
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogen atom insertion into arenols to access benzazepines†
Yi
He‡
a,
Juanjuan
Wang‡
a,
Tongtong
Zhu
a,
Zhaojing
Zheng
b and
Hao
Wei
 *a
*a
aKey Laboratory of Synthetic and Natural Functional Molecule of Ministry of the Education, College of Chemistry & Materials Science, Northwest University, Xi'an 710069, China. E-mail: haow@nwu.edu.cn
bCollege of Food Science and Technology, Northwest University, Xi'an 710069, China
First published on 16th January 2024
Abstract
Advances in site-selective molecular editing have enabled structural modification on complex molecules. However, thus far, their applications have been restricted to C–H functionalization chemistry. The modification of the underlying molecular skeleton remains limited. Here, we describe a skeletal editing approach that provides access to benzazepine structures through direct nitrogen atom insertion into arenols. Using widely available arenols as benzazepine precursors, this alternative approach allowed the streamlined assembly of benzazepines with broad functional group tolerance. Experimental mechanistic studies support a reaction pathway involving dearomatizative azidation and then aryl migration. This study further highlights the potential for carbon–nitrogen transmutation sequences through combinations with oxidative carbon atom deletion, providing an alternative for the development of N-heteroarenes and demonstrating significant potential in materials chemistry.
Introduction
Single-atom skeletal editing has been receiving increasing interest because it enables the modification of underlying molecular skeletons and provides unique opportunities to synthesize complex molecules through completely different strategies.1–9 In particular, nitrogen atom insertion into a ring skeleton presents a distinct strategy for accessing heterocyclic structures, which are highly valuable scaffolds in drug discovery, materials science, and synthetic chemistry (Fig. 1A).10 The earliest examples of this class of reactions include well-known processes such as Ciamician–Dennstedt rearrangement (1881),11 Beckmann rearrangement (1886),12 and Schmidt rearrangement (1924).13 Recent noteworthy advances include nitrogen insertion into heterocycles14–16 and cycloalkenes (Fig. 1B).17–21 However, nitrogen atom insertion into the aromatic carbocyclic skeleton is still difficult because of the inherent inertness of the carbon–carbon bonds of aromatic rings.22–25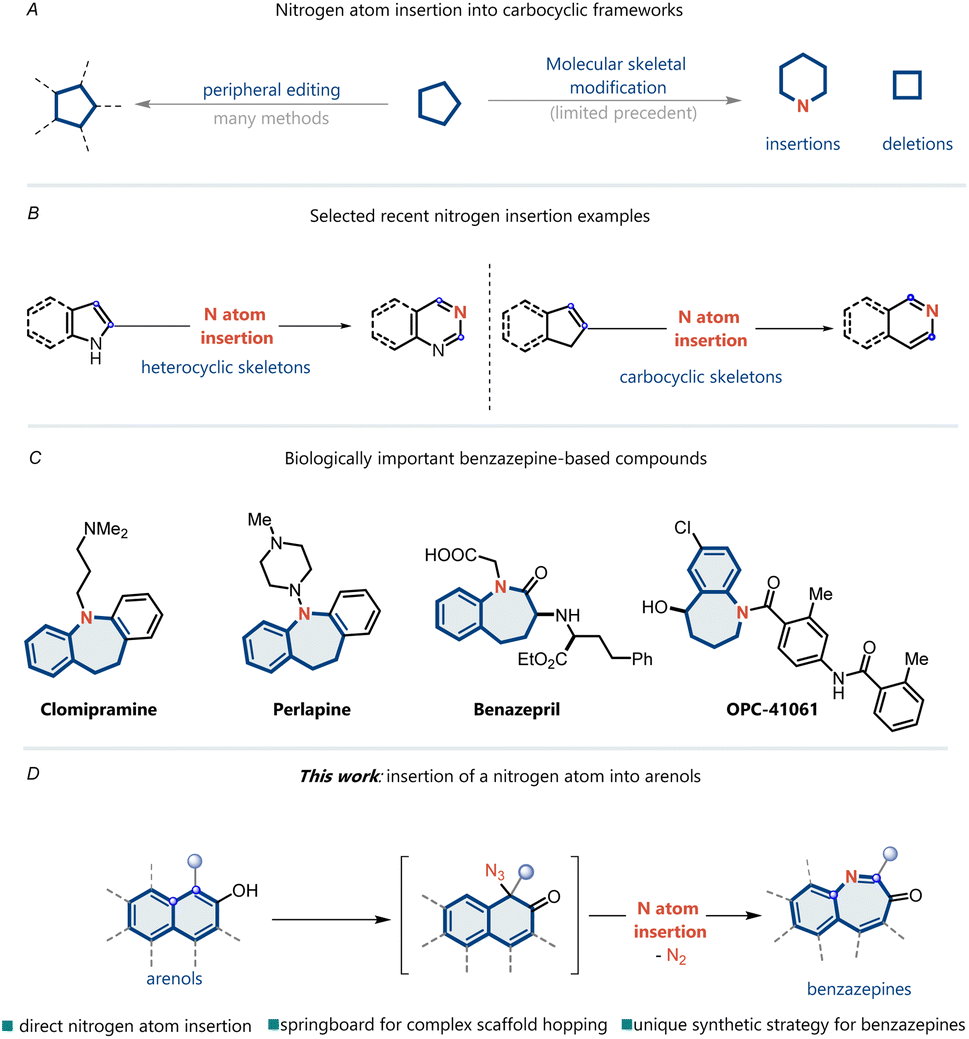 | ||
