
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUracil-Cu(I) catalyst: allylation of cyclopropanols with Morita–Baylis–Hillman alcohols under water-tolerant conditions†
Jingwei
Hou
a,
Xiaohong
Li
a,
Kaiyu
Yan
a,
Lei
Zhang
 *b,
Teck-Peng
Loh
*cd and
Peizhong
Xie
*a
*b,
Teck-Peng
Loh
*cd and
Peizhong
Xie
*a
aSchool of Chemistry and Molecular Engineering, Nanjing Tech University, Nanjing 211816, China
bSchool of Science, Tianjin Chengjian University, Tianjin 300384, China. E-mail: zhanglei-chem@tcu.edu.cn
cCollege of Advanced Interdisciplinary Science and Technology, Henan University of Technology, Zhengzhou 450001, China
dDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371. E-mail: teckpeng@ntu.edu.sg
First published on 7th December 2023
Abstract
Inspired by the high affinity of copper with DNA and RNA, a uracil-copper catalytic system was developed to promote ring-opening allylation of cyclopropanols with allylic alcohols under water-tolerant conditions. A new C–OH bond-breaking model can well resolve the trade-off between the need for acidic activators for C(allyl)–OH bond cleavage and the demand for strong basic conditions for generating homoenolates. Therefore, Morita–Baylis–Hillman alcohols, rather than their pre-activated versions, could be incorporated directly into dehydrative cross-coupling with cyclopropanols delivering water as the only by-product. A variety of functionalized δ,ε-unsaturated ketones were obtained in good-to-high yield with high E-selectivity.
Introduction
Given that copper has shown a rather high affinity for DNA and RNA,1 the interaction between copper and thymine/uracil has received great attention.2 The structure of the uracil/copper complex has been studied by Fridgen1a and Lamsabhi1b independently, while its catalytic activity has never been explored (Fig. 1). Recently, δ,ε-unsaturated ketones were identified as privileged motifs in various pharmaceutically active natural products3 and FDA-approved drugs.4 Since Cha's pioneering reported allylation of cyclopropanol5 with a stoichiometric amount of Et2Zn and CuCN·2LiCl (Fig. 1a), allylation of low-cost metal homoenolates was considered as the state of the art to access δ,ε-unsaturated ketones.6 Yin,7a Trost,7b and Sawamura7c have also paid an extensive amount of attention to addressing the asymmetric versions of related copper catalysts (Fig. 1a). The catalytic activity of other low-cost transition metals (Co and Ni) was also explored by Yoshikai and co-workers.8 Despite these impressive achievements, equivalents of a strong base and cryogenic and/or anhydrous conditions are generally inevitable for guaranteeing the generation of metal homoenolates.5–7 In addition, only activated allylic precursors5–8 were compatible for the concomitant allylation. In terms of economic and ecological concerns, the direct incorporation of the most abundant allylic alcohols9 into ring-opening allylation is highly desirable, especially in pharmaceutical industries as water will be the only by-product.10 However, the irreconcilable contradiction between the acidity activator for C(allyl)–OH bond cleavage11 and strong basic environmental5–8 requirements