
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCycl[2,2,4]azine-embedded non-alternant nanographenes containing fused antiaromatic azepine ring†
Lan
Ruan
a,
Wanhua
Luo
a,
Haifan
Zhang
a,
Peng
Liu
a,
Yong
Shi
 a and
Peng
An
*ab
a and
Peng
An
*ab
aSchool of Chemical Science and Technology, Yunnan University, Kunming 650091, P. R. China. E-mail: anp@ynu.edu.cn
bKey Laboratory of Medicinal Chemistry for Natural Resource, Ministry of Education, Yunnan University, Kunming 650091, P. R. China
First published on 14th December 2023
Abstract
The development of non-alternant nanographenes has attracted considerable attention due to their unique photophysical properties. Herein, we reported a novel aza-doped, non-alternant nanographene (NG) 1 by embedding the cycl[2,2,4]azine unit into the benzenoid NG framework. Single-crystal X-ray diffractometry suggests saddle or twisted nonplanar geometry of the entire backbone of 1 and coplanar conformation of the cycl[2,2,4]azine unit. DFT calculation together with solid structure indicates that NG 1 possesses significant local antiaromaticity in the azepine ring. By oxidative process or trifluoroacetic acid treatment, this nanographene can transform into a mono-radical cation, which was confirmed by UV/Vis absorption, 1H NMR, and electron paramagnetic resonance (EPR) spectroscopy. The antiaromaticity/aromaticity switching of the azepine ring on 1˙+ from 1 enables the high stability of this radical cation, which remained intact for over 1 day. Due to the electron-donating nature of the nitrogen and the unique electronic structure, NG 1 exhibits strong electron-donating properties, as proved by the intermolecular charge transfer towards C60 with a high association constant. Furthermore, selective modification of NG 1 was accomplished by Vilsmeier reaction, and the derivatives 7 and 8 with substituted benzophenone were obtained. The photophysical and electronic properties can be tuned by the introduction of different electronic groups in benzophenone.
Introduction
Non-alternant topological polycyclic conjugated hydrocarbons (PCHs) have recently received significant attention due to their intriguing optoelectronic properties, antiaromatic characteristics, and open-shell features.1 Among non-alternant ring structures, azulene is a prominent bicyclic unit because of its unique physicochemical properties, and it is the most commonly employed non-alternant building block for constructing non-alternant nanographenes (NGs).2 Meanwhile, various hydrocarbon-based non-alternant NGs containing three or more non-benzenoid rings have recently been developed by in-solution or on-surface chemistry.3 In comparison with their benzenoid counterparts, these non-alternant NGs normally exhibit significantly different structural conformations and electronic properties.2a,3a,4Introducing heteroatoms or non-alternant units into benzenoid polycyclic aromatic hydrocarbons (PAHs) is an efficient strategy to alter or tailor their intrinsic optoelectronic properties.2,4,5 Notably, phenalene (Fig. 1a), as an odd alternant non-Kekulé tricyclic hydrocarbon, has drawn great attention in multiple areas and is normally showed in phenalenyl form.6 Phenalenyl molecules can exist in three different redox states: cation, radical, and anion, which can be generated from phenalene by abstracting a hydride ion, hydrogen atom, and proton, respectively. In contrast, by doping nitrogen into the phenalene center, cycl[3,3,3]azine (Fig. 1a), as the nitrogen-doped phenalene, can be obtained,7 which shows stable Kekulé structure in comparison with phenalene. Due to the unique electronic structure of phenalenyl, so far, the (aza)-phenalenyl-based NGs like triangulene, aza-triangulene, and rhombene ([4]-rhombene as an example, Fig. 1b) show attractive magnetic or open-shell properties due to the formation of di- or poly-radicaloid high spin states.8
 | ||
| Fig. 1 (a) Structures of phenalene and azaphenalene (cycl[3,3,3]azine). (b) Closed-shell Kekulé and open-shell non-Kekulé structures of rhombus-shaped nanographene [4]-rhombene. (c) Structures of cycl[2,2,4]azine, cycl[2,2,4]azine anion, and cycl[2,2,4]azine cation. (d) The designed cycl[2,2,4]azine-embedded nanographene. | ||
To explore interesting nitrogen-doped, non-alternant NGs, significant efforts have so far resulted in some aesthetically pleasing nitrogen-doped non-alternant NGs by “bottom-up” strategies.9 Herein, we reported a newly cycl[2,2,4]azine-embedded NG 1 (Fig. 1d) containing 16 fused rings. Cycl[2,2,4]azine is a non-alternant, non-Kekulé building block (Fig. 1c)10 containing 12 atoms with nitrogen-centred, 5/5/7 fused ring system. Interestingly, a computational study suggested that the cycl[2,2,4]azine cation and cycl[2,2,4]azine anion (Fig. 1c), possessing 12 and 14 electrons, exhibited aromaticity and antiaromaticity, respectively.10c By the introduction of this tricyclic unit, NG 1 holds 48π electrons, the same as [4]-rhombene (Fig. 1b), and an extra lone pair of electrons on the nitrogen. The single-crystal structure demonstrates the curved structure of 1. Notably, with this unique electronic structure, NG 1 acquiring a highly antiaromatic azepine ring, can form relatively stable radical cations with switching the aromaticity of azepine ring by losing one electron. Due to the high degree of electron-rich property, NG 1, as an electron donor, can readily transfer the electron to electron-withdrawing C60 species with a significantly high association constant. Moreover, owing to the electron-donating effect of the nitrogen atom, the site-selective functionalization of NG 1 is demonstrated, and the optoelectronic properties can be tuned by introducing different benzoyl groups.
