
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective poly(vinyl ether) upcycling via photooxidative degradation with visible light†
Darren L.
Langer
a,
Sewon
Oh
 a and
Erin E.
Stache
*ab
a and
Erin E.
Stache
*ab
aDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, USA. E-mail: estache@princeton.edu
bDepartment of Chemistry, Princeton University, Princeton, New Jersey 08544, USA
First published on 27th December 2023
Abstract
Poly(vinyl ethers) (PVEs) have many applications, such as adhesives, lubricants, and anticorrosive agents, thanks to their elastic, nonirritating, and chemically inert properties. The recycling of PVEs remains largely underexplored, and current methods lack generality towards other polymer classes. Thus, the chemical upcycling of PVE into small molecule feedstocks would provide an alternative approach to combat these current issues. Here, we report a visible light-mediated method of upcycling poly(isobutyl vinyl ether) (PIBVE) into small molecules via photooxidative degradation using chlorine or bromine radicals. PIBVE can be degraded to low molecular weight oligomers within 2 h, producing good yields of alcohols, aldehydes, and carboxylic acids. Mechanistic studies suggest that hydrogen atom transfer (HAT) from the backbone or the side chain leads to small molecule generation via oxidative cleavages. Additionally, this protocol was applied to a copolymer of poly(methyl acrylate-co-isobutyl vinyl ether) to demonstrate the preference for the degradation of polymers bearing more electron-rich C–H bonds through a judicious choice of abstraction agent. Ultimately, we show that photooxidative degradation enables the selective chemical upcycling of PVEs as a method of plastic waste valorization.
Introduction
The invention of synthetic polymers has transformed modern society. Their valuable and diverse properties and low cost of production have expanded their applications from single-use, disposable packaging materials to high-performance plastics. However, their chemical inertness, which makes polymeric materials useful for diverse applications, also hampers biodegradability and recyclability, leading to environmental persistence as pollutants for many decades.1–4 One notable family of synthetic polymers includes poly(vinyl ethers) (PVEs). Known for their elasticity, thermal stability, and chemical inertness, PVEs have found numerous uses in commercial and industrial applications, including adhesives, coatings, and lubricants.5–9 However, like many other polymeric materials, most PVE samples are disposed of as waste instead of being recycled. This is likely due to their amorphous physical properties, glass transition temperatures below room temperature, and high likelihood of being found in mixed polymer material, such as a block copolymer.10,11 As such, polymer degradation is the foremost opportunity for waste remediation.12Only a few examples of PVE recycling exist in the literature, mainly centered around novel polymer design with degradable moieties built into the backbone structure.13–15 However, the sustainable recycling of PVE remains largely underexplored and lacks generality in polymer degradation methods. To address this problem, we are interested in studying the chemical upcycling of PVE into valuable commodity chemicals as a promising alternative to reduce the accumulation of its wastes.16–18 While high-intensity UV light has been utilized for photochemically degrading polymers in the past,19–21 visible light-mediated polymer degradation has gained more attention in recent decades and shown potential to initiate chemical transformations under milder conditions.22
Recent advancements in photocatalysis have greatly enhanced efforts to design novel methods to chemically upcycle polymeric materials.23 One such process, photooxidative degradation, involves using light and photocatalysts to generate radicals on the polymer backbone. These radicals can then react with atmospheric oxygen and undergo oxidative chain scissions to cleave the polymer backbone. Chain end or side chain cleavage can then produce valuable small molecules. In our and others' previous works, polystyrene (PS) has been successfully converted to benzoic acid using hydrogen atom abstraction and white light (Scheme 1a).24–28a
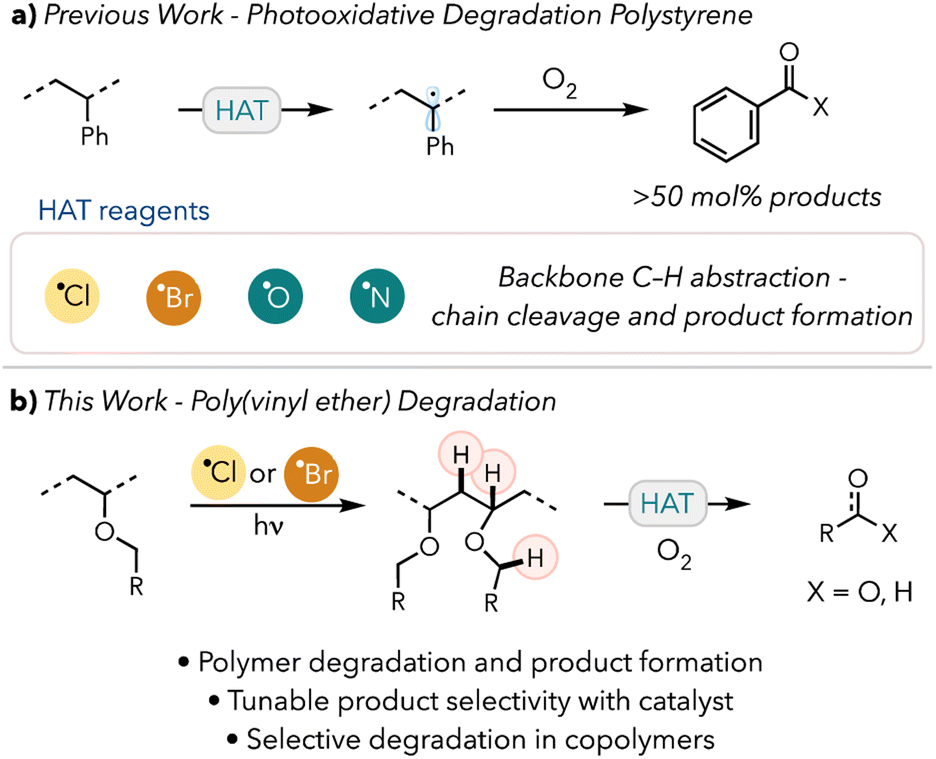 | ||
| Scheme 1 Proposed upcycling of poly(vinyl ether). | ||
We envisioned a similar photooxidative degradation pathway of PVE into commercially relevant small molecules, such as alcohols, aldehydes, or carboxylic acids (Scheme 1b). We hypothesized that the product distribution and yield would be tunable by modulating the reaction time or conditions. We have also shown that product distributions can be tuned by adjusting the thermodynamics of the HAT agent.28b In our recent work, we established that bromine radicals could convert PS preferentially into acetophenone over benzoic acid. This approach also enables selective degradation of hydridic C–H bond containing PVEs in the presence of more acidic C–H bond containing acrylates.
