while the Mn3+ species remain almost constant (Fig. S10†). The nearly unchanged ratio of Mn3+/Mn4+, despite the differing amounts of Od species in α-MnO2(1) and α-MnO2(10), may be attributed to the presence of N anionic residues. The detailed mechanisms of Od species formation are beyond the scope of the current stage of study and will be pursued in our future work.
To demonstrate the practicality of our approach, we conducted a gram-scale oxidation of vanillyl alcohol to vanillin in an aqueous environment as a model reaction, and the results are presented in Table 1. For the catalytic reaction, 0.77 g of vanillyl alcohol, 6.0 mL of H2O, and 10.0 mmol of catalyst were added to a quartz reactor and stirred at room temperature under sunlight for 10 h while exposed to air. No product is obtained in the absence of a catalyst (Table 1, entry 1). The as-prepared MnO2 catalysts, including α-, β-, ε-, and γ-MnO2 (Table 1, entries 2–9), are found to be active for the oxidation of vanillyl alcohol to vanillin. MnO2 catalysts with more abundant Od (Fig. 2d), such as α-MnO2(1) and α-MnO2(10), exhibit lower catalytic performance compared to α-MnO2 with fewer Od, indicating that Od species are not a key factor in promoting the catalytic activity of MnO2 catalysts for the oxidation under sunlight. The γ-MnO2 catalyst exhibits the highest catalytic activity and vanillin selectivity, converting vanillyl alcohol to vanillin with a yield of nearly 90% (Table 1, entry 2), outperforming the γ-MnO2(1) and γ-MnO2(10) catalysts (Table 1, entries 3,4). Since γ-MnO2, γ-MnO2(1), and γ-MnO2(10) catalysts have similar surface Od species (Fig. 2c), Brunauer–Emmett–Teller (BET) surface areas (Fig. S6†), and surface morphologies (Fig. 1c, d and S4†), the superior catalytic performance of the γ-MnO2 catalyst among them can be attributed to the most abundant Mnd species on the surface (Fig. 3a). Similarly, the best vanillin selectivity and superior catalytic performance displayed by γ-MnO2, compared to other MnO2 catalysts with diverse crystal structures (Table 1, entries 2–5 and 9–10), can be attributed to the prevalence of its Mnd species (Fig. 3). Given the significant influence of factors, such as solvent and temperature, on the catalytic oxidation reaction of vanillyl alcohol,38 we explored various conditions using the γ-MnO2 catalyst to determine the optimal reaction parameters. The γ-MnO2 catalyst exhibits remarkably superior catalytic performance at 30 °C (Table S2†) and when using H2O as a solvent (Table S3†). The significant improvement in the catalytic performance of γ-MnO2 catalyst in H2O compared to other solvents (Table S3†) may be attributed to the role of H2O in activating O2/H2O to form key active O species, as discussed in the following section on the mechanism.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Con.% | Sel.%a | Yield% |
| a Selective generation of vanillin. Reaction conditions: vanillyl alcohol (0.77 g), Solvent (H2O, 6.0 mL), catalyst 10.0 mmol, open to air, sunlight, 30 °C, 10 h. (n.d. = Not detected). | ||||
| 1 | — | n.d. | — | — |
| 2 | γ-MnO2 | 93.4 | 95.7 | 89.4 |
| 3 | γ-MnO2(1) | 66.3 | 95.5 | 63.3 |
| 4 | γ-MnO2(10) | 63.3 | 94.9 | 60.1 |
| 5 | α-MnO2 | 60.8 | 86.3 | 52.5 |
| 6 | α-MnO2(1) | 25.5 | 45.7 | 11.7 |
| 7 | α-MnO2(10) | 26.0 | 54.0 | 14.0 |
| 8 | β-MnO2 | 63.0 | 94.4 | 59.5 |
| 9 | ε-MnO2 | 61.4 | 91.3 | 56.1 |
 | ||
| Fig. 3 Catalytic performance of γ-MnO2 and comparison catalysts for oxidation of vanillyl alcohol to vanillin. (a) AOS-yield of vanillin on γ-MnO2, (b) AOS-yield of vanillin on MnO2 with various crystal structures. | ||
To elucidate its superior catalytic performance, the catalytic mechanism of the oxidation of vanillyl alcohol over the γ-MnO2 catalyst was evaluated. In comparison to the impressive catalytic performance under O2 (Fig. 4a, eqn (1)), the oxidation of vanillyl alcohol is dramatically suppressed (∼6.5% yield of vanillin) when the reaction is performed under N2 (Fig. 4a, eqn (2)), confirming that oxidation over the γ-MnO2 catalyst mainly occurs through a dissolved oxygen species-mediated route.39 After the reaction under the N2 atmosphere, the ratio of surface Mn3+/Mn4+ species increases from 1.54 to 1.63 (Fig. S11†), indicating the involvement of Mn species in the catalytic cycle.
