DOI:
10.1039/D3SC05627A
(Edge Article)
Chem. Sci., 2024,
15, 10155-10163
Molecular bowls for inclusion complexation of toxic anticancer drug methotrexate†
Received
22nd October 2023
, Accepted 13th May 2024
First published on 28th May 2024
Abstract
We describe the preparation and study of novel cavitands, molecular bowls 16+ and 26+, as good binders of the anticancer drug methotrexate (MTX). Molecular bowls are comprised of a curved tribenzotriquinacene (TBTQ) core conjugated to three macrocyclic pyridinium units at the top. The cavitands are easily accessible via two synthetic steps from hexabromo-tribenzotriquinacene in 25% yield. As amphiphilic molecules, bowls 16+ and 26+ self-associate in water by the nucleation-to-aggregation pathway (NMR). The bowls are preorganized, having a semi-rigid framework comprising a fixed bottom with a wobbling pyridinium rim (VT NMR and MD). Further studies, both experimental (NMR) and computational (DFT and MCMM), suggested that a folded MTX occupies the cavity of bowls wherein it forms π–π, C–H–π, and ion pairing intermolecular contacts but also undergoes desolvation to give stable binary complexes (μM) in water. Moreover, a computational protocol is introduced to identify docking pose(s) of MTX inside molecular bowls from NMR shielding data. Both molecular bowls have shown in vitro biocompatibility with liver and kidney cell lines (MTS assay). As bowl 26+ is the strongest binder of MTX reported to date, we envision it as an excellent candidate for further studies on the way toward developing an antidote capable of removing MTX from overdosed cancer patients.
Introduction
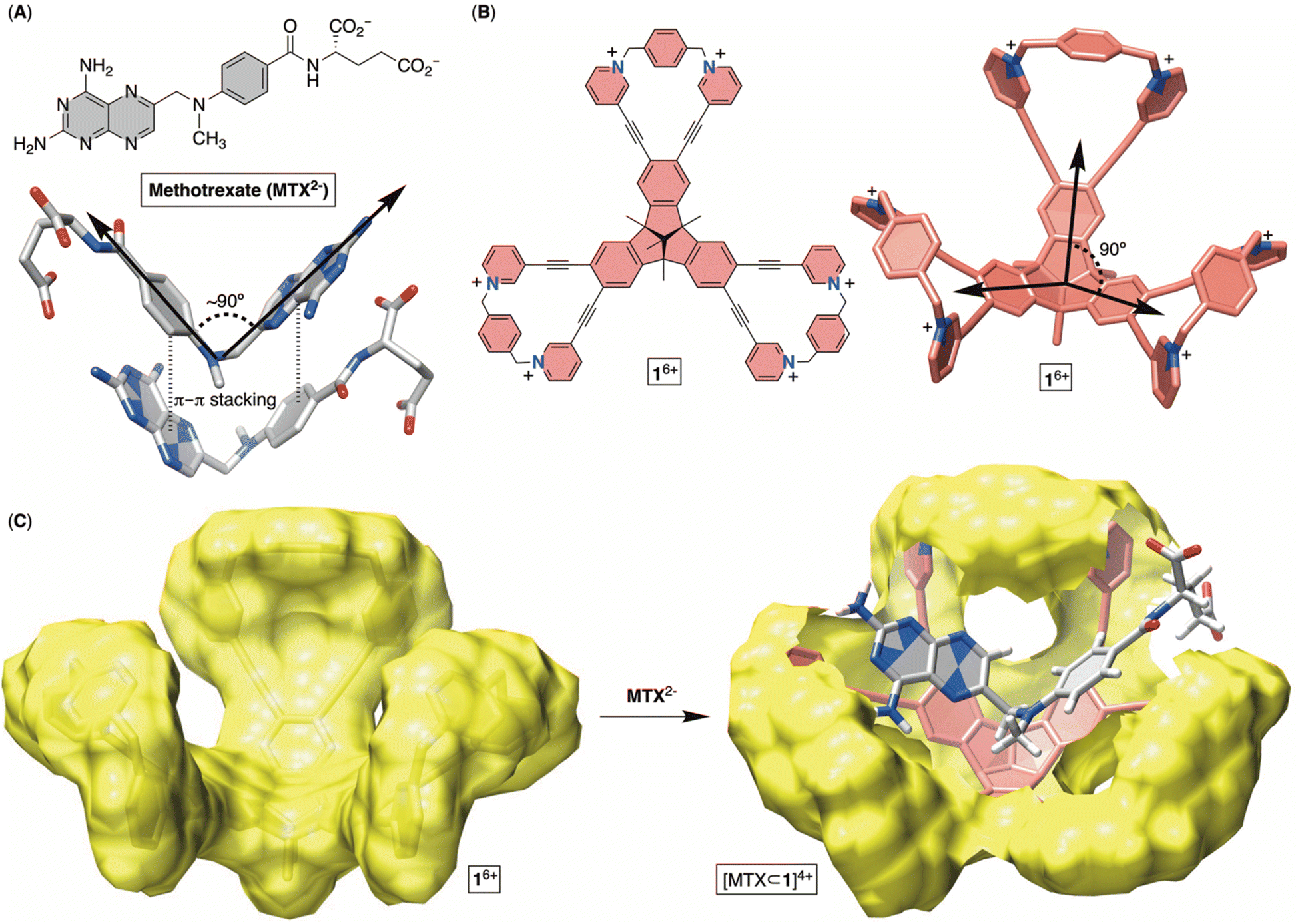
Methotrexate (MTX, Fig. 1A) is one of the world's essential drugs (World Health Organization) that the FDA has approved (in 1953) for treating neoplastic (including breast cancer, lung cancer, acute lymphocytic leukemia, osteosarcoma and non-Hodgkin's lymphoma) and autoimmune (rheumatoid arthritis and psoriasis) diseases.1 Importantly, 2–12% of cancer patients under a high-dose MTX regimen develop acute kidney injury (AKI) leading to myelosuppression, hepatotoxicity, neurotoxicity and eventually multiorgan failure.2 A standard countermeasure includes treatment with leucovorin,3 urine alkalinization and vigorous hydration to flush the molecule from the system. In more severe cases,4 or after chemotherapy medication errors,5 the enzyme glucarpidase (Voraxaze, approved by FDA in 2012), which is capable of rapidly hydrolysing MTX, is recommended.6 However, glucarpidase's distribution is limited to the cardiovascular system, so the enzyme is unable to reach cells across the intercellular matrix, requiring patients to additionally receive leucovorin. Furthermore, the enzyme does not cross the blood–brain barrier (BBB), necessitating its intrathecal injection (i.e., spine) for treating toxic effects (such as encephalopathy, seizure and stroke) in leukemia patients.7 The high cost of glucarpidase is another deficiency, along with its inability to prevent fatal toxicity in 3% of the patients. In this work, we wondered if an alternative therapy for MTX overdose can be developed in the form of an inexpensive, abiotic cavitand capable of including MTX in its cavity for pharmacokinetic drug removal.8 This inspiration comes from sugammadex9 (i.e., a derivative of γ-cyclodextrin), a cavitand that acts as a sequester of the neuromuscular relaxant rocuronium, thereby arresting the action of the relaxant and helping speed up the postoperative recovery of patients. In fact, Bridion (Merck) was approved by the FDA in 2015. Indeed, α-, β- and γ-cyclodextrin derivatives,10 cucurbit[7]uril11 and resorcinarenes12 bind to MTX by including either the pteridine or p-aminobenzoic moiety in their cylindrical cavity. However, the absence of host–guest complementarity, with only a partial desolvation of MTX, has resulted in the formation of complexes with only millimolar stability (Kd ∼ mM), so far.13 Other investigations, specifically X-ray studies of MTX in its free14 and enzyme-bound15 forms, revealed the drug folding its aromatic planes (i.e., pteridine and p-aminobenzoyl ring; Fig. 1A) at a circa 90° angle. Moreover, MTX molecules assemble in the solid-state (Fig. 1A) such that a folded drug molecule is holding onto another via π–π stacking contacts and in an anti-parallel arrangement.14b With this in mind, we wondered: can a tribenzotriquinacene (TBTQ) and bowl-shaped cavitand 16+ (Fig. 1B and C), with circa 90° angle between its fused indanes,16 host MTX with the aromatics folded at ∼90° in aqueous media? First, the electron-rich surface17 of the TBTQ moiety of 16+ (Fig. 1C) is expected to complement the electron-deficient pteridine from MTX. Second, six positively charged pyridinium units at the rim could ion-pair with the negatively charged glutamate of MTX while also enhancing the solubility of 16+ in water. The energy-minimized structure of [MTX⊂1]4+ (Fig. 1C) suggested MTX2− in its folded form occupying the cavitand with both pteridine and p-aminobenzoyl groups forming π–π stacking contacts and glutamate residing between the positively charged pyridinium moieties. While TBTQ cavitands have mostly been explored for complexing fullerenes in organic media,18 there are a handful of recent studies describing their inclusion complexation in water.19
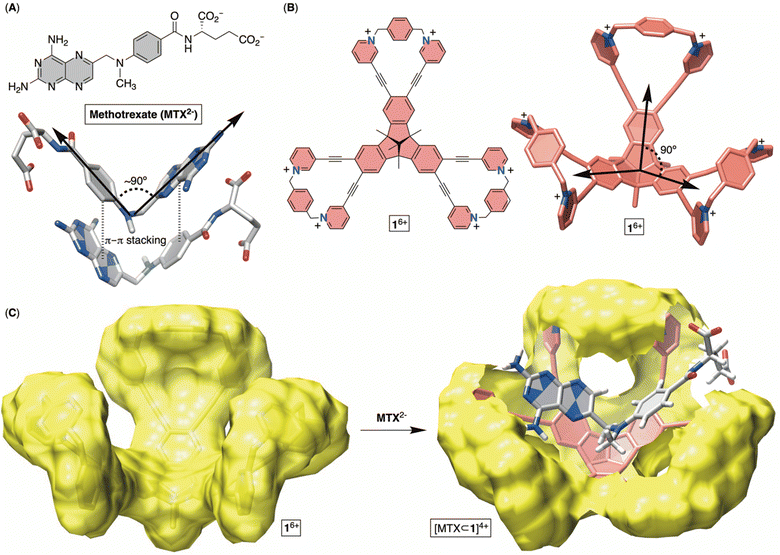 |
| | Fig. 1 (A) Chemical structure of anticancer drug methotrexate (MTX2−) with a stick representation of its folded form in the solid state. (B) Chemical structure of bowl 16+ with its stick representation. (C) Van der Waals surface of bowl 16+ (left) along with an energy-minimized structure of [MTX⊂1]4+ (OPLS3). | |
As for the main objectives of this study, we aimed to develop a facile synthetic method20 for accessing bowl-shaped 16+ in addition to its constitutional isomer 26+ (Fig. 2). Next, we set out to (a) examine conformational dynamics and assembly characteristics of molecular bowls, (b) quantify their affinity for capturing toxic anticancer drug MTX in aqueous media, (c) elucidate docking position(s) of MTX within each bowl and (d) quantify in vitro biocompatibility of these novel hosts.
