
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient synthesis of zeolite chabazite using cooperative hydration-mismatched inorganic structure-directing agents†
Adam J.
Mallette
 ,
Gabriel
Espindola
,
Nathan
Varghese
and
Jeffrey D.
Rimer
*
,
Gabriel
Espindola
,
Nathan
Varghese
and
Jeffrey D.
Rimer
*
Department of Chemical and Biomolecular Engineering, University of Houston, 4226 Martin Luther King Boulevard, Houston, TX 77204, USA. E-mail: jrimer@central.uh.edu
First published on 27th November 2023
Abstract
Chabazite (CHA type) zeolite is notoriously difficult to synthesize in the absence of organic structure-directing agents owing to long synthesis times and/or impurity formation. The ability to tailor organic-free syntheses of zeolites is additionally challenging due to the lack of molecular level understanding of zeolite nucleation and growth pathways, particularly the role of inorganic cations. In this study, we reveal that zeolite CHA can be synthesized using six different combinations of inorganic cations, including the first reported seed- and organic-free synthesis without the presence of potassium. We show that lithium, when present in small quantities, is an effective accelerant of CHA crystallization; and that ion pairings can markedly reduce synthesis times and temperatures, while expanding the design space of zeolite CHA formation in comparison to conventional methods utilizing potassium as the sole structure-directing agent. Herein, we posit the effects of cation pairings on zeolite CHA crystallization are related to their hydrated ionic radii. We also emphasize the broader implications for considering the solvated structure and cooperative role of inorganic cations in zeolite synthesis within the context of the reported findings for chabazite.
Introduction
Controlling the synthesis of zeolites is nontrivial owing to their complex growth media, which can obscure efforts to select synthesis parameters a priori to achieve materials with desired physicochemical properties.1–5 These challenges can be partially overcome by using organic structure-directing agents (OSDAs) as cations to modulate cage and/or channel formation, but the economics and environmental impacts of their use make them less desirable. Inorganic structure-directing agents (ISDAs) are frequently used as replacements for (or in combination with) organics; however, they allow for less control over zeolite structure, acid site density (i.e., Si/Al ratio), and crystal morphology.6–9The incorporation of aluminum into zeolite frameworks creates negatively-charged tetrahedral (TO4) sites balanced by extra-framework cations from OSDAs and/or ISDAs. It is well known that inorganic cations (most commonly alkali metals) can markedly alter the thermodynamics and kinetics of zeolite crystallization. Phase diagrams for zeolites that form in the presence of sodium9 or potassium10 reveal that ISDA selection is a critical determinant of the final framework type(s). Other studies have demonstrated an ability to alter the siting and/or pairing of acid sites by varying cation composition or utilizing the charge density mismatch approach.7,11,12 There are also numerous instances where ISDAs can influence the rate of zeolite crystallization or particle size/morphology.13–16 Although these mechanisms are not fully understood, hypotheses have been proposed on the basis of observed ISDA effects on colloidal (precursor) aggregation,17 water structuring around solute species,15 and aluminosilicate speciation18 and stability.19
Efforts to identify causal effects of alkali metal selection on zeolite crystallization are obfuscated by the difficulties associated with relating any outcome to a single variable. Deconvoluting these effects can be difficult owing to the coupled nature of reagents used in zeolite syntheses. For example, ISDAs are typically added in the form of hydroxide salts where the counterion (hydroxide) is the mineralizing agent; therefore, attempts to isolate the effects of alkali ions from solution pH would require the use of an alternative inorganic salt with different counterions that could potentially impact crystallization. Additional challenges associated with studying zeolite synthesis are related to the selection of silica/alumina sources and temperature, which can lead to distinct growth pathways and crystalline products via unknown mechanisms.
A rising trend in zeolite synthesis involves the use of multiple inorganic cations to control crystallization.20–22 The presence of different monovalent and/or divalent metal cations in the synthesis mixture can affect properties including crystallization rate, crystal size, morphology, and phase purity, among others.23–26 Reported cases of organic-free syntheses using multiple inorganic cations are relatively limited; however, prior studies have demonstrated that various cation combinations can accelerate zeolite crystallization.23,24 Examples include the use of two alkali metals, such as Na+ and Cs+, as a means to produce nanocrystalline RHO type zeolite, or the use of Na+ and K+ to tune the relative proportion of faujasite (FAU) and zeolite A (LTA) phases.18,24,25,27,28 Uncommon ISDAs, such as lithium, can also be used in tandem with other inorganic cations to produce zeolites including clinoptilolite (HEU) or ABW27,29 or with organic cations to produce UZM-9 (high-silica LTA).30
This study focuses on the synthesis of chabazite (CHA), which is an industrially-relevant three-dimensional (3D) zeolite. The CHA framework is composed of double-six-membered rings (d6r) and cha cages, which form sinusoidal pore networks. Organic-free syntheses employing K+ as the sole ISDA involve long synthesis times (>14 days), which often necessitates the use of zeolite seeds to accelerate crystallization.23 Recent studies have demonstrated that the binary combination23 of K+ and Sr2+ as well as the ternary combination25,31 of Na+, K+, and Cs+ are alternatives to seed-assisted (organic-free) CHA synthesis. The former leads to fully crystalline K,Sr–CHA in as little as 48 h at 100 °C, while the latter promotes nano-sized CHA (∼60 nm) when heated for 3 h at 90 °C after a period of aging at room temperature for 17 days. Here, we have developed strategies for synthesizing zeolite CHA in rapid timeframes at low synthesis temperature using different binary combinations of inorganic cations, which include K+-free growth media and the use of Li+ as an unexpected accelerant of CHA nucleation.
Results and discussion
Effect of lithium on zeolite CHA crystallization
We investigated the effects of mixed alkali metals on zeolite crystallization beginning with combinations of sodium and lithium as the major and minor ISDA, respectively. Previous studies have established a compositional space to achieve pure Na-zeolites,9 which provided a basis for this work. Several publications32–36 have reported that the presence of lithium as a primary inorganic cation can significantly alter zeolite syntheses, often in ways that inhibit crystallization. Few studies have explored the role of lithium in small quantities as a second (minor) ISDA, which we have done here using sodium as the predominant (major) ISDA. We generated growth mixtures with a molar composition of x SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y Al2O3:4 Na2O:1 Li2O:600 H2O, where the water content (H2O/OH− = 60) was selected to be consistent with other reported syntheses of Li-zeolites (Table S1†). After a 24 h period of stirring at room temperature, samples were heated for 7 days at temperatures of 65, 100, and 180 °C. The kinetic ternary phase diagram for Na-zeolite syntheses at the lowest temperature (Fig. S1 and Table S2†) exhibits two zeolite phases, FAU and LTA, in Si- and Al-rich media, respectively. For mixed sodium–lithium synthesis media (Fig. 1a), we used conditions similar to those for Na-zeolites where 80% of the sodium was retained as the major ISDA and the remaining 20% was replaced by lithium as the minor ISDA, thus keeping the total concentration of alkali metals fixed. Products of growth mixtures with Na/Li = 4 are generally comprised of two or more phases without distinct boundaries between regions. Interestingly, a small region is observed where phase-pure CHA (pink region in Fig. 1a) forms after 7 days of heating at 65 °C. To our knowledge, this is the lowest synthesis temperature that has been used to synthesize zeolite CHA, and also the first organic-free, non-seeded synthesis of CHA in the absence of potassium. Further studies enabled us to confirm that the partial substitution of lithium in potassium-based synthesis mixtures similarly promotes CHA relative to pure potassium conditions (Fig. 1b and S2†).