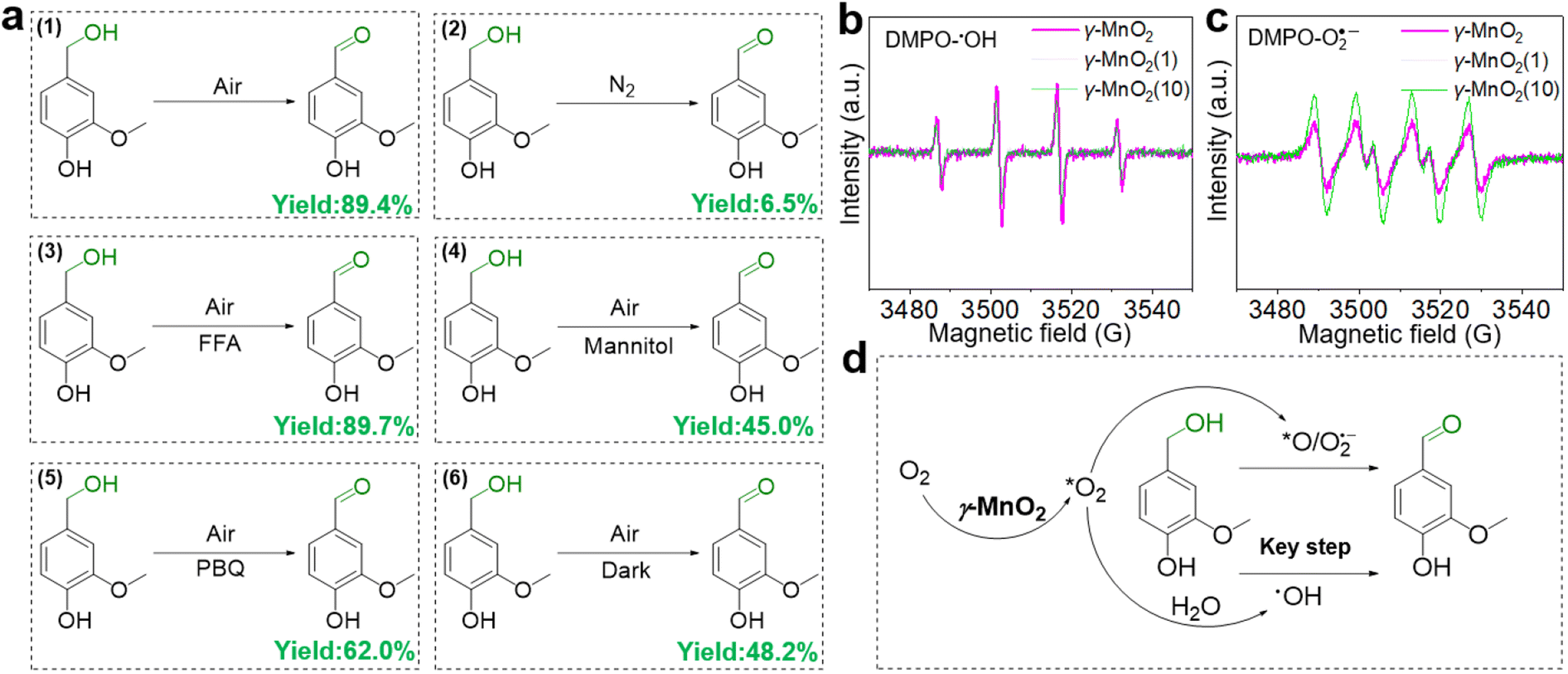 | ||
| Fig. 4 Exploring the role of oxygen species in the oxidation of vanillyl alcohol over γ-MnO2 catalyst. (a) Oxidation of vanillyl alcohol under different atmospheres or with various quenches. (b and c) EPR spectra of DMPO-˙OH and DMPO-O2˙− over various γ-MnO2 catalysts. (d) Proposed the role of oxygen species in the oxidation of vanillyl alcohol. *O2 refers to the adsorbed oxygen. | ||
Considering the importance of oxygen in determining oxidative activity, we further elucidate the role of dissolved oxygen species in sunlight oxidation through scavenger control experiments. Despite the addition of furfuryl alcohol (FFA, an efficient scavenger of 1O2) to the reactants, the yield of vanillin is 89.4% (Fig. 4a, eqn (3)), indicating that FFA does not affect the activity of the γ-MnO2 catalyst. On the other hand, mannitol (an efficient scavenger of ˙OH) causes a significant decrease in the yield of vanillin (∼45.0%) (Fig. 4a, eqn (4)). With 1,4-Benzoquinone (PBQ, an efficient scavenger of O2˙−) added during the reaction, only a normal conversion inhibition of vanillyl alcohol is observed and ∼62.0% yield of vanillin is obtained (Fig. 4a, eqn (5)). The above analyses confirm that the oxidation of vanillyl alcohol to vanillin under sunlight occurs with the assistance of O2˙− and ˙OH species derived from O2/H2O activation by the γ-MnO2 catalyst.
Electron paramagnetic resonance (EPR) was employed to confirm the presence of O2˙− and ˙OH species during the oxidation process.40 Under the reaction conditions, a quadruple peak with an intensity ratio of 1:2:2:1 is observed for the characteristic peak of pyrroline nitrogen oxide (DMPO)-˙OH (Fig. 4b).41 Another quadruple peak with an intensity ratio of 1:1:1:1 is observed for the characteristic peak of DMPO-O2˙− (Fig. 4c),42 indicating the formation of O2˙− and ˙OH species. Notably, the peak intensity of DMPO-˙OH over the γ-MnO2 catalyst with superior catalytic activity is stronger than that of γ-MnO2(1) and γ-MnO2(10) catalysts with mediocre activity. In contrast, the intensities of DMPO-O2˙− over the γ-MnO2 catalyst shows opposite trends. These results further support that ˙OH species are the key reactive oxygen species in the oxidation reaction of vanillyl alcohol via γ-MnO2 under sunlight (Fig. 4d), consistent with the results in Fig. 4a. Meanwhile, the