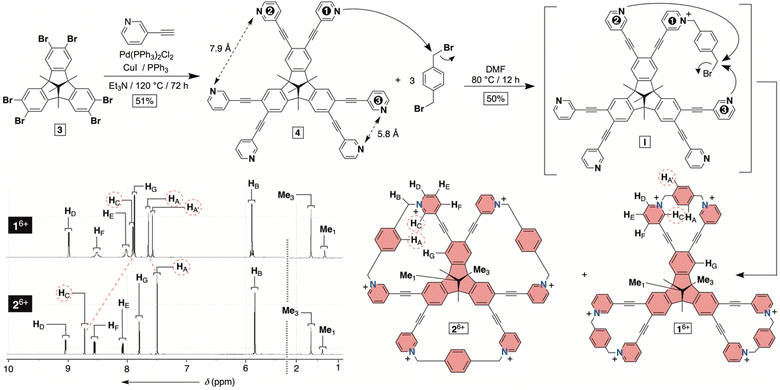 |
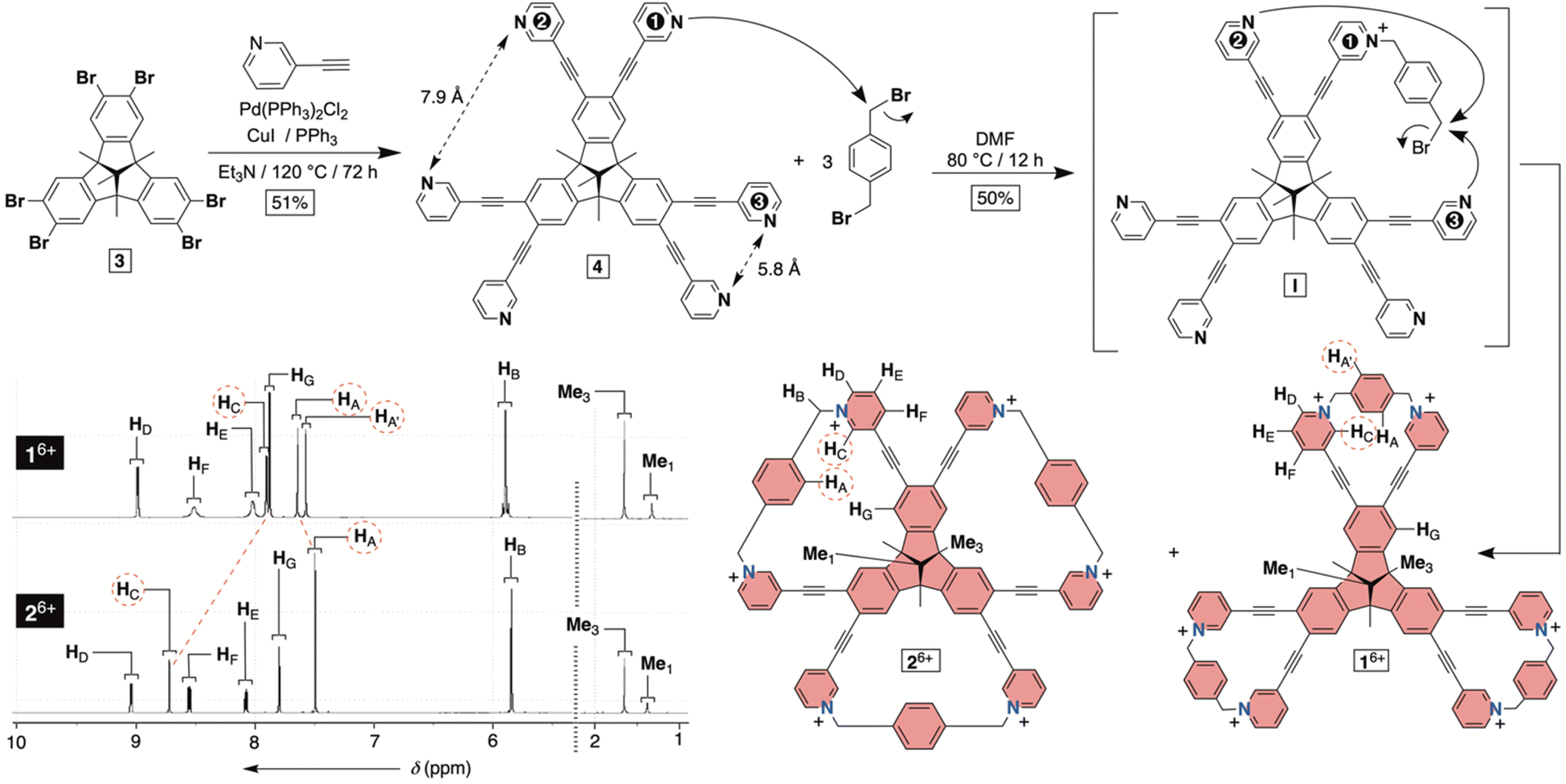
| | Fig. 2 Synthesis of molecular bowls 16+ and 26+ with their 1H NMR spectra (850 MHz, 298 K) in water (30 mM phosphate buffer at pH = 7.4). | |
Results and discussion
Syntheses of molecular bowls 16+ and 26+
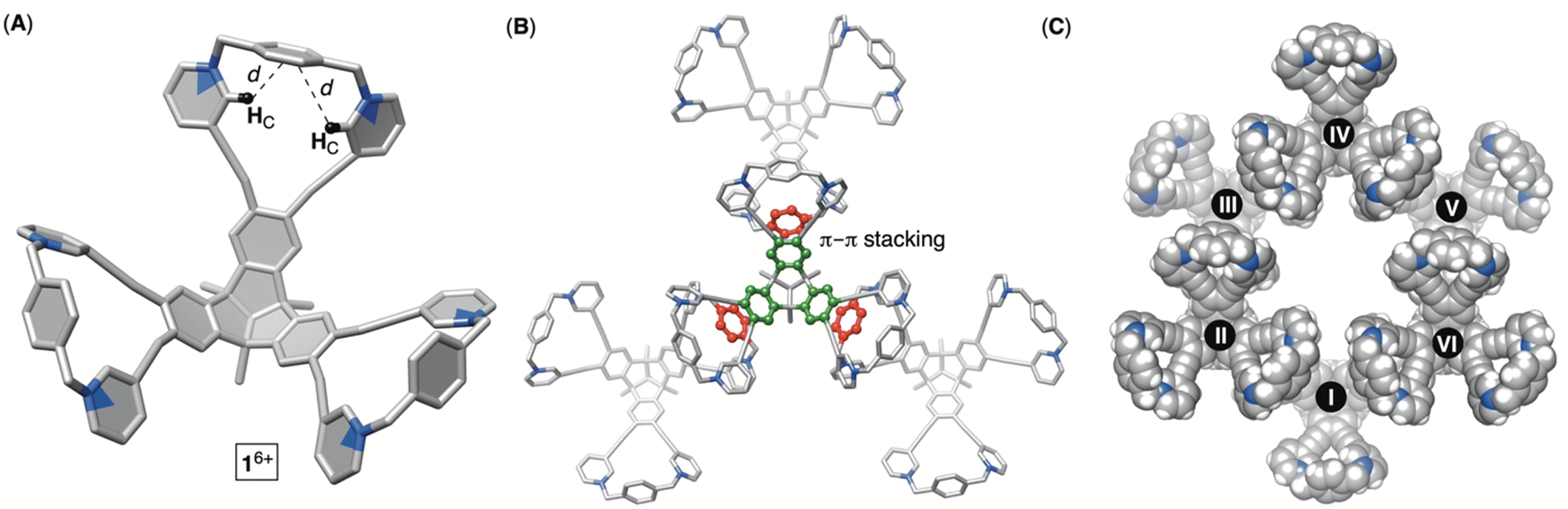
To obtain hexacationic bowl 16+ (Fig. 2), we began with Sonogashira cross-coupling of 3-ethynylpyridine to hexabromo-tribenzotriquinacene 3.21 The process, requiring six consecutive covalent-bond formations, was effective and provided hexakis-pyridine 4 in 51% yield. The alkylation of 4 with an excess of 1,4-dibromoxylene was then conducted in N,N-dimethylformamide (DMF).22 Interestingly, an HPLC chromatogram of the reaction mixture (Fig. S1†) showed the presence of two main products in the ratio of circa 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. After isolation (35 and 15% yields), 1H NMR spectra of each product in water (Fig. 2) revealed a set of signals with the integration and resonance pattern corresponding to 4 alkylated with three 1,4-dibromoxylenes. Moreover, 19F NMR spectra (Fig. S15 and S23†) showed the presence of six trifluoroacetate anions for each C3v symmetric molecule and ESI-MS corroborated their identical molecular weights (Fig. S13 and S21†). To rationalize the data, we reasoned that intermolecular alkylation of pyridine-1 with 1,4-dibromoxylene gives intermediate I (Fig. 2) which is set for intramolecular nucleophilic substitution in two distinct manners. In one case, pyridine-2 may act as a nucleophile to give the 18-membered pyridinium macrocycle on the way to the formation of 16+. Meanwhile, proximal and nucleophilic pyridine-3 (Fig. 2) can compete to give the 23-membered pyridinium macrocycle on the way to the formation of 26+. For energy-minimized 4 (AM1), the closest distance of two adjacent pyridine nitrogens is 5.8 and 7.9 Å (Fig. 2). With 6.6–7.0 Å separation of syn-periplanar pyridinium nitrogens in the solid state of related pyridinium macrocycles,23 we concluded that the formation of both 16+ and 26+ should have taken place (Fig. 2): energy-minimized 16+ and 26+ (OPLS3) have N-to-N gap of 6.6 and 7.2 Å, respectively. In the light of the above analysis, resonances from both 1H NMR spectra were fully assigned using results from 1H–1H NOESY, 13C–1H HMBC and 13C–1H HSQC spectra (Fig. S10–S12 and S18–S20†). Importantly, the more abundant product has two resonances HA/A′ corresponding to the benzene bridging pyridiniums at the rim. With hindered rotation of the benzenes, we assumed that the more shielded HA′ resides on the concave side of the cavitand. 13C NMR spectrum of the same product showed three lines from the same benzene ring (Fig. S9†). On the other hand, 1H NMR spectrum of the minor reaction product (Fig. 2B) showed a single resonance from benzene's HA protons in addition to only one 13C NMR line from carbons carrying HA nuclei (Fig. S17†). With hindered rotation of the benzene about its axis in the main product and free rotation in the minor one, we reasoned that the first molecule ought to be 16+, including the smaller 18-membered pyridinium macrocycles, while the second isomer is 26+ with the larger 23-membered pyridinium macrocycles (see also discussion below). The deduction also makes sense from the standpoint of presumably greater effective molarity24 for the annulation of smaller 18-membered rings within the major product 16+ over the larger 23-membered rings in the minor product 26+. Finally, X-ray diffraction analysis of crystals from the major product confirmed its correspondence to 16+ (Fig. 3). Each pair of pyridiniums in 16+ has its aromatic rings pointing in different directions (Fig. 3A) with a N-to-N distance of 6.6 Å. The three benzene rings that bridge the pyridinium moieties are horizontal with respect to the TBTQ, thereby residing on top of HC (d = 2.5 and 3.5 Å; Fig. 3A). With 1H-NMR spectrum of 16+ showing a magnetic shielding of HC (Fig. 2), we reason that this conformational feature is retained in solution. Furthermore, within the crystal structure, each bowl is surrounded by three other bowls holding onto it via π–π stacking contacts (Fig. 3B). Electron-rich benzene rings from TBTQ are in this way stacked with electron-deficient and pyridinium-bridging benzenes (3.5 Å of centroid-to-centroid distance). Along the crystallographic a/b plane, bowls 16+ organize into honeycomb-like (i.e., hexagonal) prismatic cells25 (Fig. 3C) whereas their vertical stacking (c axis) into columns resembles the packing of coins in a stack.