:y Al2O3:4 Na2O:1 Li2O:600 H2O, where the water content (H2O/OH− = 60) was selected to be consistent with other reported syntheses of Li-zeolites (Table S1†). After a 24 h period of stirring at room temperature, samples were heated for 7 days at temperatures of 65, 100, and 180 °C. The kinetic ternary phase diagram for Na-zeolite syntheses at the lowest temperature (Fig. S1 and Table S2†) exhibits two zeolite phases, FAU and LTA, in Si- and Al-rich media, respectively. For mixed sodium–lithium synthesis media (Fig. 1a), we used conditions similar to those for Na-zeolites where 80% of the sodium was retained as the major ISDA and the remaining 20% was replaced by lithium as the minor ISDA, thus keeping the total concentration of alkali metals fixed. Products of growth mixtures with Na/Li = 4 are generally comprised of two or more phases without distinct boundaries between regions. Interestingly, a small region is observed where phase-pure CHA (pink region in Fig. 1a) forms after 7 days of heating at 65 °C. To our knowledge, this is the lowest synthesis temperature that has been used to synthesize zeolite CHA, and also the first organic-free, non-seeded synthesis of CHA in the absence of potassium. Further studies enabled us to confirm that the partial substitution of lithium in potassium-based synthesis mixtures similarly promotes CHA relative to pure potassium conditions (Fig. 1b and S2†).
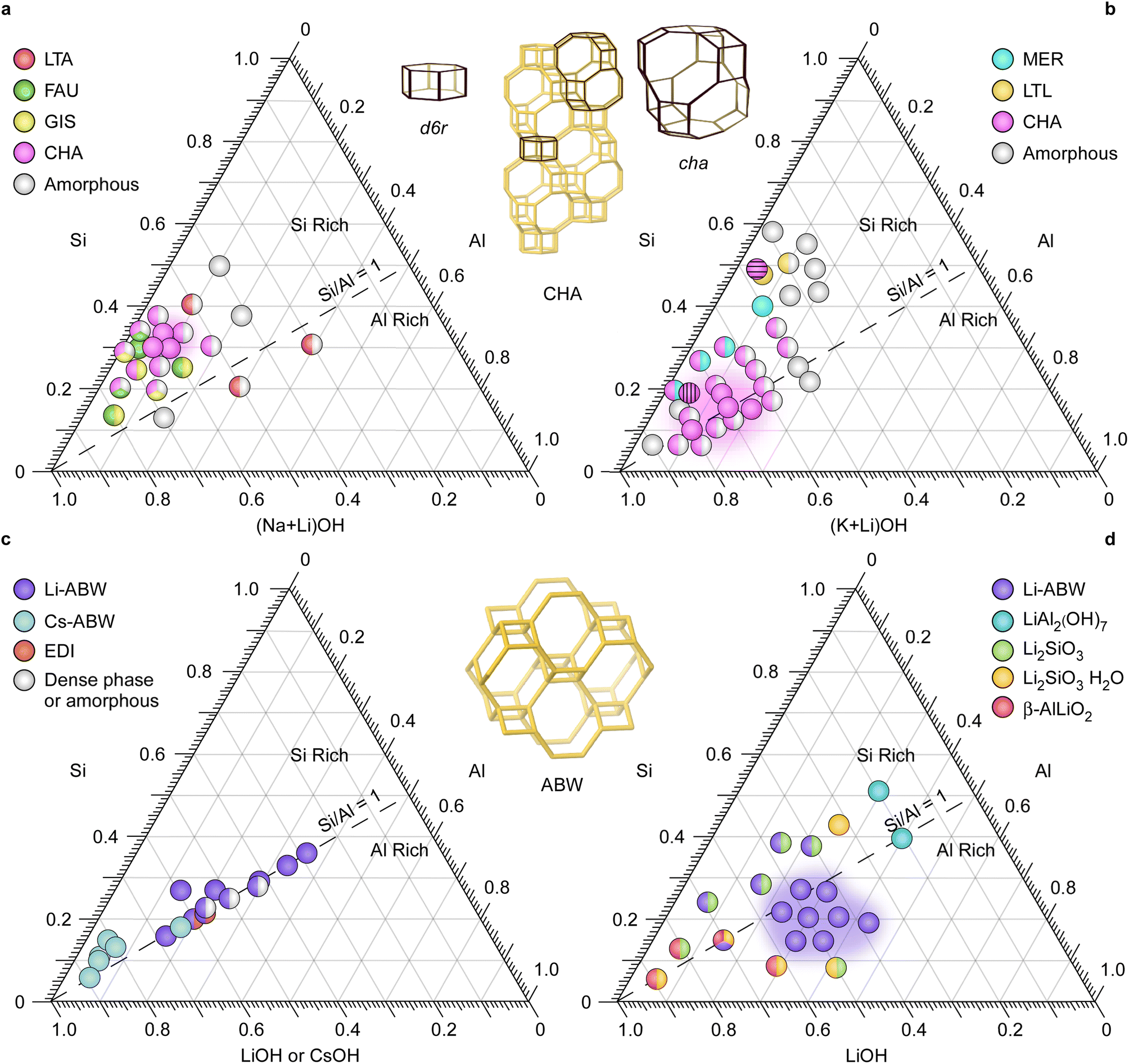 | ||
| Fig. 1 Ternary kinetic phase diagrams for zeolite syntheses using binary mixtures of Li+ with (a) Na+ (8 Na:2 Li) and (b) K+ (9 K:1 Li) heated for 7 days at 65 and 85 °C, respectively (see Tables S3 and S4† for synthesis conditions). Data points with vertical and horizontal stripes indicate syntheses reported in refs. 23 and 25 respectively. Corresponding diagrams for Li+/Na+ mixtures at synthesis temperatures of 100 and 180 °C are shown in Fig. S3.† (c) Phase diagram of zeolite ABW syntheses at temperatures ranging from 90 to 250 °C, as reported in literature (see Table S1† for synthesis conditions). (d) Ternary kinetic phase diagram for zeolite syntheses at 160 °C in this study using Li+ as the sole ISDA (see Table S5† for synthesis conditions). Insets: zeolite frameworks CHA (top) and ABW (bottom) obtained from the International Zeolite Association structure database. | ||
Synthesis of Li-zeolites in the absence of OSDAs is restricted to only a few zeolite framework types,33 with zeolite ABW as the primary structure. A review of ABW syntheses in literature reveal that most reported conditions are growth mixtures with Si/Al = 1, as depicted in Fig. 1c, with relatively few cases producing Li-ABW. In general, lithium is regarded as an inhibitor of zeolite crystallization.37 In this study, products of syntheses at 160 °C where lithium is the sole ISDA are shown in Fig. 1d (see Fig. S4† for corresponding diagrams at 65 and 100 °C). At high synthesis temperature, Li-ABW is the only zeolite phase observed. Additional phases corresponding to nonporous minerals (e.g., lithium aluminum hydroxide, lithium silicate, and β-lithium aluminate) are present and become more prevalent at lower synthesis temperatures (Table S5†).38 This is counter to trends observed in organic-free zeolite synthesis where denser phases form at higher temperatures, which is in qualitative agreement with the Ostwald rule of stages where the metastability of a zeolite generally decreases with increasing density.9 A similar line of reasoning for zeolites and nonporous minerals is difficult to make without knowledge of their relative solubility and crystallization kinetics.39 Nonporous lithium phases were observed prior to zeolite nucleation in select cases with mixtures of sodium and lithium (Fig. S5†); however, over the course of zeolite crystallization these impurities disappear from powder X-ray diffraction (PXRD) patterns.