Results and discussion
Synthesis of nitrogen-doped nanographene 1
As shown in Scheme 1, cycl[2,2,4]azine-embedded nanographene 1 was synthesized in a five-step process by taking advantage of the azepine-containing hexaphenylbenzene 2 synthesized in our previous work.11 Reaction of 2 with acetyl chloride in the presence of 4-dimethylaminopyridine (DMAP) provided the Ac-protected nanographene precursor. Scholl-type cyclodehydrogenation by the treatment of the above precursor with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ, 10.0 equiv.) in the presence of triflic acid (11.0 equiv.) in DCM in an ice-water bath resulted in the fully fused Ac-NG 3 within 12 minutes in 80% yield in two steps with the formation of five new C–C bonds. The deprotection of the acetyl group with LiAlH4 in tetrahydrofuran (THF) afforded aza-NG 4 in 55% yield. | ||
| Scheme 1 Synthesis of cycl[2,2,4]azine-embedded nanographene 1. | ||
Then, the palladium-catalyzed C–N coupling between 4 and 2,6-dichloroiodobenzene afforded 5 in 80% yield. Finally, intramolecular cyclization of 5 in the presence of [Pd(PCy3)2Cl2] and an excess amount of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in N,N-dimethylacetamide (DMAc) furnished NG 1 in 85% yield. All the purified compounds were structurally elucidated by 1H and 13C NMR spectroscopy and high-resolution MALDI-TOF-MS. The 1H NMR spectrum of NG 1 clearly showed four distinct singlets at δ from 9.1 to 8.5 and two AX spin systems (Fig. S26†), indicating a Cs symmetry.
Structural analysis of NG 1 and its radical cation NG 1˙+
The exact structure of nanographene 1 was elucidated by single-crystal X-ray analysis, which indicated its nonplanar geometry (Fig. 2a–c, Table S1†). In detail, NG 1 crystallized in a triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two conformations (1
with two conformations (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) presented in the crystal structure: a saddle-shaped, negatively curved structure (Fig. 2b) and a twisted, nonplanar structure (Fig. 2c). For the saddle-shaped conformation (Fig. 2a and b), it exhibited a nearly symmetric structure, and the depths of the saddles defined as the perpendicular distance from the center of ring A to the line across C10 and C11 and the distance between the center of ring A to the plane (C9–C12–C13) were 1.0, and 0.8 Å, respectively. Conversely, the twisted geometry of 1 revealed an asymmetrical torsional conformation, in which the torsion angles were 15.9° and 0° for C14–C15–C16–C17 and C18–C19–C20–C21, respectively (Fig. 2c). The nitrogen center is nearly planar with the three C–N–C angles summing to 356° and 353° for saddle and twisted-shaped structures, respectively, suggesting sp2 hybridization of the nitrogen center (Fig. 2a and S1b†). Moreover, the seven-membered azepine ring revealed an almost planar conformation, in which the sum of seven angles is close to 900° (893° and 894° for twisted and saddle-shaped conformations, respectively, Fig. S1†). The bond lengths of the azepine ring adopted an uneven distribution. For example, with respect to the saddle-shaped conformation, the two N–C bonds (N1–C2 and N1–C7) were 1.38 and 1.37 Å, respectively, which falls between the typical lengths for carbon–nitrogen single bonds and carbon–nitrogen double bonds, respectively, while the bonds of C2–C3 and C6–C7, close to 1.40 Å, were within the typical range for carbon–carbon bonds in benzene rings. In contrast, the bond lengths of C3–C4 and C5–C6 were 1.49 and 1.50 Å, respectively, similar to that of carbon–carbon single bonds. The alternating bond lengths indicate the low aromaticity of the azepine ring. Meanwhile, the localized orbital locator (LOL)12 of π molecular orbitals calculated based on the DFT-optimized structure revealed that the electrons were localized on the eight hexagonal rings as well as on the nitrogen, and the electrons were not fully delocalized around the cycl[2,2,4]azine unit (Fig. 2d). The valence electron density13 of NG 1 (Fig. 2e) also suggested that the electron densities of the C3–C4 and C5–C6 bonds were remarkably lower than those of the other carbon–carbon bonds due to the lack of π electrons. Although the cycl[3,3,3]azine unit in NG 1 displayed planar conformation, the X-ray, LOL-π, and valence electron density suggested a low aromaticity of the seven-membered ring. In addition, the electrostatic potential (ESP) plot of NG 1 revealed a fairly homogeneous distribution of electrons on the sp2 carbon plane with a negative potential of up to −19 kcal mol−1 (Fig. 2e). Due to the introduction of the nitrogen, an ESP maximum of −2.33 kcal mol−1 was observed on the surface above nitrogen.
:1) presented in the crystal structure: a saddle-shaped, negatively curved structure (Fig. 2b) and a twisted, nonplanar structure (Fig. 2c). For the saddle-shaped conformation (Fig. 2a and b), it exhibited a nearly symmetric structure, and the depths of the saddles defined as the perpendicular distance from the center of ring A to the line across C10 and C11 and the distance between the center of ring A to the plane (C9–C12–C13) were 1.0, and 0.8 Å, respectively. Conversely, the twisted geometry of 1 revealed an asymmetrical torsional conformation, in which the torsion angles were 15.9° and 0° for C14–C15–C16–C17 and C18–C19–C20–C21, respectively (Fig. 2c). The nitrogen center is nearly planar with the three C–N–C angles summing to 356° and 353° for saddle and twisted-shaped structures, respectively, suggesting sp2 hybridization of the nitrogen center (Fig. 2a and S1b†). Moreover, the seven-membered azepine ring revealed an almost planar conformation, in which the sum of seven angles is close to 900° (893° and 894° for twisted and saddle-shaped conformations, respectively, Fig. S1†). The bond lengths of the azepine ring adopted an uneven distribution. For example, with respect to the saddle-shaped conformation, the two N–C bonds (N1–C2 and N1–C7) were 1.38 and 1.37 Å, respectively, which falls between the typical lengths for carbon–nitrogen single bonds and carbon–nitrogen double bonds, respectively, while the bonds of C2–C3 and C6–C7, close to 1.40 Å, were within the typical range for carbon–carbon bonds in benzene rings. In contrast, the bond lengths of C3–C4 and C5–C6 were 1.49 and 1.50 Å, respectively, similar to that of carbon–carbon single bonds. The alternating bond lengths indicate the low aromaticity of the azepine ring. Meanwhile, the localized orbital locator (LOL)12 of π molecular orbitals calculated based on the DFT-optimized structure revealed that the electrons were localized on the eight hexagonal rings as well as on the nitrogen, and the electrons were not fully delocalized around the cycl[2,2,4]azine unit (Fig. 2d). The valence electron density13 of NG 1 (Fig. 2e) also suggested that the electron densities of the C3–C4 and C5–C6 bonds were remarkably lower than those of the other carbon–carbon bonds due to the lack of π electrons. Although the cycl[3,3,3]azine unit in NG 1 displayed planar conformation, the X-ray, LOL-π, and valence electron density suggested a low aromaticity of the seven-membered ring. In addition, the electrostatic potential (ESP) plot of NG 1 revealed a fairly homogeneous distribution of electrons on the sp2 carbon plane with a negative potential of up to −19 kcal mol−1 (Fig. 2e). Due to the introduction of the nitrogen, an ESP maximum of −2.33 kcal mol−1 was observed on the surface above nitrogen.