for generating homoenolates has challenged the development of the catalyst. In our continuing green chemistry efforts, we paid special attention to C–OH bond cleavage12 and related transformations. As one of the most versatile building blocks, Morita–Baylis–Hillman (MBH) alcohols have garnered widespread attention in organic synthesis,13 and seen growing interest in the field of medicine.14 We herein report a uracil-Cu catalytic system for the ring-opening allylation of cyclopropanols with MBH alcohols in a dehydrative cross-coupling manner, which affords water as the only by-product (Fig. 1b).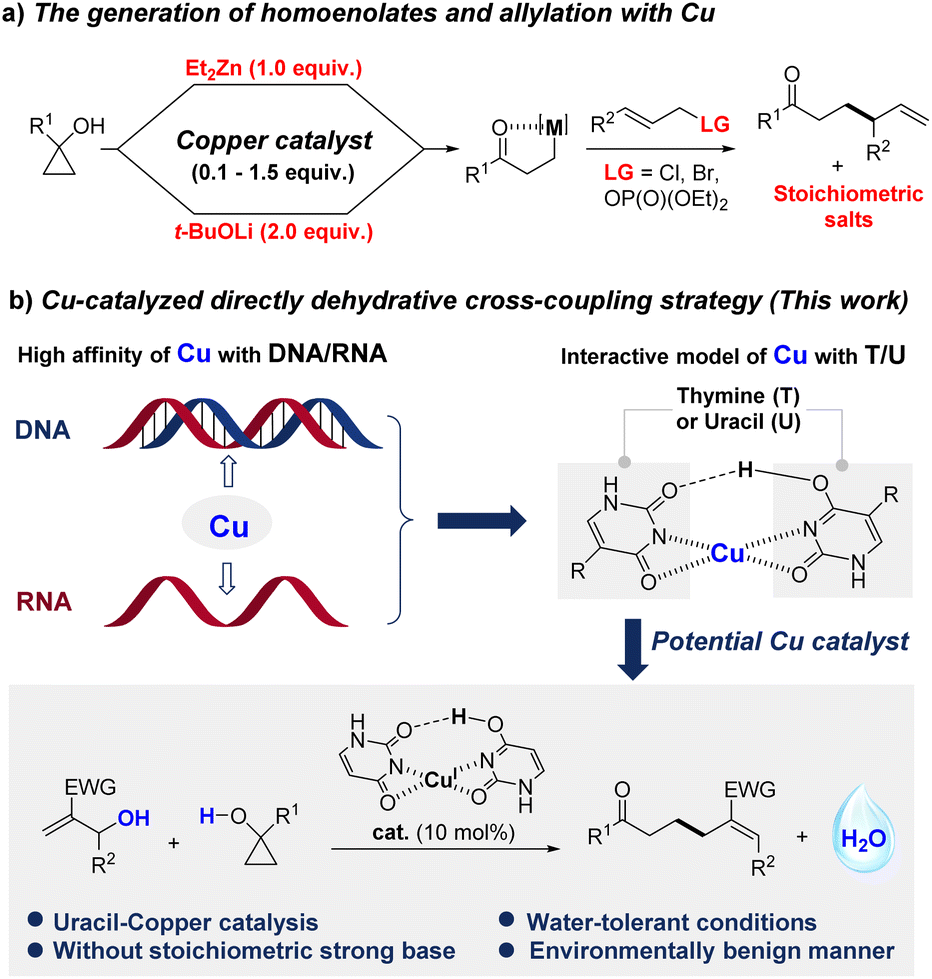 | ||
| Fig. 1 Copper-promoted allylation of cyclopropanol via homoenolates. | ||
Results and discussion
At the onset of our study, we initially evaluated the dehydrative coupling potential of 1-phenethylcyclopropan-1-ol (1a) with methyl 2-(hydroxy(phenyl)methyl)acrylate (2a) using a copper catalytic system. Notably, although MBH adducts (tert-butyl carbonates) are recognized as versatile building blocks in various fields, the allylation of cyclopropanols suffered from poor stereoselectivity or other competitive processes.15After carefully evaluating copper precursors (see Supporting Information (ESI) for details†), we found that Cu(MeCN)4PF6 provided the best results, yielding the desired 3a in 25% yield (Table 1, entry 4). Replacement of Cu(MeCN)4PF6 with CuBr (Table 1, entry 1), Cu(OAc)2 (Table 1, entry 2), or Cu(MeCN)4BF4 (Table 1, entry 3) under the same reaction conditions resulted in either trace amounts or no reaction at all. With subsequent screening of solvents and altering the ratio of 1a/2a, DMF improved the yield of 3a to 41% (Table 1, entry 5). The use of common ligands, such as 1,10-phenanthroline or 4,4′-bipyridine, had a negligible impact on the yield of 3a or even proved detrimental (Table 1, entries 6 and 7). Then, we selected uracil as the ligand to combine with Cu(MeCN)4PF6, and to our delight, the yield of 3a improved to 65% (Table 1, entry 8). In sharp contrast to the previously reported catalytic system, which was sensitive to moisture, water played an essential role in this transformation. With the assistance of 4.0 equivalents of water, the yield of 3a improved to 72% (Table 1, entry 9). This result indicated that our developed reaction was fully water-tolerant. After carefully evaluating all reaction parameters, we found that the addition of 7 mol% of Me2NH actively stabilized the yield and improved the reproducibility (Table 1, entries 10 and 11). When uracil was replaced by thymine, a slightly lower yield of 3a was obtained (Table 1, entry 12). However, only a trace amount of 3a was detected when cytosine rather than uracil was introduced into this reaction (Table 1, entry 13). These results revealed that the combination of uracil/thymine with copper was crucial for promoting the desired transformation. In addition, the reaction also proceeded smoothly even at a lower temperature (40 °C), albeit delivering 3a in a moderate yield (Table S1, entry 14†). Upon decreasing the catalyst loading to 5 mol%, 3a could also be obtained in a 45% yield (Table 1, entry 15).