:1. After isolation (35 and 15% yields), 1H NMR spectra of each product in water (Fig. 2) revealed a set of signals with the integration and resonance pattern corresponding to 4 alkylated with three 1,4-dibromoxylenes. Moreover, 19F NMR spectra (Fig. S15 and S23†) showed the presence of six trifluoroacetate anions for each C3v symmetric molecule and ESI-MS corroborated their identical molecular weights (Fig. S13 and S21†). To rationalize the data, we reasoned that intermolecular alkylation of pyridine-1 with 1,4-dibromoxylene gives intermediate I (Fig. 2) which is set for intramolecular nucleophilic substitution in two distinct manners. In one case, pyridine-2 may act as a nucleophile to give the 18-membered pyridinium macrocycle on the way to the formation of 16+. Meanwhile, proximal and nucleophilic pyridine-3 (Fig. 2) can compete to give the 23-membered pyridinium macrocycle on the way to the formation of 26+. For energy-minimized 4 (AM1), the closest distance of two adjacent pyridine nitrogens is 5.8 and 7.9 Å (Fig. 2). With 6.6–7.0 Å separation of syn-periplanar pyridinium nitrogens in the solid state of related pyridinium macrocycles,23 we concluded that the formation of both 16+ and 26+ should have taken place (Fig. 2): energy-minimized 16+ and 26+ (OPLS3) have N-to-N gap of 6.6 and 7.2 Å, respectively. In the light of the above analysis, resonances from both 1H NMR spectra were fully assigned using results from 1H–1H NOESY, 13C–1H HMBC and 13C–1H HSQC spectra (Fig. S10–S12 and S18–S20†). Importantly, the more abundant product has two resonances HA/A′ corresponding to the benzene bridging pyridiniums at the rim. With hindered rotation of the benzenes, we assumed that the more shielded HA′ resides on the concave side of the cavitand. 13C NMR spectrum of the same product showed three lines from the same benzene ring (Fig. S9†). On the other hand, 1H NMR spectrum of the minor reaction product (Fig. 2B) showed a single resonance from benzene's HA protons in addition to only one 13C NMR line from carbons carrying HA nuclei (Fig. S17†). With hindered rotation of the benzene about its axis in the main product and free rotation in the minor one, we reasoned that the first molecule ought to be 16+, including the smaller 18-membered pyridinium macrocycles, while the second isomer is 26+ with the larger 23-membered pyridinium macrocycles (see also discussion below). The deduction also makes sense from the standpoint of presumably greater effective molarity24 for the annulation of smaller 18-membered rings within the major product 16+ over the larger 23-membered rings in the minor product 26+. Finally, X-ray diffraction analysis of crystals from the major product confirmed its correspondence to 16+ (Fig. 3). Each pair of pyridiniums in 16+ has its aromatic rings pointing in different directions (Fig. 3A) with a N-to-N distance of 6.6 Å. The three benzene rings that bridge the pyridinium moieties are horizontal with respect to the TBTQ, thereby residing on top of HC (d = 2.5 and 3.5 Å; Fig. 3A). With 1H-NMR spectrum of 16+ showing a magnetic shielding of HC (Fig. 2), we reason that this conformational feature is retained in solution. Furthermore, within the crystal structure, each bowl is surrounded by three other bowls holding onto it via π–π stacking contacts (Fig. 3B). Electron-rich benzene rings from TBTQ are in this way stacked with electron-deficient and pyridinium-bridging benzenes (3.5 Å of centroid-to-centroid distance). Along the crystallographic a/b plane, bowls 16+ organize into honeycomb-like (i.e., hexagonal) prismatic cells25 (Fig. 3C) whereas their vertical stacking (c axis) into columns resembles the packing of coins in a stack.
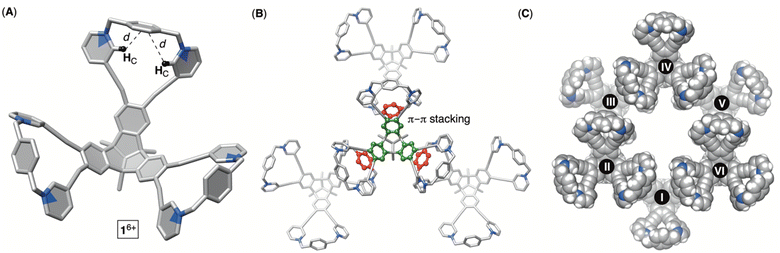 |
| | Fig. 3 Different representations of the X-ray structure of molecular bowl 16+ showing its conformation at the rim (A) and the modes of packing (B and C). | |
Conformational dynamics of bowls 16+ and 26+
Molecular dynamics (MD) simulation of bowl 16+ in a box of explicit water molecules revealed that the cavitand's TBTQ platform stays rigid while the pyridinium rim is conformationally flexible. For 200 ns of the MD simulation time, six methylene groups rocked back (out) and forth (in) thereby pulling pyridiniums to point away or toward the cavitand's inner space (Fig. 4A). For analysing the data, we identified three distinct positions of each pair of methylenes at the rim: cisin, cisout and trans (Fig. 4A). With three juxtaposed arms, there are ten possible triplets of these co-orientations. Interestingly, MD simulations showed a similar statistical and computed distribution of stereoisomers. However, energy-minimization of conformers at a higher level of theory (DFT: B3LYP/6-31+G(d)) showed a distribution in which the trans3 stereoisomer dominates (Fig. 4A). To experimentally probe the methylene rocking, variable-temperature (VT) 1H NMR spectra of 16+ revealed a singlet from diastereotopic HB nuclei (Fig. 4A) in the interval of −5 to 55 °C (CD3OD, Fig. S14†). Supposedly, a conformational in/out motion of the methylene groups at the rim of 16+ is occurring at a rapid rate to account for the observation (see also the discussion below). Additionally, the rotation of nearly horizontal bridging benzenes about their bonding axis was not taking place on the 200 ns time scale of the MD simulations. The result is supported with VT 1H NMR spectra of 16+ (Fig. S14†) showing two singlets from benzene's HA and HA′ over the entire −5 to 55 °C temperature range. To sum up, 16+ is a molecular bowl with a rigid TBTQ bottom and flexible pyridinium moieties at the top.