 | ||
| Fig. 2 (a) X-ray crystallographic structure of nanographene 1 with 50% probability of thermal ellipsoids.22 Inset: zoomed view of the nitrogen atom in the structure 1 with C–N–C dihedral angles. Side view of nanographene 1 with saddle-shaped (b) or twisted (c) conformations. (d) Isosurface map of localized orbital locator-π (LOL-π) of nanographene 1, isovalue = 0.4. (e) Isosurface map of valence electron density of nanographene 1, isovalue = 0.26. (f) Electrostatic potential (ESP) map and the energy color bar of nanographene 1. | ||
Since the azepine ring adopted 8π electrons, we sought to test the single-electron oxidation process to acquire the stable radical cation of NG 1. Chemical oxidation titrations of NG 1 in CH2Cl2 with AgSbF6 (0–10.0 equiv.) changed the solution color from yellow to brown (Fig. 3a) with a loss of fluorescence (Fig. S2a†). The absorption exhibited a gradual decrease in the absorption bands at ∼350 nm and ∼375 nm and the appearance and growth of new peaks at ∼500 nm and ∼580 nm with a tail that extended to ∼700 nm. The fluorescence emission peak at 530 nm gradually decreased upon the addition of oxidant (Fig. S2b†). The oxidized species can be ascribed to the formation of radical cation NG 1˙+. As monitored by 1H NMR spectra (Fig. S3†), 8 proton signals in the aromatic region disappeared upon adding 10.0 equiv. of AgSbF6, suggesting the generation of the open-shell species. The radical character was further evidenced by electron paramagnetic resonance (EPR) spectroscopy. The EPR spectrum of NG 1˙+ solution in CH2Cl2 at 298 K displayed a single line with a Lorentzian line shape and a g value of 2.003 (Fig. 3c). The spin density theoretical calculation of NG 1˙+ (CAM-UB3LYP/6-311G*) suggested that the spin densities were delocalized mainly at the central nitrogen and the three benzene rings fused with azepine (Fig. 3d). The radical cation 1˙+ proved to be highly stable. The UV/Vis absorption spectra indicated no detectable degradation for 5 h in open air at room temperature or 1 day under argon atmosphere at 0 °C (Fig. 3b). The half-life was determined as ∼5 days in open air at room temperature by monitoring the changes in the UV/Vis spectra (Fig. S4†). Interestingly, the radical cation 1˙+ could also form by the protonation of NG 1 with trifluoroacetic acid (TFA). A similar spectral change of the UV/Vis, fluorescence emission, and 1H NMR spectra of 1 was observed when 100 equiv. of TFA was added (Fig. S3 and S5†) in comparison with the oxidation process, which indicated that the radical formation was induced by protonation. This acid-induced radical cation formation suggested that NG 1 exhibits electron-rich properties with high reducing ability.
 | ||
| Fig. 3 (a) Oxidation titration of nanographene 1. The spectral changes in the UV/Vis absorption spectra in CH2Cl2 were recorded by gradually adding AgSbF6; inset: photographs of nanographene 1 solution before and after adding 10 equiv. of AgSbF6 under ambient conditions. (b) Time-dependent UV/Vis spectra of 10−5 mol L−1 nanographene 1˙+; inset: time-dependent UV/Vis spectra between 460 and 780 nm for nanographene 1˙+ at 0 °C in argon atmosphere. (c) EPR spectrum (ν = 9.8525 GHz, 3500 ± 200 G) of nanographene radical cation 1˙+. (d) Spin density distribution of nanographene 1˙+ (UCAM-B3LYP/6-311G*; blue: positive spin, white: negative spin; isosurface plotted at 0.003). | ||
Further insights into the aromaticity of NG 1 and its radical cation 1˙+ were obtained through DFT calculations. Nucleus-independent chemical shift (NICS)14 calculations [at the GIAO-B3LYP/6-311+G(d,p) level of theory] were first performed for 1 and 1˙+. The average of NICS(1) (determined as the average value of NICS(1) on both sides of each plane) was selected as the criterion for aromaticity because the two sides of the curved molecules are not strictly equivalent. As shown in Fig. 4a, rings A, D, E, G, and J showed typical aromatic features with NICS(1) values ranging from −7.43 to −10.97. Rings B, F, I, and H exhibited a weakened aromatic nature with corresponding NICS(1) values ranging from −5.72 to −1.6. In contrast, the NICS(1) of the seven-membered ring was 7.08, indicating local antiaromaticity.