|
|
||||
|---|---|---|---|---|
| Entry | [Cu] (10 mol%) | [L] (20 mol%) | Add. | 3a (%)b |
| a Experimental conditions: 1a (0.3 mmol), 2a (0.2 mmol), Cu (catalyst) (0.02 mmol), L (ligands) (0.04 mmol) and Add. (for entries 9–15, 4.0 equiv. of H2O was added and 7 mol% of Me2NH was also added for entries 11–15) were mixed in DMF (2.0 mL) at 60 °C (metal bath). b Determined by 1H NMR of the crude product with mesitylene as the internal standard. In parentheses are isolated yields. c 1a (0.2 mmol) and 2a (0.24 mmol) were mixed in THF (2.0 mL). d 1a (0.4 mmol) and 2a (0.2 mmol). e At 40 °C (metal bath). f Cu(CH3CN)4PF6 (0.01 mmol) and uracil (0.02 mmol) were used. In all the tested cases, the ratio of E/Z is over 99/1. | ||||
| 1c | CuBr | — | — | <5 |
| 2c | Cu(OAc)2 | — | — | n.r. |
| 3c | Cu(MeCN)4BF4 | — | — | 5 |
| 4c | Cu(MeCN)4PF6 | — | — | 25 |
| 5 | Cu(MeCN)4PF6 | — | — | 41 |
| 6 | Cu(MeCN)4PF6 | 1,10-Phen. | — | 25 |
| 7 | Cu(MeCN)4PF6 | Bipyridine | — | 32 |
| 8 | Cu(MeCN)4PF6 | Uracil | — | 65 |
| 9 | Cu(MeCN)4PF6 | Uracil | H2O | 72 |
| 10d | Cu(MeCN)4PF6 | Uracil | H2O | 80 |
| 11d | Cu(MeCN)4PF6 | Uracil | H2O and Me2NH | 82 (80) |
| 12d | Cu(MeCN)4PF6 | Thymine | H2O and Me2NH | 70 |
| 13d | Cu(MeCN)4PF6 | Cytosine | H2O and Me2NH | <5 |
| 14d,e | Cu(MeCN)4PF6 | Uracil | H2O and Me2NH | 46 |
| 15d,f | Cu(MeCN)4PF6 | Uracil | H2O and Me2NH | 45 |
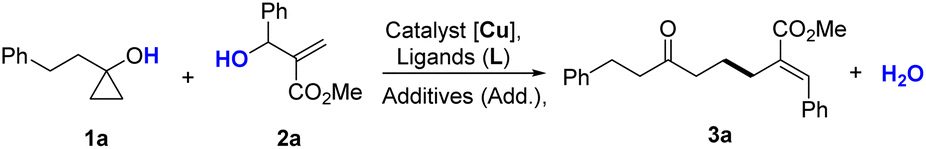
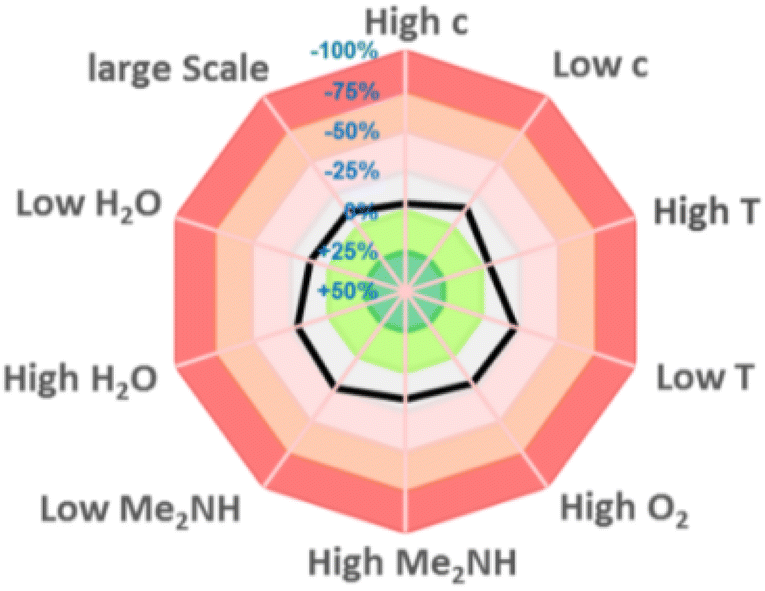
To assess the reproducibility of this transformation, we followed the methodology reported by Glorius et al.16 The parameters including concentration, levels of water and oxygen, scales, and temperature only have a limited effect on the transformation, indicating that this strategy has a good reproducibility (Fig. 2).