Kim and coworkers showed that the presence of Li+ ions in the seeded synthesis of zeolite CHA resulted in products with lower crystallinity, a higher fraction of stacking faults, and more impurities (e.g., lithium silicate hydrate) compared to syntheses employing Na+, K+, Rb+, or Cs+.40 Dusselier and coworkers also observed a dense lithium silicate phase in syntheses where Li+ was shown to promote the formation of paired Al sites in zeolite SSZ-13 (CHA type).18 According to their study and supported findings by others, K+ is more effective in stabilizing larger silicate oligomers than smaller cations, such as Li+.18,41–43 It has been postulated that K+ ions are not able to coordinate with small aluminosilicate oligomers as strongly as Li+.41,44,45 Alternative arguments posit the way cations fill and stabilize certain building units (e.g., double-4-membered rings, d4r, and d6r) more effectively than others could alter the speciation of aluminosilicate oligomers.18,32,46 It is possible that Li+ stabilizes silanol defect sites that tend to form in (alumino)silicates based on findings from Fan and coworkers who showed that syntheses using higher charge density cations led to zeolite products with a larger fraction of defect sites.47 Another aspect that should be considered is the structure of solvent (water) around ISDAs. The kosmotropic, or “order-making”, nature of lithium owing to its large hydration shell provides one possible explanation for why lithium-based growth mixtures are unfavorable for the nucleation of many zeolites.48 The mechanism(s) governing the multifaceted roles of lithium in zeolite synthesis remain elusive, but are consistent among studies in literature demonstrating that lithium can suppress, accelerate, or redirect zeolite crystallization depending on whether it is the sole inorganic, one of multiple inorganics, or used in conjunction with one or more OSDAs.
Expanding the phase space of zeolite CHA synthesis
The region of chabazite formation (phase space) in the Na+ and Li+ ternary diagram (Fig. 1a) is relatively small; therefore, we sought to identify synthesis conditions capable of expanding the range of CHA crystallization by systematically varying both lithium and water content of growth mixtures. When the Li/(Na + Li) ratio in the synthesis mixture was increased from 0.1 to 0.5, it was discovered that the crystallinity of the product progressively decreased (Fig. 2a and S6†). At relatively high amounts of lithium (Li/(Na + Li) > 0.3), we observed a large quantity of amorphous product and trace FAU impurity. A significant fraction of CHA product was observed in the pure-Na synthesis (Li/(Na + Li) = 0, Fig. 2a), which is unexpected since the direct synthesis of Na–CHA, to our knowledge, has never been reported. In contrast, pure sodium growth media tend to produce other zeolite frameworks.9 There are two significant differences between the synthesis conditions used in Fig. 2a and those of conventional Na-zeolites (Fig. S1†), which is the use of aluminum hydroxide (instead of sodium aluminate) and higher water content (H2O/OH− = 60, which is a factor of three higher than conventional syntheses).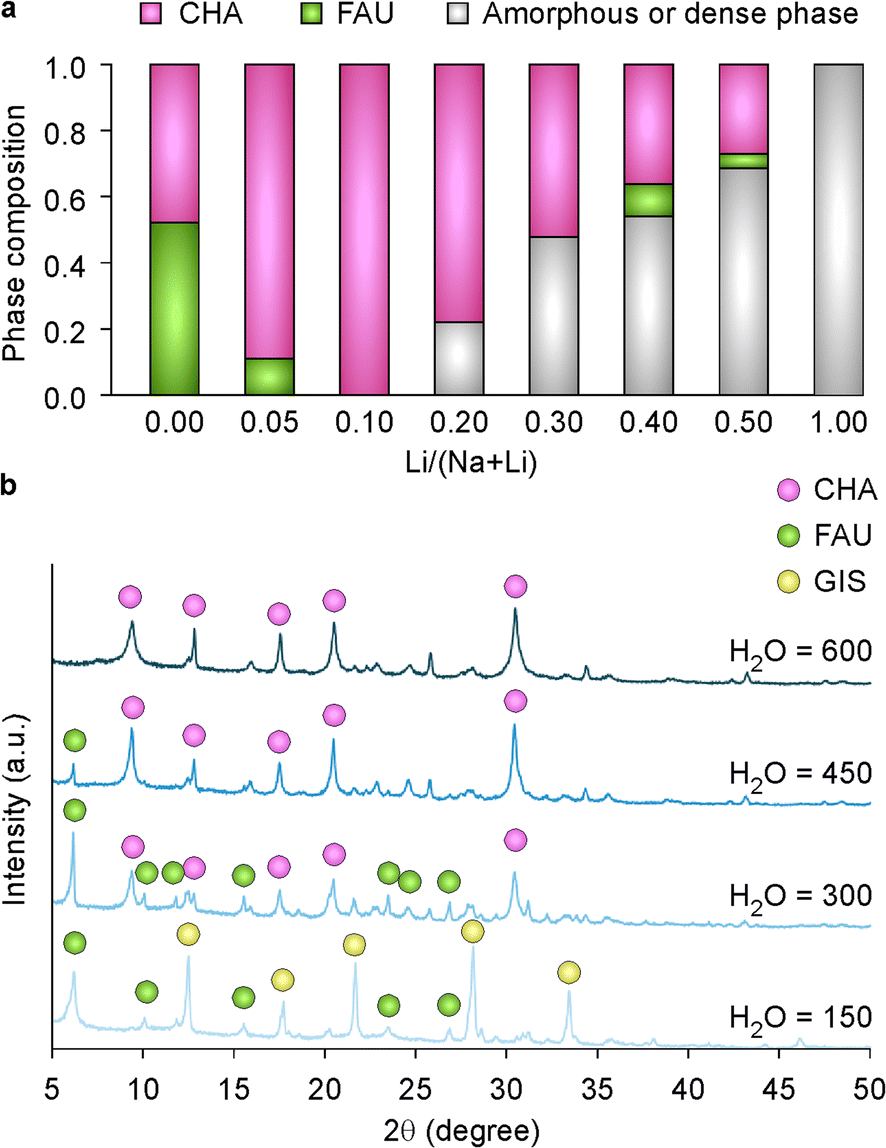 | ||
| Fig. 2 (a) Percent phase composition as a function of the Li/(Li + Na) ratio in growth mixtures synthesized for 7 days at 65 °C, as estimated from the relative peak areas in PXRD patterns. (b) Comparison of the PXRD patterns of solids extracted from synthesis mixtures with molar compositions of 4.76 SiO2:1.11 Al2O3:9 Na2O:1 Li2O:z H2O with varying water content (150 ≤ z ≤ 600) at the same synthesis conditions (7 days, 65 °C). | ||
To identify the most influential parameter, we performed a Na-FAU synthesis under conditions where sodium aluminate was replaced with aluminum hydroxide (see Fig. S1†). The resulting product was unchanged (Fig. S7†), thus indicating the aluminum source was not a significant factor in promoting zeolite CHA. To assess the effect of water content, we prepared growth mixtures with molar compositions of 4.76 SiO2:0.55 Al2O3:9 Na2O:1 Li2O:z H2O where z was varied from 150 (conventional condition, Fig. S1†) to 600 (the condition for Fig. 2a). PXRD patterns of products reveal that high water content is crucial for promoting CHA (Fig. 2b), whereas low water content leads to the formation of FAU and gismondine (GIS), a common impurity and product of FAU interzeolite transformation.13,49 This result is qualitatively consistent with general observations that more dilute synthesis mixtures tend to promote zeolite structures with higher framework densities (i.e., 15.1 and 13.3 T-sites/1000 Å3 for CHA and FAU, respectively) with an exception being the presence of GIS at very low water content (z = 150) that may arise due to accelerated crystallization kinetics at higher pH values.50
Effective pairings of alkali cations
Here we investigate how binary alkali cation selection more broadly affects CHA crystallization. First, we carried out syntheses with all possible pairings of Li+, Na+, K+, and Cs+ in a 9:1 molar ratio of major and minor ISDAs, as well as single cation syntheses. The molar ratios used in each growth mixture were based on those employed for Na,Li–CHA (4.76 SiO2:0.55 Al2O3:4.5 Q2O:0.5 W2O : 600 H2O) where Q and W are the major and minor inorganic components, respectively. Extracted solids from growth mixtures after 7 days of heating at 85 °C were characterized by PXRD (Fig. S8†) and the resulting phase(s) for each ISDA combination are presented in Fig. 3. When lithium was employed as the major cation (i.e., Q = Li), the majority of products were amorphous and/or dense nonporous crystalline phases without detectable zeolites. The lone exception was an impure zeolite analcime (ANA) phase observed when W = Cs, which is expected given that Cs+ is a known promoter of zeolite ANA.51 Consistent with results in Fig. 2a, CHA products were observed when Q = Na and W = Li. A pure-Na synthesis produced a mixture of CHA and FAU; however, the combination of sodium and potassium (Q = Na, W = K) led to phillipsite (PHI).