 | ||
| Fig. 4 (a) Calculated NICS(1) and HOMA values of nanographene 1 and nanographene 1˙+. (b) Calculated 3D ICSS map of nanographene 1 and nanographene 1˙+, isovalue = 3. The blue and green wireframes represent magnetically shielded and deshielded regions, respectively. (c) Calculated 2D ICSS(1)zz plots of nanographene 1 and nanographene 1˙+ at 1 Å above the plane. The positive and negative values represent aromatic and antiaromatic regions, respectively. | ||
The calculated harmonic oscillator measure of aromaticity (HOMA)15 provided the same tendency, whereby rings A, D, G, and J showed a high degree of aromaticity with HOMA values close to 1, indicating Clar sextets. Whereas the azepine ring gave the lowest level of aromaticity with the smallest HOMA value of 0.019. Notably, the azepine ring in NG 1 exhibited a more positive NICS(1) value (7.08 vs. 2.62) and a smaller HOMA value (0.019 vs. 0.184) than the azepine ring in compound 4 (Fig. S6†), which were in accord with the 3D iso-chemical shielding surface (ICSS) map (Fig. S7†), suggesting a higher degree of antiaromaticity of the cycl[2,2,4]azine ring pattern than the simple azepine ring in a polycyclic conjugated ring system. Compared with closed-shell NG 1, radical cation 1˙+ displayed significantly higher aromaticity with NICS(1) of −2.15 and HOMA of 0.185 for its seven-membered ring. The 3D ICSS map16 clearly revealed a deshielding green cavity in the central heptagon ring in NG 1, but almost all blue shielding surfaces for NG 1˙+ (Fig. 4b and S8†), suggesting a higher aromaticity of 1˙+ than that of 1. Likewise, the antiaromatic azepine ring was also visualized in 2D ICSS(1)zz plots with a negative value; thus, the entire structure for NG 1˙+ displayed a different degree of aromaticity with positive values (Fig. 4c). Due to the increasing aromaticity of radical cation 1˙+, NG 1 was prone to form the radical cation by losing one electron and showed relatively high stability, even without any steric hindrance.
Chemical modification of NG 1
It is always challenging to selectively functionalize the hydrocarbon nanographene due to the inert nature of the C–C or C–H bonds. Some synthetic strategies have been devoted to functionalize polycyclic aromatic hydrocarbons or nanographenes, including formylation, chlorination, alkylation, and N–C cross-coupling reactions.11,17 With the embedded electron-rich nitrogen, further modification of NG 1 was attempted using different formylation strategies. To our delight, the formylation of 1 under the Vilsmeier reaction conditions using POCl3 and DMF produced formylated product 6 (Scheme 2) as the dominant product within 10 hours by the modification of the para-position of the benzene fused with five- and seven-membered rings (Fig. S9†). The selective functionalization of ring D rather than ring A is probably because of the lower degree of aromaticity of D (Fig. 4a). Due to the deactivation of the electron-withdrawing aldehyde, the bis-formylation product was not detected under this condition. However, the Duff reaction of hexamethylenetetramine in TFA provided a complex mixture. The resulting aldehyde in 6 was directly reacted with different aryl anions, generated by lithium–halide exchange, and then subjected to Dess–Martin oxidation to afford 7 and 8 in 34% and 31% yields, respectively, over two steps (Scheme 2). The derivatives 7 and 8, with the installation of an electron-donating group (EDG)—methoxy, electron-withdrawing group (EWG)—trifluoromethane, respectively were unambiguously elucidated by 1H NMR, 13C NMR, and high-resolution MALDI-TOF-MS (Fig. S30–S33†). The tread of calculated NICS(1) values and 3D ICSS maps for derivatives 7 and 8 were similar to those of the parent NG 1 (Fig. S10 and S11†), indicating that the substituent groups barely affect the aromaticity of the nanographene backbones. Similar UV/Vis or fluorescence spectral changes for compounds 7 and 8 were observed when TFA (0–100 equiv.) was added to the CH2Cl2 solutions, suggesting the possible formation of the corresponding radical cations (Fig. S12 and S13†).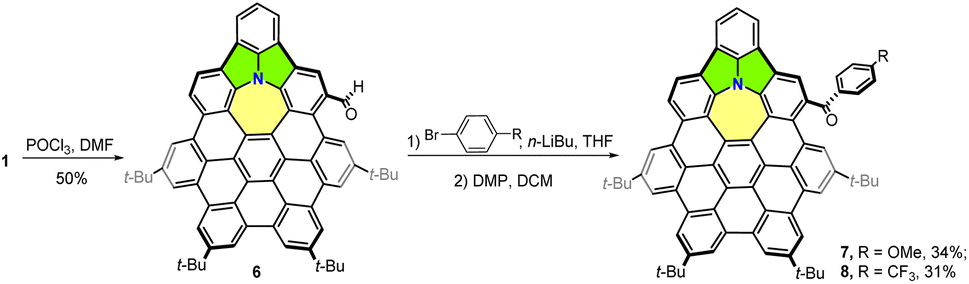 | ||
| Scheme 2 Synthesis of nanographene derivatives 7 and 8. | ||
Photophysical and electrochemical property comparison
UV/Vis absorption and fluorescence spectra for NGs 1, 7, and 8 were measured in CH2Cl2 (Fig. 5a and b). The absorption profiles of compounds 7 and 8 were almost identical to those of NG 1, with a negligible bathochromic shift of 4–5 nm for the maximum peaks. This can be attributed to the limited π-conjugation due to the perpendicular orientation of the benzophenone substituents towards the polycyclic framework (Fig. S10†). The large conjugated structures of 1, 7, and 8 resulted in a high molar absorption coefficient (ε) of 4.7–5.7 × 105 M−1 cm−1 at the absorption maxima (Table S2†). Time-dependent (TD) DFT calculations at the PBE0/def2-TZVP level of theory including CH2Cl2 as a solvent allowed us to interpret the observations in the absorption spectra (Fig. S14–S16, Tables S8–S10†). Basically, the lowest energy absorption maxima at around 440 nm with the long tail were mainly ascribed to the HOMO–LUMO transitions for 1 and 7 and HOMO–LUMO + 1 transition for 8. Furthermore, the intense absorption bands located at around 380 nm mainly originated from the HOMO–LUMO + 1 and HOMO − 1–LUMO for 1 and 7 and HOMO–LUMO + 2 and HOMO–LUMO + 3 for 8. The transition-differentiation of 8 in comparison to 1 and 7 suggested that the EWG of trifluoromethane rather than the methoxy group altered the origin of the absorptions. It is noted that the excitation of the HOMO–LUMO transition of 8 was accompanied by a significant charge redistribution: the HOMO was completely delocalized over the entire conjugated skeleton, and the majority of the LUMO was located at the benzophenone part (Fig. 5d).18 | ||
| Fig. 5 (a) UV/Vis absorption spectra for 1, 7, and 8 in CH2Cl2. (b) Fluorescence emission spectra for 1, 7, and 8 in CH2Cl2 and in ethanol, λex = 350 nm. (c) Cyclic voltammogram for 1, 7, and 8 in CH2Cl2 with tetra-n-butylammomium hexafluorophosphate as the electrolyte. (d) TD-DFT calculated HOMO and LUMO orbitals of 1, 7, and 8 with the calculated energy transition. | ||
When excited at 350 nm in CH2Cl2, 1, 7, and 8 revealed yellowish broad fluorescence with maxima at 516, 522, and 525 nm, respectively. Compounds 1 and 7 behaved similarly in the protic solvent methanol with negligible changes. However, the fluorescence maximum of 8 strongly depended on the solvents, and 8 was almost non-emissive in methanol (Fig. 5b). This is in line with the charge transfer in the excited states (Fig. S16†). Compared to 1 and 7, in which the electrons were delocalized in the conjugated backbones in the HOMO, LUMO, and LUMO + 1 (Fig. S14 and S15†), significant spatial separation of the HOMO and LUMO was observed for structure 8 (Fig. S16†). Such D–A type fluorophores usually exhibit solvatochromism, thereby leading to a substantial fluorescence quenching through twisted intramolecular charge transfer (TICT) or solvent–solute interactions.19 Electrochemical properties were investigated by cyclic voltammetry (CV) in CH2Cl2 (vs. Ag/Ag+) in the presence of Bu4NPF6 as a supporting electrolyte (Fig. 5c). Obviously, the redox properties can be modulated by introducing benzophenone substituents. Compared to the parent NG 1, structures 7 and 8 exhibited more redox couples. The reduction peaks gradually shifted higher from −1.75 V for 1 to −0.81 for 8 (Fig. 5c), suggesting that the reducing process occurred at a low reducing potential with the introduction of strong EWGs.