 | ||
| Fig. 2 Sensitivity assessment. | ||
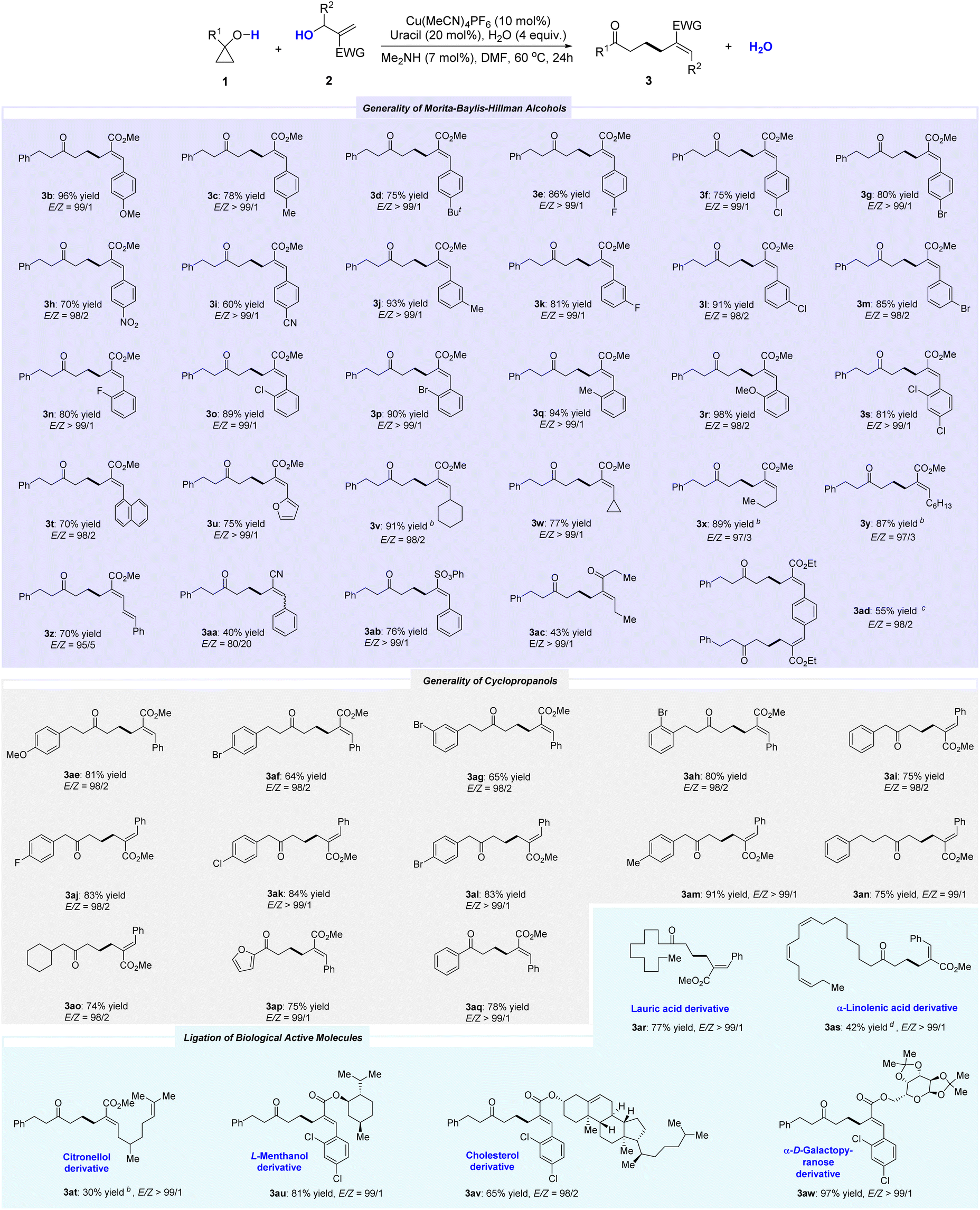
Next, we explored the generality of the reaction with Morita–Baylis–Hillman (MBH) alcohols under optimized conditions. As shown in Table 2, various MBH alcohols were successfully incorporated into the dehydrative cross-coupling with cyclopropanols, delivering corresponding products 3a–3ab in high yield. The electronic property (3b–3i) and position (3j–3s) of the substituent on the phenyl ring had limited effect on the yield. Even strong electron-donating (3b) or electron-withdrawing (3h, 3i) groups were tolerated well. Phenyl rings bearing o-substituents and poly-substituents also afforded corresponding products (3n–3s) in high yield. MBH alcohols with fused rings (3t) and hetero-aryl moiety (3u) were also amenable to this reaction. Besides aryl-substituted MBH alcohols, alkyl-substituted versions also worked well (3v–3y). MBH alcohols bearing cyclic (3v, 3w) or acyclic (3x, 3y) alkyls could also participate in this protocol, delivering the desired products in high yields. In addition, although the yield was slightly lower, methyl (E)-6-oxo-8-phenyl-2-((E)-3-phenylallylidene)octanoate (3z) can be obtained by selecting methyl (E)-3-hydroxy-2-methylene-5-phenylpent-4-enoate. 2-(Hydroxy(phenyl)methyl)acrylonitrile was identified as a suitable precursor for this reaction, although it was only 80/20. MBH alcohols derived from phenyl ethenesulfonate (3ab) or ketones (3ac) were proven to be viable coupling partners for this reaction, even carbonyl groups with adjacent acidic C–H bonds.
| a Experimental conditions: 1 (0.4 mmol), 2 (0.2 mmol), Cu(MeCN)4PF6 (0.02 mmol), uracil (0.04 mmol), H2O (0.8 mmol), and Me2NH (2.4 μL, 0.014 mmol) were mixed in DMF (2.0 mL) at 60 °C (metal bath) for 24 h. b Cu(MeCN)4PF6 (0.03 mmol), uracil (0.06 mmol). c 1a (0.8 mmol), 2 (0.2 mmol), Cu(MeCN)4PF6 (0.04 mmol), uracil (0.08 mmol), Me2NH (4.8 μL, 0.028 mmol) and H2O (1.6 mmol). d Cu(MeCN)4PF6 (0.04 mmol), uracil (0.08 mmol). |
|---|
|
By using ethyl 3-(4-(2-(ethoxycarbonyl)-1-hydroxyallyl)phenyl)-2-hydroxybut-3-enoate, the double dehydrative coupling version can also proceed well, albeit affording 3ad in slightly lower yield.