| Fig. 1 Insertion of a nitrogen atom into arenols. | ||
N-Heterocycles present the unique class of cyclic structural frameworks present in various natural and non-natural products. Big data analytics reveal that heterocycle formation has been one of the most frequently used reactions in medicinal chemistry over the past several decades.26,27 Benzazepines are among the most important N-heterocycle skeletons and have received considerable attention owing to their pharmacological properties and promising applications in organic synthesis (Fig. 1C).28 For instance, OPC-41061, a human vasopressin V2-receptor antagonist,29 clomipramine, an antidepressant agent,30 and benazepril, an ACE inhibitor,31 all contain the benzazepine skeleton. Therefore, substantial efforts have been devoted to the development of efficient methodologies for preparing benzazepine skeletons. Single-atom insertions represent one of the most intriguing methods for the synthesis of heterocycles and have established new opportunities for accessing valuable benzazepines.
Herein, we report a reaction wherein a nitrogen atom is directly inserted into an arenol to yield the corresponding benzazepine ring through an azide intermediate (Fig. 1D). To achieve nitrogen atom insertion into arenes, we propose utilizing of arenols as substrates, which can disrupt the stability of the aromatic ring through dearomatization. Moreover, arenols can function as directing groups in site-selective nitrogen insertion. Unlike classical nitrene addition into benzene rings,32–35 this strategy facilitates C–C bond cleavage and, more importantly, achieves site-selective nitrogen atom insertion.
Results and discussion
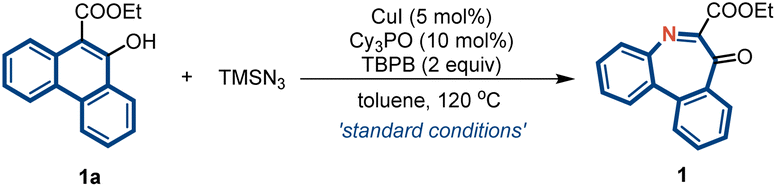
This study was initiated with the use of phenanthren-9-ol (1a) as the model substrate (Table 1). The reaction parameters, including different catalysts, ligands, solvents, and temperatures, were carefully optimized (see Tables S1–S4 in the ESI†). The desired product 2 was obtained in 80% yield using a combination of CuI/Cy3PO and tert-butyl peroxybenzoate (TBPB) as the oxidant in toluene (entry 1). Thereafter, a series of control experiments were performed to gain deeper insights into this reaction. The Cu catalyst plays a critical role in this transformation, and the desired product cannot be obtained without it (entry 2). Other Cu salts, including CuCl, CuBr, and Cu(OAc)2, could also undergo this reaction, but