
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of nitrogen sources and hydrogen adsorption on the dynamic stability of Fe–N–C catalysts in oxygen reduction reaction†
Zhou
Huang
,
Fuhua
Li
,
Yongduo
Liu
,
Siguo
Chen
,
Zidong
Wei
 * and
Qing
Tang
*
* and
Qing
Tang
*
School of Chemistry and Chemical Engineering, Chongqing Key Laboratory of Theoretical and Computational Chemistry, Chongqing University, Chongqing 401331, China. E-mail: zdwei@cqu.edu.cn; qingtang@cqu.edu.cn
First published on 15th December 2023
Abstract
Fe–N–C catalysts are promising alternatives to Pt-based electrocatalysts for the oxygen reduction reaction (ORR) in various electrochemical applications. However, their practical implementation is impeded by their instability during prolonged operation. Various degradation mechanisms have been proposed, yet the real origin of the intrinsic instability of Fe–N–C structures under ORR operations is still disputed. Herein, we observed a new type of protonation mechanism based on advanced first-principles simulations and experimental characterizations. The results revealed strong evidence of pyrrolic-N protonation in pyrrolic-type FeN4, which plays a vital role for the low kinetic barrier of Fe leaching. Conversely, the pyridinic-type FeN4 prefers protonation at the Fe site, contributing to the higher barrier of Fe leaching and relatively higher stability. The facile pyrrolic-N protonation is verified by various spectroscopy characterizations in the Nafion-treated FePc molecule. Crucially, the presence of oxygen-containing intermediates at the Fe site can further work synergistically with N protonation to promote conversion of iron atoms (Fe–N4) into ferric oxide under working potentials, and the more positive the electrode potential, the lower the kinetic barrier of Fe leaching. These findings serve as a foundation for future research endeavors on the stability issues of Fe–N–C catalysts and advancing their application in sustainable energy conversion technologies.
1. Introduction
Proton exchange membrane fuel cells (PEMFCs) are widely recognized as clean and highly efficient devices for sustainable energy utilization, particularly in the automobile industry.1–3 To enhance the efficiency of PEMFCs, platinum-based catalysts are utilized to facilitate the sluggish Oxygen Reduction Reaction (ORR) that takes place on the cathode.4,5 Nevertheless, the limited availability and high cost of platinum hinder the large-scale commercialization of PEMFCs.6 Over the past several decades, metal-nitrogen-carbon (M–N–C, particularly when M = Fe, Co, Mn) catalysts have shown great promise as potential replacements for platinum-based catalysts.7–9 Among them, Fe–N–C catalysts have demonstrated superior performance over other M–N–C catalysts,9–18 thanks to their exceptional ORR activity in an acidic milieu. Despite the impressive performance of Fe–N–C catalysts, a significant degradation (40–80%) occurs within the first 100 hours of fuel cell testing, which presents a substantial barrier to their commercial application in PEMFCs cathodes.19,20 Resolving this urgent issue is a prerequisite for the property optimization and potential commercial application of Fe–N–C catalysts. Therefore, unveiling the degradation mechanism and identifying the intrinsic unsteady structures of FeN4 moieties are essential to improve their long-term durability.To address the critical challenge of stability, substantial research efforts have been devoted to dissecting the degradation mechanisms. Numerous degradation mechanisms, broadly classified into four categories, have been proposed to explain the initial performance degradation of Fe–N–C catalysts in acidic ORR environments. (1) Protonation of nitrogen species: this mechanism concerns the protonation of highly basic nitrogen groups situated next to the active site, followed by anion adsorption. Herranz et al.21 proposed that Fe–N–C active sites would manifest high turnover frequency (TOF) values when the basic nitrogen groups adjacent to the Fe–N–X site undergo protonation, devoid of anion adsorption. Nevertheless, when anion adsorption occurs at the Fe/C site, there is a decrease in the TOF values. Herraiz et al.22 suggested that the fluorination of FeN4 active sites, possibly via oxygen electrocatalysis (interpreted as direct anion adsorption), leads to the formation of C–F and Fe–F bonds. This could trigger instability in the Fe–N–C catalyst during fuel cell processing. However, their later study23 contradicted this view, asserting that anion adsorption on FeN4 active sites doesn't affect the initial ORR activity decay, and the FeN4/NH+ site's anionic neutralization hypothesis should be dismissed. Mukerjee et al.24,25 also established that the deactivation of Fe–N–C catalysts doesn't rely on anions. Moreover, the electrochemical experiments by Sun et al.26 indicated that the pyridinic N has high ORR activity in alkaline medium, but when it is protonated in acidic medium, the catalytic activity is greatly suppressed. (2) Oxidation of carbon/metal: the active carbon/metal sites on the catalyst surface are vulnerable to radicals derived from H2O2, which can instigate a Fenton reaction or generate surface oxidation intermediates. This deactivation may be partially reversed through catalyst surface reduction. Jaouen et al.27 demonstrated the structural stability of Fe–N–C catalyst in an acidic environment, which becomes electrochemically unstable due to the presence of H2O2 and carbon surface oxidation, and results in reduced TOF that can be restored by the electrochemical reduction of the carbon surface. Their findings also indicated that the degradation of Fe–N–C catalyst is due to peroxide-derived reactive oxygen species, not the molecular H2O2. (3) Demetalation: this process entails the removal of metal from the Fe–Nx active site, directly diminishing the activity by reducing site density. Chenitz et al.28 proposed that the initial ORR activity decrease results from specific demetalation triggered by water flux into the micropores. Mayrhofer et al.29 showed that iron leaching occurs from iron particles at low potential (<0.7 V) and carbon oxidation at high potential (>0.9 V), with only the latter causing the destruction of FeNxCy species. Cherevko et al.30 find a remarkable linear correlation between the faradaic charge passed through the electrode and the amount of Fe dissolved from the electrode by varying the rate of ORR in alkaline media. (4) Water flooding: this situation occurs when water infiltrates the catalysts' pores, obstructing oxygen transportation to the cathode. Dodelet et al.23 suggested that the most active FeNX sites residing within catalyst micropores contribute to the initial swift decay of catalytic activity in fuel cells due to catalyst micropores' water flooding. Conversely, Banham et al.31 found no connection between micropore flooding and observed destruction, suggesting that the deactivation or loss of crucial active sites results in kinetic losses, with the active sites/carbon oxidation being the most likely culprit.
The endeavor to identify precise degradation mechanisms in Fe–N–C catalysts has been complicated by the elusive nature of their active sites. This complexity escalates during fuel cell stability or durability tests when multiple deactivation mechanisms may occur concurrently, resulting in a tangled degradation process and a considerable challenge in identifying the predominant mechanism. Is the rapid decrease in ORR activity due to a single mechanism or a combination of several ones? Do distinct degradation pathways exist for different active sites? How can we bolster the intrinsic stability of these catalysts? To address these questions, it becomes crucial to gain a profound understanding of the dynamic stabilities of FeN4 units under working conditions. At the computational side, however, the current evaluation of the stability of Fe–N–C materials mainly focused on the static thermodynamic calculations,32–34 which do not consider explicitly the effects of solvation, working potential, kinetics, and key intermediates on the structural stability in realistic reaction conditions. Therefore, exploring the dynamic evolution of active site structure under electrochemical conditions is crucial.
In this work, we applied the advanced constant potential hybrid solvation dynamical model with the inclusion of explicit solid–liquid interface to thoroughly explore the dynamic stability of Fe–N–C catalysts in acidic ORR environment, with specific emphasis on the roles of pyridinic-type and pyrrolic-type nitrogen. Our results indicate that the pyrrolic-type FeN4 facilitates the protonation/hydrogen adsorption at the N site, whereas the pyridinic-type FeN4 prefers the H adsorption at the Fe site. The protonation of pyrrolic N plays the key role for the rapid Fe leaching into solution at the electrochemical conditions, which can significantly reduce the kinetic barrier of Fe leaching compared to the absence of H adsorption. Our results further observed the synergistic role of hydrogen adsorption and the oxygen-containing intermediates in converting individual iron atoms (Fe–N4) into ferric oxide under differing applied potentials. The more positive the electrode potential, the lower the kinetic barrier of Fe leaching, thus accelerating the pyrrolic-type FeN4 degradation. These insights offer new atomic level understanding of the dynamic stability of Fe–N–C electrocatalysts under working conditions, which can be extended to elucidate and assess the electrochemical stability of other heterogeneous electrocatalysts and enable more promising catalyst design.