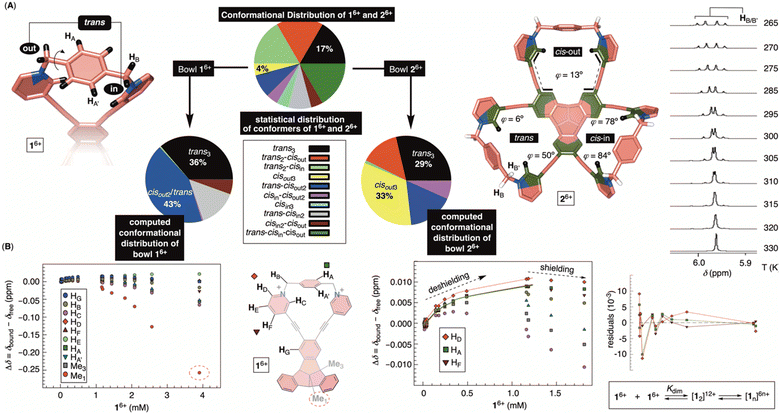 |
| | Fig. 4 (A) Energy-minimized conformers of bowls 16+ (left) and 26+ (right, trans:cisout:cisin) with pie charts showing statistical and computed (DFT: B3LYP/6-31+G(d)) distributions of conformational stereoisomers. A segment of variable-temperature 1H NMR spectra of 26+ in CD3OD showing a change in the shape of the signal from HB as a function of temperature (265–330 K). (B) A change in the chemical shift of protons from 16+ as a function of the bowl's concentration in water (30 mM PBS buffer at pH = 7.4). (Right) Dilution isotherms of HD, HF and HA protons from 16+ fit well to a dimerization model (SigmaPlot) with a random distribution of residuals. | |
As for the bowl 26+, we built its ten distinct conformers using molecular mechanics. The energy minimization in implicit water solvent (DFT: B3LYP/6-31+G(d)) showed stereoisomers having a considerable free-energy difference (Fig. 4A). In particular, the Boltzmann-weighted population showed trans3 and cisout3 states dominating the distribution. We noted a trend for dihedral angles φ (Fig. 4A) characterizing 23-membered rings of 26+. For cisin and cisout torsions, the dihedral angles φ are 80° and 13°, respectively, while the trans state appears to be a combination of the two with φ1 = 50° and φ2 = 6°. As φ denotes the degree of rotation of pyridiniums with respect to benzenes from TBTQ, it follows that by approaching φ = 0°, the extent of π-conjugation across the molecule increases. With smaller torsion angles increasing the stability of 26+, the computed paucity of cisin and the dominance of cisout/trans states makes sense. VT 1H NMR spectra of 26+ in CD3OD (Fig. 4A; Fig. S22†) showed the resonance from methylene HB protons as a singlet at higher and an AB quartet at lower temperatures. For each of the participating and exchanging conformational states (see pie chart in Fig. 4A), diastereotopic HB protons shall give one or more AB quartets that at lower temperature would be expected to give multiple signals. Since only one AB quartet was observed, we sought for an alternative explanation. If methylene groups within 26+ have similar magnetic characteristics, on both the inner or outer sides of the cavitand, then in/out movement of each CH2 will exchange the positions of its two diastereotopic CHBHB′ protons. At lower temperatures (i.e., the slow exchange regime), this will give an AB quartet coalescing into a singlet as the rocking rate increases (i.e., higher temperatures). The rate coefficient characterizing the process is, at the coalescence temperature of 315 K (Fig. 4A), estimated to 103 s−1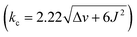 thereby corresponding to ΔG‡ = 15.6 kcal mol−1.26 In addition, VT 1H NMR spectra of 26+ (Fig. S22†) showed one singlet from benzene's HA over the entire temperature range indicating a rapid rotation of benzene about its axis. To sum up, molecular bowl 26+ has, like 16+, a rigid TBTQ platform, albeit with a less dynamic and wobbling rim.
thereby corresponding to ΔG‡ = 15.6 kcal mol−1.26 In addition, VT 1H NMR spectra of 26+ (Fig. S22†) showed one singlet from benzene's HA over the entire temperature range indicating a rapid rotation of benzene about its axis. To sum up, molecular bowl 26+ has, like 16+, a rigid TBTQ platform, albeit with a less dynamic and wobbling rim.
Self-association of bowls 16+ and 26+
Molecular bowls 16+ and 26+ are amphiphilic compounds, each possessing a nonpolar bottom and a positively charged top. With the common observation of concave amphiphiles assembling in aqueous media,27 we decided to obtain 1H NMR spectra of variously concentrated 16+ and 26+ (30 mM PBS buffer at pH = 7.4; Fig. S24 and S26†). As shown in Fig. 4B, proton resonances from 16+ showed a consistent change of their chemical shift as a function of cavitand's concentration indicating self-association. Interestingly, the proton nuclei are first getting deshielded to a small degree, with the chemical-shift change reaching a maximum point. After that, a greater degree of shielding follows with the central methyl (i.e., Me1) at the bottom of 16+ experiencing the largest perturbation. Presumably, the aggregation is characterized with two steps: nucleation in which an intermediate is accumulated (i.e., NMR deshielding) followed by its oligomerization (i.e., NMR shielding).28 NMR DOSY of variously concentrated 16+ (Fig. S25†) revealed a small change in hydrodynamic radius of the cavitand during the nucleation phase while the apparent size increased in the oligomerisation. We deduced that the nucleation could be depicted as dimerization followed by the assembly of [12]12+ into a more complex structure(s) (Fig. 4B). Indeed, the binding isotherm corresponding to nucleation fit well to a dimerization model29 with a random distribution of residuals and Kdim = 1396 ± 118 M−1 (Fig. 4B). Given the available data, it is difficult to elucidate the exact mode of the assembly, yet the observed magnetic deshielding of nuclei for dimer formation is in line with side-to-side stacking of the cavitands; in the solid-state, bowls 16+ are indeed assembling side-to-side via π–π contacts (Fig. 3B). The magnetic shielding of the central methyl points to oligomerization characterized by the bowl-in-bowl type of complexation.30 On the other hand, 1H NMR chemical shifts of 26+ as a function of concentration (Fig. S26†) resemble 16+ with DOSY NMR (Fig. S27†), indicating formation of a dimer in the nucleation phase. Interestingly, bowl 26+ forms a less stable dimer than 16+ with Ka = 299 ± 161 M−1 (Fig. S26†). With the goal of the study centered on quantifying the inclusion complexation of MTX drug by monomeric bowls 16+ and 26+ (see below), we refrained from further examining the nature of hosts' extended aggregates.
Inclusion complexation of methotrexate (MTX)
To probe the inclusion complexation of methotrexate (MTX), we began with the notion that both bowls self-associate in water. Regarding such equilibria, the results from above measurements suggested that the extent of dimerization is small (<6%) when [1]6+ or [2]6+ is less than 90 μM. Based on that but also the observed trend of chemical shifts describing the aggregation of 16+ or 26+ (Fig. 4C), we assumed that self-association equilibria can be neglected at μM concentrations. From the standpoint of MTX2−, the results of 1H NMR dilution measurements (Fig. S28†) were in line31 with the formation of its dimer in water.32 With Ka = 12 M−1, it necessitates a high (i.e., >1 M) concentration of MTX2− to prompt any appreciable self-association. Hence, we reasoned that 1H NMR spectroscopic titration33 of a standard solution of MTX2− to <90 μM solution of 16+ or 26+ shall give results for which only equilibria depicting host–guest interaction(s) would play the major role.
Incremental addition of a standard solution of MTX2− to 16+ caused a steady perturbation of 1H NMR resonances to both host and guest (Fig. 5). Interestingly, most resonances from 16+ underwent a magnetic deshielding first only to be shielded at higher proportions of the drug (Fig. 5A and S29†). With noncovalent bowl-to-drug association comprising two or more steps, we found that the binding isotherm fit well to 1:2 model of complexation (Fig. 5B and S30†). The formation of 2:1 ternary complex comprising two bowls and one drug would fit the data although with a greater covariance and less satisfactory residuals.34 Indeed, ESI mass spectrometry of a mixture of 16+ and MTX2− revealed signals corresponding to ternary [MTX2⊂1]2+ complex (Fig. S31†), in line with binding isotherms from 1H NMR titration. The stabilities of binary [MTX⊂1]4+ and ternary [MTX2⊂1]2+ complexes were, from four independent measurements in the millimolar range, K1 = 4.4 ± 3.1 × 104 M−1 and K2 = 1.9 ± 0.3 × 103 M−1. For the first binding event, the resonance from HC (pink in Fig. 5B) and HG (blue in Fig. 5B) nuclei, lining sides and bottom of the cavitand's inner space, experienced the largest magnetic perturbation. At the same time, all resonances from the drug within binary [MTX⊂1]4+ were magnetically shielded (Fig. 5A; see also Fig. 6A). As originally anticipated, we conclude that [MTX⊂1]4+ is an inclusion complex35 in which methotrexate occupies the cavity of 16+, thereby residing in the diamagnetic shielding region of the host. With the inner space occupied, binary [MTX⊂1]4+ is likely to associate with another MTX2− by holding it its outer and nonpolar surface. Perhaps, a shielding of the host's Me1 and Me3 resonances by the guest in the second binding event (Fig. 5B) provides evidence to such hypothesis. An incremental addition of MTX2− to molecular bowl 26+ resulted in perturbation of 1H NMR resonances from both host and guest (Fig. 5B and S32†). Interestingly, the first binding event was, in this case, characterized by HF (brown in Fig. 5C) and HA (green in Fig. 5C) nuclei experiencing the largest magnetic perturbation, thereby suggesting a somewhat different binding mode from the previous case. Again, ESI mass spectrometry of a mixture of 26+ and MTX2− provided evidence to the formation of ternary [MTX2⊂2]2+ (Fig. S34†) and the binding isotherm fit well to 1:2 model of complexation with K1 = 2.17 ± 0.11 × 105 M−1 and K2 = 1.88 ± 0.01 × 103 M−1 (Fig. 5B and S33†). Importantly, the stability of binary [MTX⊂2]4+ is an order of magnitude greater than [MTX⊂1]4+, thereby approaching μM range (i.e., Kd = 4.6 μM for [MTX⊂2]4+) which does correspond to micromolar concentration of MTX in the blood of actual patients.4 Moreover, the stabilities of ternary [MTX2⊂1]2+ and [MTX2⊂2]2+ are practically the same and consistent with sterically less demanding, face-to-face, noncovalent contacts of binary complexes and methotrexate.