Result and discussion
Reaction optimization
We began our degradation studies using poly(isobutyl vinyl ether) (PIBVE) due to its ease of synthesis and commercial relevancy.29,30 We identified catalysts FeCl3 and FeBr3 due to their inexpensiveness, low level of toxicity, and ability to perform direct hydrogen atom transfer (HAT) under visible light.31–33 After some initial optimization (see ESI†), we observed that PIBVE degradation in acetone at short reaction times (Fig. 1a, entries 1 & 2) was more efficient in the presence of FeBr3—in terms of number average molecular weight (Mn)—than in the presence of FeCl3. However, the yield of small molecules was higher when using FeCl3 as a catalyst. We reasoned that small molecule generation may not always accompany C–C bond cleavage along the backbone. Additionally, at longer reaction times (entries 3 & 4), chlorine radical resulted in complete degradation of polymer (no detectable oligomer in the degradation mixture) relative to the use of bromine radical. Both hydrogen atom abstraction sources produced >20 mol% products (on a per monomer basis).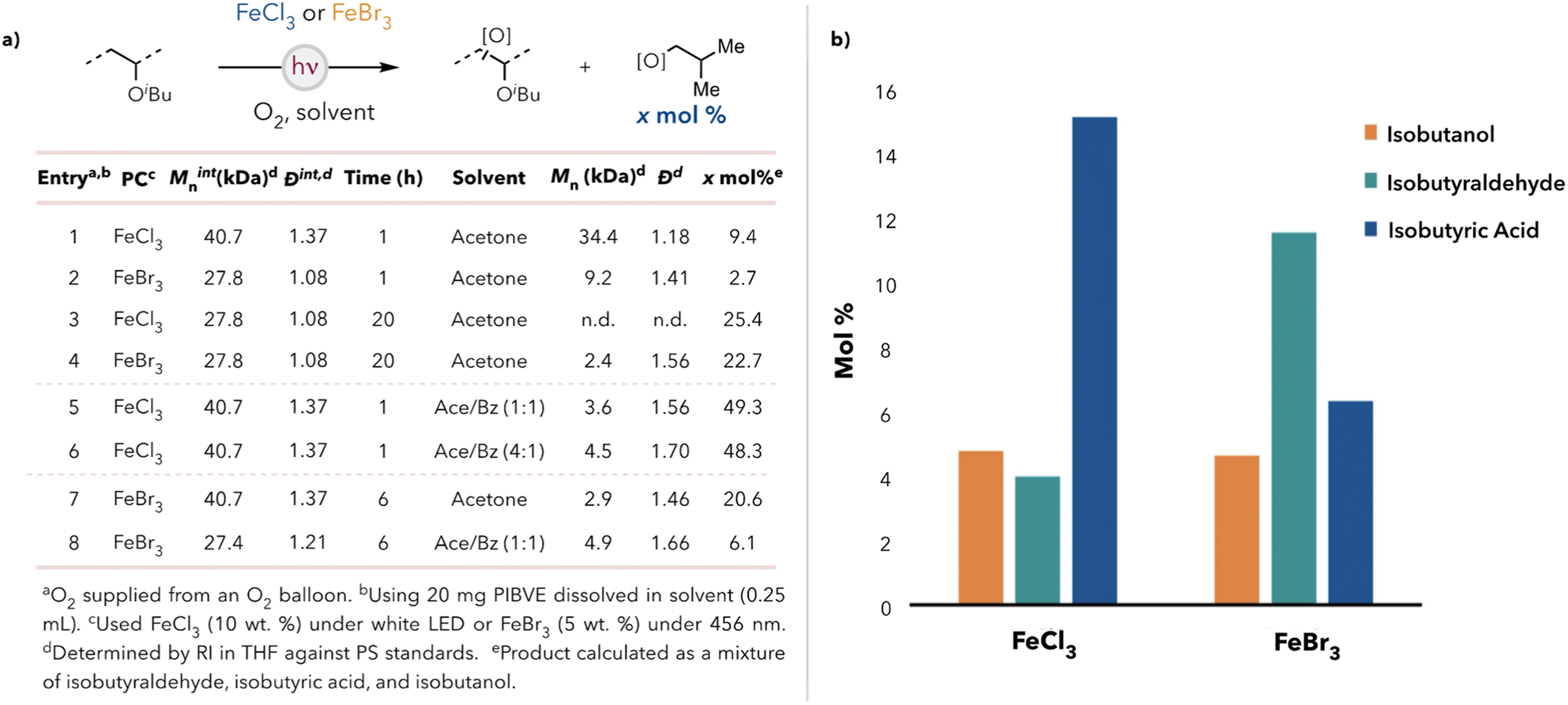 | ||
| Fig. 1 (a) Table for the PIBVE degradation results for FeCl3 and FeBr3. (b) Small molecule distributions for FeCl3 and FeBr3 catalyzed degradation in acetone after 20 h. | ||
Isobutanol, isobutyraldehyde, and isobutyric acid were identified by 1H NMR spectroscopy (see ESI† for the full spectrum) as the significant small molecule products after degradation and were quantified by gas chromatography (GC). Isobutyl formate and isobutyl isobutyrate were also detected but not quantified because of their negligible production amounts. Isobutyric acid was favored under chlorine radical generation, whereas isobutanol and isobutyraldehyde were favored using FeBr3 (Fig. 1b). These results were attributed to chlorine radicals being more reactive than bromine radicals due to the greater bond dissociation energy of HCl (103 kcal mol−1) relative to HBr (87 kcal mol−1).34 Consequently, the oxidation of isobutanol and isobutyraldehyde to isobutyric acid was more thermodynamically favorable in the presence of chlorine radicals than bromine radicals. Thus, control over the small molecule distribution can be achieved by altering the choice of photocatalyst. This is desirable as isobutanol is a known advanced biofuel, which allows for energy extraction from waste PIBVE.35