Subsequently, the generality of cyclopropanols was explored. A variety of cyclopropanols were found to readily react with MBH alcohols. For 1-phenethylcyclopropan-1-ol derivatives, the position of the substituent (ortho, para, or meta) had little effect on the overall transformations (3ae–3ah). Even 1-benzylcyclopropan-1-ol was a competent coupling partner, regardless of the electron-withdrawing (3aj–3al) or electron-donating (3am) groups on the phenyl rings. The reaction with 1-(3-phenylpropyl)cyclopropan-1-ol also proceeded smoothly to deliver 3an in 75% yield. Moreover, selecting 1-(cyclohexylmethyl)cyclopropan-1-ol as the feedstock allowed for the high-yield synthesis of methyl (E)-2-benzylidene-7-cyclohexyl-6-oxoheptanoate (3ao). In addition to 1-alkyl-substituted cyclopropanol, both 1-phenyl- and 1-furan-2-yl-substituted versions were also found to participate in this transformation, affording 3ap and 3aq in high yields, respectively. The structure and configuration of the double bond in 3aq (CCDC: 2202510) were determined by X-ray analysis.
We aimed to demonstrate the versatility of this synthetic method by applying it to several biologically active compounds. As shown in Table 2, the cyclopropanol derived from lauric acid was smoothly converted to the desired product 3ar in high yield. The skeleton of alpha-linolenic acid was also incorporated into the product (3as) in moderate yield while maintaining the Z-configuration of the double bond. Moreover, all the MBH alcohol precursors bearing the motifs of citronellol, L-menthanol, cholesterol, and alpha-D-galactopyranose could be smoothly transformed, producing corresponding 3at–3aw effectively. These results opened up possibilities for the potential utility of this protocol in pharmaceutical-related studies.
Having demonstrated the versatility of this transformation concerning biologically active molecules, we attempted to gain more insights into the reaction mechanism by conducting some related control experiments (Scheme 1, top). Under identical conditions, no corresponding product was detected when the allylic alcohol was replaced by its isomer (reaction a). These results indicated that a copper-catalyzed allylic SN2′ process was involved in this transformation, and steric hindrance at the γ-position of allylic alcohols was disfavored. In addition, only a trace amount of 3ay was detected when ethyl 2-(hydroxymethyl)acrylate was selected as the coupling partner. This is because ethyl 2-(hydroxymethyl)acrylate favored self-polymerization. Similarly, 2-methyl-1-phenylprop-2-en-1-ol could not be consumed under standard conditions (reaction b), which implied that the electron-deficient double bond was essential for this reaction. In contrast, a comparable yield of 3a was obtained when the hydroxyl group was protected (2a-OMe) (reaction c). Moreover, we conducted experiments to explore the cyclopropane ring with substitutions at the distal carbons present at the C–OH positions. As is depicted, the C–C bond cleavage has poor selectivity, with