2. Methods
2.1 Computational details
The spin-polarized geometry optimizations and ab initio molecular dynamics (AIMD) calculations were carried out using the Vienna Ab initio Simulation Package (VASP 5.4.4).35 The exchange-correlation function was modeled with the Perdew–Burke–Ernzerhof (PBE) functional, a type of generalized gradient approximation (GGA),36 while the electron–ion interaction was described using the Projector Augmented Wave (PAW) method.37 The PBE-D3 method correction with Becke–Jonson damping was used to describe the van der Waals interaction when dealing with systems with explicit water molecules and the water–slab interface.38 A supercell configuration of 5 × 3 supercell was meticulously established, characterized by a volumetric cell size of 12.40 × 12.67 × 30 Å3. The calculations were performed using a plane wave basis set with an energy cutoff of 450 eV, and a Monkhorst–Pack scheme with a 3 × 3 × 1 k-point grid was used for Brillouin zone integration. Atomic-scale simulations are carried out to study Fe leaching process at FeN4Cy–water interfaces, where the oxygen-containing adsorbates are introduced to explore the possible adsorbate effects. The catalyst/water structure was built by the incorporation of multilayer explicit water matrices, each comprised of 36H2O molecules, and maintaining a water density of 1 g cm−3. Starting from this well-equilibrated interface, 5 ps of AIMD simulations were carried out at 300 K. Fig. S1† illustrates the structural configurations, and concurrently displays the temporal progression of energy and temperature parameters throughout the simulation period. The electrode potential was determined via charging and tuning the work function.39,40 The effective surface tension parameter was assigned to 0 in VASPsol to neglect the cavitation energy contribution. The linearized Poisson–Boltzmann model with a Debye length of 3.0 Å mimics the compensating charge.41 AIMD simulations were performed within the canonical ensemble at 300 K by Nose–Hoover thermostat, and the time step was set to 0.5 fs. We used the “slow-growth” method which is implemented in VASP to get the free energy profile of the dynamic stability of the catalyst structures with a value ∂ξ of 0.0008 Å. To deal with the fluctuations of work function, we extracted at least 10 snapshots from the AIMD trajectories to determine the average work function.42–44 VASPKIT code was used for post-processing of the computational data.45 The theoretical IR calculations were implemented by the density functional theory (DFT) level of the B3LYP functional46,47 using the Gaussian 16 software package.48 The all-electron 6-311++G** basis set was used for C, N, H and Fe atoms. We utilized the Grimme's DFT-D3 method to handle van der Waals (vdW) interactions.38 The vibration frequency calculation was used to verify the optimized structure minima, i.e., zero virtual frequency, and provides free energy at 298.15 K.2.2 Experimental characterizations
3. Results and discussion
During the ORR process, the single-atom (SA) leaching of iron (Fe) leads to the formation of ferric oxides, a mechanism necessitating the weakening and eventual breaking of the Fe–N bond. To evaluate the propensity for SA Fe leaching, distinct investigations have been carried out focusing on the thermodynamics and kinetics of this process. We examined two distinct Fe moieties: pyrrolic-type FeN4 where the central Fe atom is coordinated with pyrrolic-type nitrogen, and pyridinic-type FeN4 where the Fe atom is coordinated with pyridinic-type nitrogen. The bond length of Fe–N bond in pyridinic-type FeN4 and pyrrolic-type FeN4 was determined to be 1.92 Å and 2.08 Å, indicating the slightly weaker Fe–N bonding in the pyrrolic-type nitrogen environment. The leaching process can be described as follows:| Fe–N4–C + 2H+ ↔ H2N4–C + Fe2+(aq) |
Fig. 1a presents the free energies associated with the leaching of single-atom Fe (Fe–N–C) from the surface to form Fe2+(aq) in solution. Such a highly endothermic free energy of 0.71 eV in pyridinic-type FeN4 and 1.21 eV in pyrrolic-type FeN4 indicate that the dissolution of Fe SA from the surface is thermodynamically challenging. Further evaluation of the kinetic feasibility of Fe leaching was performed using a slow-growth approach. For this, a solid–liquid interface model, depicted in Fig. S2,† was established. The introduction of an inert helium atom served as a rivet, situated directly above the Fe atom at a distance of 14.9 Å. This allowed for the calculation of the kinetic activation energy required for Fe leaching from the Fe–N–C site. In the adopted “slow-growth” approach, the reaction coordinate was selected such that the Fe–He distance was gradually reduced (Fig. S3a†). The free energy continually increases as the Fe atom moves away from the surface, reaching a maximum of 7.45 eV for pyrrolic-type FeN4 and 8.0 eV for pyridinic-type FeN4 by the end of the reaction (Fig. 1b). This suggests a high degree of dynamic stability of the four Fe–N bonds, making them difficult to break and thus hindering the Fe leaching. Fig. 1c and d track the dynamic evolution of the Fe–N bond length throughout the Fe leaching process. Therefore, both the thermodynamic and kinetic results indicate that it is very difficult for the pristine Fe–N–C systems to leach Fe center directly from the surface.
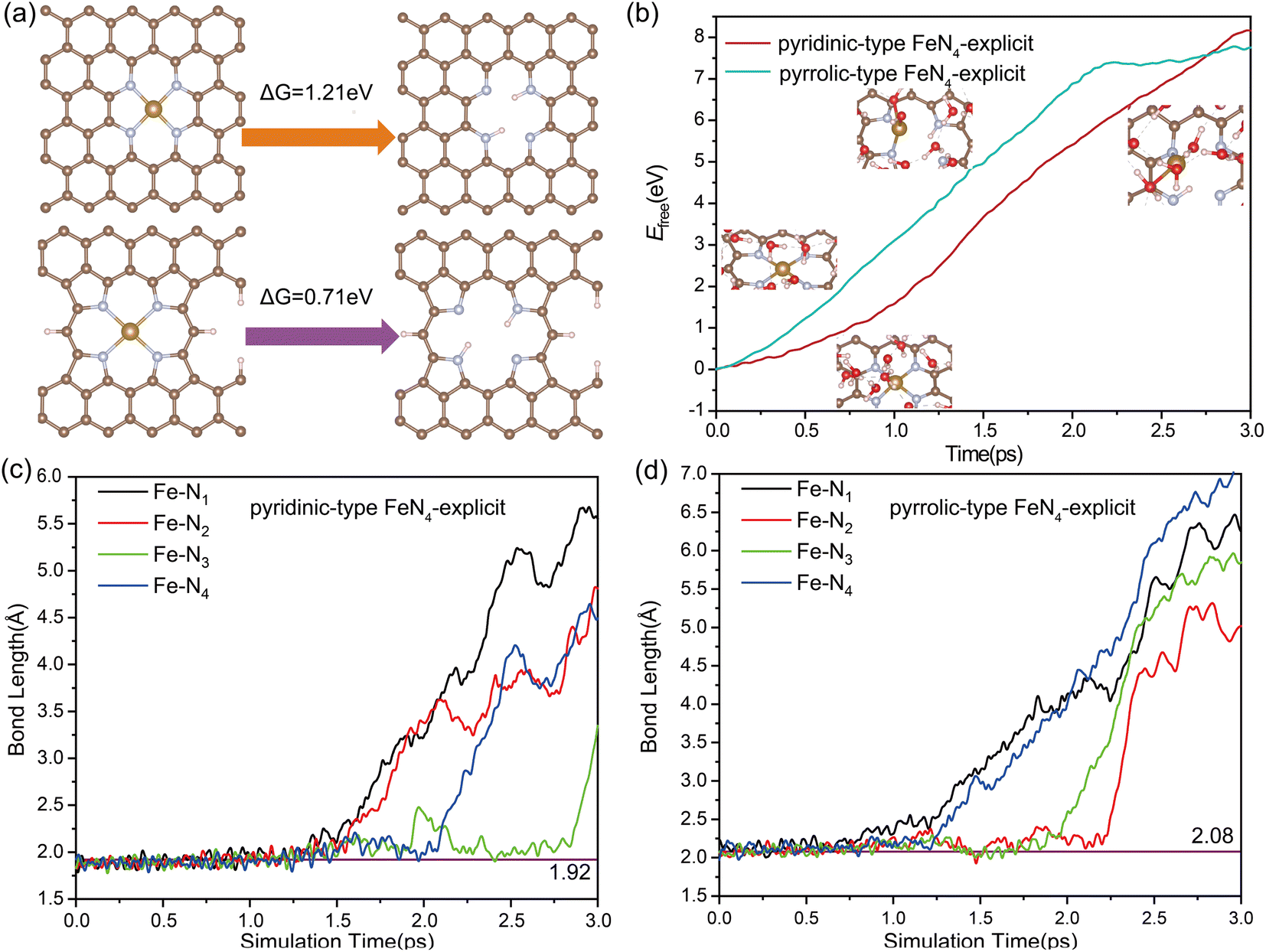 | ||
| Fig. 1 (a) The free energy (ΔG) of Fe2+(aq) formation during Fe leaching. (b) Free energy profile during the Fe leaching process. (c) and (d): dynamic evolution of the bond length of Fe–Nx (x = 1, 2, 3, and 4) during the Fe leaching process, where carbon is depicted in grey, nitrogen in blue, iron in brown, oxygen in red, and hydrogen in white. | ||
Noteworthy, from most of the experimental observations, highly active catalysts often exhibit faster degradation, particularly in the initial stages, leading to an inherent tradeoff between activity and stability.49 In the case of pyrolyzed Fe–N–C catalysts, the FeN4 sites exclusively with the pyrrolic-type FeN4 form are revealed to exhibit significantly enhanced activity compared with the pyridinic-type FeN4 counterpart.50,51 However, despite its higher electrocatalytic activity, the stability of pyrrolic-type FeN4 is shown to be much inferior to the pyridinic-type one. According to our above calculations, the high kinetic barrier of Fe leaching in pyrrolic-type FeN4 and pyridinic-type FeN4 implies that the free-standing pyrrolic-type FeN4 or pyridinic-type FeN4 itself without adsorption of any reaction intermediates may not be the main cause of Fe–N–C degradation in ORR electrocatalysis. To accurately simulate the adsorption effect on the kinetic possibility of Fe leaching involved in Fe–N–C catalysts, we have considered several potential oxygen-containing intermediates at the Fe site (Fe–O2, Fe–OH) and possible carbon oxidation at the adjacent C site (C–OH, C–O–C), as depicted in Fig. 2a. Accordingly, we have also determined the evolution of free energies in all possible FeN4 moieties when Fe moves away from the surface, and the calculated kinetic barrier of Fe leaching is shown and compared in Fig. 2b. Compared with the adsorbate-free FeN4 catalysts (Fig. 1b), the presence of oxygen intermediates at the Fe center or adjacent C site leads to decrease in the kinetic barrier of Fe leaching by about 1–2 eV. Therefore, the adsorption of oxygen intermediates is likely to promote the leaching of Fe atom from the catalytic surface. Moreover, our findings indicate that, no matter whatever adsorbate present, the kinetic barrier of Fe leaching on pyridinic-type FeN4 is always lower than that on the pyrrolic-type FeN4 counterparts. These results indicate that pyridinic-type FeN4 should be less stable than pyrrolic-type FeN4, which is, however, apparently contradictory to the experimental findings by Jaouen and co-workers,52 who identified the existence of durable FeN4C10 (pyridinic-type FeN4) moiety and nondurable high-spin FeN4C12 (pyrrolic-type FeN4) moiety of Fe–N–C catalysts in acidic ORR medium.