stronger DMPO-˙OH signal for γ-MnO2 compared to the other two samples indicates enhanced ˙OH generation due to the rich-Mnd. Interestingly, under dark conditions, the γ-MnO2 catalyst achieves a ∼48.2% yield of vanillin (Fig. 4a, eqn (6)), similar to the 45.0% yield obtained when mannitol is added to the reaction mixture (Fig. 4a, eqn (4)). Additionally, the γ-MnO2 catalyst exhibits broad light absorption from ultraviolet to visible light, effectively covering most of the solar spectrum (Fig. S12†). This characteristic enables the abundant generation of ˙OH species under natural light exposure at 30 °C.
Based on the above analysis, O2, H2O, and sunlight emerge as key factors influencing the catalytic performance of the γ-MnO2 catalyst in the oxidation of vanillyl alcohol to vanillin. Previous studies have demonstrated that ˙OH species can be generated from H2O/O2 mixtures on δ-MnO2 or Mn/Na2WO4/SiO2 catalysts under sunlight conditions.43,44 To verify the formation of ˙OH species in H2O/O2 mixtures over the γ-MnO2 catalyst, in situ IR spectroscopy was employed, and the results are depicted in Fig. 5. The stretching vibration peaks of ˙OH species at ∼2820, 3711, 3735 and 3750 cm−1 intensify with prolonged sunlight illumination (Fig. 5). However, the stretching vibration of ˙OH species is not observed in the absence of H2O (Fig. 5a, black curve) or under dark conditions (Fig. 5a, blue curve). These results indicate that both H2O and sunlight play crucial roles in the formation of ˙OH species in H2O/O2 mixtures. In the oxidation reaction, oxygen is considered to be the ideal oxidant, with H2O and H2O2 identified as by-products. Identifying these by-products is vital for elucidating the reaction mechanism, especially as ˙OH species could be produced from H2O2 species. Further investigation into the by-product of H2O2 species was conducted using the iodometry method, and the results are presented in Fig. S13.† A peak at ∼365 nm, assigned to the formation of H2O2, is detected under sunlight illustration. However, H2O2 species are not produced under dark conditions. Similar results are obtained in the absence of a reactant or γ-MnO2 catalyst. These findings confirm that H2O2 species, as a by-product, are produced during the oxidation of vanillyl alcohol to vanillin over the γ-MnO2 catalyst with the assistance of sunlight illustration.