the desired 3az and 3az′ obtained in good yields (3ax/3ax′ = 57/41) (reaction d). When a sterically demanding substance (R = CH2CH2Ph or Ph) was selected, the reaction was sluggish with most cyclopropanols being recovered and only a trace amount of the desired product was detected. These results revealed that the ring opening of cyclopropanol has poor selectivity, and the steric effect on the copper homoenolates at either α- or β-position of the carbonyl group disfavoured the following cross-coupling process. Therefore, this method appears to be primarily limited to monosubstituted cyclopropanols. To clearly outline the overall scope and limitations, we incorporate structure restriction at the bottom of Scheme 1.
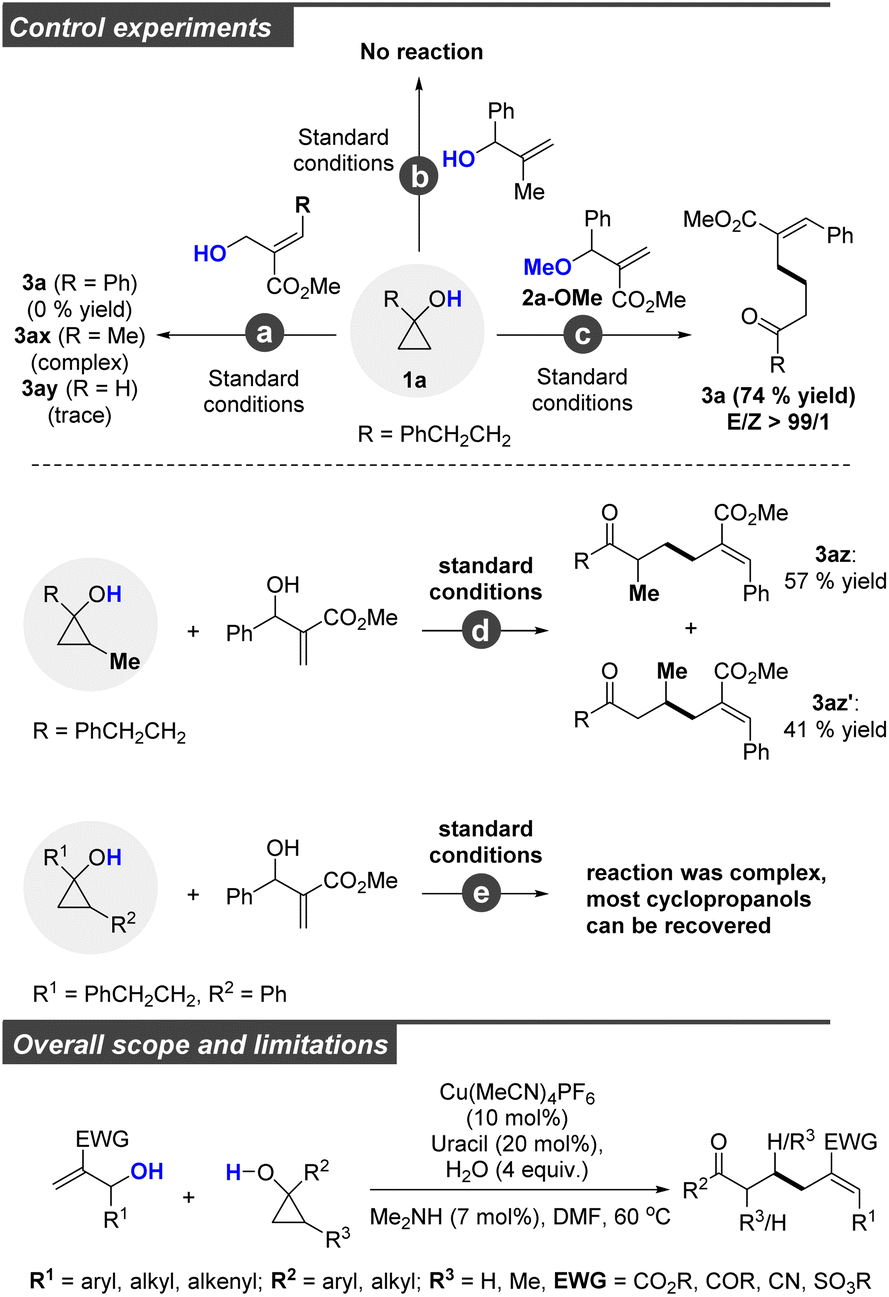 | ||
| Scheme 1 Control experiments and overall scope and limitations. | ||
The utilization of these products was preliminarily demonstrated by related derivation reactions (Scheme 2) via Wittig reaction (4), reduction (5), and Suzuki-coupling (6) reactions.
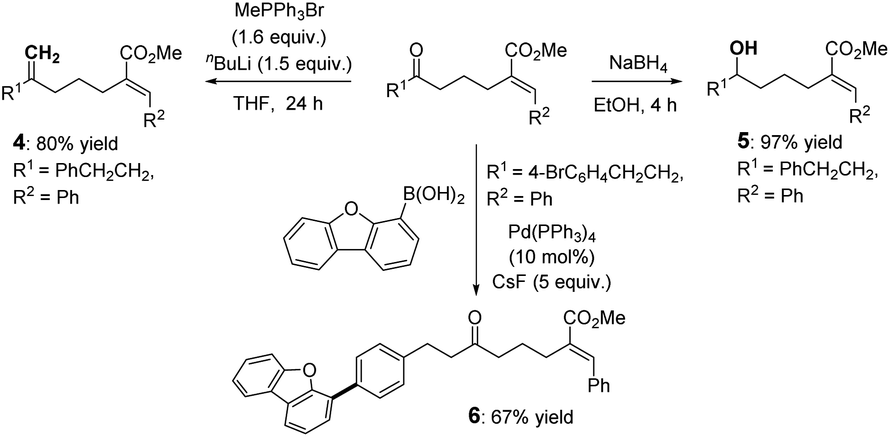 | ||
| Scheme 2 Preliminary protocol applications. | ||
The mechanism and energetic parameters of the reaction were investigated by DFT calculations at the B3LYP+D3(BJ)/def2TZVPP//B3LYP/def2svp level (Fig. 3; see ESI for computational details†). The optimized original Cu(I) catalyst17 tends to adopt a two-coordinate linear geometry. In the first step, the C–OH bond activation of 2a and addition of the terminal olefinic carbon onto copper occur in a concerted manner via transition state TS-1, delivering Cu(III)-allyl intermediate INT-1. Then, reactant 1a enters the ligand field of Cu(III)17 to form the precursor complex INT-1-1. The C–C bond activation of the cyclopropane ring is required to overcome a high-lying transition state TS-2, which delivers Cu(III) complex INT-2 with two Cu–C coordinate bonds. Finally, C–C reductive elimination via transition state TS-3 gives the desired product 3a with regeneration of the original Cu(I) catalyst. According to the calculated energy profiles, the rate-determining free-energy barrier of the reaction is 26.6 kcal mol−1, the difference between TS-2 and the initial point, which is consistent with the experimental temperature of 60 °C (Fig. 3a). Moreover, the high E-selectivity of this transformation can be attributed to the steric effects, which result in the high energy of (Z)-TS-1 (Fig. 3b). The higher energy of the rate-determining free-energy barrier (Fig. 3c) can account for the main reasons as to why some allylic alcohols cannot be consumed in the transformation (TS-2-A and TS-2-B). Although TS-2-A (27.8 kcal mol−1) is only 1.2 kcal mol−1 higher than TS-2 (26.6 kcal mol−1), the destabilization of TS-2-A might make the overall reaction difficult in the absence of EWG. This is because the present activation-free energy of 26.6 kcal mol−1 is close to the upper limit of the allowed value at 333.15 K, and an additional increase in activation free energy would prevent an efficient reaction (for details, see ESI†).