We hypothesized that the limited solubility of the polymer in acetone was limiting degradation efficiency. The reaction conditions were optimized to increase the degradation efficiency using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetone/benzene (v/v) co-solvent system instead of pure acetone to aid solubility. Using the co-solvent significantly increased the degradation efficiency with FeCl3. A 27.4 kDa PIBVE sample was observed to degrade to 3.6 kDa after 1 hour, generating 50 mol% small molecule products (Fig. 1a, entry 5). Additionally, the small molecule distribution consisted of mainly isobutanol (30 mol%), with isobutyraldehyde and isobutyric acid each nearly three times less abundant (10 mol%). Complete polymer degradation was observed after 4 hours (no detectable polymer peak in the GPC chromatogram). Reducing the benzene concentration to a 4:1 acetone/benzene co-solvent system with reduced reaction times maintained the degradation efficiency (Fig. 1a, entry 6).
:1 acetone/benzene (v/v) co-solvent system instead of pure acetone to aid solubility. Using the co-solvent significantly increased the degradation efficiency with FeCl3. A 27.4 kDa PIBVE sample was observed to degrade to 3.6 kDa after 1 hour, generating 50 mol% small molecule products (Fig. 1a, entry 5). Additionally, the small molecule distribution consisted of mainly isobutanol (30 mol%), with isobutyraldehyde and isobutyric acid each nearly three times less abundant (10 mol%). Complete polymer degradation was observed after 4 hours (no detectable polymer peak in the GPC chromatogram). Reducing the benzene concentration to a 4:1 acetone/benzene co-solvent system with reduced reaction times maintained the degradation efficiency (Fig. 1a, entry 6).
Interestingly, however, less efficient degradation was observed in the 1:1 acetone/benzene co-solvent system compared with a pure acetone solvent for the FeBr3 photocatalyst system (Fig. 1a, entries 7–8). This observation suggested polymer solvation was not the reason for the increased degradation efficiency and small molecule yield in the case of FeCl3. The literature has documented that benzene can stabilize halogen radicals, with an increased stabilizing effect with lower atomic number in the halogen series.36,37 HAT is less favorable for bromine radicals (ΔHdiss = 87 kcal mol−1) compared to chlorine radicals (ΔHdiss = 103 kcal mol−1). Given that HAT by bromine radicals is a more thermoneutral process, we hypothesized that arene-stabilized bromine radicals would experience a greater activation barrier for HAT on the polymer. In the case of chlorine radicals, we hypothesized that the arene-stabilized chlorine radicals would selectively target weaker C–H bonds than non-stabilized chlorine radicals due to the smaller thermodynamic downhill drop.38 Since the weakest C–H bonds are alpha to the oxygen atom on the side chain, HAT was localized to those positions when benzene was used as a co-solvent, in the case of FeCl3, which may explain the increased small molecule yield.