 | ||
| Fig. 3 Phase selection for syntheses at 85 °C for 7 days using binary mixtures of alkali metals with a molar ratio of 9:1 (Q:W). Symbols are color coded according to the relative proportions of each phase estimated from PXRD (Fig. S8†). The dashed line highlights syntheses using a single cation (Q = W). The dense phase (white) corresponds to a non-zeolite crystalline phase (lithium silicate). | ||
Syntheses with K+ as the major ISDA are expected to produce CHA, which was observed in all combinations with only W = Cs producing ANA as a minor impurity. One interesting observation when lithium was used as a minor ISDA (Q = K and W = Li) is an expansion of the phase space for pure CHA in the ternary diagram (Fig. 1b) compared to pure-K+ syntheses (Fig. S2†). We posit that the presence of Li+ suppresses interzeolite transformations of CHA to MER and/or EDI, which we reported in a previous study.10 We also observe that Li+ accelerates the rate of zeolite CHA crystallization (vide infra). These collective findings may be attributed to differences in alkali metal affinity for water, with some being structure-breaking cations (i.e., K+ and Cs+ are chaotropes), whereas others are structure-making cations (i.e., Li+ is a kosmotrope).52 Among all major inorganic components tested, sodium (Q = Na) was the least selective, which may be due to its moderate degree of water affinity.52 This may explain why binary mixtures where Q = Na result in the formation of a different zeolite phase for each minor cation (Fig. S9†).
It is generally assumed for OSDA-free syntheses that FAU is a “sodium-zeolite” whereas CHA is a “potassium-zeolite”. Our findings seem to indicate these labels are oversimplified. Specifically, we find that CHA crystallization does not require potassium; therefore, we posit ISDA pairings can alter syntheses in such a way that allows for a more thermodynamically stable structure, such as CHA, to nucleate instead of metastable structures (e.g., FAU) that often crystallize first during interzeolite transformations. This suggests a complex energy landscape in zeolite crystallization that can be accessed by the employment of cooperative structure direction.
Zeolite CHA crystallization kinetics
Conventional synthesis of K–CHA10 at 85 °C typically requires 2–3 weeks; however, the selection of alkali and/or alkaline earth metals can have a dramatic impact on crystallization kinetics. We conducted time-resolved syntheses for four synthesis mixtures that led to pure CHA (pink squares in Fig. 3 corresponding to K–CHA, K,Na–CHA, K,Li–CHA, and Na,Li–CHA). In all cases, the molar composition of the growth solution is identical except for the cation identity. We also studied a synthesis of K,Sr–CHA where the sole difference in conditions is a 75% reduction in the molar ratio of Sr2+ compared to other minor cations. Lastly, we investigated the synthesis of CHA using a ternary mixture of Na+, K+, and Cs+ based on a modified protocol of Mintova and coworkers25 where the room temperature aging period, Si/Al and Si/OH molar ratios were altered (see ESI†) to match the growth mixture composition used in this study. For each synthesis mixture we used time-resolved PXRD patterns to elucidate the induction time, which was estimated by the first appearance of Bragg peaks, as well as the total crystallization time, which was determined by the disappearance of the amorphous peak. Both induction time and crystallization time for each synthesis are shown in Fig. 4. The most rapid synthesis at 85 °C is K,Sr–CHA (12 h), which is shorter than our previously reported study (>48 h),45 likely owing to a different composition than the original (Fig. 1b, data point with vertical stripes). The synthesis of Na,Li–CHA is also relatively fast (<48 h), but if heated for a longer period of time the CHA phase undergoes an interzeolite transformation to zeolite GIS. The relatively small difference between induction and crystallization times for the majority of cases suggests that cation pairings have a more pronounced effect on nucleation rather than growth. | ||
| Fig. 4 Histogram depicting the induction period (cyan) and the time to reach full crystallinity (purple) for CHA samples synthesized at 85 °C with different combinations of major/minor ISDAs. Times are estimated from the analysis of PXRD patterns in Fig. S10.† | ||
The properties of CHA zeolites synthesized with different cations (Table 1) reveal slight variations in Si/Al ratios, but there is no clear correlation between this ratio and cation(s) selection. The product M/Al ratio is ∼1 for all samples, with the exception of K,Sr–CHA, where the unusually high amount of Sr2+ is due to the presence of a Sr(OH)2 phase, which is observed in the PXRD pattern (Fig. S11†). For all binary combinations of alkali metals involving potassium as the major component, elemental analysis of zeolite products (Table 1) reveals a higher percentage of extra-framework potassium compared to the major:minor cation ratio of the original growth mixture, consistent with general knowledge that K+ is an effective structure-directing agent for CHA crystallization. Conversely, the use of sodium as the major component leads to the same ratio of cations in the zeolite as that of the growth mixture (i.e., 9:1 Na:Li).
| Sample | Time (days) | Si/Al | K/Al | Na/Al | Li/Al | Sr/Al | M/Al | Fault (%) |
|---|---|---|---|---|---|---|---|---|
| a Elemental analysis was performed using ICP-OES on digested zeolite crystals (see the Material characterization section in ESI). | ||||||||
| K–CHA | 12 | 1.78 | 1.02 | — | — | — | 1.02 | 26 |
| K,Na–CHA | 7 | 2.09 | 0.97 | 0.04 | — | — | 1.01 | 20 |
| K,Li–CHA | 3 | 1.52 | 0.97 | — | 0.06 | — | 1.02 | 25 |
| K,Sr–CHA | 1 | 2.25 | 0.94 | — | — | 0.32 | 1.26 | 17 |
| Na,Li–CHA | 2 | 2.03 | — | 0.91 | 0.09 | — | 1.00 | 10 |
It is also interesting to note that the concentration of lithium in the solid phase during the transition from amorphous precursors to crystalline product decreases with synthesis time (Fig. S12†). Efforts to analyze the location of ions in CHA pores using synchrotron PXRD were unsuccessful owing to significant peak broadening (Fig. S11†) that occurs because of stacking faults.33 In our limited dataset, the percentage of stacking faults in CHA products, which is calculated according to a reported protocol33 (see Methods), is lowest for zeolites with the shortest synthesis times (Table 1); however, we were unable to correlate the amount of faulting in CHA crystals with the atomic number of the cation(s) used, as previously demonstrated by Okubo and coworkers, where heavier elements led to a lower degree of faulting in CHA crystals, purportedly because of their ability to stabilize d6r units.33
Proposed mechanism of paired inorganic cations
Small amounts of inorganic cations (<10% of the total ISDA amount) such as strontium and lithium can have a beneficial effect on zeolite crystallization kinetics when present in K–CHA growth mixtures. When lithium is used as the sole ISDA, however, ABW is the only zeolite structure that forms. Otherwise, lithium promotes dense (nonporous) crystalline phases (Fig. 1d), which also form as transient impurities when lithium is present as a minor cation (Fig. S5†). The presence of lithium appears to suppress the formation of impurities commonly observed in CHA syntheses, including MER (Fig. S2†) and FAU (Fig. 2a). The small percentage of lithium as minor ISDA leads to a small quantity of extra-framework lithium in CHA. Elemental analysis of CHA zeolites (Table 1) reveal an extra-framework cation ratio of 10:1 (Q:Li), which equates to one lithium ion (and 10 sodium or potassium ions) per cha cage, in agreement with an expected 10–14 Al atoms per cage based on the crystal Si/Al ratio.
