
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical energy storage in an organic supercapacitor via a non-electrochemical proton charge assembly†
Sanchayita
Mukhopadhyay
a,
Alagar Raja
Kottaichamy
 ad,
Mruthyunjayachari Chattanahalli
Devendrachari
a,
Rahul Mahadeo
Mendhe
a,
Harish Makri
Nimbegondi Kotresh
*b,
Chathakudath Prabhakaran
Vinod
*c and
Musthafa
Ottakam Thotiyl
*a
ad,
Mruthyunjayachari Chattanahalli
Devendrachari
a,
Rahul Mahadeo
Mendhe
a,
Harish Makri
Nimbegondi Kotresh
*b,
Chathakudath Prabhakaran
Vinod
*c and
Musthafa
Ottakam Thotiyl
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research, Pune, Dr Homi Bhabha Road, Pune 411008, India. E-mail: musthafa@iiserpune.ac.in
bDepartment of Chemistry, Acharya Institute of Technology, Soldevanahalli, Bangalore 560107, India. E-mail: harishmnk@acharya.ac.in
cCatalysis and Inorganic Chemistry Division, CSIR-NCL, Pune 411008, India. E-mail: cp.vinod@ncl.res.in
dDepartment of Chemistry, Ilse Katz Institute for Nanoscale Science and Technology, Ben-Gurion University of the Negev, Beer-Sheva 8410501, Israel
First published on 19th December 2023
Abstract
Contrary to conventional beliefs, we show how a functional ligand that does not exhibit any redox activity elevates the charge storage capability of an electric double layer via a proton charge assembly. Compared to an unsubstituted ligand, a non-redox active carboxy ligand demonstrated nearly a 4-fold increase in charge storage, impressive capacitive retention even at a rate of 900C, and approximately a 2-fold decrease in leakage currents with an enhancement in energy density up to approximately 70% via a non-electrochemical route of proton charge assembly. Generalizability of these findings is presented with various non-redox active functional units that can undergo proton charge assembly in the ligand. This demonstration of non-redox active functional units enriching supercapacitive charge storage via proton charge assembly contributes to the rational design of ligands for energy storage applications.
1. Introduction
Amidst the pressing need to address escalating global energy demands and rapid industrialization, a great deal of attention has been focused on developing high-power and high-energy-density energy storage and conversion devices.1–7 Electrochemical capacitors (ECs), or supercapacitors, have emerged as particularly promising candidates due to their superior power density, long lifetime, and high cyclic stability in comparison to secondary batteries.8–11 These ECs can bridge the power-energy trade-off between batteries (high energy) and traditional dielectric capacitors (high power).12–16Supercapacitors are classified into two categories: electrical double layer capacitors (EDLCs) and pseudocapacitors. The EDLC operates by the non-faradaic separation of charges at the electric double layer, whereas pseudocapacitors are involved with surface-confined faradaic redox reactions. EDLC-type materials such as activated carbon (AC) or carbon nanotubes undergo reversible adsorption and desorption of electrolyte ions at the electrode–electrolyte interface during charging and discharging. This process is solely dependent on the active surface area of the electrodes, and because the charge storage mechanism depends only on the physical reorientation of the ion charge assembly at the interface, the energy density is much lower in EDLCs as compared to pseudocapacitive materials.
The charge storage mechanism of pseudocapacitive materials primarily involves limited surface redox reactions.17–20 Typically, during the process of charging and discharging, there is a change in the redox state of the species involved, and that, in turn, demands intercalation and deintercalation of counter ions.21–23 Therefore, materials that can exhibit multiple redox transitions are often preferred in the development of high-energy-density capacitor devices. The conventional wisdom that multiple redox transitions within the electrode have a direct link with the attainable energy density is affirmed by classical demonstrations of charge storage in electrode materials such as RuO2, NiO, Co3O4, MnO2, and conducting polymers, and they all exhibited a very high charge storage capability in their available potential window.24–28
We show that in the process of developing high-energy supercapacitors, redox activity is not quintessential, at least in the domain of molecular systems. Even functionality without any capability for faradaic redox transitions can indeed enrich their energy storage capability. We investigated molecular systems containing functional units such as –COOH/NH2 that do not exhibit any redox activity in the available potential window, and an increase in the charge storage capability by nearly four times was revealed, with impressive performance metrics compared to non-functionalized ligands. Various physicochemical analyses affirmed that it is the proton charge assembly on the electrode that contributes to the identification of repulsive interactions of the anions, leading to dense anion storage within the EDL.
2. Results and discussion
We recently explored how a proton charge assembly can modulate the electrocatalytic behavior of Co-based phthalocyanines via the modulation of the surface charge of the molecules.29 We anticipated that this type of proton charge assembly on the molecule should increase the local density of anions residing in the electric double layer, which in turn should ensure that these molecules are a suitable platform for charge storage applications. Specifically, in this work, we show how ligand functionalization combined with non-redox active units such as –COOH/NH2 results in the metal phthalocyanine molecule becoming a potential platform for supercapacitive charge storage with enhanced energy and power capability.To demonstrate this, we initially used cobalt(II) phthalocyanine (CoPc) and tetracarboxycobalt(II) phthalocyanine (TCCoPc) molecules. It should be noted that –COOH functionality cannot result in any redox activity in the available potential window in aqueous medium. For a deeper analysis, we also used tetraaminocobalt(II) phthalocyanine (TACoPc), tetranitrocobalt(II) phthalocyanine (TNCoPc), copper(II) phthalocyanine (CuPc), and tetracarboxycopper(II) phthalocyanine (TCCuPc) molecules. We synthesized and characterized these molecules (please refer to the ESI† for details), and the structures of the unsubstituted and carboxy-substituted molecules are shown in Fig. 1a.