with lower efficiencies (entries 3–5). Furthermore, when other established metal nitrenoid formation catalysts,36,37 including iron and cobalt, were utilized, the desired product was not obtained (entries 6 and 7). This result suggests that CuI is suitable for both dearomatizative azidation and aryl migration. Among the various ligands examined, the yield drastically increased when Cy3PO was used as the ligand (entries 8–12). The reaction can proceed in the absence of the ligand (entry 13), suggesting that CuI alone can catalyze the nitrogen insertion reaction, albeit with lower efficiency. Further screening of other azide sources indicated that only TMSN3 facilitated this transformation (entries 14 and 15).|
|
||
|---|---|---|
| Entry | Variation from ‘standard conditions’ | Yieldb % |
| a Unless otherwise specified, all reactions were carried out using 1a (0.1 mmol) and TMSN3 (0.3 mmol), with CuI (5 mol%), Cy3PO (10 mol%) and TBPB (2.0 equiv.) in toluene at 120 °C for 12 h. b Isolated yields after chromatography. | ||
| 1 | None | 80 |
| 2 | w/o CuI | 0 |
| 3 | CuCl instead of CuI | 72 |
| 4 | CuBr instead of CuI | 65 |
| 5 | Cu(OAc)2 instead of CuI | 60 |
| 6 | Fe(TPP)Cl instead of CuI | 0 |
| 7 | T(p-OMe)PPCo instead of CuI | <10 |
| 8 | n Bu3PO instead of Cy3PO | 68 |
| 9 | Ph3PO instead of Cy3PO | 40 |
| 10 | bpy instead of Cy3PO | 17 |
| 11 | L1 instead of Cy3PO | 53 |
| 12 | L2 instead of Cy3PO | 56 |
| 13 | w/o Cy3PO | 30 |
| 14 | NaN3 instead of TMSN3 | <10 |
| 15 | N(nBu)4N3 instead of TMSN3 | Trace |
|
||
Having established the optimized reaction conditions, we investigated the substrate scope based on the naphthalene scaffold (Fig. 2). Various multi-arenols, including naphthol (2–33), phenanthrol (34–44), tetraphenol (45), and benzo[c]phenanthrenol (46), can effectively undergo the desired nitrogen atom insertion. Notably, the substituents at the 2-positions of the arenol proved to be significant. When the substituent was a phenyl group (3, 4, and 44) or an electron-withdrawing group such as CO2Me, the corresponding arenols underwent nitrogen atom insertion smoothly in moderate-to-good yields. The presence of an alkyl at the 2-position was found to inhibit this reaction. Substrates containing heterocycles, such as furan and thiophene (7 and 8), performed well. Various functional groups known to participate in this reaction, such as esters (2), methyl ethers (9 and 18), thioether (12), trimethylsilyl (13), aryl halides (14, 15 and 19), cyanide (16), and trifluoromethyl (17), were found to be compatible under the optimized conditions. In addition to methyl esters, other esters were evaluated. Phenyl (20), allyl (21), and cycloalkyl (22) ester derivatives were competent substrates, affording the desired azepine products in moderate-to-good yields. Bulkier esters, such as isopropyl (23) and tert-butyl (24), also worked smoothly. Moreover, 2-naphthol bearing an oxime group (25) at the 1-position was well tolerated. Given the broad substrate scope, we aimed to demonstrate this reaction in a more complex setting. Naphthols with ester-linked natural products and drug derivatives readily participated in this nitrogen atom insertion reaction. Various complex molecules with diverse structural features, such as steroids (27 and 28), N-heteroarenes (oxazole 30 and indole 32), and carbohydrate (31), were readily converted into their corresponding products.
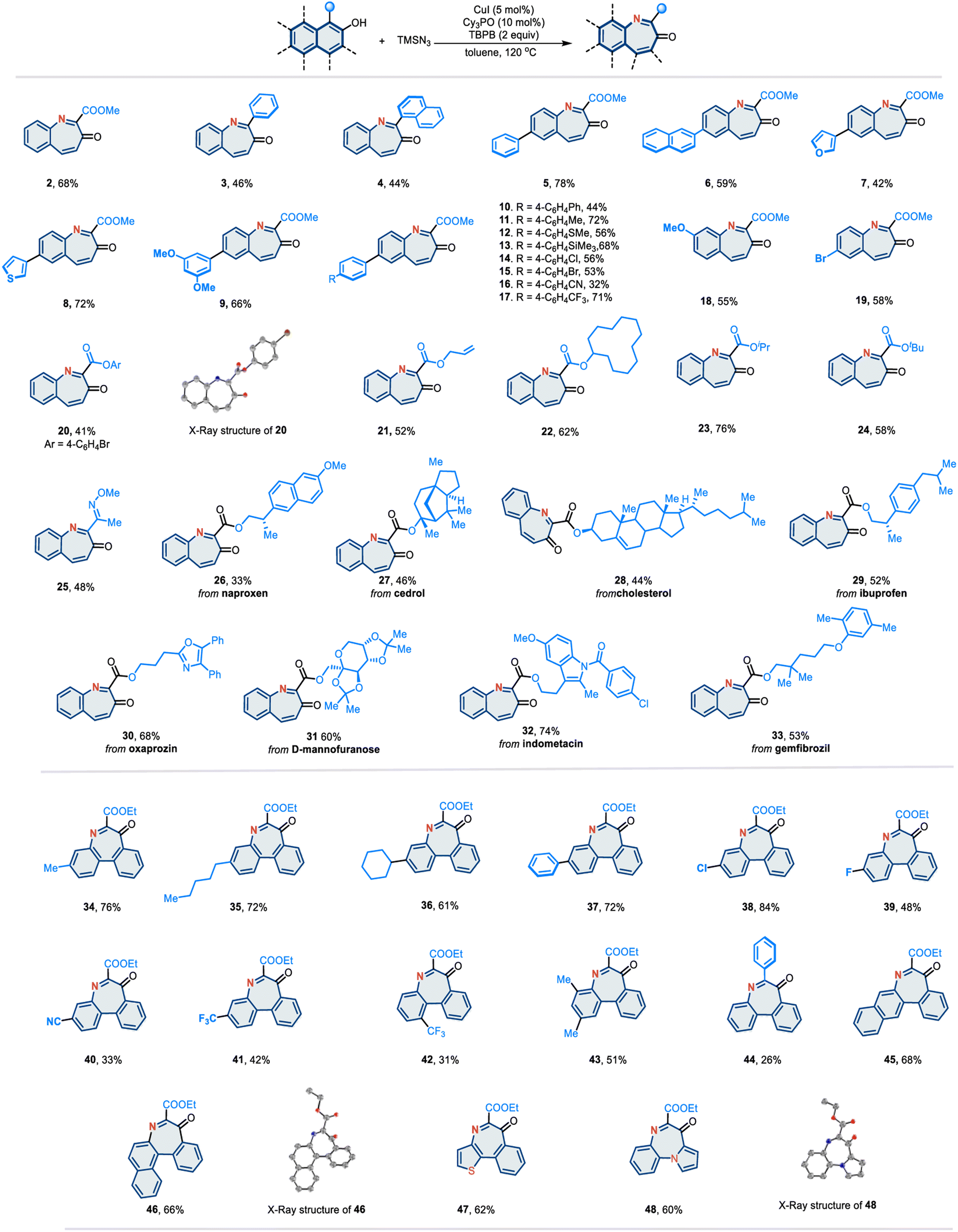 | ||
| Fig. 2 Substrate scope. | ||
In addition to the naphthol scaffold, various phenanthrol derivatives can react under standard conditions to afford the “nitrogen insertion” products (34–44). Different aliphatic chains (34–36), aromatic rings (37 and 44), halides (38 and 39), cyanide (40), and trifluoromethyl (41 and 42), are well tolerated. Moreover, fused heteroarenols such as naphtho[1,2-b]thiophene (47) and pyrrolo[1,2-a]quinoline (48) can be incorporated, providing pharmaceutically intriguing fused-ring skeletons. Notably, the structures of 20, 46 and 48 were identified by X-ray crystallography.
This nitrogen atom insertion protocol provides a basis for the development of more complex skeletal editing transformations. A longstanding challenge in skeletal editing has been the selective exchange of individual atoms, including swapping a carbon for a nitrogen atom in a ring.38–44 Based on this nitrogen atom insertion transformation, we design a ring expansion–contraction sequence to realize carbon-to-nitrogen transmutation. Various arenol derivatives can react to produce carbon-to-nitrogen transmutation products (Fig. 3A). A plausible mechanism was proposed (Fig. 3B). Initially, the mCPBA-mediated oxidation of the corresponding naphthol 2 affords oxaziridine intermediate I. Subsequently, a cleavage of the nitrogen–oxygen band occurs, generating radical intermediate II. Next, a radical-mediated rearrangement of intermediate II affords intermediate III, which in turn removes the ester to yield 61.