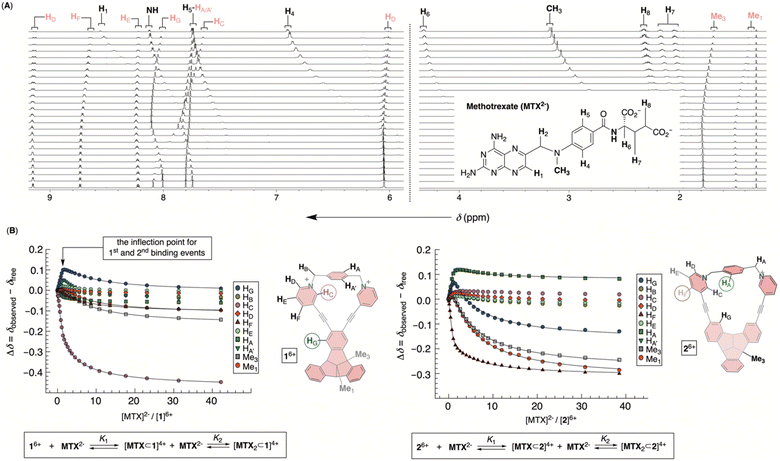 |
| | Fig. 5 (A) Segment of 1H NMR spectra of 47 μM 16+ in water (30 mM PBS buffer at pH = 7.4) obtained after incremental addition of 2.5 mM solution of MTX2−. (B) A relative change of the chemical shift of protons from 16+ (left) or 26+ (right) was fit to 1:2 model describing the formation of binary [MTX⊂1]4+ and ternary [MTX2⊂1]2+ complexes. | |
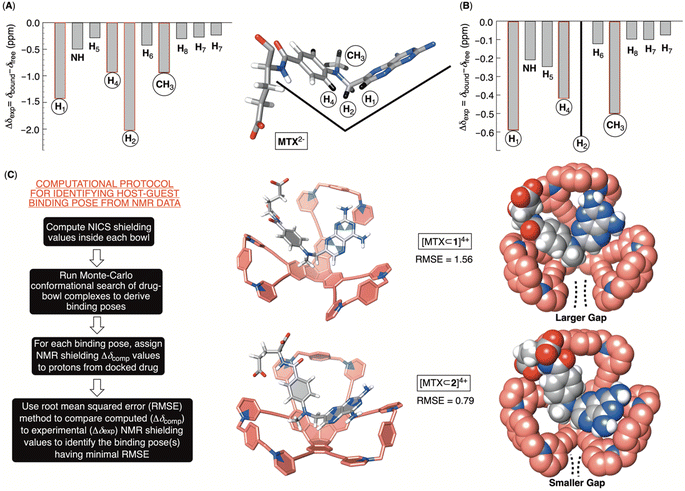 |
| | Fig. 6 Relative 1H NMR spectroscopic perturbation of resonances Δδexp from methotrexate during supramolecular titrations with bowls 16+ (A) or 26+ (B); chemical shift of H2 could not be obtained for titration with 26+. (C) The computational protocol for finding the docking pose of MTX2− inside bowls 16+ and 26+ along with binary [MTX⊂1]4+ and [MTX⊂2]4+ complexes (RMSE 1.56 and 0.79) obtained from the procedure. | |
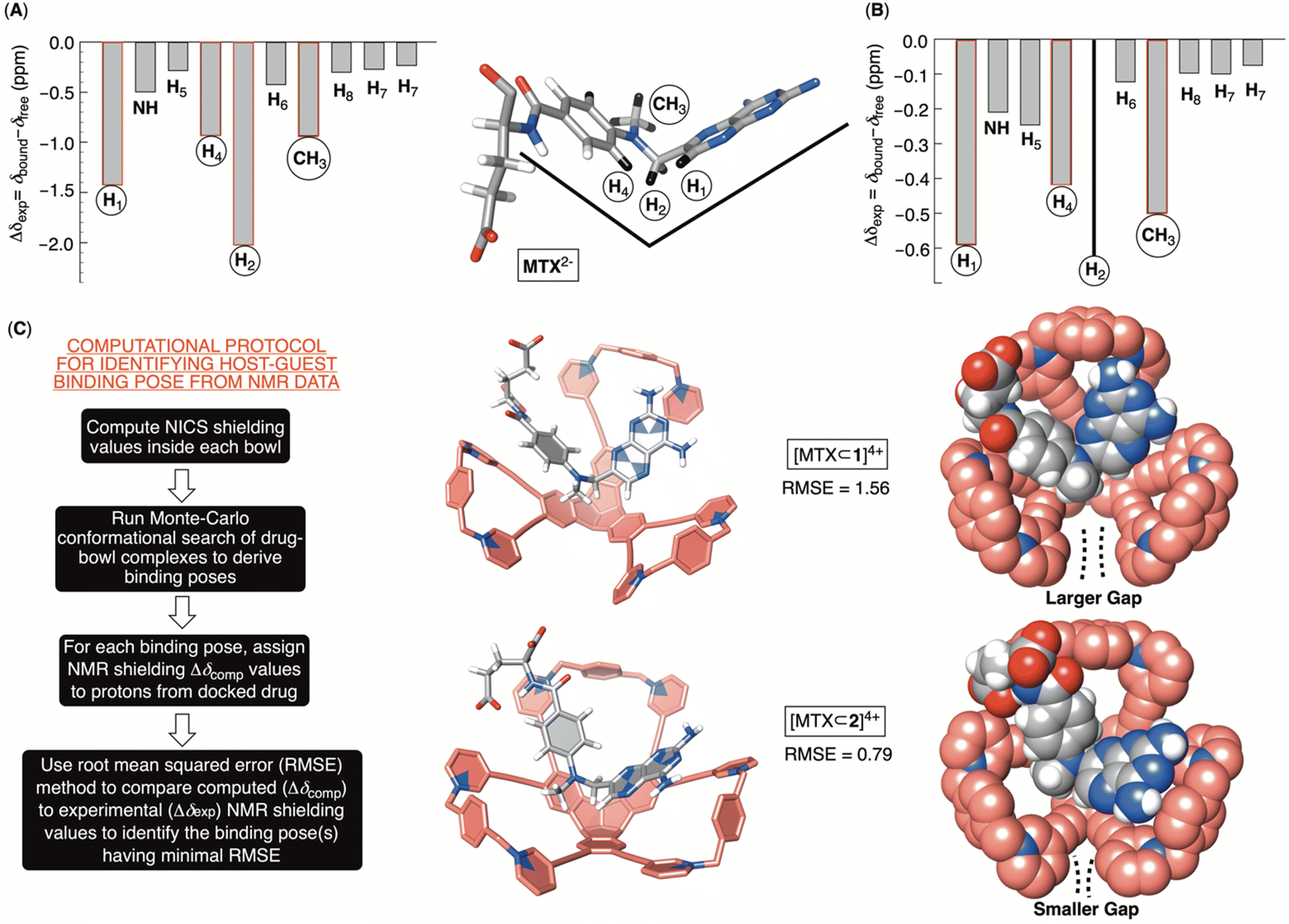
Computational and experimental protocols for elucidating docking pose of MTX2− within 16+ and 26+
The inclusion of MTX2− inside bowls 16+ and 26+ resulted in the greatest degree of diamagnetic shielding of H1, H4, H2 and CH3 from methotrexate (Fig. 6A, B, S29 and S32†). As originally anticipated, the data suggest the drug occupies each cavitand in its folded form (Fig. 6A), thereby allowing H1, H4, H2 and CH3 can reach deep inside the aromatic binding pocked. Towards the elucidation of the drug's binding pose for [MTX⊂1]4+ and [MTX⊂2]4+, we decided to compute nucleus independent chemical shifts (NICS)36 inside each bowl (in the most stable trans3 conformation) and thus map the magnetic environment of their inner space.37 Next, we aimed to run Monte Carlo conformational searches for both [MTX⊂1]4+ and [MTX⊂2]4+ using a force field suitable for organic molecules (OPLS3). By allowing the conformational change of docked MTX2− while freezing the motion of bowls, we hoped to generate a variety of binding poses between guest and host. Finally, we would assign the computed shielding value for each proton of MTX2− docked inside 16+ or 26+ (Δδcomp) by using the calculated NICS map for each host, and then we compared the computed values against the experimental ones (Δδexp; Fig. 6A and B). The pose with the lowest root mean squared error, 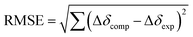 , is assumed to provide the best match with the experiment. After computing the magnetic environment of 16+ and 26+ and running the conformational search of each bowl holding a drug in its cavity, we subjected the data to RMSE analysis. Fascinatingly, the lowest RMSE value was found to correspond to conformers of [MTX⊂1]4+ and [MTX⊂2]4+ (Fig. 6C) having a folded MTX2− facing the concave surface of the bowl to form π–π and C–H–π contacts (Fig. 6C). The glutamate moiety of MTX2− sits between the cavitand's arms to create ion-pair contacts with pyridinium groups. As MTX2− binds to 16+ or 26+ in the similar manner, how do we account for the large stability difference of their binary complexes? By inspecting CPK representations of [MTX⊂1]4+ and [MTX⊂2]4+ in Fig. 6C, one can easily note that bowl 16+ has larger gaps between its macrocyclic arms than 26+. In other words, bowl 26+ is more effectively encircling the space with its concave surface capable of, we posit, more effectively desolvating the MTX guest. Indeed, the folded MTX2− is inserting into [MTX⊂2]4+ such that the pteridine moiety faces the macrocyclic arm while the p-aminobenzoic group covers the gap. In the case of [MTX⊂1]4+ though, both pteridine and p-aminobenzoic group face the gaps between macrocyclic arms of this host. A greater degree of desolvation (i.e., hydrophobic effect) is therefore suggested to contribute to the greater stability of [MTX⊂2]4+ over [MTX⊂1]4+.