 | ||
| Fig. 1 (a) Molecular structures of unsubstituted metal phthalocyanine (MPc) and tetra-carboxy-substituted metal phthalocyanine (TCMPc), where M = Co, Ni, Cu. (b) UV-visible spectra of CoPc and TCCoPc molecules. HRTEM images along with elemental mapping of (c) CoPc and (d) TCCoPc molecules. | ||
Briefly, the matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS) demonstrated that the parent ion peaks were at m/z values of approximately 571 and 746 for CoPc and TCCoPc, respectively, ensuring their successful formation (Fig. S1†). The UV-visible spectra demonstrated the characteristic phthalocyanine bands, such as the B band and Q band, for both molecules (Fig. 1b). A Q-band was found to be redshifted in the case of the TCCoPc molecule as compared to the CoPc molecule, signaling that this band is majorly originating from the ligand.30–35 This was further attributed to comparatively extensive electron delocalization in the case of the TCCoPc molecule. Fourier transform infrared (FTIR) spectroscopy revealed peaks at approximately 1110, 1280, 1411, and 1640 cm−1, corresponding to the υC–H, υC–C, υC–N, and υC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C macrocyclic ring vibration, respectively. Phthalocyanine skeletal vibrations were observed at approximately 724, 872, 913, and 1083 cm−1.30–37 The peak at approximately 1700 cm−1 in TCCoPc corresponds to υCO of the –COOH group, as shown in Fig. S2a.† The Raman spectra of both molecules provided characteristic Raman shifts, as detailed in Fig. S2b and Table S1,† which confirms successful molecular formation. The morphology of the molecules was analyzed with the assistance of high-resolution transmission electron microscopy (HRTEM). The TEM images show a rod-like morphology for the CoPc molecule and a sheet-like morphology for the TCCoPc molecules, as shown in Fig. 1c and d, indicating a difference in the self-assembly of the molecule. The morphology of CoPc was found to be mostly that of a nanowire in nature.38,39 We also observed a similar nanowire morphology for the CoPc system (Fig. 1c). However, for the TCCoPc system, the morphology was found to be sheet-like and aggregated (Fig. 1d). This may be because of the presence of intermolecular H-bonding between the –COOH groups in this molecule. Similarly, a randomly aggregated morphology was previously reported for carboxy-substituted cobalt phthalocyanine.33,36,40,41
C macrocyclic ring vibration, respectively. Phthalocyanine skeletal vibrations were observed at approximately 724, 872, 913, and 1083 cm−1.30–37 The peak at approximately 1700 cm−1 in TCCoPc corresponds to υCO of the –COOH group, as shown in Fig. S2a.† The Raman spectra of both molecules provided characteristic Raman shifts, as detailed in Fig. S2b and Table S1,† which confirms successful molecular formation. The morphology of the molecules was analyzed with the assistance of high-resolution transmission electron microscopy (HRTEM). The TEM images show a rod-like morphology for the CoPc molecule and a sheet-like morphology for the TCCoPc molecules, as shown in Fig. 1c and d, indicating a difference in the self-assembly of the molecule. The morphology of CoPc was found to be mostly that of a nanowire in nature.38,39 We also observed a similar nanowire morphology for the CoPc system (Fig. 1c). However, for the TCCoPc system, the morphology was found to be sheet-like and aggregated (Fig. 1d). This may be because of the presence of intermolecular H-bonding between the –COOH groups in this molecule. Similarly, a randomly aggregated morphology was previously reported for carboxy-substituted cobalt phthalocyanine.33,36,40,41
We carried out elemental mapping to increase our understanding regarding the composition of the molecules. There was a homogenous distribution of carbon, nitrogen, and cobalt for CoPc, whereas TCCoPc demonstrated a uniform distribution of carbon, nitrogen, cobalt, and oxygen, as seen in Fig. 1c and d. 1H NMR of these molecules further confirmed their successful formation, as shown in Fig. S3.† There were 1H NMR peaks at 8.3 ppm, 9.37 ppm, 10.22 ppm, and 12.48 ppm for TCCoPc, which corresponded to the four types of H atoms. The broad peak at 12.48 ppm corresponded to the acidic proton. CoPc demonstrated peaks at chemical shift values of 8.29 ppm and 9.47 ppm, indicating the presence of only two different types of protons in it, as shown in Fig. S3.† Characterizations of TNCoPc, TACoPc, CuPc, and TCCuPc are given in Fig. S4.†
After successful formation of the molecules, we evaluated their electrochemical behavior in acidic medium. They were anchored on a glassy carbon electrode as a monolayer via a self-assembly process, and to ensure that a monolayer properly formed, we carried out quartz crystal microbalance (QCM) studies (details are given in the Experimental section of the ESI†). It was observed that the value of surface coverage (obtained with the assistance of Saurbrey's equation) was in accordance with reported values for a flat monolayer (see Fig. S5† and the associated text in the ESI†).42–44 The surface coverage values were 9.12 × 10−11 mol cm−2 and 8.02 × 10−11 mol cm−2 for the CoPc and TCCoPc molecules, respectively, suggesting that the electrode was modified with a similar number of molecules. Following this, cyclic voltammograms were recorded for the monolayer-modified glassy carbon electrodes in 0.5 M H2SO4 solution. Although there are a similar number of redox active centers in both molecules, upon functionalization with a non-redox active –COOH functionality, the double-layer current was found to be noticeably enhanced, as illustrated in Fig. S6a.† The galvanostatic charge–discharge data (Fig. S6b†) also indicated that there was a much higher charge storage capability for the TCCoPc molecule as compared to the unsubstituted CoPc molecule.
After ensuring their pristine behavior in acidic medium, we produced composites of these molecules with a YP-50 porous carbon support to confirm their application in real-life supercapacitor devices. The composites were synthesized with the assistance of a previously reported procedure (see the Experimental section in the ESI†). They were characterized by various physicochemical techniques to confirm their successful formation. The Raman and FT-IR spectra demonstrated the respective phthalocyanine features, although they were suppressed because of the presence of the carbon material, as shown in Fig. S7.† The FESEM and HRTEM images further show the presence of molecular systems as a part of the composites, as shown in Fig. S8.† To confirm the homogenous distribution of all the elements in the YP-50 composites, we carried out elemental analysis as well as energy dispersive X-ray spectroscopy (EDX) mapping (Fig. S9–S11†), which confirmed the homogenous distribution of the corresponding elements on YP-50 in both composites.