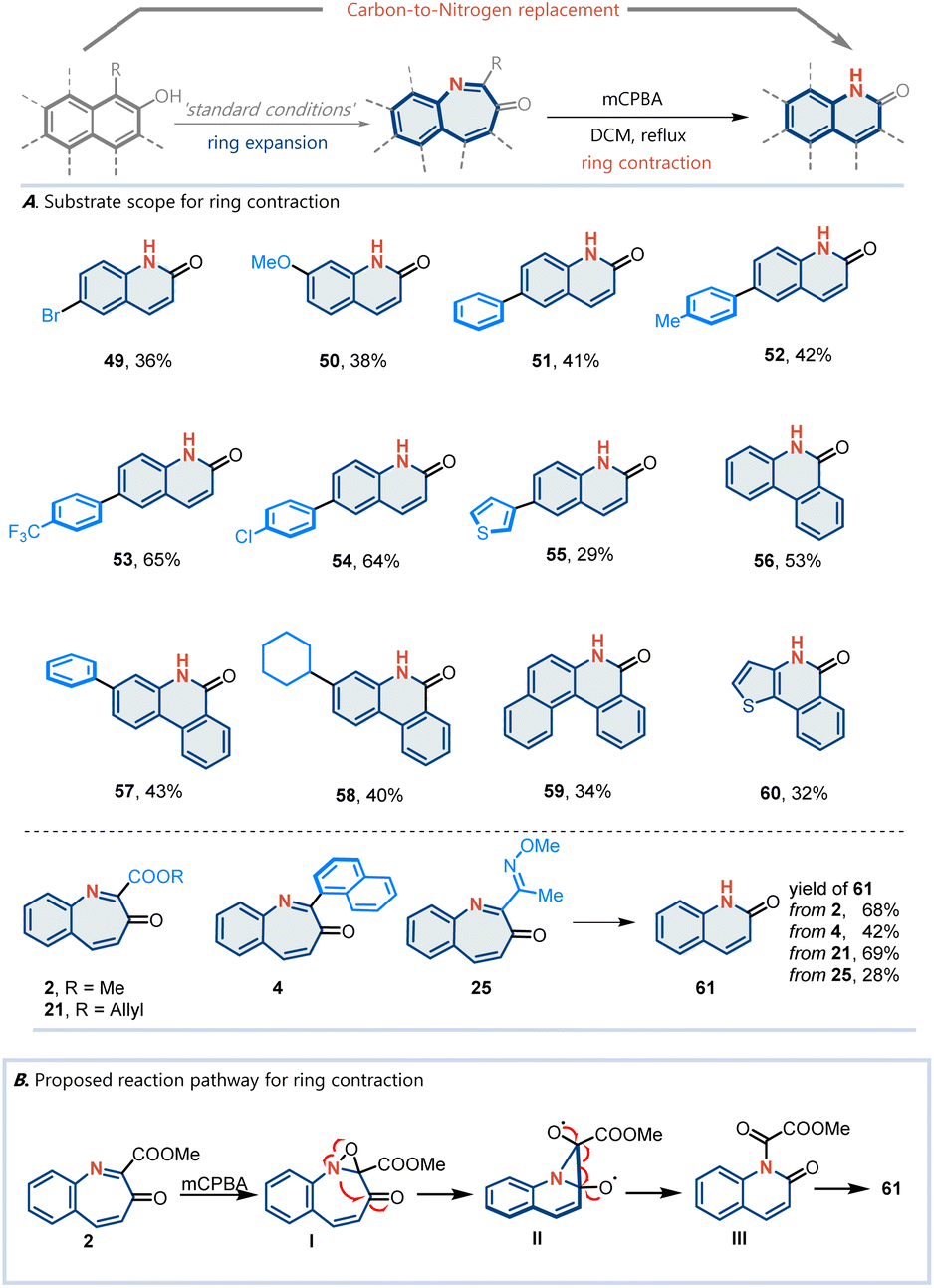 | ||
| Fig. 3 Application potential of the atom-swapping process. | ||
A series of experiments were performed to gain insights into the reaction mechanism (Fig. 4). First, reactions in the presence of a radical inhibitor (BHT) under standard conditions did not furnish any nitrogen atom products, which was consistent with a radical mechanism (Fig. 4A). By changing the reaction temperature to 80 °C, we were able to separate the reactive quaternary azide intermediate 2c. In addition, thermal activation allowed for the smooth initiation of subsequent aryl migration to furnish product 2 in excellent yield (Fig. 4B). Control experiments indicated that the copper catalyst did not affect the rate of the migration step. Further, the phenolic OH group of 2a was protected by a methyl group and subjected to standard conditions (Fig. 4C). This reaction failed to furnish either the azidated intermediate or the nitrogen insertion product, indicating that the phenolic OH groups of arenols played an important role in this reaction. Furthermore, the reaction performed using stoichiometric amounts of CuI and Cy3PO in the absence of TBHP did not yield the desired product. This finding indicates that TBHP plays a key role in initiating the catalytic cycle.