Mechanistic studies
Using our optimized reaction conditions, time course experiments were performed to quantify the amount of small molecule generation at each hour time point for 6 hours. The small molecule yield peaked at 2 hours (60 mol%), then gradually decreased and leveled off around 35 mol% at longer reaction times (Fig. 2a). Additionally, the yield of isobutanol and isobutyraldehyde peaked early in the reaction and then gradually converted to isobutyric acid at longer reaction times. We hypothesized that the major products formed during the degradation were isobutanol and isobutyraldehyde, which were then oxidized to isobutyric acid as the degradation continued. The decrease in the total small molecule yield after 2 hours was attributed to the potential oxidation of isobutyric acid to formic acid. To test these hypotheses, we subjected pure samples of the small molecules to the optimized reaction conditions. Indeed, the conversion of isobutanol and isobutyraldehyde to isobutyric acid was observed (see ESI† for more details). Additionally, when pure isobutyric acid was subjected to the reaction conditions, the percent recovery was approximately 80%, and formic acid was detected by 1H NMR spectroscopy.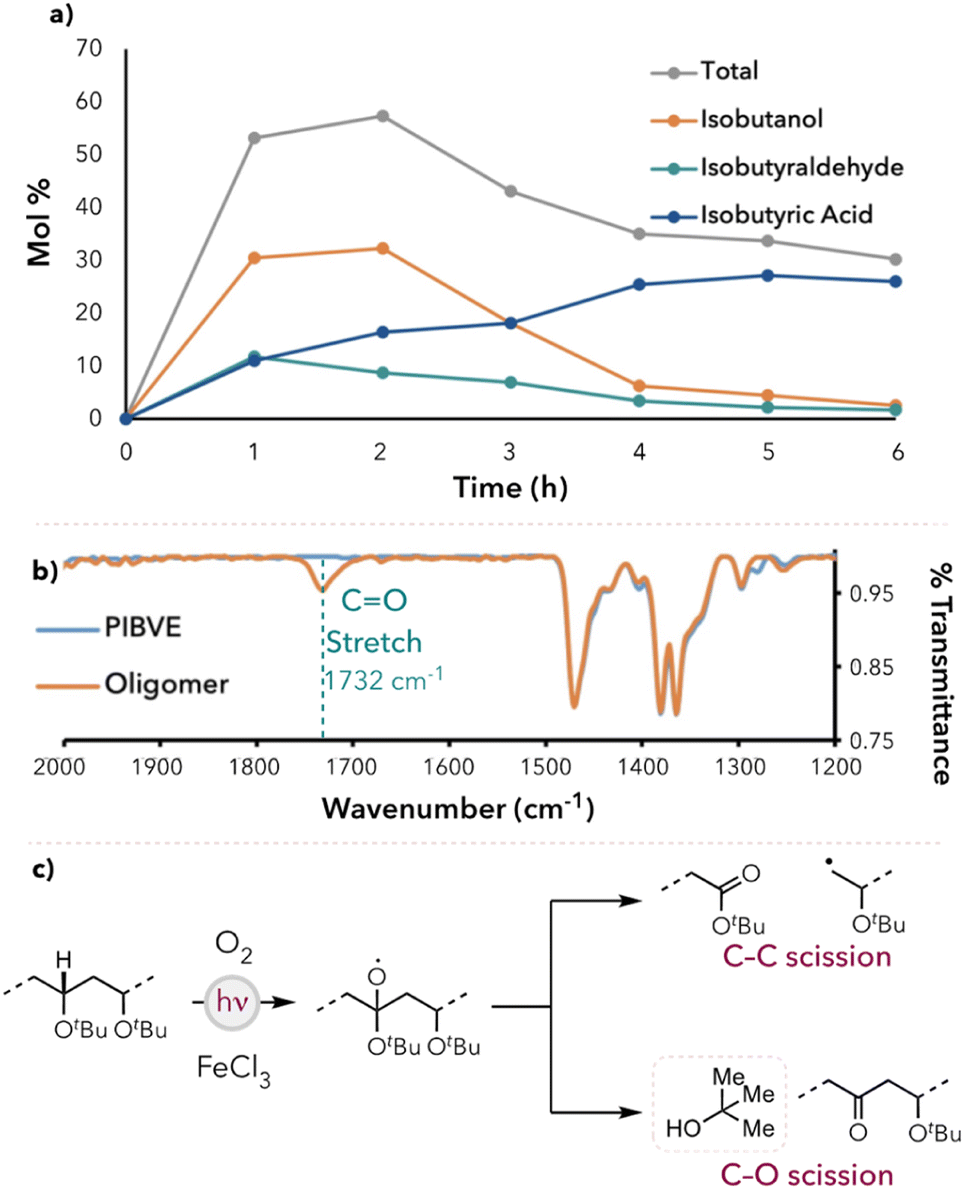 | ||
| Fig. 2 (a) Kinetic study of PIBVE degradation under FeCl3 in (1:1) acetone/benzene co-solvent system. (b) IR spectrum of PIBVE and partially degraded oligomer. (c) Degradation study of poly(tert-butyl vinyl ether). | ||
To characterize the partially degraded oligomers, a PIBVE sample was degraded for 3 hours, precipitated, and dried (see ESI† for more detailed procedures). IR studies revealed carbonyl groups (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O = 1732 cm−1) within the degraded oligomers, which indicated an oxidative degradation pathway (Fig. 2b).39–41 We also found similar observations in our previous work on polystyrene photooxidative degradation.28a
O = 1732 cm−1) within the degraded oligomers, which indicated an oxidative degradation pathway (Fig. 2b).39–41 We also found similar observations in our previous work on polystyrene photooxidative degradation.28a
To further investigate the mechanism of degradation and small molecule formation, the degradation of poly(tert-butyl vinyl ether) (PTBVE) was studied under the optimized reaction conditions. PTBVE is structurally similar to PIBVE but lacks an electron-rich hydrogen atom on the polymer side chain. This prevented the HAT agent from abstracting hydrogens from the side chain and directed HAT toward the polymer backbone. When degraded with FeCl3 in 1:1 acetone/benzene (v/v), tert-butyl alcohol was detected as the primary component in the degradation mixture (Fig. 2c). Additionally, the polymer molecular weight did not decrease below 2 kDa after 6 hours, indicating a decrease in degradation efficiency (relative to PIBVE degradation). We hypothesized that this may be attributed to the mechanism of tert-butyl alcohol formation, resulting in ketone moieties within the polymer backbone. Chlorine radicals are known to be electrophilic and thus selectively abstract electron-rich hydrogens.42,43 As more tert-butyl alcohol is generated, more carbonyl groups may be introduced into the backbone, causing the alpha-hydrogens to become less hydridic and thus result in less favorable HAT at those positions.