, is assumed to provide the best match with the experiment. After computing the magnetic environment of 16+ and 26+ and running the conformational search of each bowl holding a drug in its cavity, we subjected the data to RMSE analysis. Fascinatingly, the lowest RMSE value was found to correspond to conformers of [MTX⊂1]4+ and [MTX⊂2]4+ (Fig. 6C) having a folded MTX2− facing the concave surface of the bowl to form π–π and C–H–π contacts (Fig. 6C). The glutamate moiety of MTX2− sits between the cavitand's arms to create ion-pair contacts with pyridinium groups. As MTX2− binds to 16+ or 26+ in the similar manner, how do we account for the large stability difference of their binary complexes? By inspecting CPK representations of [MTX⊂1]4+ and [MTX⊂2]4+ in Fig. 6C, one can easily note that bowl 16+ has larger gaps between its macrocyclic arms than 26+. In other words, bowl 26+ is more effectively encircling the space with its concave surface capable of, we posit, more effectively desolvating the MTX guest. Indeed, the folded MTX2− is inserting into [MTX⊂2]4+ such that the pteridine moiety faces the macrocyclic arm while the p-aminobenzoic group covers the gap. In the case of [MTX⊂1]4+ though, both pteridine and p-aminobenzoic group face the gaps between macrocyclic arms of this host. A greater degree of desolvation (i.e., hydrophobic effect) is therefore suggested to contribute to the greater stability of [MTX⊂2]4+ over [MTX⊂1]4+.
Cytotoxicity of molecular bowls 16+ and 26+
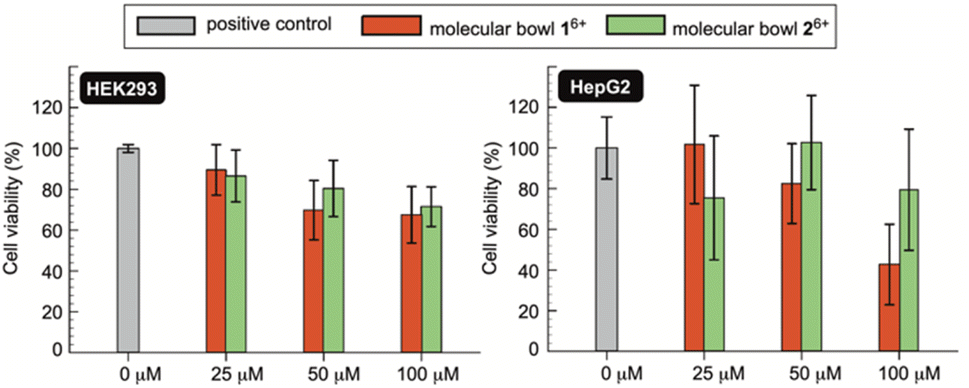
If novel hosts 16+ and 26+, or their future variants, are to act as sequesters of toxic methotrexate (MTX) in blood circulation,4 they need to be innocuous to human cells.38 To examine in vitro biocompatibility of 16+ and 26+, we completed MTS assays39 using human kidney (HEK 293) and human liver cancer (HepG2) cell lines. MTS assay is a quantitative colorimetric method in which NAD(P)H-dependent dehydrogenase enzymes from healthy cells reduce 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) into colored and water-soluble formazan dye.40 By measuring UV-Vis absorbance of the formazan produced by metabolically active cells, cellular viability can be quantified.41 Importantly, as methotrexate is known to cause renal2b (kidney) and hepatotoxicity42 (liver), we decided to probe the effect of molecular bowls 16+ and 26+ on the viability of HEK293 and HepG2 cell lines. In this regard, a positive control was established by incubating wells of untreated cell media in both the HEK293 and HepG2 lines (Fig. 7). When HEK293 cells were incubated in the presence of 25–100 μM of bowls 16+ and 26+, the viability dropped to circa 80% to be in line with both hosts having negligible toxic effect on the kidney cell line. On the other side, bowl 16+ revealed a somewhat greater cytotoxicity toward HepG2 with the viability dropping as a function of its concentration (Fig. 7). Bowl 26+ showed acceptable biocompatibility with the viability fluctuating around 80% (Fig. 7); note that this bowl is the more effective sequester of toxic MTX. With IC50 (half maximal inhibitory concentration)25 of 16+ and 26+ greater than 100 μM (Fig. 7) we conclude that molecular bowls are biocompatible hosts and promising candidates for further examining the sequestration of toxic MTX, for which IC50 = 78 nM.43
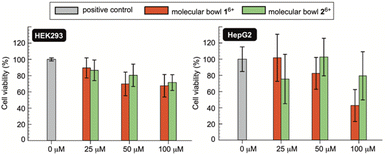 |
| | Fig. 7 The cell viability (%) of HEK293 (left) and HepG2 (right) cells were quantified using MTS assays without (positive control, gray bars) and with molecular bowls 16+ (red bars) and 26+ (green bars) at their respective 25, 50 and 100 μM concentrations. Each measurement was completed three times (48 h incubation time) with values reported as median ± standard deviation. | |
Conclusions
In conclusion, molecular bowls 16+ and 26+ are novel, modular, and accessible hosts. Their curved nonpolar platform with a positively charged and macrocyclic rim makes amphiphilic 16+ and 26+ undergo self-association in water. Moreover, molecular bowls are relatively rigid and preorganized molecules capable of including aromatic methotrexate (MTX): the curved platform of 16+/26+ has three fused indane rings, arranged at 90° angles, that are complementary to anticancer MTX having two aromatic moieties folded at circa 90°. Both experimental and computational studies suggested that the folded drug occupies the cavity of bowls wherein it forms π–π, C–H–π, and ion pairing intermolecular contacts but also undergoes desolvation to give stable binary complexes. In this regard, we described a computational protocol for identifying docking pose(s) of MTX inside molecular bowls that may be applied toward elucidation of binding of other host–guest systems in supramolecular chemistry. Finally, bowl 26+ is, to our knowledge, the strongest binder of MTX reported to date, thus holding promise as a potential sequestering agent capable of removing the drug from overdosed cancer patients. In this vein, both 16+ and 26+ showed minimal adverse effects on the growth of human kidney (HEK 293) and liver cancer (HepG2) cells thereby attesting to the biocompatibility of this class of hosts.
Our next objectives center on tuning the structure of molecular bowls to minimize their self-association but also to permit selective recognition of MTX in the presence of similarly sized and shaped leucovorin and folic acid. This will set the stage for probing the action of this intriguing family of hosts in biological systems.
Data availability
The data that support our findings are available on request from the corresponding author.
Author contributions
Pratik Karmakar synthesized hosts, studied their aggregation and binding to MTX. Tyler J. Finnegan completed computational studies. Darian C. Rostam assisted with synthesis of hosts and study of their biocompatibility. Sagarika Taneja completed biocompatibility studies. Sefa Uçar worked on synthesis of hosts. Alexandar L. Hansen assisted with NMR studies. Curtis E. Moore collected X-ray data and processed them. Christopher M. Hadad assisted with interpretation of computational data. Kornkanya Pratumyot worked on the synthesis of hosts and interpretation of data. Jon R. Parquette directed biocompatibility studies and interpretation of data. Jovica D. Badjić wrote the paper, directed all experimental and computational studies and interpretation of data.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported with funds from the NSF under CHE-2304883. We acknowledge generous computational resources from the Ohio Supercomputer Center. S. U. is grateful for the postdoctoral fellowship received from the Turkish National Science Foundation (TUBITAK BIDEB 2219).
Notes and references
- E. S. L. Chan and B. N. Cronstein, Nat. Rev. Rheumatol., 2010, 6, 175–178 CrossRef CAS PubMed.
-
(a) B. C. Widemann, F. M. Balis, A. R. Kim, M. Boron, N. Jayaprakash, A. Shalabi, M. O'Brien, M. Eby, D. E. Cole, R. F. Murphy, E. Fox, P. Ivy and P. C. Adamson, J. Clin. Oncol., 2010, 28, 3979–3986 CrossRef CAS PubMed;
(b) P. T. Condit, R. E. Chanes and W. Joel, Cancer, 1969, 23, 126–131 CrossRef CAS PubMed.
- W. A. Bleyer, Cancer, 1989, 63, 995–1007 CrossRef CAS PubMed.
- M. A. Rattu, N. Shah, J. M. Lee, Q. Antony and N. Marzella, Pharm. Ther., 2013, 38, 732–744 Search PubMed.
- S. N. Weingart, L. Zhang, M. Sweeney and M. Hassett, Lancet Oncol., 2018, 19, e191–e199 CrossRef PubMed.