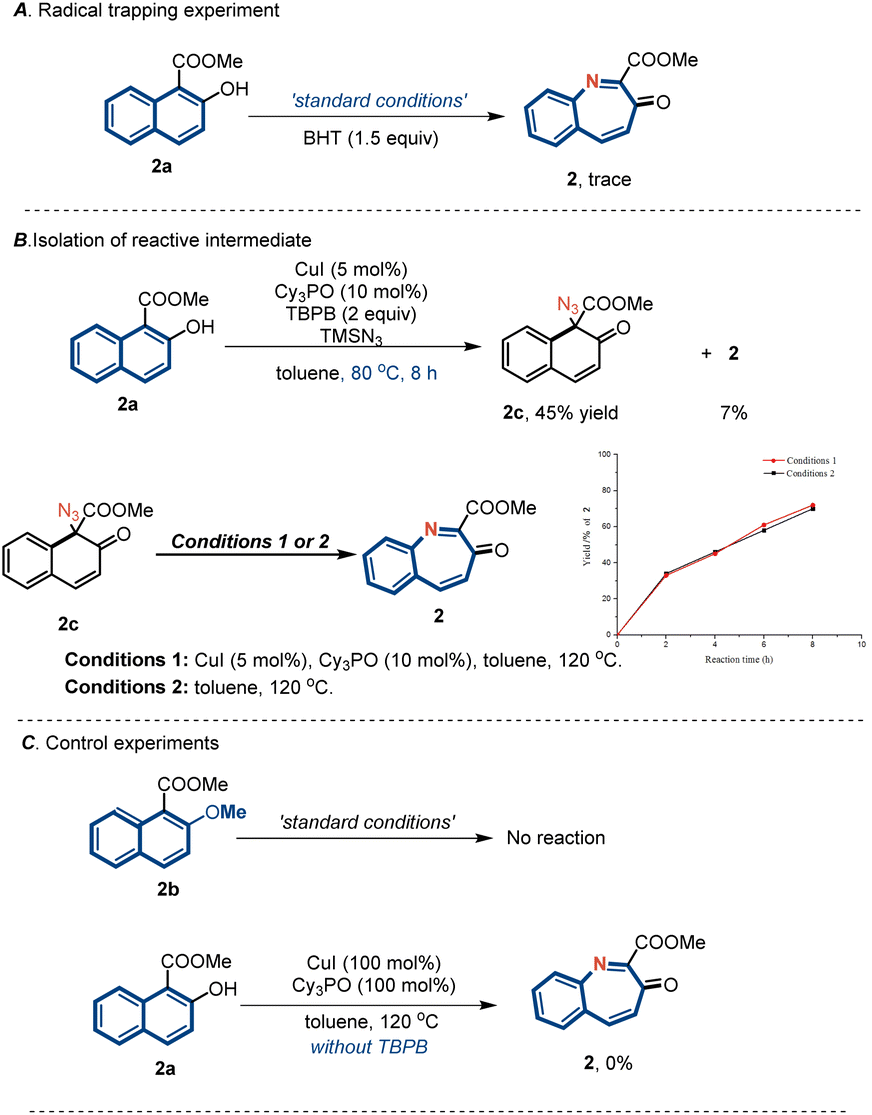 | ||
| Fig. 4 Mechanistic studies. | ||
A plausible mechanism based on literature reports and our observations was proposed (Fig. 5).45–48 Initially, a single-electron transfer between Cu(I) and TBPB initiates the reaction by generating a tert-butoxyl radical and Cu(II) species. A radical relay process then occurs between 2a and the tert-butoxyl radical to afford radical intermediate A.49,50 Subsequently, ligand exchange delivers Cu(II)N3, which then reacts with the internal radical to deliver the azidation intermediate B and regenerate the Cu(I) catalyst. Finally, intermediate B undergoes aryl migration to eventually yield product 2 through the extrusion of N2.51,52
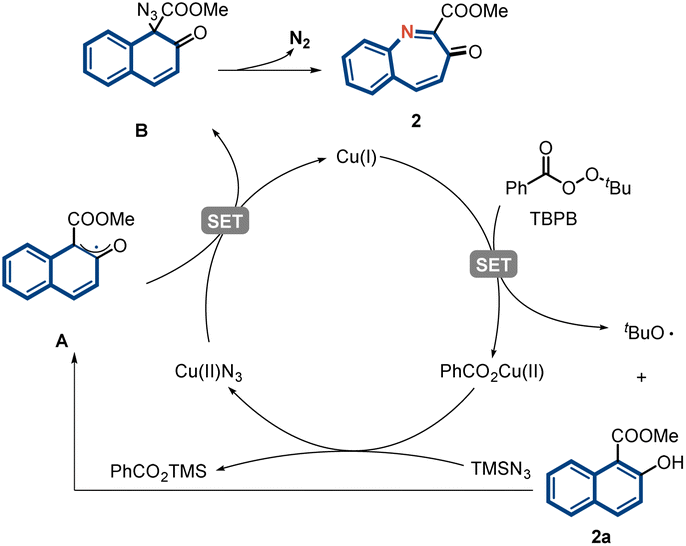 | ||
| Fig. 5 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we reported a unique strategy for the straightforward insertion of nitrogen atoms into arenols. Nitrogen atom insertion exhibits reasonable site-selectivity, whereas copper catalysis disrupts the aromaticity of arenols. A carbon-to-nitrogen atom-swapping process was achieved via a combination of oxidative carbon-atom deletions. This protocol exhibited a wide substrate scope, encompassing a variety of multi-arenols, including naphthol, phenanthrol, benzo[c]phenanthrenol, and tetraphenol, along with various functional groups. It can produce the corresponding benzazepines in a highly site-selective manner. This report establishes a framework for creating new skeletal editing processes for aromatic molecules via an azido intermediate.Data availability
Detailed synthetic procedures and complete characterization data for all new compounds can be found in the ESI.†Author contributions
H. W. conceived and designed the project and composed the manuscript. Y. H., J. W. and T. Z. conducted the experiments and analyzed the data. J. W. and Z. Z. discussed the experimental results and commented on the manuscript. H. W. conducted general guidance, project directing, and manuscript revisions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (21971205 and 22271231), the Natural Science Basic Research Plan for Distinguished Young Scholars in Shaanxi Province of China (2022JC-08), and the Key Research and Invention Program in Shaanxi Province of China (2021SF-299).Notes and references
- J. B. Roque, Y. Kuroda, L. T. Göttemann and R. Sarpong, Nature, 2018, 564, 244–248 CrossRef CAS PubMed
.