Proposed mechanism
Based on our studies, we propose the following degradation mechanism of PIBVE. For both iron photocatalysts, the halogen radical generation mechanism has been well documented in the literature (see ESI† for a more detailed photocatalytic cycle).44 The halogen radicals abstract hydric hydrogens either on the backbone or on the side chain to generate carbon-centered radicals, which are subsequently quenched by O2. In the case of the backbone C–H bond activation, β-scission occurs through two pathways: (1) C–C bond scission, resulting in backbone cleavage, or (2) C–O bond scission, forming a ketone moiety on the backbone while concurrently generating isobutanol (Fig. 3a). Conversely, the side chain C–H bond abstraction generates isobutyraldehyde, resulting in an oxygen-centered radical that eventually drives polymer chain cleavage (Fig. 3b). We hypothesized that, unlike PTBVE, PIBVE degradation is more efficient due to the significant number of available pathways leading to backbone cleavage.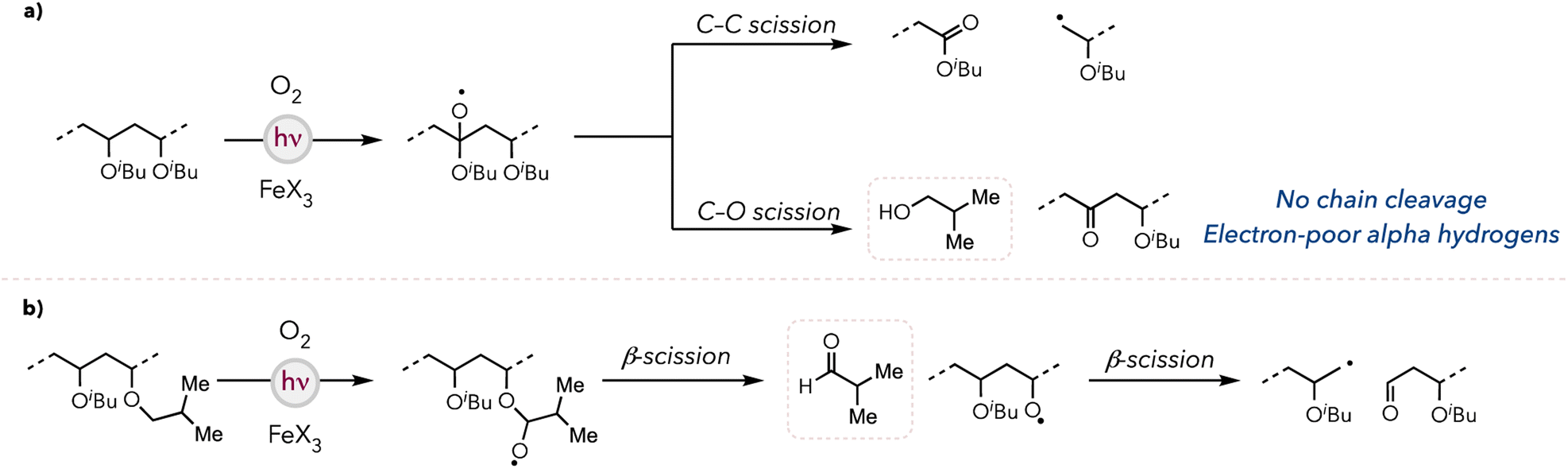 | ||
| Fig. 3 Proposed mechanism of (a) backbone C–H activation and (b) side chain C–H activation. | ||
Degradation selectivity
To assess the substrate scope of our optimized reaction conditions, we synthesized other polymers like poly(cyclohexyl vinyl ether) (PCyVE) for degradation studies (see the ESI† for detailed syntheses of polymers). The Mn and dispersities of the degraded oligomers and the small molecule generation are shown in Fig. 4. PCyVE was reduced in molecular weight and provided near equimolar amounts of cyclohexanol and cyclohexanone using chlorine radical (Fig. 4a). When changing the catalyst to FeBr3, degradation was slightly less efficient and favored cyclohexanone formation over cyclohexanol. As shown earlier, PTBVE is degraded to tert-butyl alcohol as the primary product in 44 and 30 mol% with chlorine and bromine radical, respectively (Fig. 4b).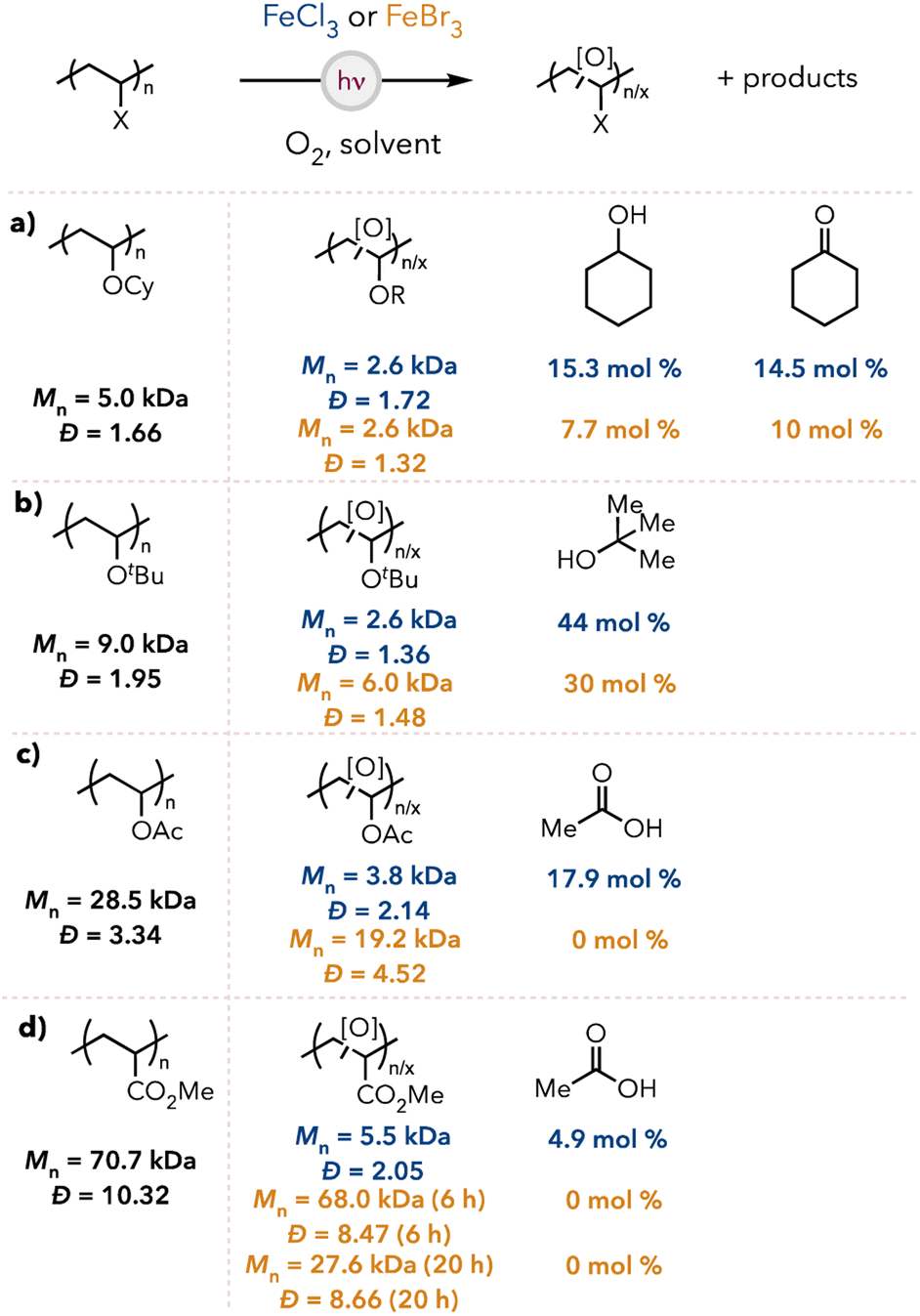 | ||
| Fig. 4 Expanded substrate scope degradation results for (a) PCyVE, (b) PTBVE, (c) PVAc, (d) PMA. FeCl3 runs in 1:1 acetone/benzene solvent for 2 hours and FeBr3 runs in acetone solvent (except PCyVE in 1:1 acetone/benzene) for 20 hours. PMA degradation with FeBr3 was run for 6 and 20 h. O2 supplied from an O2 balloon, using 20 mg polymer dissolved in solvent (0.25 mL). | ||