Intermolecular interactions with C60
As a result of the incorporation of nitrogen and antiaromaticity of the backbones, these nanographenes should display high electron-rich properties as electron donors. Thus, we evaluated the intermolecular interaction between NGs 1, 7, and 8 and the electron-deficient π-electronic system C60. The addition of 0–10 equiv. of C60 into a 1,2-dichlorobenzene solution of NGs caused a change in the UV/Vis absorption and emission spectra. As shown in Fig. 6a, the appearance of broad absorption bands between 500 nm and 660 nm upon the addition of 0–10 equiv. of C60 into a solution of 1 indicated intermolecular charge-transfer interaction and similar spectral changes were observed for 7 and 8 (Fig. S17 and S18†). The association behavior was also monitored by 1H NMR analysis. The gradual upfield shift of specific aromatic proton signals suggested this supramolecular interaction (Fig. S19†). Additionally, the supramolecular charge transfer in solution was confirmed by the observation of fluorescence quenching when C60 was titrated into solutions of NG 1. Based on the nonlinear Stern–Volmer equation,20 the association constant K1 was determined to be 1.1 × 105 M−1 by fitting the data from the plots of I0/I vs. the concentration of C60, and the same range of association constants was observed for 7 and 8. These values are much higher than the reported curved nanographenes.9g,21 Such strong association constants indicate the high-level electron-donating properties of these nanographenes. | ||
| Fig. 6 (a) UV/Vis absorption spectra for the addition of 0–10 equiv. of C60 into a 1,2-dichlorobenzene solution of nanographene 1. (b) Fluorescence quenching spectra for the addition of 0–10 equiv. of C60 into a 1,2-dichlorobenzene solution of nanographene 1. Inset: data fit of concentration of C60vs. fluorescence intensity (I0/I) using the non-linear Stern–Volmer equation to determine the association constant K. | ||
Conclusions
We have developed a novel nitrogen-doped, non-alternant nanographene 1, in which a cycl[2,2,4]azine unit is conceptually embedded into the benzenoid nanographene backbone. The saddle and twisted three-dimensional structures of nanographene 1 and the planar conformation of the cycl[2,2,4]azine fragment were revealed by single-crystal X-ray diffraction analysis. Processing planar conformation, significant antiaromaticity for the azepine ring was determined by the DFT calculation study. The single-electron oxidation of 1 was accomplished to generate radical cation 1˙+ by using AgSbF6 or trifluoroacetic acid. The open-shell structure of 1˙+ was evidenced by UV/Vis absorption, 1H NMR spectroscopy, and electron paramagnetic resonance spectroscopy. Due to the higher level of aromaticity, 1˙+ exhibits high stability with a half-life of 5 days in ambient condition and stayed intact at 0 °C for 1 day. Due to the inherent nitrogen, the selective formylation of nanographene 1 was proved by Vilsmeier reaction, and the derivatives 7 and 8 with substituted benzophenone were obtained. The photophysical and electronic properties of derivatives 7 and 8 together with parent compound 1 were studied by UV/Vis absorption, fluorescence emission, cyclic voltammetry, and time-dependent DFT calculation. The properties can be efficiently modulated by switching the electronic group in the benzophenone for derivatives 7 and 8 in comparison with 1. Moreover, the intermolecular charge transfer from 1, 7, and 8 to C60 was investigated, and high association constants of over 1.0 × 105 M−1 were detected, suggesting the significant electron-donating properties of these structures.Overall, this work illustrates how the embedded cycl[2,2,4]azine unit modulates the properties of hydrocarbon nanographenes. The structures reported in this work displays some interesting properties, such as antiaromaticity, open-shell characteristic, and electron-donating nature. By means of the regio-selective functionalization, it should be possible to manipulate these structures for real applications in the future.