- B. D. Dherange, P. Q. Kelly, J. P. Liles, M. S. Sigman and M. D. Levin, J. Am. Chem. Soc., 2021, 143, 11337–11344 CrossRef CAS PubMed
- J. Jurczyk, M. C. Lux, D. Adpressa, S. F. Kim, Y.-H. Lam, C. S. Yeung and R. Sarpong, Science, 2021, 373, 1004–1012 CrossRef CAS PubMed
- S. H. Kennedy, B. D. Dherange, K. J. Berger and M. D. Levin, Nature, 2021, 593, 223–227 CrossRef CAS PubMed
- J. Woo, A. H. Christian, S. A. Burgess, Y. Jiang, U. F. Mansoor and M. D. Levin, Science, 2022, 376, 527–532 CrossRef CAS PubMed
- G. L. Bartholomew, F. Carpaneto and R. Sarpong, J. Am. Chem. Soc., 2022, 144, 22309–22315 CrossRef CAS PubMed
- J. Jurczyk, J. Woo, S. F. Kim, B. D. Dherange, R. Sarpong and M. D. Levin, Nat. Synth., 2022, 1, 352–364 CrossRef PubMed
- F. Liu, L. Anand and M. Szostak, Chem.–Eur. J., 2023, 29, e202300096 CrossRef CAS PubMed
- B. A. Wight, A. Matviitsuk, M. J. Black, P. García-Reynaga, L. Hanna, A. T. Herrmann, M. K. Ameriks, R. Sarpong and T. P. Lebold, J. Am. Chem. Soc., 2023, 145, 10960–10966 CrossRef PubMed
- A. Sattler and G. Parkin, Nature, 2010, 463, 523–526 CrossRef CAS PubMed
- G. L. Ciamician and M. Dennstedt, Ber. Dtsch. Chem. Ges., 1881, 14, 1153–1163 CrossRef
- E. Beckmann, Ber. Dtsch. Chem. Ges., 1886, 19, 988–993 CrossRef
- K. F. Schmidt, Ber. Dtsch. Chem. Ges., 1924, 57, 704–706 CrossRef
- J. C. Reisenbauer, O. Green, A. Franchino, P. Finkelstein and B. Morandi, Science, 2022, 377, 1104–1109 CrossRef CAS PubMed
- R. S. Atkinson, B. D. Judkins, D. R. Russell and L. J. S. Sherry, J. Chem. Soc., Perkin Trans. 1, 1985, 1967–1969 RSC
- T. Yang, X. Fan, X. Zhao and W. Yu, Org. Lett., 2018, 20, 1875–1879 CrossRef CAS PubMed
- K. Narasimhan and P. R. Kumar, Heterocycles, 1984, 22, 1369–1375 CrossRef CAS
- S. Liu and X. Cheng, Nat. Commun., 2022, 13, 425 CrossRef CAS PubMed
- P. Q. Kelly, A. S. Filatov and M. D. Levin, Angew. Chem., Int. Ed., 2022, 61, e202213041 CrossRef CAS PubMed
- J. Wang, H. Lu, Y. He, C. Jing and H. Wei, J. Am. Chem. Soc., 2022, 144, 22433–22439 CrossRef CAS PubMed
- P. Finkelstein, J. C. Reisenbauer, B. B. Botlik, O. Green, A. Florin and B. Morandi, Chem. Sci., 2023, 14, 2954–2959 RSC
- V. M. Benitez, J. M. Grau, J. C. Yori, C. L. Pieck and C. R. Vera, Energy Fuels, 2006, 20, 1791–1798 CrossRef CAS
- M. Jokoobi, Y. Tian, R. Boulatov and A. G. Sergeev, J. Am. Chem. Soc., 2019, 141, 6048–6053 CrossRef PubMed
- Y. Tian, M. Jokoobi, R. Boulatov and A. G. Sergeev, Chem. Sci., 2021, 12, 3568–3579 RSC
- X. Qiu, Y. Sang, H. Wu, X.-S. Xue, Z. Z. Yan, Y. Wang, Z. Cheng, X. Wang, H. Tan, S. Song, G. Zhang, X. Zhang, K. N. Houk and N. Jiao, Nature, 2021, 597, 64–69 CrossRef CAS PubMed