 | ||
| Fig. 5 In situ diffuse reflectance IR spectra (DRIFTS) of activation of O2/H2O over γ-MnO2 catalyst. (a) Activation of O2/H2O under various conditions. (b) Activation of O2/H2O under sunlight illustration. | ||
Density functional theory (DFT) calculations were employed to elucidate the absorption sites on the γ-MnO2 catalyst, comprising a perfect (120) facet and defective (120) facets (Fig. S14†). The DFT calculations (Fig. 6a and b) reveal that vanillyl alcohol and H2O preferentially adsorb on the Mnd species (Mn3+ species) of defective (120) facets, regardless of the presence of Od species for both models. The preference of vanillyl alcohol for adsorption on the Mnd species is further supported by DFT calculations in Fig. 6a and S15,† illustrating the interaction between Mn3+ species of MnO2 and vanillyl alcohol. This interaction results in an adsorption energy of −217.3 kJ mol−1 (Fig. S15†), significantly exceeding the adsorption energy of −131.5 kJ mol−1 observed for MnO2 featuring only Od sites. Similarly, H2O exhibits a preference for adsorption on the Mnd species rather than Od sites, as depicted in Fig. 6b. Furthermore, oxygen molecules from the air can also be absorbed by the Mnd. Previous studies have reported the adsorption of oxygen molecules on Mnd in MnO2 catalysts.45
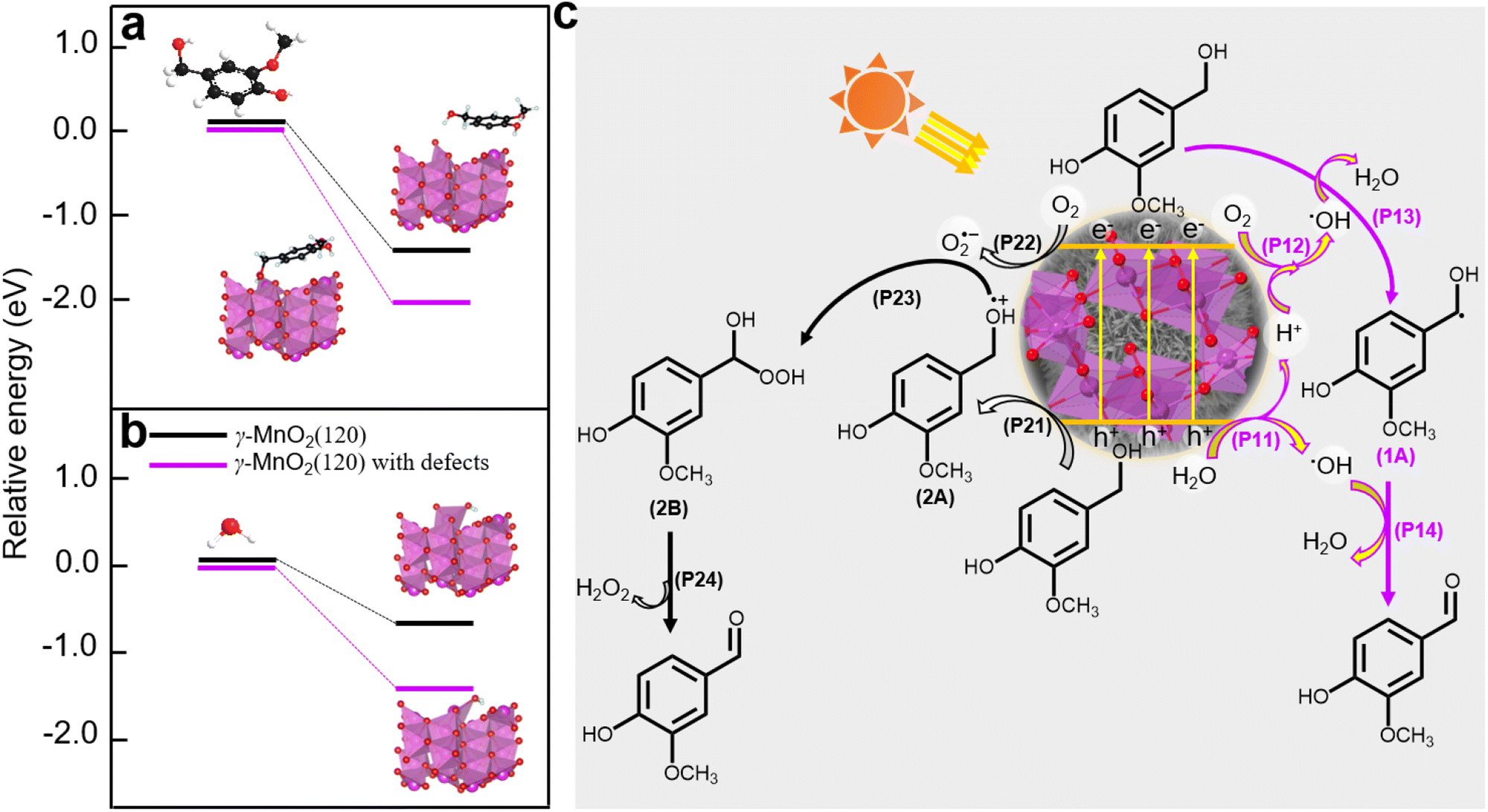 | ||
| Fig. 6 (a) DFT calculations for vanillyl alcohol adsorbed on γ-MnO2 catalyst with/without defects. (b) DFT calculations for H2O adsorbed on γ-MnO2 catalyst with/without defects. (c) A plausible mechanism of oxidation of vanillyl alcohol to vanillin over γ-MnO2 catalyst under air atmosphere. | ||