After successfully confirming the formation of the composites, their electrochemical behavior was monitored in 0.5 M H2SO4 solution. We optimised the ratio of YP-50 to phthalocyanine molecule, and a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (YP-50 to phthalocyanine) was found to furnish the highest capacitance. The data for the different weight ratios along with their capacitance are shown in Fig. S12 and Table S2.† The alteration in capacitance with respect to the weight ratio of individual counterparts in the composite electrode was attributed to the interplay between the electronic conductivity of the composite electrode and the availability of phthalocyanine molecules for charge storage. It should be noted that the electronic conductivity of phthalocyanine molecules is lower than that of carbon-based materials.45,46
:1 (YP-50 to phthalocyanine) was found to furnish the highest capacitance. The data for the different weight ratios along with their capacitance are shown in Fig. S12 and Table S2.† The alteration in capacitance with respect to the weight ratio of individual counterparts in the composite electrode was attributed to the interplay between the electronic conductivity of the composite electrode and the availability of phthalocyanine molecules for charge storage. It should be noted that the electronic conductivity of phthalocyanine molecules is lower than that of carbon-based materials.45,46
With a surplus amount of YP-50 (4:1 ratio), the number of phthalocyanine molecules accessible for charge storage decreased, which in turn led to lower capacitance. When the amount of phthalocyanine was increased (as in a 1:1 ratio), the overall capacitance decreased because of a decrease in the electronic conductivity of the composite system. The enhanced capacitance in the 2:1 ratio composite system could be due to the overall enhancement in the electronic conductivity coupled with the sufficient availability of phthalocyanine molecules for charge storage. The double-layer current obtained from the cyclic voltammograms was found to be enhanced nearly 4 times for the TCCoPc molecule as compared to that for the CoPc molecule (Fig. 2a), which is nearly in accordance with the monolayer data, as shown in Fig. S6.† The scan rate dependence study ranging from 5 mV s−1 to 100 mV s−1 with all three systems is shown in Fig. S13.† The calculated specific capacitance (Csp) from the cyclic voltammograms at various scan rates suggests the maintenance of high capacitance by TCCoPc as compared to CoPc at all the studied scan rates, as shown in Fig. S14.†
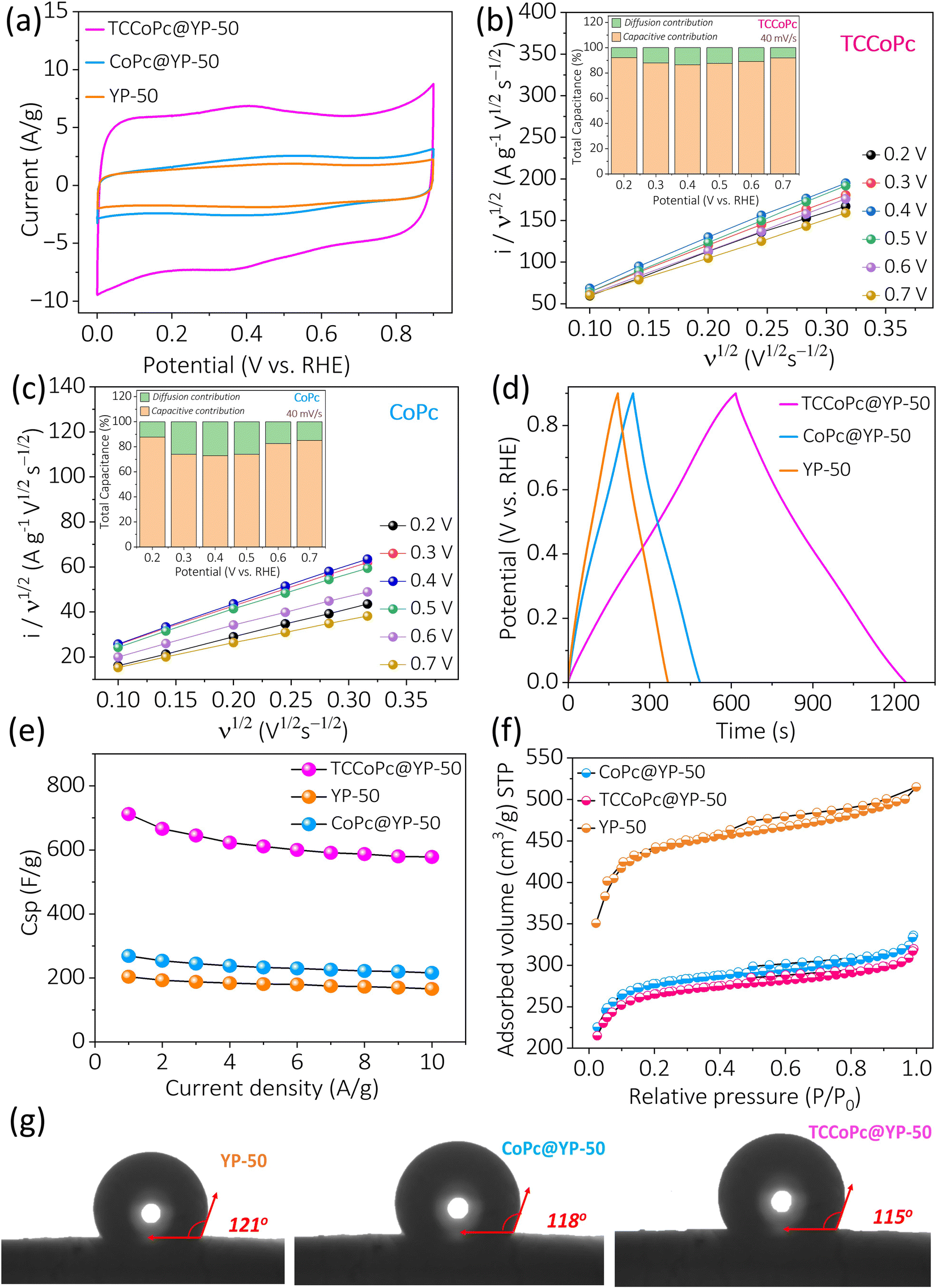 | ||
| Fig. 2 (a) Cyclic voltammograms for the composite electrodes with YP-50 at a scan rate of 5 mV s−1. (b and c) Faradaic and capacitive contributions extracted from the CVs for TCCoPc and CoPc, respectively. The inset shows their percentage contributions at different potentials. (d) Galvanostatic charge–discharge profiles and (e) rate profiles extracted from charge discharge plots of the YP-50, CoPc@YP-50, and TCCoPc@YP-50 electrodes. (f) BET N2 adsorption isotherm along with the (g) contact angle measurement for the YP-50, CoPc@YP-50, and TCCoPc@YP-50 electrodes. | ||
To check the contribution of pseudocapacitance to the overall capacitance, the faradaic and capacitive contributions were extracted from the cyclic voltammograms. Fig. 2b and c show that the capacitive contribution is predominant for the TCCoPc molecule as compared to the CoPc molecule even in the potential range of 0.3–0.5 V where faradaic redox peaks appear for both molecules. Even at low scan rates from 0.5 mV s−1 to 0.1 mV s−1, the capacitive contribution dominated the charge storage as compared to the faradaic contribution in CoPc as well as TCCoPc (Fig. S15†). A galvanostatic charge–discharge (GCD) study with these molecules at various current densities ranging from 1 A g−1 to 10 A g−1 (Fig. S16†) demonstrated that the TCCoPc molecule excels in terms of rate capability as compared to the CoPc molecule (Fig. 2d). The Csp values extracted from these GCD studies were found to be in accordance with scan rate-dependent studies, with TCCoPc maintaining a higher value of capacitance over the CoPc molecule at all the studied rates, as seen in Fig. 2e. Table S3† compiles the specific capacitance of all the systems at 5 mV s−1 and 1 A g−1, and it suggests that functionalization by a non-redox active –COOH functionality greatly enhances the double-layer charge storage.