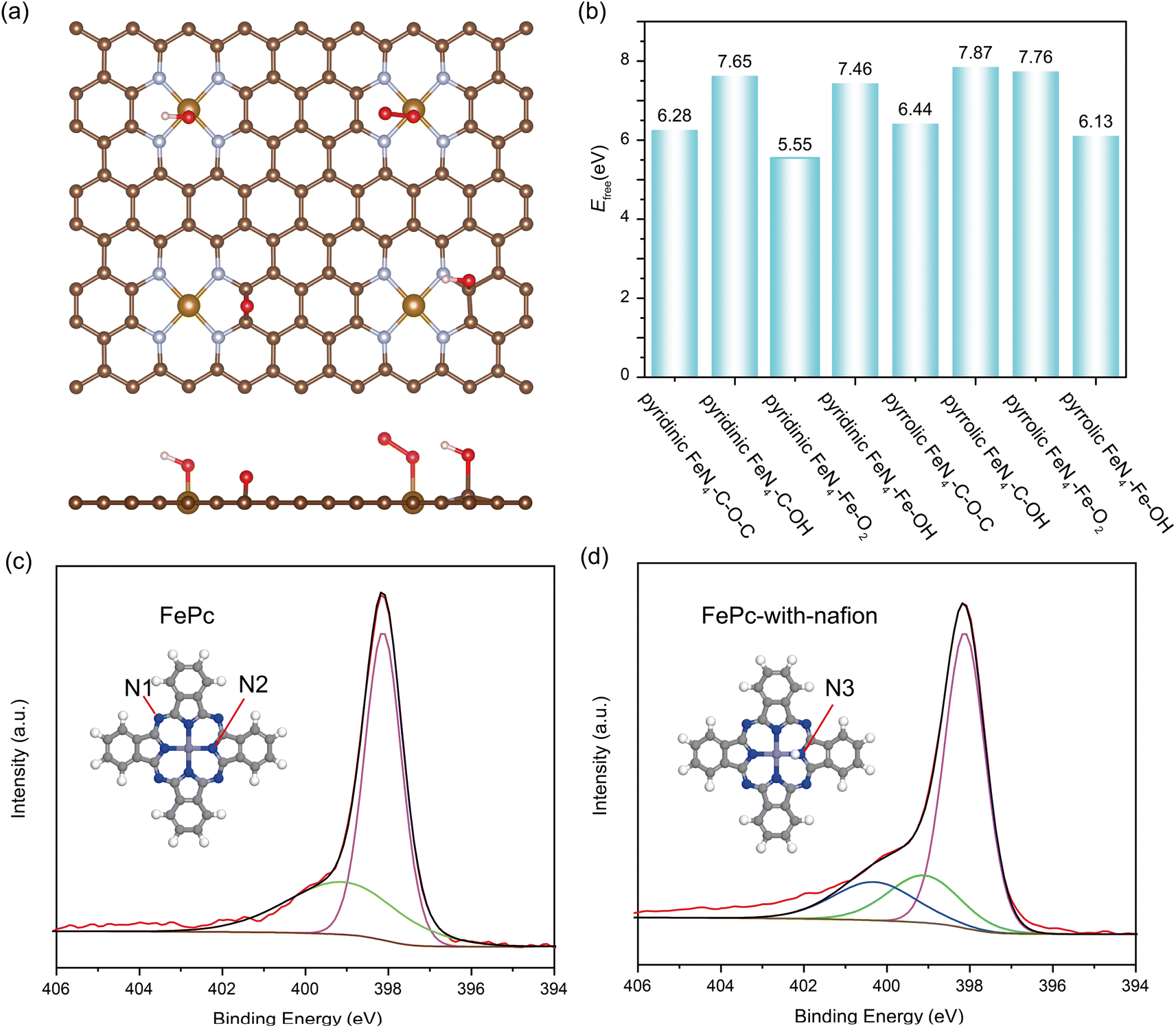 | ||
| Fig. 2 (a) Schematic of FeN4 moieties with adsorbed intermediates at the Fe site and carbon oxidation at the adjacent C site. (b) The kinetic activation energies of Fe SA leaching within FeN4 with various adsorbates at the Fe or C sites. The N 1s XPS spectrum of the FePc molecule (c) and the FePc molecule with Nafion (d). | ||
To resolve this ambiguity, we speculate that the difference may be attributed to the different degradation mechanism of FeN4 moiety sites, which may not be fully understood without considering the influence of the acidic medium. In other words, the protonation at the FeN4 active site might play a vital role. To test this hypothesis, the transformation in FePc's molecular structure before and after the post-Nafion treatment was probed using X-ray photoelectron spectroscopy (XPS). The Nafion is a perfluorosulfonic acid polymer with free H+ ion used in fuel cells, which has low volatility and can tolerate ultrahigh vacuum conditions during the XPS test. The high-resolution N 1s spectra of FePc and FePc-Nafion are compared in Fig. 2c and d. The pristine FePc molecule displays two pronounced peaks: an initial peak at 398.2 eV denoted as N1, representing the mesobridging aza nitrogens in –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– form not bonded to the Fe center, and a secondary peak at 399.2 eV, representing pyrrolic-type N bonded with the Fe center (N2). After the post-Nafion treatment, a new peak emerges (blue line) at 400.4 eV, which is associated with the pyrrole nitrogen bonded to hydrogen atoms in –NH– form (N3).53 The actual condition might involve a mixture of N–H and 2N–2H, as inferred from the Fe 2p XPS spectrum of FePc with Nafion (Fig. S4†). The Fe 2p spectrum of FePc was deconvoluted into two peaks, corresponding to the typical Fe2+ 2p3/2 (710.01 eV) and Fe2+ 2p1/2 (723.20 eV). However, the Fe 2p spectrum of FePc with Nafion does not fit these peaks, suggesting the possible coexistence of N–H and 2N–2H, which may cause a left shift in the Fe binding energy. Therefore, the XPS characterizations of the pristine and acid-treated FePc molecule shows clear evidence of possible protonation of the pyrrolic N.
C– form not bonded to the Fe center, and a secondary peak at 399.2 eV, representing pyrrolic-type N bonded with the Fe center (N2). After the post-Nafion treatment, a new peak emerges (blue line) at 400.4 eV, which is associated with the pyrrole nitrogen bonded to hydrogen atoms in –NH– form (N3).53 The actual condition might involve a mixture of N–H and 2N–2H, as inferred from the Fe 2p XPS spectrum of FePc with Nafion (Fig. S4†). The Fe 2p spectrum of FePc was deconvoluted into two peaks, corresponding to the typical Fe2+ 2p3/2 (710.01 eV) and Fe2+ 2p1/2 (723.20 eV). However, the Fe 2p spectrum of FePc with Nafion does not fit these peaks, suggesting the possible coexistence of N–H and 2N–2H, which may cause a left shift in the Fe binding energy. Therefore, the XPS characterizations of the pristine and acid-treated FePc molecule shows clear evidence of possible protonation of the pyrrolic N.