Several observations suggest that the hydrated cation size, a property that is often ignored in mechanistic discussions, is an important factor in zeolite crystallization, where cations are generally fully hydrated throughout the entire synthesis process. For syntheses with the major cation being sodium, which is borderline between a kosmotrope (water structure maker) and chaotrope (water structure breaker),52 the product shifts depending on the selection of minor cation (Fig. 3). For syntheses where Li+ is the minor cation, we observe an accelerated rate of CHA formation. In Fig. 5a, we depict the approximate sizes of hydrated cations in this study compared to trimethyladamantyl ammonium (TMAda+), a common OSDA for zeolite SSZ-13 (CHA). When steric considerations of ISDAs within zeolite pores include ion hydration, the idealized packing of solvated species within a single cha cage (Fig. 5b) helps visualize a possible connection between the role of cooperative ISDAs. Indeed, in K+-containing media where K+ is either the sole ISDA or the major component of binary mixtures, there is a strong correlation between hydrated ionic radius and the rate of CHA formation (Fig. 5c), which suggests that ion effects are related to water structuring. Notably, we observe monotonic trends for crystallization time and crystal size with changes in the hydrated radius of the minor cation (Fig. 5c), but not with the anhydrous ionic radius (Fig. S13†).
 | ||
| Fig. 5 (a) Illustrations of alkali metals with their hydration shell compared to trimethyladamantyl ammonium (TMAda+). The hydrated cation radii were obtained from ref. 48 and the size of the TMAda+ cation was estimated from data in refs. 53 and 54. (b) Idealized schematic of a cha cage filled with 10 hydrated potassium ions and 1 hydrated lithium ion in the center (according to elemental compositions in Table 1). Inset: idealized adsorption of an extra-framework cation with a negatively-charged Al framework site (dashed lines are overlapping hydration shells). (c) Correlation between CHA crystallization time (blue circles, left) and crystal size (orange squares, right) with the hydrated ionic radii of cations partially substituting K+ in the synthesis mixture (each cation is labelled above its data point). Crystal size was determined from SEM images (Fig. S14†). Symbols are the average of 30 points and error bars span two standard deviations. (d) Volcano plot of the free energy of solution for alkali bromides (assuming framework Al− is a chaotrope similar to Br−) and the difference in hydration energy for bromine and the selected alkali cation. This figure was recreated from Collins' theory in ref. 55 for kosmotrope (K) and chaotrope (C) cation–anion pairs. | ||
Based on the findings in this study and those of other research groups, two mechanisms for the effects of hydration shells on zeolite crystallization are proposed: (1) electrostatic interactions between cations and aluminosilicate oligomers are weaker for ions with a larger hydration shell, which can affect local pH56 and the distribution of (alumino)silicate species;50,57 and (2) super-ions58 (e.g., cations linked by water and/or hydroxide ligands) and/or hydrated cations59 can function as structure-directing agents that facilitate the formation of certain cages or building units. It has been demonstrated that certain cations are preferentially sited at particular locations within the CHA framework,38,49 thus suggesting that hydrated cations may promote or stabilize the formation of specific structural units.
We posit the strength of interactions between extra-framework cations and negatively-charged framework sites depend on the sizes of their respective hydration shells, as depicted in the inset of Fig. 5b. The exact nature of these interactions, however, remains unclear. One aspect that is particularly interesting is the potential for cation hydration shells to shield their electrostatic interactions with the negatively-charged crystal structure and/or fill pores and cages to facilitate crystallization. We propose that an effect similar to Collins' law of matching water affinities60 may occur, where the thermodynamic driving force to stabilize cation–anion pairs is based on whether the cation and anion are kosmotropes (K) or chaotropes (C). The former are ions that strongly attract immobilized water, while the latter are ions that loosely bind water. Collins developed this theory for alkali (M+) and halide (X−) pairs using the following reaction,
 | (1) |
The most strongly hydrated cation in this study, lithium, is known to promote the formation of 4-membered ring species in zeolites, which are crucial to the formation of CHA crystals.61 The fact that lithium generally promotes the formation of dense (non-zeolitic) crystal phases57 may be due to high entropic barriers to remove water from this kosmotropic cation,59 or may be attributed to the putatively low energetic barrier for the dissociation of Li+ – (Si–O–Al)− pairings (Fig. 5d). Lithium is known to promote zincosilicate zeotypes, where it can be either a framework or extra-framework species.35,62–68 In contrast, the least hydrated cation, Cs+, is rather limited in its role as an ISDA for zeolite CHA. It was recently demonstrated by Wakihara and coworkers that Cs+ ions function as a template for building units of zeolites RHO and ANA, which aligns with our findings described above (Fig. 3).69
Our findings suggest that the judicious selection of positively-charged species (i.e., extra-framework ISDAs and/or OSDAs) and negatively-charged species (i.e., Al or heteroatom sites) impact zeolite crystallization on the basis of their relative hydration spheres. A recent study by Yu and coworkers70 demonstrated that the addition of inorganic anions into silicalite-1 growth mixtures altered crystallization kinetics in a manner consistent with the Hofmeister series.71 These findings support the hypothesis that the hydration of cationic species is an aspect of zeolite crystallization that significantly affects the stabilization energy of aluminosilicate-cation pairings. One of the biggest challenges with experimentally determining the nature and location of water in zeolite crystals is the facile migration of H2O molecules, particularly during temperature changes and dehydration72,73 associated with post-synthesis extraction and analysis of zeolite samples. To this end, ongoing studies by our group and others collectively seek to advance the understanding of molecular mechanisms governing hydration effects and cooperative structure-direction in zeolite crystallization with the objective of verifying our hypothesis of hydration mismatch.