Data availability
The full experimental details, synthetic procedures, characterization data, UV/Vis, fluorescence, and NMR spectra, computational details, and supplementary discussions associated with this article are provided in the ESI.†Author contributions
L. Ruan performed the major synthesis work and property studies. W. Luo and H. Zhang helped with NMR and photophysical property measurements. P. Liu helped with the UV/Vis/NIR measurements. P. An conceived the concept and performed the DFT calculations. P. An and Y. Shi prepared and revised the manuscript. All the authors analyzed and interpreted the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support provided by the National Natural Science Foundation of China (22061046 and 21901226). L. Ruan thanks the Postgraduate Scientific Research Innovation Project of Yunnan University (KC-22222493). We thank the Advanced Analysis and Measurement Center of Yunnan University for the assistance with instrumentation. Particularly, we thank Dr Jie Zhou from the Advanced Analysis and Measurement Center of Yunnan University for the X-ray analysis.Notes and references
- (a) V. M. Tsefrikas and L. T. Scott, Chem. Rev., 2006, 106, 4868–4884 CrossRef CAS PubMed; (b) Y. Tobe, Chem. Rec., 2015, 15, 86–96 CrossRef CAS PubMed; (c) S. H. Pun and Q. Miao, Acc. Chem. Res., 2018, 51, 1630–1642 CrossRef CAS.
- (a) B. Pigulski, K. Shoyama and F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 15908–15912 CrossRef CAS; (b) A. Diaz-Andres, J. Marín-Beloqui, J. Wang, J. Liu, J. Casado and D. Casanova, Chem. Sci., 2023, 14, 6420–6429 RSC; (c) Y. Fei and J. Liu, Adv. Sci., 2022, 9, 2201000 CrossRef CAS; (d) J. Ma, Y. Fu, E. Dmitrieva, F. Liu, H. Komber, F. Hennersdorf, A. A. Popov, J. J. Weigand, J. Liu and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 5637–5642 CrossRef CAS; (e) A. Costa and A. López-Castillo, Diamond Relat. Mater., 2021, 112, 108235 CrossRef CAS.
- (a) J. Wang, F. G. Gámez, J. Marín-Beloqui, A. Diaz-Andres, X. Miao, D. Casanova, J. Casado and J. Liu, Angew. Chem., Int. Ed., 2023, 62, e202217124 CrossRef CAS; (b) Y. Han, G. Li, Y. Gu, Y. Ni, S. Dong and C. Chi, Angew. Chem., Int. Ed., 2020, 59, 9026–9031 CrossRef CAS; (c) N. Ogawa, Y. Yamaoka, H. Takikawa, K.-i. Yamada and K. Takasu, J. Am. Chem. Soc., 2020, 142, 13322–13327 CrossRef CAS PubMed; (d) T. G. Lohr, J. I. Urgel, K. Eimre, J. Liu, M. D. Giovannantonio, S. Mishra, R. Berger, P. Ruffieux, C. A. Pignedoli, R. Fasel and X. Feng, J. Am. Chem. Soc., 2020, 142, 13565–13572 CrossRef CAS; (e) I. C.-Y. Hou, Q. Sun, K. Eimre, M. D. Giovannantonio, J. I. Urgel, P. Ruffieux, A. Narita, R. Fasel and K. Müllen, J. Am. Chem. Soc., 2020, 142, 10291–10296 CrossRef CAS; (f) Q. Fan, D. Martin-Jimenez, D. Ebeling, C. K. Krug, L. Brechmann, C. Kohlmeyer, G. Hilt, W. Hieringer, A. Schirmeisen and J. M. Gottfried, J. Am. Chem. Soc., 2019, 141, 17713–17720 CrossRef CAS; (g) L. Qin, Y.-Y. Huang, B. Wu, J. Pan, J. Yang, J. Zhang, G. Han, S. Yang, L. Chen, Z. Yin, Y. Shu, L. Jiang, Y. Yi, Q. Peng, X. Zhou, C. Li, G. Zhang, X. S. Zhang, K. Wu and D. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304632 CrossRef CAS PubMed; (h) C. Zhu, K. Shoyama and F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 21505–21509 CrossRef CAS PubMed; (i) V. Akhmetov, M. Feofanov, O. Papaianina, S. Troyanov and K. Amshatov, Chem.–Eur. J., 2019, 25, 11609–11613 CrossRef CAS PubMed; (j) Y. Fei, Y. Fu, X. Bai, L. Du, Z. Li, H. Komber, K.-H. Low, S. Zhou, D. L. Pjillips, X. Feng and J. Liu, J. Am. Chem. Soc., 2021, 143, 2353–2360 CrossRef CAS; (k) K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739–744 CrossRef CAS; (l) Q. Fan, L. Yan, M. W. Tripp, O. Krejčí, S. Dimosthenous, S. R. Kachel, M. Chen, A. S. Foster, U. Koert, P. Lijeroth and J. M. Gottfried, Science, 2021, 372, 852–856 CrossRef CAS PubMed; (m) T. Kirschbaum, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2020, 59, 270–274 CrossRef CAS; (n) T. Guo, A. Li, J. Xu, K. K. Baldridge and J. Siegel, Angew. Chem., Int. Ed., 2021, 60, 25809–25814 CrossRef CAS.
- (a) A. Ong, T. Tao, Q. Jiang, Y. Han, Y. Ou, K.-W. Huang and C. Chi, Angew. Chem., Int. Ed., 2022, 61, e202209286 CrossRef CAS; (b) L. Chen, B. Wu, L. Qin, Y.-Y. Huang, W. Meng, R. Kong, X. Yu, K. ChenChai, C. Li, G. Zhang, X.-S. Zhang and D. Zhang, Chem. Commun., 2022, 58, 5100–5103 RSC; (c) P. Liu, X.-Y. Chen, J. Cao, L. Ruppenthal, J. M. Gottfried, K. Müllen and X.-Y. Wang, J. Am. Chem. Soc., 2021, 143, 5314–5318 CrossRef CAS; (d) K. Horii, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, A. Konishi and M. Yasuda, J. Am. Chem. Soc., 2022, 144, 3370–3375 CrossRef CAS PubMed; (e) K. Yamamoto, Y. le, N. Tohnai, F. Kakiuchi and Y. Aso, Sci. Rep., 2018, 8, 17663 CrossRef PubMed.
- (a) M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS; (b) A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565–788 CrossRef CAS; (c) V. Barát and M. C. Stuparu, Chem.–Asian J., 2021, 16, 20–29 CrossRef.