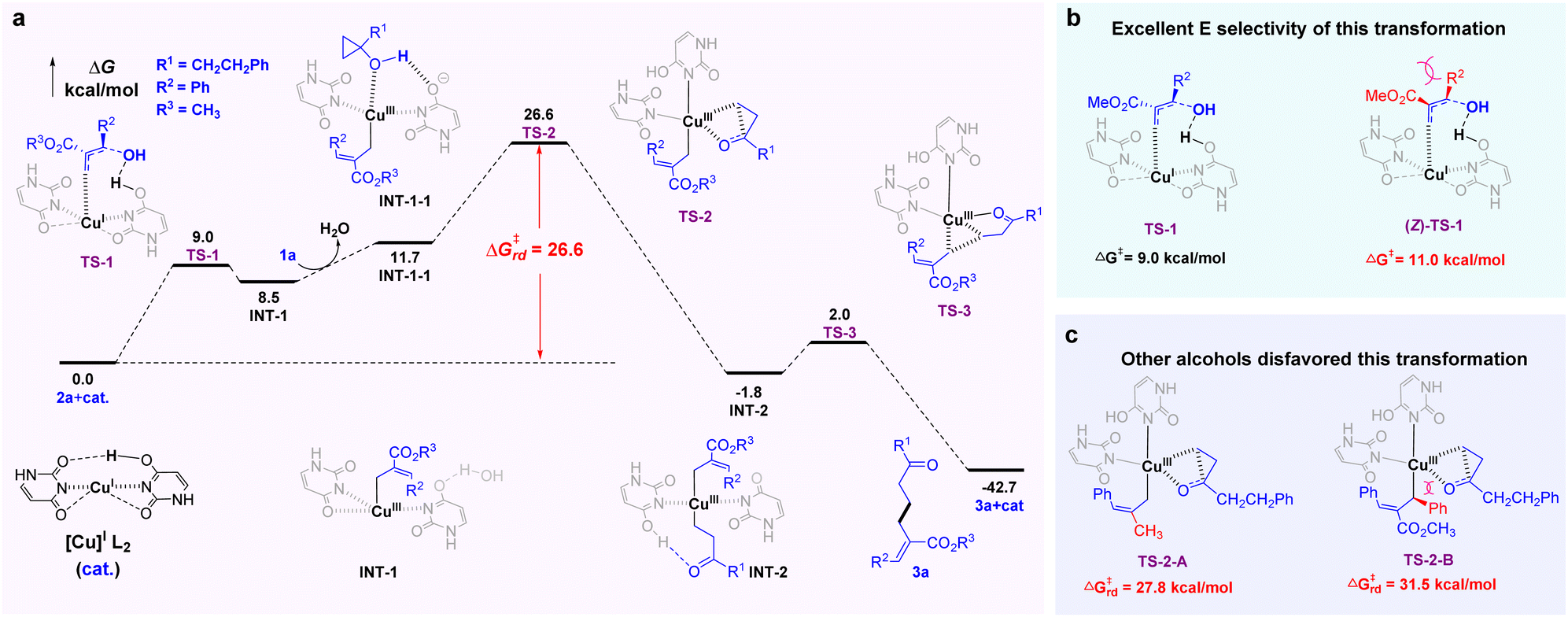 | ||
| Fig. 3 Mechanism investigation with the assistance of DFT calculation. | ||
Conclusions
In summary, we have developed a direct dehydrative cross-coupling protocol by selecting readily available MBH alcohols with cyclopropanols, which produce water as the only by-product. The Cu-uracil catalytic system has resolved the trade-off between the need for acidic activators for C(allyl)–OH bond cleavage and the demand for strong basic conditions for generating homoenolates. The protocol has demonstrated a broad substrate scope, delivering δ,ε-unsaturated ketones in high yields with exceptional stereoselectivity. Significantly, the usefulness of the methodology has been demonstrated by incorporating biologically active molecules. This study has also opened up possibilities for other metal-uracil catalytic processes for dehydrative allylation, although only MBH alcohols have been investigated thus far.Data availability
All of the necessary data have been included in the ESI.†Author contributions
J. H. performed the laboratory experiments (optimization, substrate scope, application, and mechanistic studies) and analyzed the experimental data. X. L. performed the laboratory experiments for substrate scope. K. Y. performed the laboratory experiments for mechanistic studies. L. Z. conducted the DFT calculations. T.-P. L. directed the investigations and prepared the manuscript. P. X. conceived and directed the investigations, analyzed the experimental data, and prepared the manuscript. All authors interpreted the results and contributed to the development of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (21702108, 22003045), the Natural Science Foundation of Jiangsu Province, China (BK20211257), and the Six Talent Peaks Project in Jiangsu Province (YY-033).Notes and references
-
(a) O. Y. Ali and T. D. Fridgen, Structures and Fragmentation of [Cu(Uracil-H)(Uracil)]+ in the Gas Phase, ChemPhyChem, 2012, 13, 588–596 CrossRef CAS
; (b) O. Brea, M. Yáñez, O. Mó and A. M. Lamsabhi, On the stability of [(uracil)2-Cu]2+ complexes in the gas phase. Different pathways for the formation of [(uracil-H)(uracil)-Cu]+ monocations, Org. Biomol. Chem., 2013, 11, 3862–3870 RSC
-
(a) B. Kumar, T. Das, S. Das, W. Maniukiewicz, D. S. Nesterov, A. M. Kirillov and S. Das, Coupling 6-chloro-3-methyluracil with copper: structural features, theoretical analysis, and biofunctional properties, Dalton Trans., 2021, 50, 13533–13542 RSC
-
(a) K. Tadpetch, B. Kaewmee, K. Chantakaew, K. Kantee, V. Rukachaisirikul and S. Phongpaichit, Synthesis and cytotoxic activities of semisynthetic zearalenone analogues, Bioorg. Med. Chem. Lett., 2016, 26, 3612–3616 CrossRef CAS
-