Conclusions
We have shown that distinct combinations of inorganic cations can promote CHA crystallization. In this study, we examined the effects of cooperative ISDAs on zeolite formation. Our findings revealed that the judicious selection of binary (major/minor) ISDAs can significantly accelerate the rate of CHA crystallization, while also impacting crystal size and defects (faults) relative to conventional organic-free CHA syntheses using K+ as the sole ISDA. Although Li+ has been traditionally viewed as an inhibitor of zeolite formation, we have shown that its use in small quantities (ca. 90 mM) can promote zeolite CHA crystallization, similar to the previously reported effects of Sr2+. We posit that the large hydrated radius of Li+ poses steric constraints for its packing in CHA cages, which offers a potential explanation for its effectiveness in promoting CHA crystallization at low concentrations. Moreover, we hypothesize the reduced affinity of Li+ for negatively-charged Al sites, consistent with Collins' theory, imparts greater mobility as a structure-directing agent.Here, we hypothesize that the effects of co-cation additives are related to their respective hydrated radii, which is a parameter that is often overlooked in studies of zeolite structure direction. Recent investigations have emphasized, however, that ion hydration57–59,74 and their electrostatic interactions with aluminosilicate species (e.g., oligomers)75 may be crucial phenomena to consider for developing a more holistic understanding of zeolite precursor evolution and crystal phase selection. To our knowledge, there is only one reported example where Collins' theory was applied to ion–crystal interactions to describe the effect of alkali metals as crystal growth modifiers.76 Extending this concept to zeolite synthesis will require computational efforts to verify the hypothesis put forth in this study and to assess whether it is more broadly applicable to other framework types, such as Na-zeolites where the primary cation (Na+) is paired with other alkali or alkaline earth metals. Future studies employing synergistic combination of experiments and modeling have the potential to provide a more molecular-level understanding of cooperative ISDA mechanisms in zeolite crystallization, as well as their effects on physicochemical properties that are crucial to numerous commercial applications.
Data availability
The data supporting this article have been uploaded as part of the ESI.† Additional data is available upon requests sent to the corresponding author.Author contributions
Conceptualization: JDR and AJM; data curation: AJM, GE, and NV; formal analysis: AJM; writing – original draft: JDR and AJM; project administration: JDR.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was primarily funded by the Department of Energy, Office of Basic Energy Sciences, Materials Science Division (award DE-SC0021384). Use of the Advanced Photon Source at Argonne National Laboratory was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Additional financial support for laboratory supplies was provided by the Welch Foundation (award E-1794). We thank Dr S. Seo for assistance with analysis of synchrotron PXRD data. We thank Dr Y. Gao for sample digestion and analysis of zeolite samples using ICP.References
- S. Li, J. Li, M. Dong, S. Fan, T. Zhao, J. Wang and W. Fan, Strategies to control zeolite particle morphology, Chem. Soc. Rev., 2019, 48(3), 885–907 RSC.
- J. Grand, H. Awala and S. Mintova, Mechanism of zeolites crystal growth: new findings and open questions, CrystEngComm, 2016, 18(5), 650–664 RSC.
- J. D. Rimer, M. Kumar, R. Li, A. I. Lupulescu and M. D. Oleksiak, Tailoring the physicochemical properties of zeolite catalysts, Catal. Sci. Technol., 2014, 4(11), 3762–3771 RSC.
- L. Tosheva and V. P. Valtchev, Nanozeolites: synthesis, crystallization mechanism, and applications, Chem. Mater., 2005, 17(10), 2494–2513 CrossRef CAS.
- R. Jain, A. J. Mallette and J. D. Rimer, Controlling nucleation pathways in zeolite crystallization: seeding conceptual methodologies for advanced materials design, J. Am. Chem. Soc., 2021, 143(51), 21446–21460 CrossRef CAS PubMed.
- M. Moliner, F. Rey and A. Corma, Towards the rational design of efficient organic structure-directing agents for zeolite synthesis, Angew. Chem., Int. Ed., 2013, 52(52), 13880–13889 CrossRef CAS.
- J. R. Di Iorio and R. Gounder, Controlling the isolation and pairing of aluminum in chabazite zeolites using mixtures of organic and inorganic structure-directing agents, Chem. Mater., 2016, 28(7), 2236–2247 CrossRef CAS.
- M. D. Oleksiak and J. D. Rimer, Synthesis of zeolites in the absence of organic structure-directing agents: factors governing crystal selection and polymorphism, Rev. Chem. Eng., 2014, 30(1), 1–49 CAS.
- M. Maldonado, M. D. Oleksiak, S. Chinta and J. D. Rimer, Controlling crystal polymorphism in organic-free synthesis of Na-zeolites, J. Am. Chem. Soc., 2013, 135(7), 2641–2652 CrossRef CAS PubMed.
- A. Chawla, A. J. Mallette, R. Jain, N. Le, F. C. R. Hernández and J. D. Rimer, Crystallization of potassium-zeolites in organic-free media, Microporous Mesoporous Mater., 2022, 341, 112026 CrossRef CAS.
- M. B. Park, S. H. Ahn, C. P. Nicholas, G. J. Lewis and S. B. Hong, Charge density mismatch synthesis of zeolite beta in the presence of tetraethylammonium, tetramethylammonium, and sodium ions: influence of tetraethylammonium decomposition, Microporous Mesoporous Mater., 2017, 240, 159–168 CrossRef CAS.
- M. B. Park, S. J. Cho and S. B. Hong, Synthesis of aluminosilicate and gallosilicate zeolites via a charge density mismatch approach and their characterization, J. Am. Chem. Soc., 2011, 133(6), 1917–1934 CrossRef CAS.
- O. Larlus and V. P. Valtchev, Crystal morphology control of LTL-type zeolite crystals, Chem. Mater., 2004, 16(17), 3381–3389 CrossRef CAS.
- M. Goepper, H.-X. Li and M. E. Davis, A possible role of alkali metal ions in the synthesis of pure-silica molecular sieves, J. Chem. Soc. Chem. Commun., 1992,(22), 1665–1666 RSC.
- C. Liu, W. Gu, D. Kong and H. Guo, The significant effects of the alkali-metal cations on ZSM-5 zeolite synthesis: from mechanism to morphology, Microporous Mesoporous Mater., 2014, 183, 30–36 CrossRef CAS.
- A. Tuel and Y. B. Taarit, Synthesis of TS-1 from titanosilicate gels containing TPAOH/TEAOH and TPAOH/NH4OH mixtures, Microporous Mater., 1993, 1(3), 179–189 CrossRef CAS.
- V. Pashkova, P. Klein, J. Dedecek, V. Tokarová and B. Wichterlová, Incorporation of Al at ZSM-5 hydrothermal synthesis. Tuning of Al pairs in the framework, Microporous Mesoporous Mater., 2015, 202, 138–146 CrossRef CAS.
- S. Robijns, J. Devos, T. Donckels, R. de Oliveira-Silva, N. De Witte, D. Sakellariou, T. R. C. Van Assche and M. Dusselier, Steering interzeolite conversion with alkali metal cations: lithium maximizes Al proximity in SSZ-13 zeolite genesis, Cryst. Growth Des., 2022, 23(1), 289–299 CrossRef.
- M. Debost, P. B. Klar, N. Barrier, E. B. Clatworthy, J. Grand, F. Laine, P. Brázda, L. Palatinus, N. Nesterenko and P. Boullay, Synthesis of discrete CHA zeolite nanocrystals without organic templates for selective CO2 capture, Angew. Chem., Int. Ed., 2020, 59(52), 23491–23495 CrossRef CAS.
- H. Lee, J. Shin and S. B. Hong, Tetraethylammonium-mediated zeolite synthesis via a multiple inorganic cation approach, ACS Mater. Lett., 2021, 3(4), 308–312 CrossRef.
- J. Shin, D. Jo and S. B. Hong, Rediscovery of the importance of inorganic synthesis parameters in the search for new zeolites, Acc. Chem. Res., 2019, 52(5), 1419–1427 CrossRef CAS PubMed.
- H. Imai, N. Hayashida, T. Yokoi and T. Tatsumi, Direct crystallization of CHA-type zeolite from amorphous aluminosilicate gel by seed-assisted method in the absence of organic-structure-directing agents, Microporous Mesoporous Mater., 2014, 196, 341–348 CrossRef CAS.