Employing DMSO-d6 as the NMR solvent, we observed distinct proton signals in FePc and FePc-with-Nafion (Fig. S5†). DMSO-d6 was used as the NMR solvent (2.50 ppm) and H2O also remained in the polymer sample (3.33 ppm). The peaks at 7.07, 7.59 and 7.81 ppm are the proton signals of the aromatic ring. The most significant observation was the peak at 11.3 ppm in the FePc-Nafion spectrum, attributed to nitrogen protonation, a feature absent in the FePc. The deuterium exchangeable inner core protons could not be observed in the 1H NMR spectrum of H2Pc due to the high aggregation ability.54 Fourier-Transform Infrared Spectroscopy (FT-IR) analyses (Fig. S6†) provided crucial insights into the molecular structures of iron(II) phthalocyanine (FePc), metal-free phthalocyanine (H2Pc), and the FePc-Nafion composite. Notable observations include the distinct C–H stretching vibrations near 3046 cm−1 across these spectra,55 with a pronounced peak at 1000 cm−1 in H2Pc, indicative of metal-free phthalocyanine.56 The absence of this peak in the FePc and FePc-Nafion spectra supports the retention of Fe2+ within its coordination sphere in these complexes. Furthermore, the N–H bond stretching frequencies observed at different wavenumbers (3288 cm−1 in H2Pc and 3198 cm−1 in FePc-Nafion) align with our DFT results, suggesting varied N–H bonding environments. These comprehensive analyses not only confirm the presence of protonated nitrogen in FePc-Nafion but also demonstrate that iron atoms have not leached out from the compound.
To specify the factors of acidic media, we further investigated the kinetic possibility of H adsorption at the N site of FeN4 under the working potential of ORR. Previously, the electrochemical measurements and in situ infrared reflectance spectroscopy experiments by Léger et al.57 suggested that the poor stability of FePc in acid medium occurred via the substitution of the central Fe metal of the macrocycle by two protons, leading to the formation of a free base phthalocyanine that is inactive for oxygen reduction catalysis. Herein, we conducted unconstrained AIMD simulations and captured the snapshots of nitrogen protonation in pyridinic-type FeN4 and pyrrolic-type FeN4 (Fig. 3a). Initially, we observed an optimized pyridinic-type FeN4 structure with one H atom adsorbed on the pyridinic N site (Fig. 3a(i)). Interestingly, during the unconstrained AIMD simulations at 300 K, the *H species spontaneously shifted from the pyridinic N site to the Fe site at 0.2 ps (Fig. 3b), consistent with previous research.33 However, when the coordinated N changes from pyridinic N to pyrrolic N, as illustrated in Fig. 3a(ii) and (iii), the *H species (protonation at one or two adjacent N sites) remained stably bonded with the N site instead of shifting to the Fe site even after 5 ps AIMD simulations. This indicates that the stable nitrogen protonation is directly related to the different N varieties, which in turn directly affects the Fe–N bond lengths. From Fig. 3c, the N protonation in pyrrolic-type FeN4 results in the inequivalent distribution of Fe–N bonds: one shortened Fe–N bond (2.04 Å for one N protonation and 1.98 Å for two N protonation) and one elongated Fe–N bond (2.12 Å for one N protonation and 2.38 Å for two N protonation) as compared with the bare pyrrolic-type FeN4 (2.08 Å). This indicates the slightly weakening of the Fe–N bond in pyrrolic-type FeN4 after protonation. The similar results can be identified in AIMD simulations of protonated pyridinic-type FeN4 and pyrrolic-type FeN4 executed with explicit water environments (Fig. S7†). Therefore, nitrogen protonation in the FeN4 moieties is likely to occur on the pyrrolic-type FeN4, which in turn leads to the inequivalent distribution and weakening of in-plane Fe–N bonds, while in pyridinic-type FeN4, the N protonation is unstable, and the proton prefers to bind with the Fe site instead, which has minimal effect on the structure of the FeN4 moiety. In our following discussion, we would mainly focus on the protonation effect on the dynamic instability of the pyrrolic-type FeN4 system.
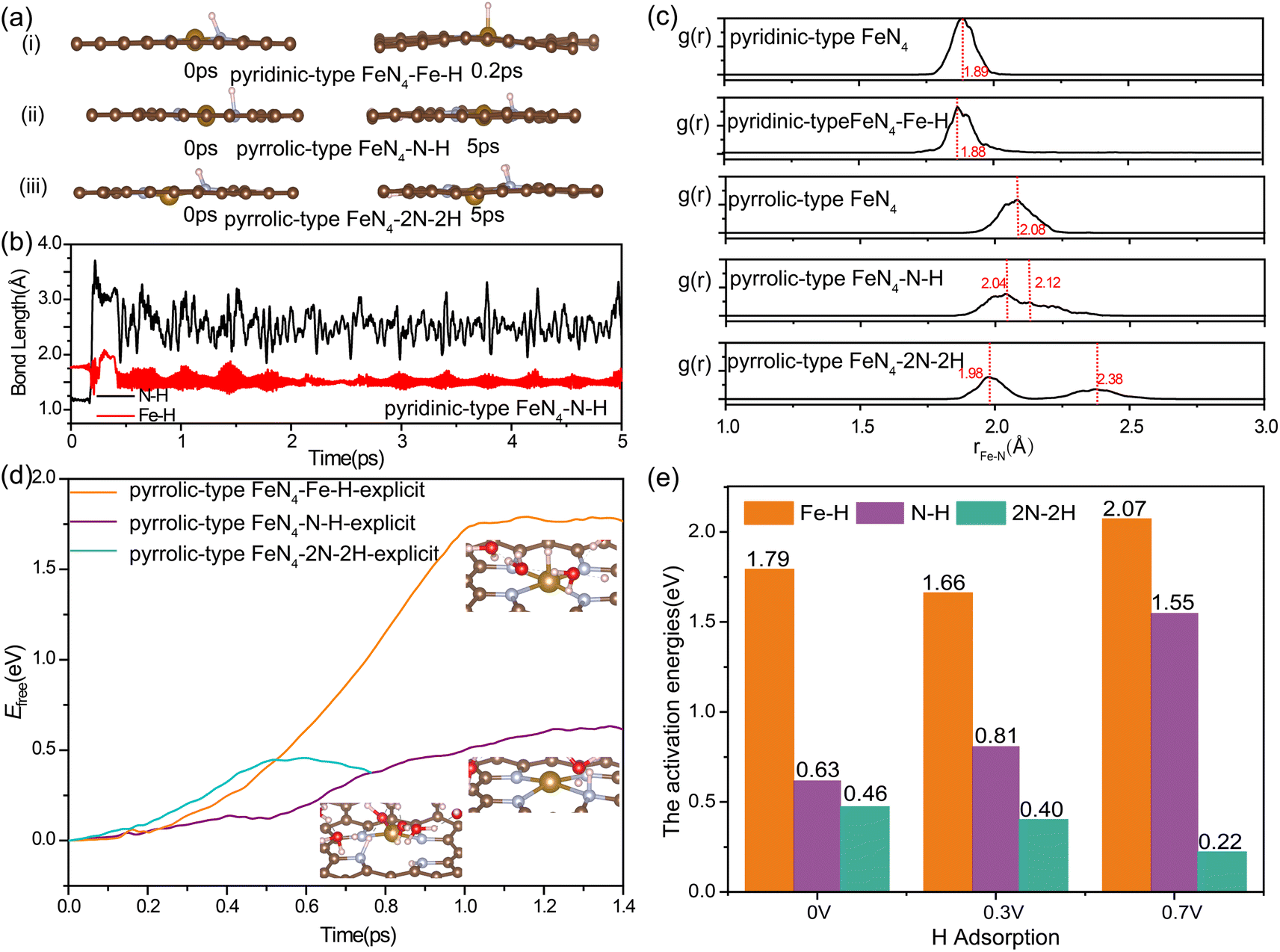 | ||
| Fig. 3 (a) AIMD simulation snapshots of nitrogen protonation in pyridinic-type FeN4 and pyrrolic-type FeN4. (b) An unconstrained 5 ps AIMD simulation shown the bond lengths of N–H and Fe–H in pyridinic-type FeN4–N–H. (c) Radial Distribution Functions (RDFs) showcasing the Fe–N distance in bare and protonated pyridinic-type FeN4 and pyrrolic-type FeN4, within a 5 ps time frame of AIMD simulation at 300 K. (d) Free energy profile of pyrrolic-type FeN4 during the protonation to form Fe–H and N–H bonds under URHE = 0 V. (e) The kinetic activation barrier for forming the Fe–H and N–H bonds in pyrrolic-type FeN4 under different working potentials. | ||
In a detailed examination of the kinetics of H adsorption behavior, the kinetic barrier for the transformation of hydronium species (H3O+) from solution into adsorbed H* at pyrrolic-type FeN4 and H2O is then determined under different working potentials. The criterion for the successful completion of this reaction is indicated by an N–H bond length of 1.00 Å or a Fe–H bond length of 1.5 Å, as demonstrated in Fig. S8 and S9.† Specifically, in the case of pyrrolic-type FeN4 (Fig. 3d), the kinetic barrier at potential of 0.0 V is computed to be 0.62 eV for protonation at the N site (the purple line) and 1.79 eV for protonation at the Fe site (the orange line). Interestingly, after the first N–H formation, the adsorption of the second H at the adjacent N site needs much lower barrier of 0.46 eV at 0.0 V (the green line in Fig. 3d), indicating significant enhancement of H adsorption and the nitrogen protonation reaction can easily occur at room temperature. Note that at the dynamic electrochemical pyrrolic-type FeN4/water interfaces, adsorption of 2H at adjacent N sites would cause the breaking of two Fe–N bonds, and meantime, the protruded under-coordinated Fe atom has strong affinity to adsorb and dissociate the H2O molecule from the solution, leading to adsorbed OH* species at the Fe site (inset structure in Fig. 3d). As the applied potential becomes more positive (Fig. 3e), the kinetic barrier for the initial nitrogen protonation is increased (from 0.63 eV at 0.0 V, to 0.81 eV at 0.3 V and 1.55 eV at 0.7 V), while the barrier for the second nitrogen protonation remains significantly lower (0.2–0.5 eV). The facilitated formation of 2N–2H species at 0.7 V RHE, compared to 0 V RHE, can be attributed to the potential-dependent alteration in the electronic state of nitrogen. This is evidenced by the calculated Bader charge of nitrogen in pyrrolic-type FeN4–N–H, which is −1.15e at 0 V and −1.18e at 0.7 V. At all the investigated working potentials, the formation barrier for the Fe–H bond is considerably much higher in comparison to the formation of N–H bond, again confirming the greater kinetic preference for nitrogen protonation at pyrrolic-type FeN4. Particularly, the highly facile nitrogen protonation at lower potentials could be an important factor affecting Fe desorption from catalysts during actual ORR operations, which will be discussed later.