- (a) D. H. Reid, Q. Rev., Chem. Soc., 1965, 19, 274–302 RSC; (b) J. Ahmed and S. K. Mandal, Chem. Rev., 2022, 122, 11369–11431 CrossRef CAS PubMed; (c) A. Mukherjee, S. C. Sau and S. K. Mandal, Acc. Chem. Res., 2017, 50, 1679–1691 CrossRef CAS PubMed; (d) T. Kubo, Chem. Rec., 2015, 15, 218–232 CrossRef CAS; (e) Q. Du, X. Sun, Y. Liu, Y. Jiang, C. Li, K. Yan, R. Ortiz, T. Frederiksen, S. Wang and P. Yu, Nat. Commun., 2023, 14, 4802 CrossRef CAS.
- (a) M. J. S. Dewar and N. Trinajstić, J. Chem. Soc. A, 1969, 1754–1755 RSC; (b) D. Farquhar and D. Leaver, J. Chem. Soc. D, 1969, 24–25 RSC.
- (a) E. Clar and D. G. Stewart, J. Am. Chem. Soc., 1953, 75, 2667–2672 CrossRef CAS; (b) N. Pavliček, A. Mistry, Z. Majzik, N. Moll, G. Meyer, D. J. Fox and L. Gross, Nat. Nanotechnol., 2017, 12, 308–311 CrossRef PubMed; (c) S. Mishra, D. Beyer, K. Eimre, J. Liu, R. Berger, O. Gröning, C. A. Pignedoli, K. Müllen, R. Fasel, X. Feng and P. Ruffieux, J. Am. Chem. Soc., 2019, 141, 10621–10625 CrossRef CAS; (d) J. Su, M. Telychko, P. Hu, G. Macam, P. Mutombo, H. Zhang, Y. Bao, F. Cheng, Z.-Q. Huang, Z. Qiu, S. J. R. Tan, H. Lin, P. Jelínek, F.-C. Chuang, J. Wu and J. Lu, Sci. Adv., 2019, 5, aav7717 CrossRef PubMed; (e) T. Wang, A. Berdonces-Layunta, N. Friedrich, M. Vilas-Varela, J. P. Calupitan, J. I. Pascual, D. Peña, D. Casanova, M. Corso and D. G. de Oteyze, J. Am. Chem. Soc., 2022, 114, 4522–4529 CrossRef; (f) M. Vilas-Varela, F. Eomero-Lara, A. Vegliante, J. P. Calupitan, A. Martínez, L. Meyer, U. Uriarte-Amiano, N. Friedrich, D. Wang, F. Schulz, N. E. Koval, M. E. Sandoval-Salinas, D. Casanova, M. Corso, E. Artacho, D. Peña and J. I. pascual, Angew. Chem., Int. Ed., 2023, 62, e202307884 CrossRef CAS PubMed; (g) S. Mishra, X. Yao, Q. Chen, K. Eimre, O. Gröning, R. Ortiz, M. D. Giovannantonio, J. C. Sancho-García, J. Fernández-Rossier, C. A. Pignedoli, K. Müllen, P. Ruffieux, A. Narita and R. Fasel, Nat. Chem., 2021, 13, 581–586 CrossRef CAS PubMed; (h) M. E. Sandoval-Salinas, R. Bernabeu-Cabañero, A. J. Pérez-Jiménez, E. San-Fabián and J. C. Sancho-García, Phys. Chem. Chem. Phys., 2023, 25, 11697–11706 RSC; (i) S. Moles Quintero, M. M. Haley, M. Kertesz and J. Casado, Angew. Chem., Int. Ed., 2022, 61, e202209138 CrossRef CAS; (j) S. Arikawa, A. Shimizu, D. Shiomi, K. Sato and R. Shintani, J. Am. Chem. Soc., 2021, 143, 19599–19605 CrossRef CAS; (k) H. Wei, X. Hou, T. Xu, Y. Zou, G. Li, S. Wu, Y. Geng and J. Wu, Angew. Chem., Int. Ed., 2022, 61, e202210386 CrossRef CAS.
- (a) M. Krzeszewski, Ł. Dobrzycki, A. L. Sobolewski, M. K. Cyrański and D. T. Gryko, Angew. Chem., Int. Ed., 2021, 60, 14998–15005 CrossRef CAS PubMed; (b) C. Wang, Z. Deng, D. L. Phillips and J. Liu, Angew. Chem., Int. Ed., 2023, 62, e202306890 CrossRef CAS; (c) M. Krzeszewski, Ł. Dobrzycki, A. L. Sobolewski, M. K. Cyrański and D. T. Gryko, Chem. Sci., 2023, 14, 2353–2360 RSC; (d) K. Oki, M. Takase, S. Mori, A. Shiotari, Y. Sugimoto, K. Ohara, T. Okujima and H. Uno, J. Am. Chem. Soc., 2018, 140, 10430–10434 CrossRef CAS; (e) K. Oki, M. Takase, S. Mori and H. Uno, J. Am. Chem. Soc., 2019, 141, 16255–16259 CrossRef CAS; (f) P. An, R. Li, B. Ma, R.-Y. He, Y.-K. Zhang, M.-J. Xiao and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 24478–24483 CrossRef CAS; (g) H. Yokoi, Y. Hiraoka, S. Hiroto, D. Sakamaki, S. Seki and H. Shinokubo, Nat. Commun., 2015, 6, 8215 CrossRef; (h) S. Ito, Y. Tokimaru and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 7256–7260 CrossRef CAS PubMed; (i) F. Gan, C. Shen, W. Cui and H. Qiu, J. Am. Chem. Soc., 2023, 145, 5952–5959 CrossRef CAS PubMed; (j) J. Nebauer, C. Neiß, M. Krug, A. Vogel, D. Fehn, S. Ozaki, F. Rominger, K. Meyer, K. Kamada, D. M. Guldi, A. Görling and M. Kivala, Angew. Chem., Int. Ed., 2022, 61, e202205287 CrossRef CAS PubMed; (k) Z.-L. Qiu, X.-W. Chen, Y.-D. Huang, R.-J. Wei, K.-S. Chu, X.-J. Zhao and Y.-Z. Tan, Angew. Chem., Int. Ed., 2022, 61, e202116955 CrossRef CAS; (l) T. Kirschbaum, F. Rominger and M. Mastalerz, Chem.–Eur. J., 2020, 26, 14560–14564 CrossRef CAS PubMed; (m) Y. Tokimaru, S. Ito and K. Nozaki, Angew. Chem., Int. Ed., 2018, 57, 9818–9822 CrossRef CAS; (n) K. Nakamura, Q.-Q. Li, O. Krejčí, A. S. Foster, K. Sun, S. Kawai and S. Ito, J. Am. Chem. Soc., 2020, 142, 11363–11369 CrossRef CAS PubMed.