To rule out the possibility of factors such as wettability and surface area of the composite electrodes (CoPc@YP-50 and TCCoPc@YP-50) contributing to charge storage, these parameters were extracted by Brunauer–Emmett–Teller (BET) surface area and contact angle measurements. The surface area extracted from the N2 adsorption isotherm was found to be 1029 m2 g−1, 1014 m2 g−1, and 1682 m2 g−1 for CoPc@YP-50, TCCoP@YP-50, and YP-50, respectively (Fig. 2f), suggesting that there are nearly similar surface areas for both composite electrodes, and they are lower than that of pure YP-50. This is expected because the pores on YP-50 may become blocked while forming composites with phthalocyanine molecules. Fig. S17† shows the pore size distribution for the composites, which was also in a similar range. The pore volumes for CoPc@YP-50 and TCCoPc@YP-50 were found to be 0.449 cc g−1 and 0.427 cc g−1, respectively, suggesting that these parameters are similar in range for the composite materials. Thus, wettability and surface area cannot be factors responsible for the alterations in the double-layer current, as shown in Fig. 2.
To determine the reason why the TCCoPc molecule exhibited greater charge storage than the CoPc molecule, we carried out UV-visible acid titration, as shown in Fig. 3a. It was observed that after addition of the same concentration of acid into the DMSO solution containing the molecules, there was significant redshifting of the Q-band for the TCCoPc molecule as compared to the CoPc molecule. The redshift in the Q-band is an indication of protonation of the molecule.47–51 Therefore, these results suggest that the extent of protonation was more pronounced for the TCCoPc molecule than for the unsubstituted CoPc molecule. To substantiate this claim and to verify the nature of this surface charge, cyclic voltammograms for these two molecules were recorded in the presence of positively charged [Ru(NH3)6]3+/[Ru(NH3)6]2+ redox probes in acidic medium. With the positively charged redox species in the medium, a relative decrease in current was observed for the TCCoPc molecule, suggesting electrostatic repulsion at play in the interfacial region, as shown in Fig. S18.†
 | ||
| Fig. 3 (a) UV-visible spectra for the CoPc and TCCoPc molecules with and without acid treatment. (b) Zeta potential measurement for the CoPc and TCCoPc molecules in acidic medium. (c and d) XPS analysis of CoPc and TCCoPc with and without acid treatment. (e) Comparative cyclic voltammograms for CoPc, TACoPc, and TCCoPc at a scan rate of 5 mV s−1 in 0.5 M H2SO4 medium. | ||
To further verify this, we performed zeta potential measurements in the presence of acid, and the surface charge of TCCoPc molecule was more positive than that of CoPc (Fig. 3b) in acidic medium. To unambiguously confirm this, XPS spectra of the molecules were recorded (Fig. 3c), and they showed a significantly upshifted O 1s binding energy (BE) in the TCCoPc molecule upon acid treatment. The N 1s spectra for both molecules demonstrated an upshift in BE upon acid treatment. However, the shift was much more pronounced in the TCCoPc molecule than the unsubstituted molecule, as shown in Fig. 3d.52–57 It should be noted that the peak intensity of –CN noticeably increased after protonation, as shown in Fig. 3c. Generally, the peak intensity in the XPS spectra is related to the surface concentration of the species in question, and it provides information regarding how much of that species is exposed at the surface. There are studies where the increase in intensity of a certain peak in the XPS spectra was attributed to the higher surface exposure of that species.58–60 Thus, the increase in the peak intensity of CN after protonation was attributed to its dominant surface exposure after the proton charge assembly. This behaviour was consistently observed in CoPc as well as TCCoPc molecules. Nevertheless, no variation in intensity was observed for the Co–N peak because the N is deeply embedded in the macrocyclic ligand.