Our above analysis has adequately demonstrated that the nitrogen protonation easily occurs on the pyrrolic-type N rather than the pyridinic-type N, which is a key factor that has not been revealed in previous studies. How does the adsorption of H affect the transformation of the pyrrolic-type FeN4 structure during ORR? The N protonation would weaken the binding strength between Fe and the substrate, which is expected to promote the leaching process of Fe atom. To verify this assumption, we re-evaluated the kinetic barrier of the Fe leaching from the pyrrolic-type FeN4 surface, where one and two N sites are adsorbed by H. The criterion for the end of this reaction is that the N–H bond length reaches 1.08 Å and the O–H bond length reaches 1.80 Å. Upon adsorption of one H, an activation barrier of 4.36 eV and 4.22 eV is respectively obtained for Fe leaching at 0.7 V and 1.2 V (Fig. 4a), where the Fe atom is completely detached from the surface, dissolved in the aqueous solution. Under the adsorption of 2H, the Fe center of pyrrolic-type FeN4 would spontaneously adsorb one OH species along with the breaking of two Fe–N bonds under 0.7 V and 1.2 V. In this case, the Fe leaching becomes much easier and the activation barrier is dramatically reduced to 1.56 eV at 0.7 V and 1.12 eV at 1.2 V (Fig. 4b). Fig. S10† shows the corresponding dynamic evolution of bond length of Fe–N(x) (x = 1, 2, 3 and 4). Compared with the Fe leaching barrier in pure pyrrolic-type FeN4 surface (7.45 eV), the adsorption of H at the N site significantly promotes the leaching of Fe atom. For comparison, we also examined the kinetics of Fe leaching in pyridinic-type FeN4 with H adsorption at the Fe site, an activation barrier of 4.25 eV and 1.71 eV is respectively obtained at 0.7 V and 1.2 V (Fig. S11†). This indicates that protonation at the Fe site can also significantly facilitate the Fe leaching in the pyridinic type FeN4 system, yet the kinetic barrier is comparatively much higher compared with the protonated pyrrolic-type FeN4 with adsorption of two H atoms.
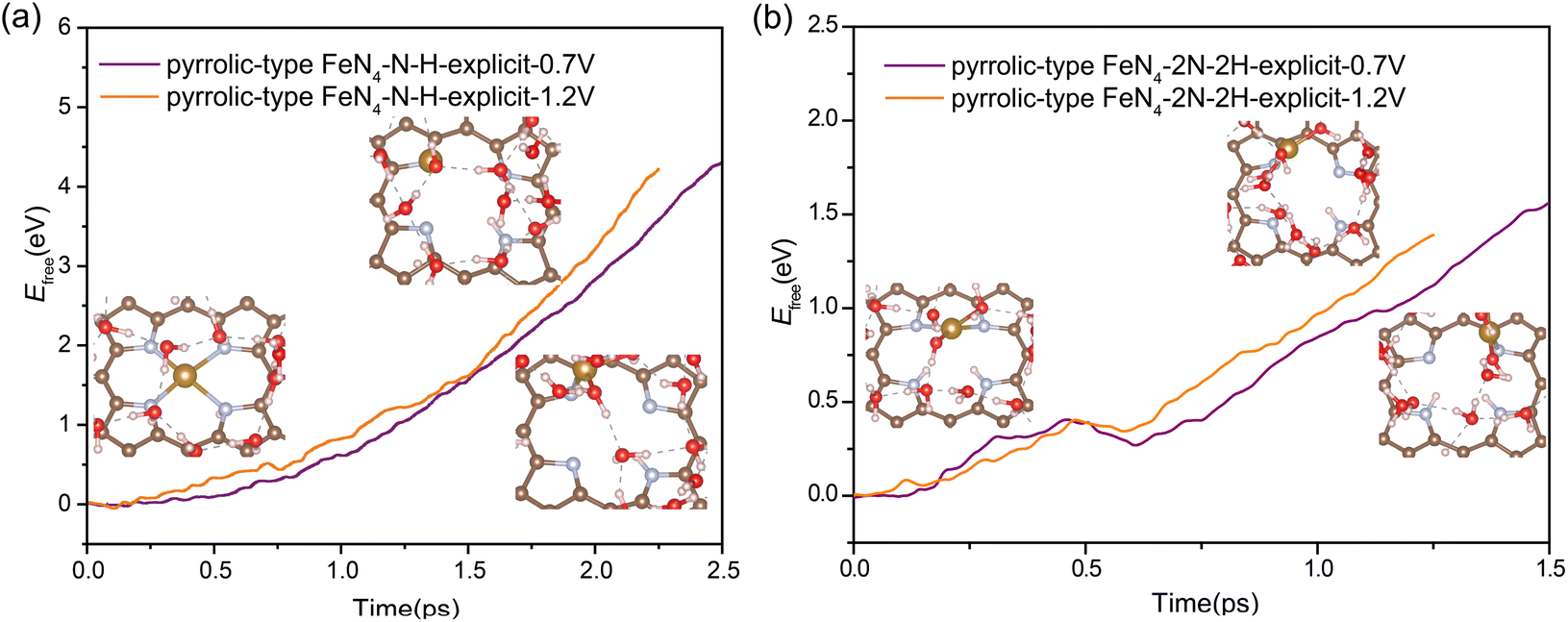 | ||
| Fig. 4 The free energy profiles during Fe leaching process in pyrrolic-type FeN4–N–H (a) and pyrrolic-type FeN4–2N–2H (b) at URHE = 0.7 V and 1.2 V. | ||
In addition to the protonation on the N site, the oxygen adsorbates also have an important effect on the stability of Fe atom. We found that the presence of oxygen-containing species at the Fe active center coupled with the protonation effect at the N site play a synergistic effect to facilitate the Fe leaching process. As shown in Fig. 5, when only one N site is protonated, the kinetic activation barrier for Fe leaching can be further reduced to 3.75 eV (2.66 eV) and 2.99 eV (2.03 eV) at potential of 0.7 V (1.2 V) when the Fe site is adsorbed with 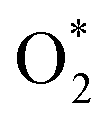 and OH* intermediates. The dynamic evolution of bond length of Fe–N(x) (x = 1, 2, 3 and 4) are shown in Fig. S12a–d.† When two adjacent N sites are protonated, the adsorption of
and OH* intermediates. The dynamic evolution of bond length of Fe–N(x) (x = 1, 2, 3 and 4) are shown in Fig. S12a–d.† When two adjacent N sites are protonated, the adsorption of 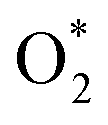 species at the Fe site leads to significantly reduced barrier of 1.3 eV at 0.7 V and 0.9 eV at 1.2 V, which means that the leaching of Fe becomes very favorable and the dissolution reaction can easily take place under mild conditions. Fig. S12e and f† show the dynamic evolution of bond length of Fe–N(x) (x = 1, 2, 3 and 4), indicating that the pyrrolic Fe–N–C catalyst spontaneously leaches from the surface in a short time (∼1.25 ps) under the condition of 2H coadsorption on N and O2 coadsorption on Fe. Therefore, our results demonstrate that the facile N protonation dictates the intrinsic instability of pyrrolic-type FeN4 structure, while the attachment of oxygen-containing species during realistic ORR process further promotes the leaching dynamics of active Fe atom.
species at the Fe site leads to significantly reduced barrier of 1.3 eV at 0.7 V and 0.9 eV at 1.2 V, which means that the leaching of Fe becomes very favorable and the dissolution reaction can easily take place under mild conditions. Fig. S12e and f† show the dynamic evolution of bond length of Fe–N(x) (x = 1, 2, 3 and 4), indicating that the pyrrolic Fe–N–C catalyst spontaneously leaches from the surface in a short time (∼1.25 ps) under the condition of 2H coadsorption on N and O2 coadsorption on Fe. Therefore, our results demonstrate that the facile N protonation dictates the intrinsic instability of pyrrolic-type FeN4 structure, while the attachment of oxygen-containing species during realistic ORR process further promotes the leaching dynamics of active Fe atom.