(a) Y. Ji, S. Chen, J. Zhou, K. Yang, X. Zhang, B. Xiang, T. Qiu, X. Gong, Z. Zhang, Z. Lan, F. Hu, F. Kong, Q. Qiu and Y. Zhang, Sirolimus plus prednisolone vs sirolimus monotherapy for kaposiform hemangioendothelioma: a randomized clinical trial, Blood, 2022, 139, 1619–1630 CrossRef CAS
- P. P. Das, K. Belmore and J. K. Cha, SN2’ Alkylation of Cyclopropanols via Homoenolates, Angew. Chem., Int. Ed., 2012, 51, 9517–9520 CrossRef CAS
- For selected recent reviews, see:
(a) T. R. McDonald, L. R. Mills, M. S. West and S. A. L. Rousseaux, Selective. Carbon–Carbon Bond Cleavage of Cyclopropanols, Chem. Rev., 2021, 121, 3–79 CrossRef CAS
-
(a) Q. Zhang, S.-W. Zhou, C.-Y. Shi and L. Yin, Catalytic Asymmetric Allylic Substitution with Copper(I) Homoenolates Generated from Cyclopropanols, Angew. Chem., Int. Ed., 2021, 60, 26351–26356 CrossRef CAS
-
(a) J. Yang, Y. Sekiguchi and N. Yoshikai, Cobalt-Catalyzed Enantioselective and Chemodivergent Addition of Cyclopropanols to Oxabicyclic Alkenes, ACS Catal., 2019, 9, 5638–5644 CrossRef CAS
-
(a) R. Kumar and E. V. Van der Eycken, Recent approaches for C–C bond formation via direct dehydrative coupling strategies, Chem. Soc. Rev., 2013, 42, 1121–1146 RSC
-
(a) R. Jiang, L. Ding, C. Zheng and S.-L. You, Iridium-catalyzed Z-retentive asymmetric allylic substitution reactions, Science, 2021, 371, 380–386 CrossRef CAS PubMed
-
(a)
M. B. Smith and J. March, March's Advanced Organic Chemistry, Wiley, New York, 2001 Search PubMed
-
(a) W. Gao, D. Zhang, X. Zhang, X. Cai, P. Xie and T.-P. Loh, One-Pot and Unsymmetrical Bis-Allylation of Malononitrile with Conjugated Dienes and Allylic Alcohols, Org. Lett., 2022, 24, 9355–9360 CrossRef CAS
-
(a) For some selected recent review, see: H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Phosphine Organocatalysis, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS PubMed
- For the selected review, see: C. G. Lima-Junior and M. L. A. A. Vasconcellos, Morita–Baylis–Hillman adducts: Biological activities and potentialities to the discovery of new cheaper drugs, Bioorg. Med. Chem., 2012, 20, 3954–3971 CrossRef CAS PubMed
-
(a) Y. Sekiguchi and N. Yoshikai, Zinc-Catalyzed β-Functionalization of Cyclopropanols via Enolized Homoenolate, J. Am. Chem. Soc., 2021, 143, 18400–18405 CrossRef CAS PubMed
- L. Pitzer, F. Schäfers and F. Glorius, Rapid Assessment of the Reaction-Condition-Based Sensitivity of Chemical Transformations, Angew. Chem., Int. Ed., 2019, 58, 8572–8576 CrossRef CAS
- Y. Luo, Y. Li, J. Wu, X.-S. Xue, J. F. Hartwig and Q. Shen, Oxidative addition of an alkyl halide to form a stable Cu(III) product, Science, 2023, 381, 1072–1079 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2202510. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04890j |
| This journal is © The Royal Society of Chemistry 2024 |