Based on the analysis and insights gleaned from previous studies,10,46,47 we propose a plausible mechanism for the oxidation of vanillyl alcohol to vanillin using the γ-MnO2 catalyst in air (Fig. 6c). In this mechanism, O2, H2O, and vanillyl alcohol are simultaneously adsorbed onto the catalyst. Under sunlight illumination, electrons (e−) and holes (h+) are separated from the surface of the γ-MnO2 catalyst. During this step, adsorbed H2O accepts holes (h+) to form ˙OH species, releasing H+ species (P11, Fig. 6c). Simultaneously, vanillyl alcohol accepts holes (h+) to generate alcohol radical species (2A) (P21, Fig. 6c). Adsorbed oxygen can accept e− from the γ-MnO2 catalyst surface to form O2˙− species (P22, Fig. 6c), or it can combine with protons generated from P11 to form ˙OH species (P12, Fig. 6c). The formation of ˙OH species from O2 and H2O in oxide-based catalysts, including MnO2, has also been proposed44,48 Vanillyl alcohol radical (1A, Fig. 6c) is selectively obtained by combining the adsorbed vanillyl alcohol with ˙OH species (P13, Fig. 6c), which further converts to vanillin through a subsequent reaction with ˙OH species (P14, Fig. 6c). The positive radical of vanillyl alcohol (2A, Fig. 6c) reacts with O2˙− species to produce the alcohol peroxo species (2B) (P23, Fig. 6c), eventually converting to vanillin and releasing H2O2 (P24, Fig. 6c). Under these conditions, the catalytic cycle is completed, and the γ-MnO2 catalyst is ready for the next catalytic process.