To further prove that the proton charge assembly contributes to enhanced charge storage, we evaluated the performance of CoPc substituted with functional units such as –NH2, which can also undergo protonation without exhibiting any redox activity. As expected, the cyclic voltammograms suggested that there was enhanced charge storage for tetraamino-substituted CoPc (TACoPc) as compared to unsubstituted CoPc, with the former molecule exhibiting a charge storage value similar to that of the TCCoPc molecule, as seen in Fig. 3e. The pH of the electrolyte (0.5 M H2SO4) was measured to be 0.2. The pKa value of aromatic Ph-COOH is approximately 4.2, and that for Ph-NH2 is approximately 4.6.61–63 According to the Henderson–Hasselbalch equation, it is known that if the pKa of a compound is higher than the pH of the solution where it is present, the compound will exist in its protonated form.64 Therefore, because the pKa values of the amino substituent and carboxy substituent are greater than the pH of the 0.5 M H2SO4 medium (at nearly 0.2), both functional units should undergo proton charge assembly in this medium. Furthermore, a nitro functionality that may not undergo protonation such as that by TACoPc/TCCoPc demonstrated similar double-layer features, such as an unsubstituted CoPc molecule, as seen in Fig. S19.† Upon changing the central metal ion from Co to Cu, a similar disparity in double-layer features was evidently present between the –COOH functionalized ligand and the unsubstituted ligand, which in turn indicates that this enhanced double-layer charge storage definitely stems from the ligand, as shown in Fig. S20 and Table S4.†
| Co3+TAPc−2 + e− ⇄ Co2+TAPc−2 | (1) |
| Co3+TAPc−1 + e− ⇄ Co3+TAPc−2 | (2) |
![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CN– + 2e− + 2H+ ⇄ CH–NH– CN– + 2e− + 2H+ ⇄ CH–NH– | (3) |
All these data indicate that the proton charge assembly in the ligand, upon functionalization with non-redox active functional units such as –COOH and –NH2, is extensive when compared to the unsubstituted ligand. This in turn results in former ligands without any additional redox active functionality becoming potential platforms for charge-storage applications. Based on these, a schematic representation for the proton charge assembly in CoPc and TCCoPc is provided in Scheme S1.† It is assumed that in an ideal situation, TCCoPc can hold up to 8 protons, which is twice that on unsubstituted CoPc (with 4 protons), as seen in Scheme S1.† It should be noted that the redox peaks in the cyclic voltammetry (CV) curve of TACoPc (Fig. 3e) correspond to the faradaic reactions of the metal centre and phthalocyanine ligand, as shown in eqn (1)–(3):
The redox peak at a lower potential range is attributed to eqn (1) and (3), whereas the redox peak at a higher potential is attributed to eqn (2).65,66 However, for TCCoPc, only a broad redox peak pair was observed, as shown in Fig. 3e, which was attributed to the merging of redox peaks corresponding to eqn (1)–(3). Although the reason for this behaviour between TCCoPc and TACoPc is unclear at the moment, it is likely that it is related to the nature of the intermolecular interactions in the TACoPc and TCCoPc molecules.
To specifically determine the contribution of the proton charge assembly to pseudocapacitance, we compared the faradaic contributions for TCCoPc as well as CoPc in the potential range from 0.3 V to 0.5 V, where a metal–nitrogen redox transition was observed, as shown in Fig. 2a. Fig. S21† shows that at 0.4 V, the pseudocapacitance was similar in magnitude for the TCCoPc and CoPc molecules. The capacitive and diffusional contributions further suggest that the non-faradaic contributions in the voltage range of 0.3 V to 0.5 V are noticeably enriched in TCCoPc as compared to CoPc, as shown in Fig. S21.† All these indicate that the proton charge assembly does not greatly influence the faradaic contribution. However, it greatly contributes to non-faradaic capacitance.
To demonstrate a practical supercapacitor device, we assembled a two-electrode asymmetric device using composite electrodes and activated carbon (AC) as the positive and negative electrodes, respectively. The two-electrode fabrication procedure is explained in the Experimental section of the ESI.† CV and galvanostatic charge–discharge measurements (Fig. 4a and b) using the two-electrode configuration suggest an enhanced charge storage in the TCCoPc system as compared to unsubstituted CoPc, as demonstrated in the case of the three-electrode systems (Fig. 2). We carried out rate capability and cycling tests with the TCCoPc/AC two-electrode device because it is the system that provided the most optimal performance. Specific capacity and GCD measurements demonstrated the excellent charge storage capability of the TCCoPc/AC device, as shown in Fig. S22.†
 | ||
| Fig. 4 (a) Cyclic voltammogram of a two-electrode split cell in asymmetric configuration, with TCCoPc and CoPc as the (+) electrodes and activated carbon as the (−) electrodes at a scan rate of 5 mV s−1. The current was normalized with respect to the total mass of the materials in the (+) and (−) electrodes. (b) Galvanostatic charge–discharge profiles of the device. (c) Capacity retention plot for the TCCoPc/AC split cell with increasing C rates. The rate was lowered to 8C after cycling it to a high rate of 900C. (d) Specific capacitance and coulombic efficiency of the split cell with the TCCoPc/AC system over 10000 cycles at a rate of 257C (20 A g−1). (e) Ragone plots for the asymmetric two-electrode split cell. (f) Plot of the imaginary part of capacitance (C′′) vs. frequency acquired in the frequency range of 100 kHz to 5 mHz, with a 10 mV (peak to peak) AC excitation signal at the open circuit voltage (OCV). (g) Photographs showing the architectural components of the supercapacitor along with the assembled device powering a 2 V LED. | ||
The rate capability with the TCCoPc/AC device was stable even at a very high current rating of 900C (50 A g−1), while maintaining a capacity of 270 F g−1. The device demonstrated a swift return to nearly its original performance when the current rating was lowered to 8C, without compromising its initial capacity, as shown in Fig. 4c. For the TCCoPc molecule, a stable profile and a capacity retention of nearly 99.5% even after 10000 cycles was observed during long-term cycling at a current rate of 20 A g−1, as seen in Fig. 4d. The Ragone plot in Fig. 4e suggests that functionalization by a non-redox active unit such as the –COOH group can dramatically influence the charge storage by enhancing the energy density without compromising the power capability.