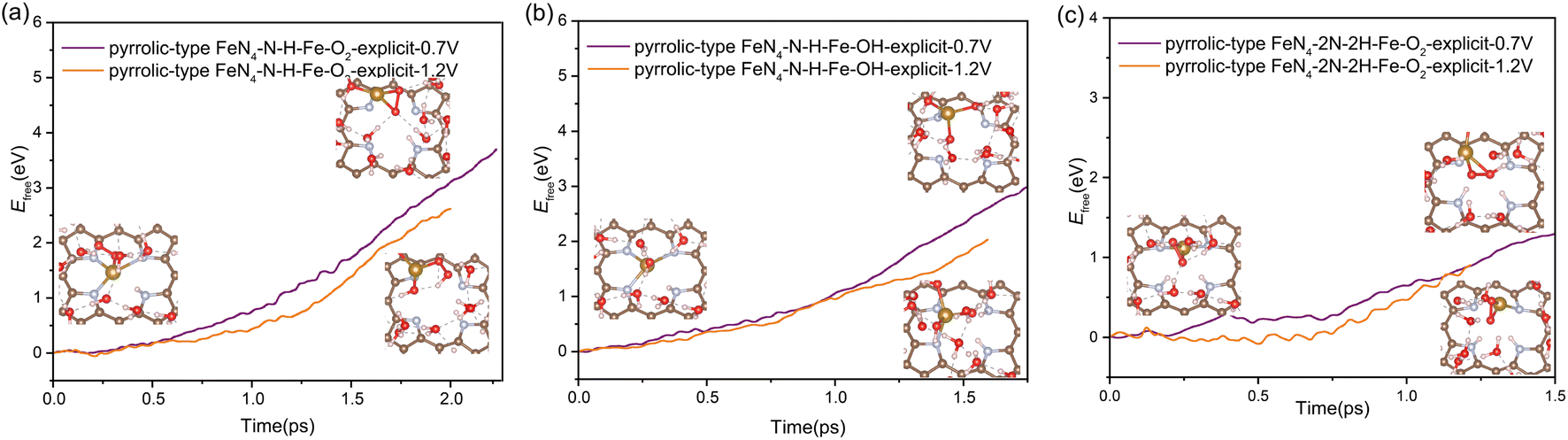 | ||
| Fig. 5 The free energy profiles during Fe leaching process in pyrrolic-type FeN4–N–H–Fe–O2 (a), pyrrolic-type FeN4–N–H–Fe–OH (b) and pyrrolic-type FeN4–2N–2H–Fe–O2 (c) at URHE = 0.7 V and 1.2 V. | ||
To get deeper insight into the bonding interactions, we performed calculations on the projected crystal orbital Hamilton population (pCOHP)58,59 to assess the bond strength between the central iron atom and the nitrogen atom in various configurations (Fig. 6a): pyridinic-type FeN4, pyrrolic-type FeN4 (pyrrolic-type N), pyrrolic-type FeN4–N–H with one N protonation, and pyrrolic-type FeN4–2N–2H with two N protonation. The pCOHP analysis provides valuable insights into the bonding characteristics.60 Moreover, the integrated pCOHP (IpCOHP) offers quantitative information on the bond strength. Notably, we observed that the central iron atom in the bare pyridinic-type FeN4 exhibits the strongest Fe–N bonding, with an IpCOHP value of −1.31 eV. This is followed by pyrrolic-type FeN4 (−0.99 eV), pyrrolic-type FeN4–N–H (−0.55 eV), and pyrrolic-type FeN4–2N–2H (−0.37 eV). These findings suggest the weaker pyrrolic-type Fe–N bond than the pyridinic-type Fe–N bond and the further stepwise weakening of the pyrrolic-type Fe–N bond with N protonation, resulting in the transition from unfavorable to favorable leaching of the Fe atom as the number of adsorbed H increases, consistent with our above dynamic results. Additionally, the IpCOHP values for the N–H bond in pyrrolic-type FeN4–N–H and pyrrolic-type FeN4–2N–2H are calculated to be −3.40 eV and −3.51 eV (Fig. S13†), respectively, indicating the formation of stabilized N–H bond. Especially, analysis via the Electron Localization Function (ELF)61 reveals a significant alteration in the electron delocalization within the FeN4 complex, particularly within a defined area (herein referred to as the “white circle”, Fig. 6b). This change is notably instigated by the presence of pyrrole N within the structure, which appears to reduce the delocalization. The impact is even more profound upon the adsorption of H, which leads to a further reduction in electron delocalization within the FeN4 complex. These findings suggest that the pyrrole N and the adsorbed hydrogen exert a significant influence on the electronic structure and, in turn, the reactivity of the FeN4 complex. Hence, the adsorption of H with pyrrolic-type N plays a crucial role as a driving force in the leaching of the Fe single atom from the Fe–N–C catalyst surface.
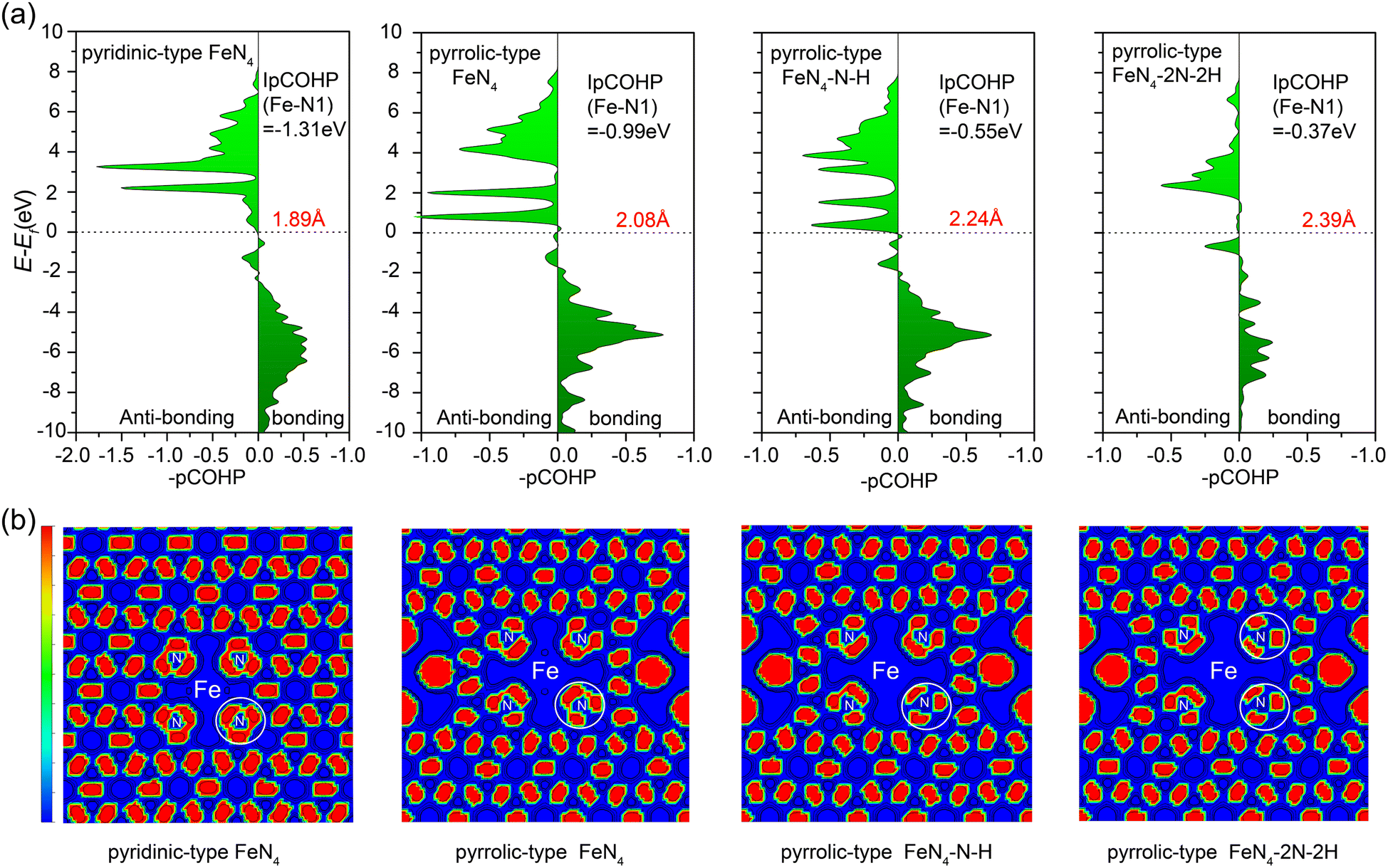 | ||
| Fig. 6 (a) Projected crystal orbital Hamilton population (pCOHP) between the central iron atom and the nitrogen atom as changes occur in N-type and hydrogen adsorption. The related IpCOHP values (depicted in black) and Fe–N bond lengths (depicted in red) are displayed in the diagrams. (b) The electron localization function for Fe–N–C as changes occur in N-type and hydrogen adsorption. | ||
4. Conclusions
The FeN4 catalyst is a good alternative to platinum based catalysts for ORR in fuel cells, but the major problem of this catalyst is its poor long-term stability in acid medium. The pyrrolic-type FeN4 has been experimentally demonstrated to be much more active for ORR than the pyridinic-type counterpart, however, the stability of the pyrrolic-type FeN4 structure is much inferior. The origin of the deactivation process remains thus far a mystery. In this work, we have systematically examined the dynamic stability of FeN4 catalysts under the acid ORR working conditions, with a particular focus on the impact of pyridinic-type and pyrrolic-type nitrogen in the Fe–N–C catalysts. Our findings revealed that the pyrrolic nitrogen of FeN4 can be easily protonated, while conversely, the pyridinic-type FeN4 prefers the protonation at the Fe site instead. The protonation at the N site plays the key role for the facile leaching of Fe single atom in pyrrolic-type FeN4, with a lower kinetic barrier of Fe leaching as the number of adsorbed H increases. In addition to the protonation effect at the pyrrolic N site, the presence of oxygen intermediates at the Fe site also has an important effect on the kinetics of Fe leaching, which work synergistically to promote conversion of iron atoms (Fe–N4) into ferric oxide, and the more positive the working potential, the lower the kinetic barrier. These observations contribute to a fundamental understanding of the structural evolution of pyrrolic-type FeN4 catalysts under operating conditions. This knowledge would inform the future rational design and optimization of Fe–N–C catalysts for better performance and durability in various electrochemical applications.Data availability
All the data in this study are provided in the main text and ESI.†Author contributions
Q. T. conceived the idea. Z. H. developed the theoretical models and performed the theoretical calculation and F. L. developed the theoretical models. Y. L. performed the experiment and analyzed the data under the guidance of S. C. and Z. W. Z. H. wrote the manuscript and with support from Z. W. and Q. T. finalized it. All authors discussed and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Q. T. acknowledges the support by the National Natural Science Foundation of China (No. 21903008), the Chongqing Science and Technology Commission (cstc2020jcyj-msxmX0382), Z. W acknowledges the support by the National Key Research and Development Program of China (No. 22090030, No. 52021004, No. 2021YFB4000300).References
- M. Grätzel, Photoelectrochemical cells, Nature, 2001, 414, 338–344 CrossRef.