When we pivoted to less activated polymer backbones, such as poly(vinyl acetate) (PVAc), we still observed efficient degradation with chlorine radical and acetic acid formed as the primary product (Fig. 4c). However, bromine radical was not an efficient abstraction agent, with little observed degradation and no formation of acetic acid. We next examined poly(methyl acrylate) (PMA) due to the lack of hydridic C–H bonds in the polymer backbone. PMA underwent oxidative degradation using chlorine radical, forming oligomers and producing nearly 5 mol% of acetic acid. Bromine radical was ineffective for degrading PMA under standard conditions and resulted in only a few chain cleavages after extended reaction times (Fig. 4d).
Based on our observations with bromine radical-mediated HAT, we were interested if degradation selectivity could be achieved between polymers with electronically different C–H bonds. Indeed, when a mixture of PIBVE and PMA was subjected to the optimized reaction conditions of our FeBr3 system, mainly PIBVE degradation was observed, whereas PMA remained largely intact (Fig. 5a). Additionally, isobutanol, isobutyraldehyde, and isobutyric acid were detected as the major small molecule products (13 mol% yield). This indicated that selective degradation could be achieved within a polymer mixture with our FeBr3 system. Furthermore, when a poly(IBVE-co-MA) block copolymer was subjected to the FeBr3 optimized reaction conditions, degradation of the copolymer was observed, albeit slower than the degradation of pure PIBVE (Fig. 5). Furthermore, isobutanol, isobutyraldehyde, and isobutyric acid were detected as the small molecule products after degradation (12 mol% after 6 hours and 15 mol% after 20 hours, relative to IBVE repeat units in the block copolymer). Furthermore, the GPC trace of the degraded oligomers showed a bimodal peak, which became more pronounced at longer reaction times, suggesting differing degradation efficiencies between the MA and IBVE blocks (see ESI† for the GPC traces). These observations indicated that degradation was mainly localized to the IBVE block of the copolymer (the PMA block had some IBVE units substituted within the chain, which may have allowed for cleavage of the PMA block at these sites), which corroborated our hypothesis that selective degradation could be achieved with our system in a copolymer.