Electrochemical impedance spectroscopy (EIS) with the composite electrodes (in a three-electrode configuration) and the corresponding Bode plot revealed a high-frequency resistance to capacitive switching in TCCoPc@YP-50F; however, this switching occurred at a lower frequency in CoPc@YP-50F (Fig. S23†). These further reflect the superior supercapacitor characteristics of TCCoPc versus the CoPc molecule. From the frequency corresponding to the maximum of the imaginary part of the capacitance (C′′) in the C′′ versus frequency plot, which generally reflects the transition boundary between purely capacitive and purely resistive behavior,18,67–71 the knee frequency (fo) reflecting the power capability of a supercapacitor was extracted, as seen in Fig. S23.† The fo was highest for the TCCoPc (49 Hz) molecule as compared to unsubstituted CoPc (10 Hz) and YP-50 (3 Hz), suggesting the higher power capability of the former molecule. The relaxation time, which is the inverse of the knee frequency (τ0 = 1/fo) and indicates the minimum time required to fully discharge the capacitor, was shorter for the TCCoPc molecule as compared to the CoPc molecule, as shown in Fig. 4f and Table S5.†
The TCCoPc system as compared to the unsubstituted system demonstrated remarkably decreased self-discharge rates and leakage currents, as shown in Fig. S24 and Table S6.† Practical viability of the device was demonstrated by powering a 2 V LED using a series of configurations of the asymmetric capacitor, as shown in Video 1 (ESI†) and Fig. 4g. To position the work with respect to the literature, we have compared the results obtained with molecules containing non-redox active functional units with various phthalocyanine molecule-based supercapacitors. As shown in Table S7,† the performance metrics of molecule-based supercapacitors can be noticeably enhanced with a non-redox active functionality that can undergo a non-faradaic proton charge assembly. The approach detailed here shows that the EDL structure can be altered by a non-redox active functionality via a non-faradaic proton charge assembly that in turn can be exploited for designing ligands for potential molecule-based supercapacitor devices.
3. Conclusions
We demonstrated how supercapacitive performance can be enhanced with the assistance of a functional unit that cannot undergo any redox activity. We showed how a non-redox active substituent can alter the population of anions in an electric double layer (EDL) via a proton charge assembly over the molecule. It was observed that the double-layer charge storage was enhanced to similar magnitudes for carboxylic-substituted as well as amine-substituted molecules as compared to the unsubstituted ligand. However, for the nitro-substituted molecule, the double-layer storage was not increased compared to the unsubstituted ligand.Various physicochemical analyses indicated that the –COOH and –NH2 groups can undergo protonation in acidic medium, whereas the –NO2 group cannot. This in turn leads to a higher local density of anions in the EDL that will enhance the energy density of the supercapacitor device without compromising the power capability. This concept of a non-redox active functional unit contributing to supercapacitive charge storage via a non-electrochemical proton charge assembly can be harnessed for designing ligands for charge storage applications.
Data availability
Experimental data are available from the corresponding author upon reasonable request.Author contributions
S. M. performed characterization of the molecules, electrochemical experiments, and supercapacitor performance measurements. A. R. K. assisted in data analysis. M. C. D. recorded the video and synthesized the molecules along with H. M. N. K. R. M. M. assisted in data curation. C. P. V. assisted with the XPS analysis. M. O. T. supervised the work and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. O. T. is indebted to DST-SERB (CRG/2020/002549) and DST-WTI (DST/TMD-EWO/WTI/2K19/EWFH/2019/272) for financial support. S. M. thanks IISER Pune for their financial support.Notes and references
- Y. Zhu, J. Deng and O. Fontaine, Nat. Energy, 2023, 8, 643–644 CrossRef.
- X. Wang, C. Zhang, M. Sawczyk, J. Sun, Q. Yuan, F. Chen, T. C. Mendes, P. C. Howlett, C. Fu, Y. Wang, X. Tan, D. J. Searles, P. Král, C. J. Hawker, A. K. Whittaker and M. Forsyth, Nat. Mater., 2022, 21, 1057–1066 CrossRef CAS.
- S. F. Linnell, E. J. Kim, Y. S. Choi, M. Hirsbrunner, S. Imada, A. Pramanik, A. F. Cuesta, D. N. Miller, E. Fusco, B. E. Bode, J. T. S. Irvine, L. C. Duda, D. O. Scanlon and A. R. Armstrong, J. Mater. Chem. A, 2022, 10, 9941–9953 RSC.
- N. Mahne, B. Schafzahl, C. Leypold, M. Leypold, S. Grumm, A. Leitgeb, G. A. Strohmeier, M. Wilkening, O. Fontaine, D. Kramer, C. Slugovc, S. M. Borisov and S. A. Freunberger, Nat. Energy, 2017, 2, 17036 CrossRef CAS.
- S. Nikman, D. Zhao, V. Gonzalez-Perez, H. E. Hoster and S. F. L. Mertens, Electrochim. Acta, 2021, 386, 138373 CrossRef CAS.
- J. M. Becker, A. Lielpetere, J. Szczesny, J. R. C. Junqueira, P. Rodríguez-Maciá, J. A. Birrell, F. Conzuelo and W. Schuhmann, ACS Appl. Mater. Interfaces, 2022, 14, 46421–46426 CrossRef CAS PubMed.
- G. S. Phun, R. Bhide and S. Ardo, Energy Environ. Sci., 2023, 16, 4593–4611 RSC.