Conclusions
In summary, we have successfully fabricated manganese oxide-based catalysts with different Mn defects on MnO2 with various crystal structures and on γ-MnO2 with different amounts of urea. Comprehensive structural characterization highlights the significance of urea in tuning the coordination environment of Mn species in γ-MnO2 and the O species in α-MnO2. In the gram-scale oxidation of vanillyl alcohol to vanillin, the Mn-defected γ-MnO2 catalyst demonstrates excellent photocatalytic performance compared to the O-defected α-MnO2 catalyst and other type (β, ε)-MnO2 catalysts. DFT calculations and control experiments demonstrate that the defected-Mn species in the γ-MnO2 catalyst facilitate the adsorption of vanillyl alcohol and H2O, which converts to the corresponding ˙OH species under natural light illustration. The synergistic effect of the Mn-defected species and ˙OH species plays a crucial role in enhancing the photocatalytic performance in the aerobic oxidation of vanillyl alcohol to vanillin over the γ-MnO2 catalyst. This study not only presents effective methods for tuning the coordination environments of Mn atoms in MnO2 for the selective oxidation of vanillyl alcohol to vanillin but also provides valuable insights for the development of advanced transition metal oxide photocatalysts.Author contributions
Q. Ke, Y. Zhang and C. Wan: conceptualization, methodology, and writing – original draft. Y. Zhang, J. Tang, S. Li, X. Guo, T. Hamada, and S. M. Osman: data curation, chemical experiments, and formal analysis. Q. Ke, and C. Wan: funding acquisition and investigation. C. Wan, M. Han, Y. Kang, and Y. Yamauchi: supervision and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Y. K. thanks the support from JSPS Postdoctoral Fellowships for Research in Japan. This work was financially supported by the National Natural Science Foundation of China (22108238, 22302001), Key Projects of the Department of Education of Anhui Province of China (RZ2000003450, 2022AH050314), the Anhui Provincial Natural Science Foundation of China (2008085MB47), the China Postdoctoral Science Foundation (2019M662060, 2020T130580, PC2022046), the Scientific Research Training Program for College Students of Anhui University of Technology (202110360040, S202110360213), the JST-ERATO Yamauchi Materials Space-Tectonics Project (JPMJER2003), the Researchers Supporting Project RSP2023R405 (King Saud University, Saudi Arabia), and the UQ-Yonsei International Research Project (via UQ). The authors also thank Shiyanjia Lab (https://www.shiyanjia.com/) for the support of the XRD, XPS, and BET tests. This work used the Queensland node of the NCRIS-enabled Australian National Fabrication Facility (ANFF). We express our gratitude for English editing software, such as Grammarly and ChatGPT, for refining language and checking grammatical errors in our manuscript.References
- R. Singathi, R. Raghunathan, R. Krishnan, S. K. Rajendran, S. Baburaj, M. P. Sibi, D. C. Webster and J. Sivaguru, Angew. Chem., Int. Ed., 2022, 61, e202203353 CrossRef CAS PubMed.
- B. Schaefer, Natural Products in the Chemical Industry, Springer, Berlin, 2014 Search PubMed.
- X. Shen, Q. Meng, Q. Mei, H. Liu, J. Yan, J. Song, D. Tan, B. Chen, Z. Zhang, G. Yang and B. Han, Chem. Sci., 2020, 11, 1347–1352 RSC.
- Z. Xiang, W. Han, J. Deng, W. Zhu, Y. Zhang and H. Wang, ChemSusChem, 2020, 13, 4199–4213 CrossRef CAS PubMed.
- D. Zhu, L. Xu, S. Sethupathy, H. Si, F. Ahmad, R. Zhang, W. Zhang, B. Yang and J. Sun, Green Chem., 2021, 23, 9554–9570 RSC.
- W. Sun, S. Wu, Y. Lu, Y. Wang, Q. Cao and W. Fang, ACS Catal., 2020, 10, 7699–7709 CrossRef CAS.
- Z. Pan, A. Puente-Urbina, A. Bodi, J. A. van Bokhoven and P. Hemberger, Chem. Sci., 2021, 12, 3161–3169 RSC.
- M. Fache, B. Boutevin and S. Caillol, ACS Sustainable Chem. Eng., 2016, 4, 35–46 CrossRef CAS.
- M. B. Figueirêdo, I. Hita, P. J. Deuss, R. H. Venderbosch and H. J. Heeres, Green Chem., 2022, 24, 4680–4702 RSC.