- P. Prabhu and J.-M. Lee, Metallenes as functional materials in electrocatalysis, Chem. Soc. Rev., 2021, 50, 6700–6719 RSC.
- F. T. Wagner, B. Lakshmanan and M. F. Mathias, Electrochemistry and the future of the automobile, J. Phys. Chem. Lett., 2010, 1, 2204–2219 CrossRef CAS.
- L. Chong, J. Wen, J. Kubal, F. G. Sen, J. Zou, J. Greeley, M. Chan, H. Barkholtz, W. Ding and D.-J. Liu, Ultralow-loading platinum-cobalt fuel cell catalysts derived from imidazolate frameworks, Science, 2018, 362, 1276–1281 CrossRef CAS PubMed.
- X. Tian, X. Zhao, Y.-Q. Su, L. Wang, H. Wang, D. Dang, B. Chi, H. Liu, E. J. Hensen and X. W. Lou, Engineering bunched Pt-Ni alloy nanocages for efficient oxygen reduction in practical fuel cells, Science, 2019, 366, 850–856 CrossRef CAS PubMed.
- N. Tian, Z.-Y. Zhou, S.-G. Sun, Y. Ding and Z. L. Wang, Synthesis of tetrahexahedral platinum nanocrystals with high-index facets and high electro-oxidation activity, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- W. Zhang, J. Li and Z. Wei, Carbon-based catalysts of the oxygen reduction reaction: Mechanistic understanding and porous structures, Chin. J. Catal., 2023, 48, 15–31 CrossRef CAS.
- Y. Wang, J. Li and Z. Wei, Recent Progress of Carbon-Based Materials in Oxygen Reduction Reaction Catalysis, ChemElectroChem, 2018, 5, 1764–1774 CrossRef CAS.
- X. Zhang, A. Chen, L. Chen and Z. Zhou, 2D materials bridging experiments and computations for electro/photocatalysis, Adv. Energy Mater., 2022, 12, 2003841 CrossRef CAS.
- Y. Deng, J. Luo, B. Chi, H. Tang, J. Li, X. Qiao, Y. Shen, Y. Yang, C. Jia and P. Rao, Advanced atomically dispersed metal-nitrogen-carbon catalysts toward cathodic oxygen reduction in PEM fuel cells, Adv. Energy Mater., 2021, 11, 2101222 CrossRef CAS.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Understanding catalytic activity trends in the oxygen reduction reaction, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS.
- C. X. Zhao, B. Q. Li, J. N. Liu and Q. Zhang, Intrinsic electrocatalytic activity regulation of M-N-C single-atom catalysts for the oxygen reduction reaction, Angew. Chem., Int. Ed., 2021, 60, 4448–4463 CrossRef CAS.
- M. J. Workman, A. Serov, L.-k. Tsui, P. Atanassov and K. Artyushkova, Fe-N-C catalyst graphitic layer structure and fuel cell performance, ACS Energy Lett., 2017, 2, 1489–1493 CrossRef CAS.
- Q. Liu, X. Liu, L. Zheng and J. Shui, The solid-phase synthesis of an Fe-N-C electrocatalyst for high-power proton-exchange membrane fuel cells, Angew. Chem., 2018, 130, 1218–1222 CrossRef.
- X. Wan, W. Chen, J. Yang, M. Liu, X. Liu and J. Shui, Synthesis and Active Site Identification of Fe-N-C Single-Atom Catalysts for the Oxygen Reduction Reaction, ChemElectroChem, 2019, 6, 304–315 CrossRef CAS.
- Y. Li, X. Liu, L. Zheng, J. Shang, X. Wan, R. Hu, X. Guo, S. Hong and J. Shui, Preparation of Fe-N-C catalysts with FeNx (x= 1, 3, 4) active sites and comparison of their activities for the oxygen reduction reaction and performances in proton exchange membrane fuel cells, J. Mater. Chem. A, 2019, 7, 26147–26153 RSC.
- L. Chen, X. Wan, X. Zhao, W. Li, X. Liu, L. Zheng, Q. Liu, R. Yu and J. Shui, Spatial porosity design of Fe-N-C catalysts for high power density PEM fuel cells and detection of water saturation of the catalyst layer by a microwave method, J. Mater. Chem. A, 2022, 10, 7764–7772 RSC.
- L. Li, Y. Wen, G. Han, Y. Liu, Y. Song, W. Zhang, J. Sun, L. Du, F. Kong, Y. Ma, Y. Gao, J. Wang, C. Du and G. Yin, Tailoring the stability of Fe-NC via pyridinic nitrogen for acid oxygen reduction reaction, Chem. Eng. J., 2022, 437, 135320 CrossRef CAS.
- E. Proietti, F. Jaouen, M. Lefèvre, N. Larouche, J. Tian, J. Herranz and J.-P. Dodelet, Iron-based cathode catalyst with enhanced power density in polymer electrolyte membrane fuel cells, Nat. Commun., 2011, 2, 416 CrossRef PubMed.
- Y. Yang, L. Lai, L. Wei and Y. Chen, Degradation: a critical challenge for M-N-C electrocatalysts, J. Energy Chem., 2021, 63, 667–674 CrossRef CAS.
- J. Herranz, F. Jaouen, M. Lefèvre, U. I. Kramm, E. Proietti, J.-P. Dodelet, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach and P. Bertrand, Unveiling N-protonation and anion-binding effects on Fe/N/C catalysts for O2 reduction in proton-exchange-membrane fuel cells, J. Phys. Chem. C, 2011, 115, 16087–16097 CrossRef CAS.
- G. Zhang, X. Yang, M. Dubois, M. Herraiz, R. Chenitz, M. Lefevre, M. Cherif, F. Vidal, V. P. Glibin and S. Sun, Non-PGM electrocatalysts for PEM fuel cells: effect of fluorination on the activity and stability of a highly active NC_Ar+NH3 catalyst, Energy Environ. Sci., 2019, 12, 3015–3037 RSC.
- G. Zhang, R. Chenitz, M. Lefèvre, S. Sun and J.-P. Dodelet, Is iron involved in the lack of stability of Fe/N/C electrocatalysts used to reduce oxygen at the cathode of PEM fuel cells?, Nano Res., 2016, 29, 111–125 CAS.
- K. Strickland, R. Pavlicek, E. Miner, Q. Jia, I. Zoller, S. Ghoshal, W. Liang and S. Mukerjee, Anion resistant oxygen reduction electrocatalyst in phosphoric acid fuel cell, ACS Catal., 2018, 8, 3833–3843 CrossRef CAS.
- U. Tylus, Q. Jia, H. Hafiz, R. Allen, B. Barbiellini, A. Bansil and S. Mukerjee, Engendering anion immunity in oxygen consuming cathodes based on Fe-Nx electrocatalysts: Spectroscopic and electrochemical advanced characterizations, Appl. Catal., B, 2016, 198, 318–324 CrossRef CAS.