- J. M. Green, Ther. Clin. Risk Manage., 2012, 8, 403–413 CrossRef CAS PubMed.
- A. M. Bradley, L. W. Buie, A. Kuykendal and P. M. Voorhees, Clin. Lymphoma, Myeloma Leuk., 2013, 166–170 CrossRef CAS PubMed.
-
(a) C.-L. Deng, S. L. Murkli and L. D. Isaacs, Chem. Soc. Rev., 2020, 49, 7516–7532 RSC;
(b) M. A. Beatty and F. Hof, Chem. Soc. Rev., 2021, 50, 4812–4832 RSC;
(c) N. Bertrand, M. A. Gauthier, C. Bouvet, P. Moreau, A. Petitjean, J.-C. Leroux and J. Leblond, J. Controlled Release, 2011, 155, 200–210 CrossRef CAS PubMed;
(d) H. Yin, X. Zhang, J. Wei, S. Lu, D. Bardelang and R. Wang, Theranostics, 2021, 11, 1513–1526 CrossRef CAS PubMed.
- A. Bom, M. Bradley, K. Cameron, J. K. Clark, J. Van Egmond, H. Feilden, E. J. MacLean, A. W. Muir, R. Palin, D. C. Rees and M.-Q. Zhang, Angew. Chem., Int. Ed., 2002, 41, 265–270 CrossRef CAS.
-
(a) A. Kritskiy, R. Kumeev, T. Volkova, D. Shipilov, N. Kutyasheva, M. Grachev and I. Terekhova, New J. Chem., 2018, 42, 14559–14567 RSC;
(b) A. Aykac, M. C. Martos-Maldonado, J. M. Casas-Solvas, L. Garcia-Fuentes and A. Vargas-Berenguel, J. Drug Delivery Sci. Technol., 2012, 22, 270–272 CrossRef CAS;
(c) F. Pattarino, L. Giovannelli, G. B. Giovenzana, M. Rinaldi and M. Trotta, J. Drug Delivery Sci. Technol., 2005, 15, 465–468 CrossRef CAS.
- Y.-X. Chang, X.-M. Zhang, X.-C. Duan, F. Liu and L.-M. Du, Spectrochim. Acta, Part A, 2017, 183, 131–137 CrossRef CAS PubMed.
- Z. Preisz, Z. Nagymihaly, B. Lemli, L. Kollar and S. Kunsagi-Mate, Int. J. Mol. Sci., 2020, 21, 4345 CrossRef CAS PubMed.
- W. Liu, S. Bobbala, C. L. Stern, J. E. Hornick, Y. Liu, A. E. Enciso, E. A. Scott and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 3165–3173 CrossRef CAS PubMed.
-
(a) T. W. Hambley, H. K. Chan and I. Gonda, J. Am. Chem. Soc., 1986, 108, 2103–2105 CrossRef CAS;
(b) P. A. Sutton, V. Cody and G. D. Smith, J. Am. Chem. Soc., 1986, 108, 4155–4158 CrossRef CAS.
-
(a) J. T. Bolin, D. J. Filna, D. A. Matthews, R. C. Hamlin and J. Kraut, J. Biol. Chem., 1982, 257, 13650–13662 CrossRef CAS PubMed;
(b) D. A. Matthews, J. T. Bolin, J. M. Burridge, D. J. Filman, K. W. Volz, B. T. Kaufman, C. R. Beddell, J. N. Champness, D. K. Stammers and J. Kraut, J. Biol. Chem., 1985, 260, 381–391 CrossRef CAS PubMed.
-
(a) B. P. Benke, T. Kirschbaum, J. Graf, J. H. Gross and M. Mastalerz, Nat. Chem., 2023, 15, 413–423 CrossRef CAS PubMed;
(b) J. Struebe, B. Neumann, H.-G. Stammler and D. Kuck, Chem.–Eur. J., 2009, 15, 2256–2260 CrossRef CAS PubMed;
(c) S. Klotzbach and F. Beuerle, Angew. Chem., Int. Ed., 2015, 54, 10356–10360 CrossRef CAS PubMed.
- T.-Y. Hung, D. Kuck and H.-F. Chow, Chem.–Eur. J., 2023, 29, e202203749 CrossRef CAS PubMed.
-
(a) B. Bredenkotter, S. Henne and D. Volkmer, Chem.–Eur. J., 2007, 13, 9931–9938 CrossRef CAS PubMed;
(b) V. Leonhardt, S. Fimmel, A.-M. Krause and F. Beuerle, Chem. Sci., 2020, 11, 8409–8415 RSC;
(c) P. E. Georghiou, L. N. Dawe, H.-A. Tran, J. Strube, B. Neumann, H.-G. Stammler and D. Kuck, J. Org. Chem., 2008, 73, 9040–9047 CrossRef CAS PubMed;
(d) T. Wang, Z.-Y. Li, A.-L. Xie, X.-J. Yao, X.-P. Cao and D. Kuck, J. Org. Chem., 2011, 76, 3231–3238 CrossRef CAS PubMed;
(e) S. Henne, B. Bredenkoetter, A. A. Dehghan Baghi, R. Schmid and D. Volkmer, Dalton Trans., 2012, 41, 5995–6002 RSC.
-
(a) M.-P. Li, N. Yang and W.-R. Xu, Beilstein J. Org. Chem., 2022, 18, 539–548 CrossRef CAS PubMed;
(b) S.-Y. Liu, X.-R. Wang, M.-P. Li, W.-R. Xu and D. Kuck, Beilstein J. Org. Chem., 2020, 16, 2551–2561 CrossRef CAS PubMed.
-
(a) A. Dhara and F. Beuerle, Synthesis, 2018, 50, 2867–2877 CrossRef CAS;
(b) E. U. Mughal and D. Kuck, Eur. J. Org Chem., 2012, 2012, 3416–3423 CrossRef CAS;
(c) G. Markopoulos, L. Henneicke, J. Shen, Y. Okamoto, P. G. Jones and H. Hopf, Angew. Chem., Int. Ed., 2012, 51, 12884–12887 CrossRef CAS PubMed.
- D. Kuck, A. Schuster, R. A. Krause, J. Tellenbroker, C. P. Exner, M. Penk, H. Bogge and A. Muller, Tetrahedron, 2001, 57, 3587–3613 CrossRef CAS.
- N. Hafezi, J. M. Holcroft, K. J. Hartlieb, E. J. Dale, N. A. Vermeulen, C. L. Stern, A. A. Sarjeant and J. F. Stoddart, Angew. Chem., Int. Ed., 2015, 54, 456–461 CrossRef CAS PubMed.
-
(a) O. Anamimoghadam, J. A. Cooper, M. T. Nguyen, Q.-H. Guo, L. Mosca, I. Roy, J. Sun, C. L. Stern, L. Redfern, O. K. Farha and J. F. Stoddart, Angew. Chem., Int. Ed., 2019, 58, 13778–13783 CrossRef CAS PubMed;
(b) X. Meng, S.-Y. Song, X.-Z. Song, M. Zhu, S.-N. Zhao, L.-L. Wu and H.-J. Zhang, Inorg. Chem. Front., 2014, 1, 757–760 RSC.
- R. Cacciapaglia, S. Di Stefano and L. Mandolini, Acc. Chem. Res., 2004, 37, 113–122 CrossRef CAS PubMed.
- V. W. Liyana Gunawardana, C. Ward, H. Wang, J. H. Holbrook, E. R. Sekera, H. Cui, A. B. Hummon and J. D. Badjic, Angew. Chem., Int. Ed., 2023, 62, e202306722 CrossRef CAS PubMed.
- L. Hu, S. Polen, A. M. Hardin, Y. Pratumyot, C. M. Hadad and J. D. Badjic, Eur. J. Org Chem., 2015, 2015, 6832–6840 CrossRef CAS.
-
(a) S. Chen, Y. Ruan, J. D. Brown, J. Gallucci, V. Maslak, C. M. Hadad and J. D. Badjic, J. Am. Chem. Soc., 2013, 135, 14964–14967 CrossRef CAS PubMed;
(b) T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401–1443 CrossRef CAS PubMed.
- J. E. A. Webb, M. J. Crossley, P. Turner and P. Thordarson, J. Am. Chem. Soc., 2007, 129, 7155–7162 CrossRef CAS PubMed.
-
(a) P. Thordarson, Chem. Soc. Rev., 2012, 2, 239–274 CAS;
(b) H. Xie, L. Zhiquan, R. Z. Pavlovic, J. Gallucci and J. D. Badjic, Chem. Commun., 2019, 55, 5479–5482 RSC.
-
(a) L. Zhiquan, S. Polen, C. M. Hadad, T. V. RajanBabu and J. D. Badjic, J. Am. Chem. Soc., 2016, 138, 8253–8258 CrossRef CAS PubMed;
(b) L. Zhiquan, S. M. Polen, C. M. Hadad, T. V. RajanBabu and J. D. Badjic, Org. Lett., 2017, 19, 4932–4935 CrossRef CAS PubMed.
- M. A. Khaled and C. L. Krumdieck, Biochem. Biophys. Res. Commun., 1985, 130, 1273–1280 CrossRef CAS PubMed.
- M. Poe, J. Biol. Chem., 1973, 248, 7025–7032 CrossRef CAS PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- D. Brynn Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- H.-J. Kim, W. S. Jeon, Y. H. Ko and K. Kim, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5007–5011 CrossRef CAS PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- R. Z. Pavlovic, S. E. Border, T. J. Finnegan, L. Zhiquan, M. J. Gunther, E. Munoz, C. E. Moore, C. M. Hadad and J. D. Badjic, J. Am. Chem. Soc., 2019, 141, 16600–16604 CrossRef CAS PubMed.