- Y. Liang, A. J. Jacobson and J. D. Rimer, Strontium ions function as both an accelerant and structure-directing agent of chabazite crystallization, ACS Mater. Lett., 2021, 3(2), 187–192 CrossRef CAS.
- E. B. Clatworthy, A. A. Paecklar, E. Dib, M. Debost, N. Barrier, P. Boullay, J.-P. Gilson, N. Nesterenko and S. Mintova, Engineering RHO nanozeolite: controlling the particle morphology, Al and cation content, stability, and flexibility, ACS Appl. Energy Mater., 2022, 5(5), 6032–6042 CrossRef CAS.
- S. Ghojavand, E. B. Clatworthy, A. Vicente, E. Dib, V. Ruaux, M. Debost, J. El Fallah and S. Mintova, The role of mixed alkali metal cations on the formation of nanosized CHA zeolite from colloidal precursor suspension, J. Colloid Interface Sci., 2021, 604, 350–357 CrossRef CAS.
- E. I. Basaldella and J. C. Tara, Synthesis of LSX zeolite in the NaK system: influence of the NaK ratio, Zeolites, 1995, 15(3), 243–246 CrossRef CAS.
- D. Zhao, R. Szostak and L. Kevan, Role of alkali-metal cations and seeds in the synthesis of silica-rich heulandite-type zeolites, J. Mater. Chem., 1998, 8(1), 233–239 RSC.
- M. D. Oleksiak, J. A. Soltis, M. T. Conato, R. L. Penn and J. D. Rimer, Nucleation of FAU and LTA zeolites from heterogeneous aluminosilicate precursors, Chem. Mater., 2016, 28(14), 4906–4916 CrossRef CAS.
- J. M. Newsam, Synthesis and structural characterization of a lithium gallosilicate with the zeolite ABW-framework, J. Phys. Chem., 1988, 92(2), 445–452 CrossRef CAS.
- M. B. Park, D. Jo, H. C. Jeon, C. P. Nicholas, G. J. Lewis and S. B. Hong, Zeolite synthesis from a charge density perspective: the charge density mismatch synthesis of UZM-5 and UZM-9, Chem. Mater., 2014, 26(23), 6684–6694 CrossRef CAS.
- M. Debost, E. B. Clatworthy, J. Grand, N. Barrier, N. Nesterenko, J. P. Gilson, P. Boullay and S. Mintova, Direct synthesis of nanosized CHA zeolite free of organic template by a combination of cations as structure directing agents, Microporous Mesoporous Mater., 2023, 358, 112337 CrossRef CAS.
- A. Inayat, C. Schneider and W. Schwieger, Organic-free synthesis of layer-like FAU-type zeolites, Chem. Commun., 2015, 51(2), 279–281 RSC.
- K. Iyoki, Y. Kamimura, K. Itabashi, A. Shimojima and T. Okubo, Synthesis of MTW-type zeolites in the absence of organic structure-directing agent, Chem. Lett., 2010, 39(7), 730–731 CrossRef CAS.
- M. Yoshikawa, S. I. Zones and M. E. Davis, Synthesis of VPI-8: I. The effects of reaction components, Microporous Mater., 1997, 11(3–4), 127–136 CrossRef CAS.
- M. Yoshikawa, S. I. Zones and M. E. Davis, Synthesis of VPI-8. II. mechanism of crystallization, Microporous Mater., 1997, 11(3–4), 137–148 CrossRef CAS.
- N. Wang, Z. He, B. Wang, B. Liu, W. Xing and R. Zhou, Zirconia-supported all-silica zeolite CHA membrane with unprecedentedly high selectivity in humidified CO2/CH4 mixture, J. Membr. Sci., 2023, 673, 121452 CrossRef CAS.
- A. J. Mallette, J. T. Reiser, G. Mpourmpakis, R. K. Motkuri, J. J. Neeway and J. D. Rimer, The effect of metals on zeolite crystallization kinetics with relevance to nuclear waste glass corrosion, NPJ Mater. Degrad., 2023, 7(1), 1–7 CrossRef.
- Y. Duan, H. Pfeiffer, B. Li, I. C. Romero-Ibarra, D. C. Sorescu, D. R. Luebke and J. W. Halley, CO2 capture properties of lithium silicates with different ratios of Li2O/SiO2: an ab initio thermodynamic and experimental approach, Phys. Chem. Chem. Phys., 2013, 15(32), 13538–13558 RSC.
- P. T. Cardew, Ostwald rule of stages-Myth or reality?, Cryst. Growth Des., 2023, 23(6), 3958–3969 CrossRef CAS.
- J. Kim and D. H. Kim, Synthesis of faulted CHA-type zeolites with controllable faulting probability, Microporous Mesoporous Mater., 2018, 256, 266–274 CrossRef CAS.
- R. Aiello, F. Crea, A. Nastro, B. Subotić and F. Testa, Influence of cations on the physicochemical and structural properties of aluminosilicate gel precursors. Part 1. Chemical and thermal properties, Zeolites, 1991, 11(8), 767–775 CrossRef CAS.
- P. Wijnen, T. P. M. Beelen, J. W. De Haan, L. J. M. Van De Ven and R. A. Van Santen, The structure directing effect of cations in aqueous silicate solutions. A 29Si-NMR study, Colloids Surf., 1990, 45, 255–268 CrossRef CAS.
- A. V. McCormick, A. T. Bell, and C. J. Radke, Effect of Alkali Metal Cations on Silicate Structures in Aqueous Solution, ACS Publications, 1988 Search PubMed.
- I. I. Ivanova, R. Aiello, J. B. Nagy, F. Crea, E. G. Derouane, N. Dumont, A. Nastro, B. Subotic and F. Testa, Influence of cations on the physicochemical and structural properties of aluminosilicate gel precursors: II. Multinuclear magnetic resonance characterization, Microporous Mater., 1994, 3(3), 245–257 CrossRef CAS.
- M. Iwama, Y. Suzuki, J. Plévert, K. Itabashi, M. Ogura and T. Okubo, Location of alkali ions and their relevance to crystallization of low silica x zeolite, Cryst. Growth Des., 2010, 10(8), 3471–3479 CrossRef CAS.
- S. Ghojavand, B. Coasne, E. B. Clatworthy, R. Guillet-Nicolas, P. Bazin, M. Desmurs, L. Jacobo Aguilera, V. Ruaux and S. Mintova, Alkali metal cations influence the CO2 adsorption capacity of nanosized chabazite: modeling vs. experiment, ACS Appl. Nano Mater., 2022, 5(4), 5578–5588 CrossRef CAS.
- V. Vattipalli, A. M. Paracha, W. Hu, H. Chen and W. Fan, Broadening the scope for fluoride-free synthesis of siliceous zeolites, Angew. Chem., Int. Ed., 2018, 57(14), 3607–3611 CrossRef CAS PubMed.
- B. E. Conway, Ionic Hydration in Chemistry and Biophysics, Netherlands, 1981 Search PubMed.
- Y. Hu, C. Liu, Y. Zhang, N. Ren and Y. Tang, Microwave-assisted hydrothermal synthesis of nanozeolites with controllable size, Microporous Mesoporous Mater., 2009, 119(1), 306–314 CrossRef CAS.
- G. Sastre, A. Pulido, R. Castañeda and A. Corma, Effect of the germanium incorporation in the synthesis of EU-1, ITQ-13, ITQ-22, and ITQ-24 zeolites, J. Phys. Chem. B, 2004, 108(26), 8830–8835 CrossRef CAS.