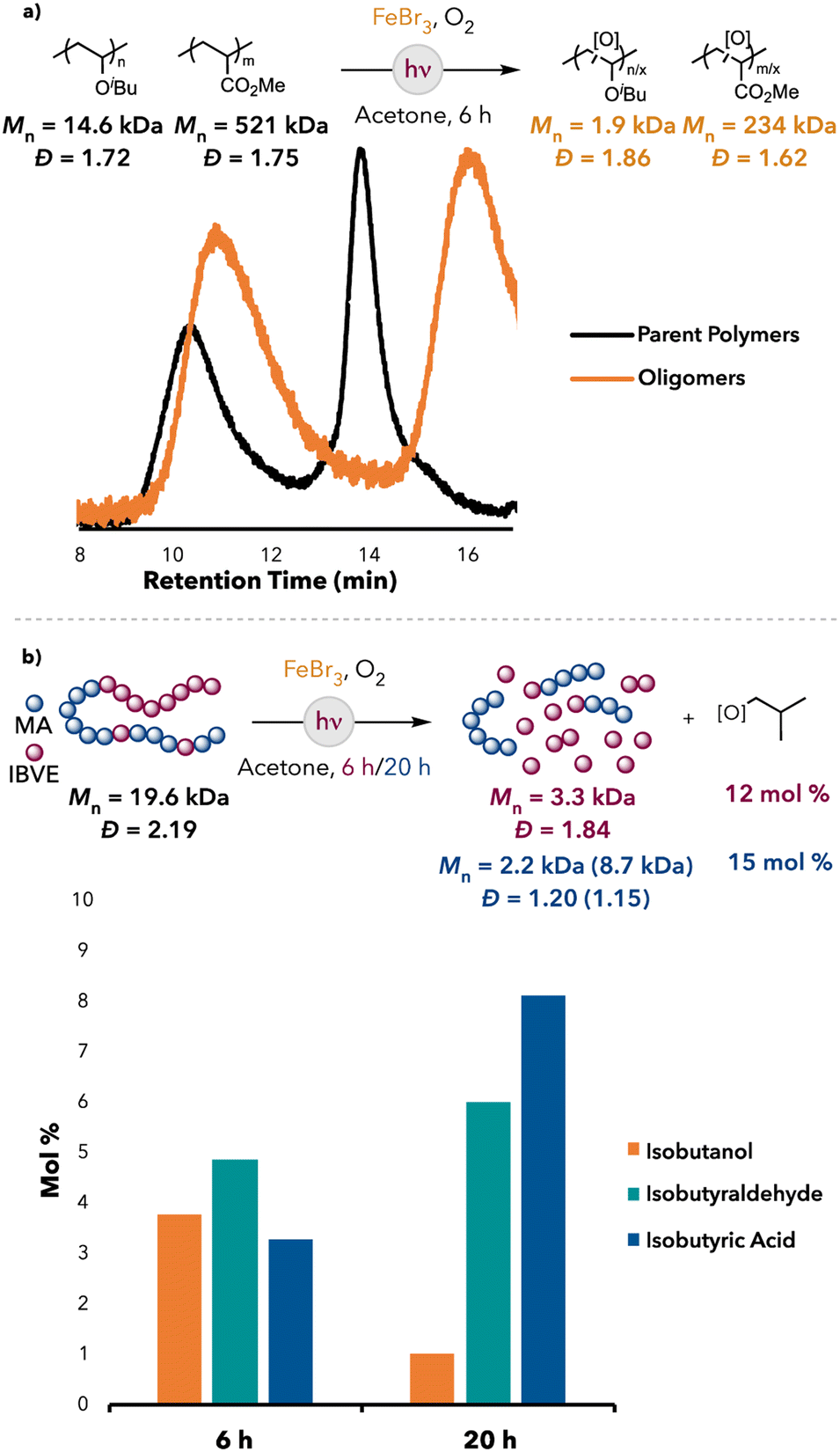 | ||
| Fig. 5 (a) PIBVE and PMA mixture degradation with FeBr3. (b) Poly(IBVE-co-MA) degradation results: Lower Mn peak (higher Mn peak). | ||
Conclusions
In summary, we report a sustainable, low-energy method of PVE upcycling to valuable small molecule feedstock. Additionally, control over the small molecule distribution can be achieved by altering the reaction conditions—FeCl3 favors oxidation to the carboxylic acid, whereas FeBr3 is less oxidizing, allowing for higher yields of alcohols and aldehydes. The addition of benzene as a co-solvent provides for an increase in small molecule yield, as well as a decrease in the reaction time. Lastly, we have shown the potential for our optimized system to selectively degrade polymers with different steric and electronic properties.Data availability
All experimental and characterization details are available in the ESI.†Author contributions
D. L. L. conducted experiments. D. L. L., S. O., and E. E. S. discussed the results and wrote the manuscript. E. E. S. supervised and led this project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported in part by Cornell University. E.E.S. thanks the Department of Energy, Office of Science, Office of Basic Energy Sciences, Catalysis Science Early Career Award (DE-SC0024412). We thank the NMR Facility at Cornell University, supported, in part, by the National Science Foundation under Award CHE-1531632. We also thank Cornell Center for Materials Research Facilities supported by the National Science Foundation under Award DMR-1719875. We thank the Fors group for use of their GPC and the Milner group for their FT-IR and GC-MS.Notes and references
- R. Geyer, R. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- A. Albertsson and M. Hakkarinen, Science, 2017, 358, 872–873 CrossRef CAS PubMed.
- B. Singh and N. Sharma, Polym. Degrad. Stab., 2008, 93, 561–584 CrossRef CAS.
- W. Kaminsky and I. J. N. Zorriqueta, J. Anal. Appl. Pyrolysis, 2007, 79, 368–374 CrossRef CAS.
- H. Kalita, S. Selvakumar, A. Jayasooriyamu, S. Fernando, S. Samanta, J. Bahr, S. Alam, M. Sibi, J. Vold, C. Ulven and B. J. Chisholm, Green Chem., 2014, 16, 1974–1986 RSC.
- H. Kammiyada, M. Ouchi and M. Sawamoto, Macromol, 2017, 50, 841–848 CrossRef CAS.
- T. Hashimoto, A. Takahashi, M. Urushisaki and T. Sakaguchi, J. Polym. Sci., 2010, 48, 1641–1648 CrossRef CAS.
- E. Goethals, W. Reyntijens and S. Lievens, Macromol. Symp., 2011, 132, 57–64 CrossRef.
- H. Akiyama, M. Mori, M. Yoshida and H. Kihara, J. Polym. Sci., 2020, 58, 568–577 CrossRef.