- B.-C. Li, N. N. Huy, J.-Y. Lin, S. Phattarapattamawong, G. Lisak, H. Wang and K.-Y. A. Lin, J. Environ. Chem. Eng., 2021, 9, 106092 CrossRef CAS.
- R. Zhang, R. Maltari, M. Guo, J. Kontro, A. Eronen and T. Repo, Ind. Crops Prod., 2020, 145, 112095 CrossRef CAS.
- J. Dai, A. F. Patti, G. N. Styles, S. Nanayakkara, L. Spiccia, F. Arena, C. Italiano and K. Saito, Green Chem., 2019, 21, 2005–2014 RSC.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS PubMed.
- C.-R. Chang, X.-F. Yang, B. Long and J. Li, ACS Catal., 2013, 3, 1693–1699 CrossRef CAS.
- X. Cao, A. Huang, C. Liang, H.-C. Chen, T. Han, R. Lin, Q. Peng, Z. Zhuang, R. Shen, H. M. Chen, Y. Yu, C. Chen and Y. Li, J. Am. Chem. Soc., 2022, 144, 3386–3397 CrossRef CAS PubMed.
- J. Li, H. Yuan, Q. Zhang, K. Luo, Y. Liu, W. Hu, M. Xu and S. Xu, Phys. Chem. Chem. Phys., 2020, 22, 27272–27279 RSC.
- D. A. Kitchaev, S. T. Dacek, W. Sun and G. Ceder, J. Am. Chem. Soc., 2017, 139, 2672–2681 CrossRef CAS PubMed.
- X. Zeng, G. Cheng, Q. Liu, W. Yu, R. Yang, H. Wu, Y. Li, M. Sun, C. Zhang and L. Yu, Ind. Eng. Chem. Res., 2019, 58, 13926–13934 CrossRef CAS.
- J. C. Hunter, J. Solid State Chem., 1981, 39, 142–147 CrossRef CAS.
- X. Luo, X. Tang, J. Ni, B. Wu, C. Li, M. Shao and Z. Wei, Chem. Sci., 2023, 14, 1679–1686 RSC.
- X. Yan, T. Gan, S. Shi, J. Du, G. Xu, W. Zhang, W. Yan, Y. Zou and G. Liu, Catal. Sci. Technol., 2021, 11, 6369–6373 RSC.
- K.-Y. A. Lin, W.-D. Oh, M.-W. Zheng, E. Kwon, J. Lee, J.-Y. Lin, X. Duan and F. Ghanbari, J. Colloid Interface Sci., 2021, 592, 416–429 CrossRef CAS PubMed.
- S. Ndayiragije, Y. Zhang, Y. Zhou, Z. Song, N. Wang, T. Majima and L. Zhu, Appl. Catal., B, 2022, 307, 121168 CrossRef CAS.
- C. Wang, Y. Zeng, X. Xiao, S. Wu, G. Zhong, K. Xu, Z. Wei, W. Su and X. Lu, J. Energy Chem., 2020, 43, 182–187 CrossRef.
- L. Ni, Z. Wu, G. Zhao, C. Sun, C. Zhou, X. Gong and G. Diao, Small, 2017, 13, 1603466 CrossRef PubMed.
- F. Lai, J. Feng, R. Yan, G. C. Wang, M. Antonietti and M. Oschatz, Adv. Funct. Mater., 2018, 28, 1801298 CrossRef.
- Y. Li, X. Wei, S. Han, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 60, 21464–21472 CrossRef CAS PubMed.
- A. Singh, O. Sel, H. Perrot, V. Balland, B. Limoges and C. Laberty-Robert, J. Mater. Chem. A, 2021, 9, 1500–1506 RSC.
- Z. Zhou, X. Zheng, M. Liu, P. Liu, S. Han, Y. Chen, B. Lan, M. Sun and L. Yu, ChemSusChem, 2022, 15, e202200612 CrossRef CAS PubMed.
- Q. Zhang, G. Xie, M. Duan, Y. Liu, Y. Cai, M. Xu, K. Zhao, H. Tai, Y. Jiang and Y. Su, ACS Appl. Nano Mater., 2023, 6, 17445–17456 CrossRef CAS.