- M. Rauf, Y.-D. Zhao, Y.-C. Wang, Y.-P. Zheng, C. Chen, X.-D. Yang, Z.-Y. Zhou and S.-G. Sun, Insight into the different ORR catalytic activity of Fe/N/C between acidic and alkaline media: Protonation of pyridinic nitrogen, Electrochem. Commun., 2016, 73, 71–74 CrossRef CAS.
- C. H. Choi, H.-K. Lim, M. W. Chung, G. Chon, N. R. Sahraie, A. Altin, M.-T. Sougrati, L. Stievano, H. S. Oh, E. S. Park, F. Luo, P. Strasser, G. Dražić, K. J. J. Mayrhofer, H. Kim and F. D. R. Jaouen, The Achilles' heel of iron-based catalysts during oxygen reduction in an acidic medium, Energy Environ. Sci., 2018, 11, 3176–3182 RSC.
- R. Chenitz, U. I. Kramm, M. Lefèvre, V. Glibin, G. Zhang, S. Sun and J.-P. Dodelet, A specific demetalation of Fe-N4 catalytic sites in the micropores of NC_Ar+ NH3 is at the origin of the initial activity loss of the highly active Fe/N/C catalyst used for the reduction of oxygen in PEM fuel cells, Energy Environ. Sci., 2018, 11, 365–382 RSC.
- C. H. Choi, C. Baldizzone, J. P. Grote, A. K. Schuppert, F. Jaouen and K. J. Mayrhofer, Stability of Fe-N-C catalysts in acidic medium studied by operando spectroscopy, Angew. Chem., Int. Ed., 2015, 54, 12753–12757 CrossRef CAS PubMed.
- Y.-P. Ku, K. Ehelebe, A. Hutzler, M. Bierling, T. Böhm, A. Zitolo, M. Vorokhta, N. Bibent, F. D. Speck, D. Seeberger, I. Khalakhan, K. Mayrhofer, S. Thiele, F. Thiele and S. Cherevko, Oxygen reduction reaction in alkaline media causes iron leaching from Fe-N-C electrocatalysts, J. Am. Chem. Soc., 2022, 144, 9753–9763 CrossRef CAS.
- J.-Y. Choi, L. Yang, T. Kishimoto, X. Fu, S. Ye, Z. Chen and D. Banham, Is the rapid initial performance loss of Fe/N/C non precious metal catalysts due to micropore flooding?, Energy Environ. Sci., 2017, 10, 296–305 RSC.
- E. F. Holby, G. Wang and P. Zelenay, Acid stability and demetalation of PGM-free ORR electrocatalyst structures from density functional theory: a model for “single-atom catalyst” dissolution, ACS Catal., 2020, 10, 14527–14539 CrossRef CAS.
- N. Yang, L. Peng, L. Li, J. Li, Q. Liao, M. Shao and Z. Wei, Theoretically probing the possible degradation mechanisms of an FeNC catalyst during the oxygen reduction reaction, Chem. Sci., 2021, 12, 12476–12484 RSC.
- X. Tan, H. A. Tahini and S. C. Smith, Unveiling the role of carbon oxidation in irreversible degradation of atomically-dispersed FeN 4 moieties for proton exchange membrane fuel cells, J. Mater. Chem. A, 2021, 9, 8721–8729 RSC.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- L. Yu, X. Pan, X. Cao, P. Hu and X. Bao, Oxygen reduction reaction mechanism on nitrogen-doped graphene: A density functional theory study, J. Catal., 2011, 282, 183–190 CrossRef CAS.
- X. Zhao and Y. Liu, Unveiling the active structure of single nickel atom catalysis: critical roles of charge capacity and hydrogen bonding, J. Am. Chem. Soc., 2020, 142, 5773–5777 CrossRef CAS.
- X. Hu, S. Chen, L. Chen, Y. Tian, S. Yao, Z. Lu, X. Zhang and Z. Zhou, What is the Real Origin of the Activity of Fe-N-C Electrocatalysts in the O2 Reduction Reaction? Critical Roles of Coordinating Pyrrolic N and Axially Adsorbing Species, J. Am. Chem. Soc., 2022, 144, 18144–18152 CrossRef CAS PubMed.
- T. Cheng, H. Xiao and W. A. Goddard III, Reaction mechanisms for the electrochemical reduction of CO2 to CO and formate on the Cu (100) surface at 298 K from quantum mechanics free energy calculations with explicit water, J. Am. Chem. Soc., 2016, 138, 13802–13805 CrossRef CAS.
- T. Cheng, L. Wang, B. V. Merinov and W. A. Goddard III, Explanation of dramatic pH-dependence of hydrogen binding on noble metal electrode: Greatly weakened water adsorption at high pH, J. Am. Chem. Soc., 2018, 140, 7787–7790 CrossRef CAS PubMed.
- K. Xie, F. Wang, F. Wei, J. Zhao and S. Lin, Revealing the origin of nitrogen electroreduction activity of molybdenum disulfide supported iron atoms, J. Phys. Chem. C, 2022, 126, 5180–5188 CrossRef CAS.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, Gaussian 16 Rev. C. 01, Wallingford, CT, 2016 Search PubMed.
- S. Liu, Q. Shi and G. Wu, Solving the activity–stability trade-off riddle, Nat. Catal., 2021, 4, 6–7 CrossRef.
- L. Jiao, J. Li, L. L. Richard, Q. Sun, T. Stracensky, E. Liu, M. T. Sougrati, Z. Zhao, F. Yang and S. Zhong, Chemical vapour deposition of Fe–N–C oxygen reduction catalysts with full utilization of dense Fe-N4 sites, Nat. Mater., 2021, 20, 1385–1391 CrossRef CAS PubMed.
- N. Zhang, T. Zhou, M. Chen, H. Feng, R. Yuan, W. Yan, Y. Tian, X. Wu, W. Chu and C. Wu, High-purity pyrrole-type FeN 4 sites as a superior oxygen reduction electrocatalyst, Energy Environ. Sci., 2020, 13, 111–118 RSC.
- J. Li, M. T. Sougrati, A. Zitolo, J. M. Ablett, I. C. Oğuz, T. Mineva, I. Matanovic, P. Atanassov, Y. Huang, I. Zenyuk, A. Di Cicco, K. Kumar, L. Dubau, F. Maillard, G. Dražić and F. Jaouen, Identification of durable and non-durable FeN x sites in Fe–N–C materials for proton exchange membrane fuel cells, Nat. Catal., 2021, 4, 10–19 CrossRef CAS.
- Y. Alfredsson, B. Brena, K. Nilson, J. Åhlund, L. Kjeldgaard, M. Nyberg, Y. Luo, N. Mårtensson, A. Sandell and C. Puglia, Electronic structure of a vapor-deposited metal-free phthalocyanine thin film, J. Chem. Phys., 2005, 122, 214723 CrossRef CAS PubMed.
- C. F. van Nostrum, S. J. Picken, A.-J. Schouten and R. J. Nolte, Synthesis and supramolecular chemistry of novel liquid crystalline crown ether-substituted phthalocyanines: toward molecular wires and molecular ionoelectronics, J. Am. Chem. Soc., 1995, 117, 9957–9965 CrossRef CAS.
- H. Shurvell and L. Pinzuti, Sur Les Spectres Infrarouges Des Phtalocyanines, Can. J. Chem., 1966, 44, 125–136 CrossRef CAS.
- X. X. Zhang, M. Bao, N. Pan, Y. X. Zhang and J. Z. Jiang, IR and Raman Vibrational Assignments for Metal-free Phthalocyanine from Density Functional B3LYP/6-31G (d) Method, Chin. J. Chem. Phys., 2004, 22, 325–332 CrossRef CAS.
- S. Baranton, C. Coutanceau, C. Roux, F. Hahn and J.-M. Léger, Oxygen reduction reaction in acid medium at iron phthalocyanine dispersed on high surface area carbon substrate: tolerance to methanol, stability and kinetics, J. Electroanal. Chem., 2005, 577, 223–234 CrossRef CAS.
- Z. Huang and Q. Tang, Axial Coordination Effect on the Oxygen Reduction Reaction of FeN4 Electrocatalysts Based on Grand Canonical Density Functional Theory, J. Phys. Chem. C, 2022, 126, 21606–21615 CrossRef CAS.
- Y. Chen, F. Sun and Q. Tang, Computational Insights and Design of Promising Ultrathin PdM Bimetallenes for Oxygen Reduction Electrocatalysis, Small Methods, 2023, 2300276 CrossRef CAS.
- M. Zhu, W. Song, P. M. Konze, T. Li, B. Gault, X. Chen, J. Shen, S. Lv, Z. Song and M. Wuttig, Direct atomic insight into the role of dopants in phase-change materials, Nat. Commun., 2019, 10, 3525 CrossRef.
- A. D. Becke and K. E. Edgecombe, A simple measure of electron localization in atomic and molecular systems, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05378d |
| This journal is © The Royal Society of Chemistry 2024 |