- A. T. Brockett, W. Xue, D. King, C.-L. Deng, C. Zhai, M. Shuster, S. Rastogi, V. Briken, M. R. Roesch and L. Isaacs, Chem, 2023, 9, 881–900 CAS.
- G. Malich, B. Markovic and C. Winder, Toxicology, 1997, 124, 179–192 CrossRef CAS PubMed.
- D. D. Dunigan, S. B. Waters and T. C. Owen, BioTechniques, 1995, 19, 640–649 CAS.
- K. M. L. Taylor, J. S. Kim, W. J. Rieter, H. An, W. Lin and W. Lin, J. Am. Chem. Soc., 2008, 130, 2154–2155 CrossRef CAS PubMed.
- R. Conway and J. J. Carey, World J. Hepatol., 2017, 9, 1092–1100 CrossRef PubMed.
- R. E. Norris and P. C. Adamson, Cancer Chemother. Pharmacol., 2010, 65, 1125–1130 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For additional experimental and computational data. CCDC 2288891. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05627a |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Kornkanya
Pratumyot
b,
Jon R.
Parquette
a and
Jovica D.
Badjić
a,
Kornkanya
Pratumyot
b,
Jon R.
Parquette
a and
Jovica D.
Badjić

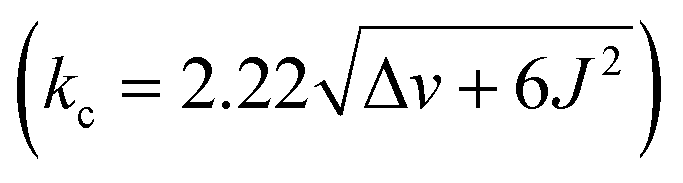 thereby corresponding to ΔG‡ = 15.6 kcal mol−1.26 In addition, VT 1H NMR spectra of 26+ (Fig. S22†) showed one singlet from benzene's HA over the entire temperature range indicating a rapid rotation of benzene about its axis. To sum up, molecular bowl 26+ has, like 16+, a rigid TBTQ platform, albeit with a less dynamic and wobbling rim.
thereby corresponding to ΔG‡ = 15.6 kcal mol−1.26 In addition, VT 1H NMR spectra of 26+ (Fig. S22†) showed one singlet from benzene's HA over the entire temperature range indicating a rapid rotation of benzene about its axis. To sum up, molecular bowl 26+ has, like 16+, a rigid TBTQ platform, albeit with a less dynamic and wobbling rim.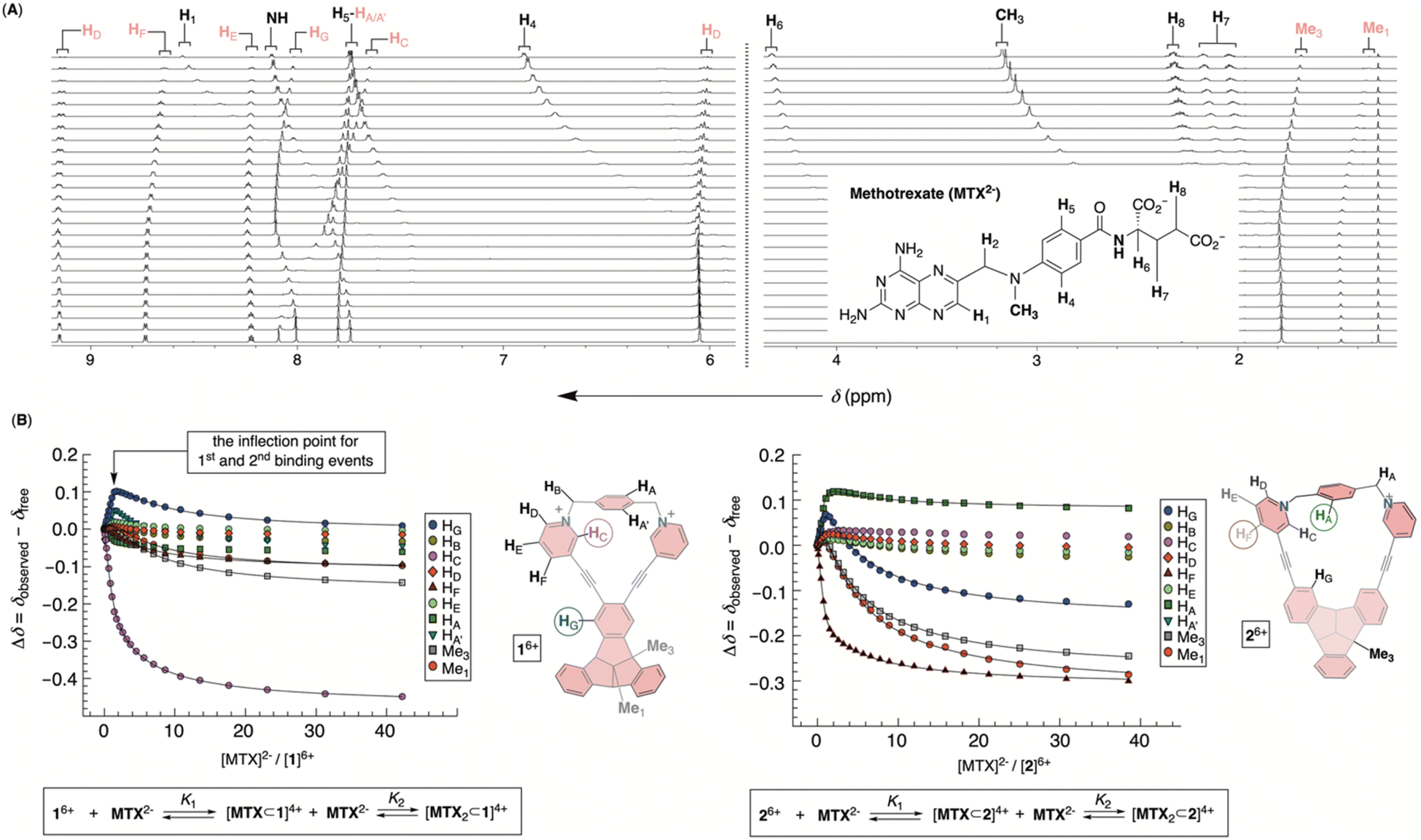
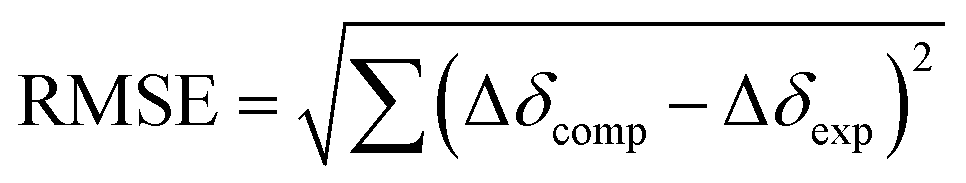 , is assumed to provide the best match with the experiment. After computing the magnetic environment of 16+ and 26+ and running the conformational search of each bowl holding a drug in its cavity, we subjected the data to RMSE analysis. Fascinatingly, the lowest RMSE value was found to correspond to conformers of [MTX⊂1]4+ and [MTX⊂2]4+ (Fig. 6C) having a folded MTX2− facing the concave surface of the bowl to form π–π and C–H–π contacts (Fig. 6C). The glutamate moiety of MTX2− sits between the cavitand's arms to create ion-pair contacts with pyridinium groups. As MTX2− binds to 16+ or 26+ in the similar manner, how do we account for the large stability difference of their binary complexes? By inspecting CPK representations of [MTX⊂1]4+ and [MTX⊂2]4+ in Fig. 6C, one can easily note that bowl 16+ has larger gaps between its macrocyclic arms than 26+. In other words, bowl 26+ is more effectively encircling the space with its concave surface capable of, we posit, more effectively desolvating the MTX guest. Indeed, the folded MTX2− is inserting into [MTX⊂2]4+ such that the pteridine moiety faces the macrocyclic arm while the p-aminobenzoic group covers the gap. In the case of [MTX⊂1]4+ though, both pteridine and p-aminobenzoic group face the gaps between macrocyclic arms of this host. A greater degree of desolvation (i.e., hydrophobic effect) is therefore suggested to contribute to the greater stability of [MTX⊂2]4+ over [MTX⊂1]4+.
, is assumed to provide the best match with the experiment. After computing the magnetic environment of 16+ and 26+ and running the conformational search of each bowl holding a drug in its cavity, we subjected the data to RMSE analysis. Fascinatingly, the lowest RMSE value was found to correspond to conformers of [MTX⊂1]4+ and [MTX⊂2]4+ (Fig. 6C) having a folded MTX2− facing the concave surface of the bowl to form π–π and C–H–π contacts (Fig. 6C). The glutamate moiety of MTX2− sits between the cavitand's arms to create ion-pair contacts with pyridinium groups. As MTX2− binds to 16+ or 26+ in the similar manner, how do we account for the large stability difference of their binary complexes? By inspecting CPK representations of [MTX⊂1]4+ and [MTX⊂2]4+ in Fig. 6C, one can easily note that bowl 16+ has larger gaps between its macrocyclic arms than 26+. In other words, bowl 26+ is more effectively encircling the space with its concave surface capable of, we posit, more effectively desolvating the MTX guest. Indeed, the folded MTX2− is inserting into [MTX⊂2]4+ such that the pteridine moiety faces the macrocyclic arm while the p-aminobenzoic group covers the gap. In the case of [MTX⊂1]4+ though, both pteridine and p-aminobenzoic group face the gaps between macrocyclic arms of this host. A greater degree of desolvation (i.e., hydrophobic effect) is therefore suggested to contribute to the greater stability of [MTX⊂2]4+ over [MTX⊂1]4+.