- J. Tang, J. Chen, Z. Zhang, Q. Ma, X. Hu, P. Li, Z. Liu, P. Cui, C. Wan, Q. Ke, L. Fu, J. Kim, T. Hamada, Y. Kang and Y. Yamauchi, Chem. Sci., 2023, 14, 13402–13409 RSC.
- H. Liu, W. Jia, X. Yu, X. Tang, X. Zeng, Y. Sun, T. Lei, H. Fang, T. Li and L. Lin, ACS Catal., 2021, 11, 7828–7844 CrossRef CAS.
- Q. Ke, Y. Jin, F. Ruan, M. N. Ha, D. Li, P. Cui, Y. Cao, H. Wang, T. Wang, V. N. Nguyen, X. Han, X. Wang and P. Cui, Green Chem., 2019, 21, 4313–4318 RSC.
- D. Sun, L. Peng, Y. Yang, Y. Fang, S. P. Jiang and Z. Shao, J. Catal., 2022, 409, 48–58 CrossRef CAS.
- C. Chen, G. Xie, J. Dai, W. Li, Y. Cai, J. Li, Q. Zhang, H. Tai, Y. Jiang and Y. Su, Nano Energy, 2023, 116, 108788 CrossRef CAS.
- A. V. Emeline, N. V. Sheremetyeva, N. V. Khomchenko, V. K. Ryabchuk and N. Serpone, J. Phys. Chem. C, 2007, 111, 11456–11462 CrossRef CAS.
- Y. Liu, M.-S. Niu, X. Yi, G. Li, H. Zhou and W. Gao, Appl. Surf. Sci., 2021, 561, 150081 CrossRef CAS.
- J. Estrada-Pomares, S. Ramos-Terrón, G. Lasarte-Aragonés, R. Lucena, S. Cárdenas, D. Rodríguez-Padrón, R. Luque and G. de Miguel, J. Mater. Chem. A, 2022, 10, 11298–11305 RSC.
- F. Li, J. Tang, Q. Ke, Y. Guo, M. N. Ha, C. Wan, Z. Lei, J. Gu, Q. Ling, V. N. Nguyen and W. Zhan, ACS Catal., 2021, 11, 11855–11866 CrossRef CAS.
- J. Li, G. Xie, J. Jiang, Y. Liu, C. Chen, W. Li, J. Huang, X. Luo, M. Xu, Q. Zhang, M. Yang and Y. Su, Nano Energy, 2023, 108, 108234 CrossRef CAS.
- S. Mu, H. Lu, Q. Wu, L. Li, R. Zhao, C. Long and C. Cui, Nat. Commun., 2022, 13, 3694 CrossRef CAS PubMed.
- Y. Yang, B. Cheng, J. Yu, L. Wang and W. Ho, Nano Res., 2023, 16, 4506–4514 CrossRef CAS.
- S. Das, A. Samanta and S. Jana, ACS Sustainable Chem. Eng., 2017, 5, 9086–9094 CrossRef CAS.
- K. Takanabe and E. Iglesia, Angew. Chem., Int. Ed., 2008, 47, 7689–7693 CrossRef CAS PubMed.
- J. Shi, T. Qi, B.-C. Sun, G.-W. Chu and J.-F. Chen, Chem. Eng. J., 2022, 440, 135802 CrossRef CAS.
- Q. Zhu, P. Zhan, C. Zhang, R. Chen, C. Ren, H. Zhao, W. Ren, J. Zhang, P. Qin and D. Cai, ChemPhotoChem, 2023, 7, e202200337 CrossRef CAS.
- M. Bellardita, S. Yurdakal, B. S. Tek, Ç. Değirmenci, G. Palmisano, V. Loddo, L. Palmisano, J. Soria, J. Sanz and V. Augugliaro, J. Environ. Chem. Eng., 2021, 9, 105308 CrossRef CAS.
- S. Zhao, Y. Wen, X. Liu, X. Pen, F. Lü, F. Gao, X. Xie, C. Du, H. Yi D. Kang and X. Tang, Nano Res., 2020, 13, 1544–1551 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05654f |
| This journal is © The Royal Society of Chemistry 2024 |