- Y. Takahashi, H. Suzuki, Y. Nakagawa, M. Yamaguchi and I. Noda, Polym. J., 1991, 23, 1333–1337 CrossRef CAS.
- I. A. Ignatyev, W. Thielemans and B. V. Beke, ChemSusChem, 2014, 7, 1579–1593 CrossRef CAS PubMed.
- H. Zhou, Y. Wang, Y. Ren, Z. Li, X. Kong, M. Shao and H. Duan, ACS Catal., 2022, 12, 9307–9324 CrossRef CAS.
- M. Uchiyama, Y. Murakami, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2023, 62, e202215021 CrossRef CAS PubMed.
- V. Deplace and J. Nicolas, Nat. Chem., 2015, 7, 771–784 CrossRef PubMed.
- E. Elizondo, A. Córdoba, S. Sala, N. Ventosa and J. Veciana, J. Supercrit. Fluids, 2010, 53, 108–114 CrossRef CAS.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- T. Kimura and M. Ouchi, Angew. Chem., Int. Ed., 2023, 135, e20230525 CrossRef.
- C. Li, X. Y. Kong, M. Lyu, X. T. Tay, M. Ðokić, K. F. Chin, C. T. Yang, E. K. X. Lee, J. Zhang, C. Y. Tham, W. X. Chan, W. J. Lee, T. T. Lim, A. Goto, M. B. Sullivan and H. A. Soo, Chem, 2023, 9, 2683–2700 CAS.
- E. Yousif and R. Haddad, SpringerPlus, 2013, 2, 398 CrossRef PubMed.
- P. Gijsman and M. Diepens, ACS Symp. Ser., 2009, 1004, 287–306 CrossRef CAS.
- P. Gijsman, G. Meijers and G. Vitarelli, Polym. Degrad. Stab., 1999, 65, 433–441 CrossRef CAS.
- K. Hakobyan, T. Gegenhuber, C. S. P. McErlean and M. Müllner, Angew. Chem., Int. Ed., 2019, 58, 1828–1832 CrossRef CAS PubMed.
- Y. Zhang, M. Qi, Z. Tang and Y. Xu, ACS Catal., 2023, 13, 3575–3590 CrossRef CAS.
- G. Zhang, Z. Zhang and R. Zeng, Chin. J. Chem., 2021, 39, 3225–3230 CrossRef CAS.
- Z. Huang, M. Shanmugam, Z. Liu, A. Brookfield, E. L. Bennett, R. Guan, D. E. V. Herrera, J. A. Lopez-Sanchez, A. G. Slater, E. J. L. McInnes, X. Qi and J. Xiao, J. Am. Chem. Soc., 2022, 144, 6532–6542 CrossRef CAS PubMed.
- M. Wang, J. Wen, Y. Huang and P. Hu, ChemSusChem, 2021, 14, 5049–5056 CrossRef CAS PubMed.
- T. Li, A. Vijeta, C. Casavevall, A. S. Gentleman, T. Euser and E. Reisner, ACS Catal., 2022, 12, 8155–8163 CrossRef CAS PubMed.
- (a) S. Oh and E. E. Stache, J. Am. Chem. Soc., 2022, 144, 5745–5749 CrossRef CAS PubMed; (b) S. Oh and E. E. Stache, ACS Catal., 2023, 13, 10968–10975 CrossRef CAS.
- A. J. Teator and F. A. Leibfarth, Science, 2019, 363, 1439–1443 CrossRef CAS PubMed.
- V. Kottisch, J. Jermaks, J. Mak, R. A. Woltornist, T. H. Lambert and B. P. Fors, Angew. Chem., Int. Ed., 2020, 60, 4535–4539 CrossRef PubMed.
- P. Wardman and L. P. Candeias, Radiat. Res., 1996, 145, 523–531 CrossRef CAS PubMed.
- X. Wang, C. Shi, M. Yang, Y. Ma, Y. Chen, T. Lu, W. Tang and J. Feng, Asian J. Org. Chem., 2023, 12, e202300077 CrossRef CAS.
- Z. Dai, S. Zhang, X. Hong, P. Wang and L. Gong, Chem Catal., 2022, 2, 1211–1222 CrossRef CAS.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- A. M. Brownstein, Renewable Motor Fuels: The Past, The Present and The Future, Butterworth-Heinemann, Oxford, 2014, ch. 5, p. 47 Search PubMed.
- M. Tsao, C. M. Hadad and M. S. Platz, J. Am. Chem. Soc., 2003, 125, 8390–8399 CrossRef CAS PubMed.
- G. Russell, J. Am. Chem. Soc., 1958, 80, 4987–4996 CrossRef CAS.
- M. I. Gonzalez, D. Gygi, Y. Qin, Q. Zhu, E. J. Johnson, Y. Chen and D. G. Nocera, J. Am. Chem. Soc., 2022, 144, 1464–1472 CrossRef CAS PubMed.
- E. Pelizzetti and C. Minero, Electrochim. Acta, 1993, 38, 47–55 CrossRef CAS.
- X. Tao, W. Ma, T. Zhang and J. Zhao, Angew. Chem., Int. Ed., 2001, 40, 3014–3016 CrossRef CAS PubMed.
- O. Chiantore, L. Trossarelli and M. Lazzari, Polym. J., 2000, 41, 1657–1668 CAS.
- B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25–35 RSC.
- F. D. Vleeschouwer, V. V. Speybroeck, M. Waroquier, P. Geerlings and F. D. Proft, Org. Lett., 2007, 9, 2721–2724 CrossRef PubMed.
- J. Rabek, Compr. Chem. Kinet., 1975, 14, 425–538 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05613a |
| This journal is © The Royal Society of Chemistry 2024 |