- A. Nearchou and A. Sartbaeva, Influence of alkali metal cations on the formation of zeolites under hydrothermal conditions with no organic structure directing agents, CrystEngComm, 2015, 17(12), 2496–2503 RSC.
- Y. Marcus, Effect of ions on the structure of water: structure making and breaking, Chem. Rev., 2009, 109(3), 1346–1370 CrossRef CAS PubMed.
- D. H. Aue, H. M. Webb and M. T. Bowers, A thermodynamic analysis of solvation effects on the basicities of alkylamines. An electrostatic analysis of substituent effects, J. Am. Chem. Soc., 1976, 98(2), 318–329 CrossRef CAS.
- F. Jimenez-Cruz and J. L. García-Gutiérrez, Molecular size and shape properties of diamondoid molecules occurring in crude oil, Arab. J. Chem., 2020, 13(12), 8592–8599 CrossRef CAS.
- K. D. Collins, Charge density-dependent strength of hydration and biological structure, Biophys. J., 1997, 72(1), 65–76 CrossRef CAS PubMed.
- B. N. Ruggiero, K. M. S. Gutierrez, J. D. George, N. M. Mangan, J. M. Notestein and L. C. Seitz, Probing the relationship between bulk and local environments to understand impacts on electrocatalytic oxygen reduction reaction, J. Catal., 2022, 414, 33–43 CrossRef CAS.
- K. Asselman, N. Pellens, B. Thijs, N. Doppelhammer, M. Haouas, F. Taulelle, J. A. Martens, E. Breynaert and C. E. A. Kirschhock, Ion-pairs in aluminosilicate-alkali synthesis liquids determine the aluminum content and topology of crystallizing zeolites, Chem. Mater., 2022, 34(16), 7150–7158 CrossRef CAS.
- K. Asselman, N. Pellens, S. Radhakrishnan, C. V. Chandran, J. A. Martens, F. Taulelle, T. Verstraelen, M. Hellström, E. Breynaert and C. E. A. Kirschhock, Super-ions of sodium cations with hydrated hydroxide anions: inorganic structure-directing agents in zeolite synthesis, Mater. Horiz., 2021, 8(9), 2576–2583 RSC.
- K. Asselman, M. Haouas, M. Houlleberghs, S. Radhakrishnan, W. Wangermez, C. E. A. Kirschhock, and E. Breynaert, Does Water Enable Porosity in Aluminosilicate Zeolites? Porous Frameworks versus Dense Minerals, Crystal Growth & Design, 2023 Search PubMed.
- K. D. Collins, G. W. Neilson and J. E. Enderby, Ions in water: characterizing the forces that control chemical processes and biological structure, Biophys. Chem., 2007, 128(2), 95–104 CrossRef CAS PubMed.
- A. Minami, P. Hu, Y. Sada, H. Yamada, K. Ohara, Y. Yonezawa, Y. Sasaki, Y. Yanaba, M. Takemoto and Y. Yoshida, Tracking sub-nano-scale structural evolution in zeolite synthesis by in situ high-energy x-ray total scattering measurement with pair distribution function analysis, J. Am. Chem. Soc., 2022, 144(51), 23313–23320 CrossRef CAS PubMed.
- S. H. Park, P. Daniels and H. Gies, RUB-23: a new microporous lithosilicate containing spiro-5 building units, Microporous Mesoporous Mater., 2000, 37(1), 129–143 CrossRef CAS.
- K. Iyoki, K. Itabashi, W. Chaikittisilp, S. P. Elangovan, T. Wakihara, S. Kohara and T. Okubo, Broadening the applicable scope of seed-directed, organic structure-directing agent-free synthesis of zeolite to zincosilicate components: a case of VET-type zincosilicate zeolites, Chem. Mater., 2014, 26(5), 1957–1966 CrossRef CAS.
- Y. Sakamoto, H. Zhao, H. Gies, K. Yamamoto, U. Kolb and T. Ikeda, A new microporous 12-ring zincosilicate THK-2 with many terminal silanols characterized by automated electron diffraction tomography, Dalton Trans., 2020, 49(37), 12960–12969 RSC.
- N. Koike, K. Iyoki, S. H. Keoh, W. Chaikittisilp and T. Okubo, Synthesis of new microporous zincosilicates with CHA zeolite topology as efficient platforms for ion-exchange of divalent cations, Chem.–A Eur. J., 2018, 24(4), 808–812 CrossRef CAS PubMed.
- N. Koike, K. Iyoki, W. Chaikittisilp and T. Okubo, Synthesis of microporous zincosilicate *BEA molecular sieves from zincosilicate gels co-precipitated in the presence of an organic structure-directing agent, Chem. Lett., 2018, 47(7), 897–900 CrossRef CAS.
- D. P. Serrano, R. van Grieken, M. E. Davis, J. A. Melero, A. Garcia and G. Morales, Mechanism of CIT-6 and VPI-8 crystallization from zincosilicate gels, Chem.–A Eur. J., 2002, 8(22), 5153–5160 CrossRef CAS.
- T. Takewaki, L. W. Beck and M. E. Davis, Zincosilicate CIT-6: a precursor to a family of *BEA-type molecular sieves, J. Phys. Chem. B, 1999, 103(14), 2674–2679 CrossRef CAS.
- H. Yamada, H. Horikawa, C. Anand, K. Ohara, T. Ina, A. Machida, S. Tominaka, T. Okubo, Z. Liu, K. Iyoki and T. Wakihara, Atom-selective analyses reveal the structure-directing effect of cs cation on the synthesis of zeolites, J. Phys. Chem. Lett., 2023, 3574–3580 CrossRef CAS.
- C. Sun, Z. Liu, S. Wang, H. Pang, R. Bai, Q. Wang, W. Chen, A. Zheng, W. Yan and J. Yu, Anionic tuning of zeolite crystallization, CCS Chem., 2021, 3(12), 189–198 CrossRef CAS.
- W. Kunz, J. Henle and B. W. Ninham, ‘Zur Lehre von der Wirkung der Salze’ (about the science of the effect of salts): franz Hofmeister's historical papers, Curr. Opin. Colloid Interface Sci., 2004, 9(1–2), 19–37 CrossRef CAS.
- X. Du, H. Zhang, X. Li, Z. Tan, H. Liu and X. Gao, Cation location and migration in lanthanum-exchanged NaY zeolite, Chin. J. Catal., 2013, 34(8), 1599–1607 CrossRef CAS.
- J. Song, L. Liu and S. Cai, Water desalination through FAU zeolite studied by using molecular dynamics simulations, J. Mol. Liq., 2023, 380, 121683 CrossRef CAS.
- K. Asselman, D. Vandenabeele, N. Pellens, N. Doppelhammer, C. E. A. Kirschhock and E. Breynaert, Structural aspects affecting phase selection in inorganic zeolite synthesis, Chem. Mater., 2022, 34(24), 11081–11092 CrossRef CAS.
- J. Devos, M. A. Shah and M. Dusselier, On the key role of aluminium and other heteroatoms during interzeolite conversion synthesis, RSC Adv., 2021, 11(42), 26188–26210 RSC.
- S. Farmanesh, B. G. Alamani and J. D. Rimer, Identifying alkali metal inhibitors of crystal growth: a selection criterion based on ion pair hydration energy, Chem. Commun., 2015, 51(73), 13964–13967 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The authors have cited additional references within the ESI. See DOI: https://doi.org/10.1039/d3sc05625b |
| This journal is © The Royal Society of Chemistry 2024 |