- J. Bisquert, Phys. Rev. Appl., 2023, 20, 44022 CrossRef CAS.
- A. H. Faqeeh and M. D. Symes, Electrochim. Acta, 2023, 444, 142030 CrossRef CAS.
- J. Lei, Y. Yao, Y. Huang and Y. C. Lu, ACS Energy Lett., 2023, 8, 429–435 CrossRef CAS.
- M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P.-L. Taberna, C. P. Grey, B. Dunn and P. Simon, Nat. Energy, 2016, 1, 16070 CrossRef CAS.
- P. Lannelongue, R. Bouchal, E. Mourad, C. Bodin, M. Olarte, S. le Vot, F. Favier and O. Fontaine, J. Electrochem. Soc., 2018, 165, A657–A663 CrossRef CAS.
- J. Oni and K. I. Ozoemena, J. Porphyrins Phthalocyanines, 2012, 16, 754–760 CrossRef CAS.
- T. Li, Y. Wang, Z. Yin, J. Li, X. Peng and M. Zeng, Chem. Sci., 2022, 13, 10786–10791 RSC.
- E. Mourad, L. Coustan, P. Lannelongue, D. Zigah, A. Mehdi, A. Vioux, S. A. Freunberger, F. Favier and O. Fontaine, Nat. Mater., 2017, 16, 446–454 CrossRef CAS.
- J. N. Lekitima, K. I. Ozoemena, C. J. Jafta, N. Kobayashi, Y. Song, D. Tong, S. Chen and M. Oyama, J. Mater. Chem. A, 2013, 1, 2821–2826 RSC.
- J. Yuan, M. Qiu, X. Hu, Y. Liu, G. Zhong, H. Zhan and Z. Wen, ACS Nano, 2022, 16, 14807–14818 CrossRef CAS PubMed.
- A. T. Chidembo, K. I. Ozoemena, B. O. Agboola, V. Gupta, G. G. Wildgoose and R. G. Compton, Energy Environ. Sci., 2010, 3, 228–236 RSC.
- O. Gorduk, M. Gencten, S. Gorduk, M. Sahin and Y. Sahin, J. Energy Storage, 2021, 33, 102049 CrossRef.
- J. J. Chen, J. C. Ye, X. G. Zhang, M. D. Symes, S. C. Fan, D. L. Long, M. Sen Zheng, D. Y. Wu, L. Cronin and Q. F. Dong, Adv. Energy Mater., 2018, 8, 1–6 Search PubMed.
- D. Sarkar, D. Das, S. Das, A. Kumar, S. Patil, K. K. Nanda, D. D. Sarma and A. Shukla, ACS Energy Lett., 2019, 4, 1602–1609 CrossRef CAS.
- G. Åvall, G. A. Ferrero, K. A. Janßen, M. Exner, Y. Son and P. Adelhelm, Adv. Energy Mater., 2023, 2301944, 1–13 Search PubMed.
- S. He, Z. Mo, C. Shuai, W. Liu, R. Yue, G. Liu, H. Pei, Y. Chen, N. Liu and R. Guo, Appl. Surf. Sci., 2022, 577, 151904 CrossRef CAS.
- Q. Z. Zhang, D. Zhang, Z. C. Miao, X. L. Zhang and S. L. Chou, Small, 2018, 14, 1702883 CrossRef.
- F. F. Alharbi, M. S. Waheed, S. Aman, N. Ahmad, H. M. T. Farid and T. A. Mohaymen Taha, Energy Fuels, 2023, 37, 12225–12235 CrossRef CAS.
- W. Zhang, S. Yu, H. Hu, Y. Fei, L. Chen and T. Zhang, Appl. Surf. Sci., 2023, 640, 158285 CrossRef CAS.
- S. Halder, S. Roy, S. Roy and C. Chakraborty, J. Phys. Chem. C, 2023, 127, 18279–18290 CrossRef CAS.
- S. Arun Kumar, I. Sarasamreen, C. Balaji, A. Gowdhaman, R. Ramesh and P. M. Anbarasan, Inorg. Chem. Commun., 2023, 158, 111506 CrossRef CAS.
- S. Mukhopadhyay, A. R. Kottaichamy, P. V. Chame, P. Ghosh, C. P. Vinod, H. Makri Nimbegondi Kotresh, S. C. Kanade and M. O. Thotiyl, J. Phys. Chem. Lett., 2023, 14, 5377–5385 CrossRef CAS PubMed.
- I. Mohammed, M. Nemakal, S. Aralekallu, V. A. Sajjan, T. R. Divakara, M. Palanna, C. P. Keshavananda Prabu and L. K. Sannegowda, J. Electroanal. Chem., 2020, 871, 114292 CrossRef CAS.
- X. Sun, L. Wang and Z. Tan, Catal. Lett., 2015, 145, 1094–1102 CrossRef CAS.
- B. N. Achar, G. M. Fohlen, J. A. Parker and J. Keshavayya, Indian J. Chem., 1988, 27, 411–416 Search PubMed.
- I. S. Hosu, Q. Wang, A. Vasilescu, S. F. Peteu, V. Raditoiu, S. Railian, V. Zaitsev, K. Turcheniuk, Q. Wang, M. Li, R. Boukherroub and S. Szunerits, RSC Adv., 2015, 5, 1474–1484 RSC.
- T. Mugadza and T. Nyokong, J. Colloid Interface Sci., 2011, 354, 437–447 CrossRef CAS.
- M. P. Malathesh, N. Y. P. Kumara, B. S. Jilani, C. D. Mruthyunjayachari and K. R. V. Reddy, Heliyon, 2019, 5, e01946 CrossRef.
- P. T. Mafuwe, M. Moyo, T. Mugadza, M. Shumba and S. Nyoni, J. Solid State Electrochem., 2019, 23, 285–294 CrossRef CAS.
- G. P. Shaposhnikov, V. E. Maizlish and V. P. Kulinich, Russ. J. Gen. Chem., 2005, 75, 1480–1488 CrossRef CAS.