- (a) R. J. Windgassen, W. H. Saunders and V. Boekelheide, J. Am. Chem. Soc., 1958, 81, 1459–1465 CrossRef; (b) O. Ceder and B. Beijer, J. Heterocycl. Chem., 1976, 13, 1029–1031 CrossRef CAS; (c) E. Kleinpeter and A. Koch, Eur. J. Org Chem., 2022, e202101362 CrossRef CAS; (d) J. Wagner, P. Z. Crocomo, M. A. Kochman, A. Kubas, P. Data and M. Lindner, Angew. Chem., Int. Ed., 2022, 61, e202202232 CrossRef CAS PubMed.
- B. Zhang, L. Ruan, Y.-K. Zhang, H. Zhang, R. Li and P. An, Org. Lett., 2023, 25, 732–737 CrossRef CAS.
- H. L. Schmider and A. D. Becke, J. Mol. Struct., 2020, 527, 51–61 CrossRef.
- T. Lu and Q. Chen, Acta Phys.-Chim. Sin., 2018, 34, 0001–0009 Search PubMed.
- (a) P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed; (b) H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v R. Schleyer, Org. Lett., 2006, 8, 863–866 CrossRef CAS PubMed.
- (a) J. Kruszewski and T. M. Krygowski, Tetrahedron Lett., 1972, 13, 3839–3842 CrossRef; (b) T. M. Krygowski, J. Chem. Inf. Comput. Sci., 1993, 33, 70–78 CrossRef CAS; (c) T. M. Krygowski and M. K. Cyrański, Chem. Rev., 2001, 101, 1385–1420 CrossRef CAS.
- (a) S. Klod and E. Kleinpeter, J. Chem. Soc., Perkin Trans. 2, 2001, 1893–1898 CAS; (b) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- (a) V. Rajeshkumar, Y. T. Lee and M. C. Stuparu, Eur. J. Org Chem., 2016, 1, 36–40 CrossRef; (b) V. Rajeshkumar and M. C. Stuparu, Chem. Commun., 2016, 52, 9957–9960 RSC; (c) J. Holzwarth, K. Y. Amsharov, D. I. Sharapa, D. Reger, K. Roshchyna, D. Lungerich, N. Jux, F. Hauke, T. Clark and A. Hirech, Angew. Chem., Int. Ed., 2017, 56, 12184–12190 CrossRef CAS; (d) D. Halilovic, M. Budanović, Z. R. Wong, R. D. Webster, J. Huh and M. C. Stuparu, J. Org. Chem., 2018, 83, 3529–3536 CrossRef CAS; (e) D. Halilovic, V. Rajeshkumar and M. C. Stuparu, Org. Lett., 2021, 23, 1468–1472 CrossRef CAS PubMed; (f) J. Stanojkovic, R. William, Z. Zhang, I. Fernández, J. Zhou, R. D. Webster and M. C. Stuparu, Nat. Commun., 2023, 14, 803 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- (a) J. P. Kurtz, T. Grusenmeyer, L. Tong, G. Kosgei, R. H. Schmehl, J. T. Mague and R. A. Pascal, Tetrahedron, 2011, 67, 7211–7216 CrossRef CAS; (b) C. A. Hoelzel, H. Hu, C. H. Wolstenholme, B. A. Karim, K. T. Munson, K. H. Jung, H. Zhang, Y. Liu, H. P. Yennawar, J. B. Asbury, X. Li and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 4785–4792 CrossRef CAS; (c) S. Singha, D. Kim, B. Roy, S. Sambasivan, H. Moon, A. S. Rao, J. Y. Kim, T. Joo, J. W. Park, Y. M. Rhee, T. Wang, K. H. Kim, Y. H. Shin, J. Jung and K. H. Ahn, Chem. Sci., 2015, 6, 4335–4342 RSC; (d) E. Yamaguchi, C. Wang, A. Fukazawa, M. Taki, Y. Sato, T. Sasaki, M. Ueda, N. Sasaki, T. Higashiyama and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 4539–4543 CrossRef CAS; (e) M. Kawashiro, T. Mori, M. Ito, N. Ando and S. Yamaguchi, Angew. Chem., Int. Ed., 2023, 62, e202303725 CrossRef CAS.
- K. Campbell, A. Zappas, U. Bunz, Y. S. Thio and D. G. Bucknall, J. Photochem. Photobiol., A, 2012, 249, 41–46 CrossRef CAS.
- (a) Y. Sun, X. Wang, B. Yang, M. Chen, Z. Guo, Y. Wang, J. Li, M. Xu, Y. Zhang, H. Sun, J. Dang, J. Fan, J. Li and J. Wei, Nat. Commun., 2023, 14, 3446 CrossRef CAS; (b) S. Zank, J. M. Fernández-García, A. J. Stasyuk, A. A. Voityuk, M. Krug, M. Solà, D. M. Guldi and N. Martín, Angew. Chem., Int. Ed., 2023, 62, e202112834 Search PubMed; (c) H. Yokoi, S. Hiroto, D. Sakamaki, S. Seki and H. Shinokubo, Chem. Sci., 2018, 9, 819–824 RSC; (d) Y.-Y. Xu, H.-R. Tian, S.-H. Li, Z.-C. Chen, Y.-R. Yao, S.-S. Wang, X. Zhang, Z.-Z. Zhu, S.-L. Deng, Q. Zhang, S. Yang, S.-Y. Xie, R.-B. Huang and L.-S. Zheng, Nat. Commun., 2019, 10, 485 CrossRef CAS PubMed.
- Deposition numbers 2301272 for compound 1 contain the supplementary crystallographic data for this paper.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2301272. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05515a |
| This journal is © The Royal Society of Chemistry 2024 |