- X. Ji, T. Zou, H. Gong, Q. Wu, Z. Qiao, W. Wu and H. Wang, Cryst. Res. Technol., 2016, 51, 154–159 CrossRef CAS.
- A. Zhang, W. Zhang, J. Lu, G. G. Wallace and J. Chen, Electrochem. Solid-State Lett., 2009, 12, 16–19 CrossRef.
- C. Chen, Z. Ma, S. Zhou, T. Li and X. Sun, Catal. Lett., 2017, 147, 2399–2409 CrossRef CAS.
- A. Kumar, P. K. Prajapati, M. S. Aathira, A. Bansiwal, R. Boukherroub and S. L. Jain, J. Colloid Interface Sci., 2019, 543, 201–213 CrossRef CAS PubMed.
- B. D. Fleming, S. Praporski, A. M. Bond and L. L. Martin, Langmuir, 2008, 24, 323–327 CrossRef CAS PubMed.
- M. P. Somashekarappa, J. Keshavayya and S. Sampath, Pure Appl. Chem., 2002, 74, 1609–1620 CrossRef CAS.
- Z. O. Makinde, M. Louzada, P. Mashazi, T. Nyokong and S. Khene, Appl. Surf. Sci., 2017, 425, 702–712 CrossRef CAS.
- A. Wang, J. Ye, M. G. Humphrey and C. Zhang, Adv. Mater., 2018, 30, 1–9 Search PubMed.
- Y. Chen, S. Qu, W. Shi, Q. Yao and L. Chen, Carbon, 2020, 159, 471–477 CrossRef CAS.
- A. Ogunsipe and T. Nyokong, J. Mol. Struct., 2004, 689, 89–97 CrossRef CAS.
- A. Beeby, S. FitzGerald and C. F. Stanley, Photochem. Photobiol., 2001, 74, 566 CrossRef CAS.
- Y. Yamada, S. Yoshida, T. Honda and S. Fukuzumi, Energy Environ. Sci., 2011, 4, 2822–2825 RSC.
- E. Tokunaga, S. Mori, Y. Sumii and N. Shibata, ACS Omega, 2018, 3, 10912–10917 CrossRef CAS PubMed.
- F. Bächle, C. Maichle-Mössmer and T. Ziegler, Chempluschem, 2019, 84, 1081–1093 CrossRef.
- A. Gładysiak, T. N. Nguyen, S. L. Anderson, P. G. Boyd, R. G. Palgrave, J. Bacsa, B. Smit, M. J. Rosseinsky and K. C. Stylianou, Inorg. Chem., 2018, 57, 1888–1900 CrossRef.
- T. Mthethwa, E. Antunes and T. Nyokong, Dalton Trans., 2014, 43, 8230–8240 RSC.
- J. V Rojas, M. Toro-Gonzalez, M. C. Molina-Higgins and C. E. Castano, Mater. Sci. Eng., B, 2016, 205, 28–35 CrossRef.
- J. A. Quezada-Renteria, C. O. Ania, L. F. Chazaro-Ruiz and J. R. Rangel-Mendez, Carbon, 2019, 149, 722–732 CrossRef CAS.
- S. Sotoma, K. Akagi, S. Hosokawa, R. Igarashi, H. Tochio, Y. Harada and M. Shirakawa, RSC Adv., 2015, 5, 13818–13827 RSC.
- M. Pedrosa, E. S. Da Silva, L. M. Pastrana-Martínez, G. Drazic, P. Falaras, J. L. Faria, J. L. Figueiredo and A. M. T. Silva, J. Colloid Interface Sci., 2020, 567, 243–255 CrossRef CAS.
- K. Juodkazis, J. Juodkazyt≐, V. Jasulaitien≐, A. Lukinskas and B. Šebeka, Electrochem. Commun., 2000, 2, 503–507 CrossRef CAS.
- A. Kumar, A. Ganguly and P. Papakonstantinou, J. Phys.: Condens.Matter, 2012, 24, 235503 CrossRef.
- H. B. Motejadded Emrooz, M. Maleki, A. Rashidi and M. Shokouhimehr, Biomass Convers. Biorefin., 2021, 11, 943–954 CrossRef CAS.
- A. Gomez, J. Mullens and P. Huyskens, J. Phys. Chem., 1972, 76, 4011–4014 CrossRef CAS.
- A. Bryson, J. Am. Chem. Soc., 1960, 82(18), 4858–4862 CrossRef CAS.
- M. Guillaume, E. Botek, B. Champagne, F. Castet and L. Ducasse, Int. J. Quantum Chem., 2002, 90, 1396–1403 CrossRef.
- H. N. Po and N. M. Senozan, J. Chem. Educ., 2001, 78(11), 1499 CrossRef CAS.
- K. P. Madhuri and N. S. John, Appl. Surf. Sci., 2018, 449, 528–536 CrossRef CAS.
- F. Qi, Y. Wang, J. Xu, X. Wang, J. Wang, H. Shan, M. Li and J. Xu, Synth. Met., 2023, 293, 117284 CrossRef CAS.
- T. Purkait, G. Singh, D. Kumar, M. Singh and R. S. Dey, Sci. Rep., 2018, 8, 1–13 CAS.
- M. Shrestha, I. Amatya, K. Wang, B. Zheng, Z. Gu and Q. H. Fan, J. Energy Storage, 2017, 13, 206–210 CrossRef.
- K. Sheng, Y. Sun, C. Li, W. Yuan and G. Shi, Sci. Rep., 2012, 2, 3–7 Search PubMed.
- P. L. Taberna, P. Simon and J. F. Fauvarque, J. Electrochem. Soc., 2003, 150, A292 CrossRef CAS.
- Y. Yoo, M. S. Kim, J. K. Kim, Y. S. Kim and W. Kim, J. Mater. Chem. A, 2016, 4, 5062–5068 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05639b |
| This journal is © The Royal Society of Chemistry 2024 |