- N. Schneider, D. M. Lowe, R. A. Sayle, M. A. Tarselli and G. A. Landrum, J. Med. Chem., 2016, 59, 4385–4402 CrossRef CAS PubMed
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed
- Y. Yamamura, S. Nakamura, S. Itoh, T. Hirano, T. Onogawa, T. Yamashita, Y. Yamada, K. Tsujimae, M. Aoyama, K. Kotosai, H. Ogawa, H. Yamashita, K. Kondo, M. Tominaga, G. Tsujimoto and T. Mori, J. Pharmacol. Exp. Ther., 1998, 287, 860–867 CAS
- D. McTavish and P. Benfield, Drugs, 1990, 39, 136–153 CrossRef CAS PubMed
-
J. C. Towson and S.-C. Wong, US Pat., US20150315195, 2013 Search PubMed
- R. A. Abramovitch, T. D. Baily, T. Takaya and V. Uma, J. Org. Chem., 1974, 39, 340–345 CrossRef CAS
- F. D. Marsh and H. E. Simmons, J. Am. Chem. Soc., 1965, 87, 3529–3530 CrossRef CAS
- O. Subba Rao and W. Lwowski, Tetrahedron Lett., 1980, 21, 727–730 CrossRef
- I. Sailia, B. Kashyap and P. Phukan, Chem. Commun., 2011, 47, 2967–2969 RSC
- N. P. Van Leest, M. A. Tepaske, J. P. H. Oudsen, B. Venderbosch, N. R. Rietdijk, M. A. Siegler, M. Tromp, J. I. Van Der Vlugt and B. De Bruin, J. Am. Chem. Soc., 2020, 142, 552–563 CrossRef CAS PubMed
- Y. Liu, K.-P. Shing, V. K.-Y. Lo and C.-M. Che, ACS Catal., 2023, 13, 1103–1124 CrossRef CAS
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed
- K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef CAS PubMed
- S. C. Patel and N. Z. Burns, J. Am. Chem. Soc., 2022, 144, 17797–17802 CrossRef CAS PubMed
- Q. H. Luu and J. Li, Chem. Sci., 2022, 13, 1095–1100 RSC
- H. Zhong, T. Effer, V. C. M. Gasser, P. Finkelstein, L. Keim, N. Trapp and B. Morandi, Nat. Commun., 2023, 14, 5273 CrossRef CAS PubMed
- T. J. Pearson, R. Shimazumi, J. L. Driscoll, B. D. Dherange, D. Park and M. D. Levin, Science, 2023, 381, 1474–1479 CrossRef CAS PubMed
- J. Woo, C. Stein, A. H. Christian and M. D. Levin, Nature, 2023, 623, 77–82 CrossRef CAS PubMed
- D. Sarkar, M. K. Ghosh, N. Rout and P. Kuila, New J. Chem., 2017, 41, 3715–3718 RSC
- P. Wang, J. Wang, L. Wang, D. Li, K. Wang, Y. Liu, H. Zhu, X. Liu, D. Yang and W. Rui, Adv. Synth. Catal., 2018, 360, 401–405 CrossRef CAS
- Y. Xiang, Y. Sun and G. Zhang, Org. Lett., 2018, 20, 6250–6254 CrossRef PubMed
- S.-J. Shen, C.-L. Zhu, D.-F. Lu and H. Xu, ACS Catal., 2018, 8, 4473–4482 CrossRef CAS PubMed
- J. Dhineshkumar, P. Samaddar and K. R. Prabhu, Chem. Commun., 2016, 52, 11084–11087 RSC
- C.-J. Wang, J. Sun, W. Zhou, J. Xue, B.-T. Ren, G.-Y. Zhang, Y.-L. Mei and Q.-H. Deng, Org. Lett., 2019, 21, 7315–7319 CrossRef CAS PubMed
- W. Von E. Doering and R. A. Odum, Tetrahedron, 1966, 22, 81–93 CrossRef CAS
- B. Iddon, O. Meth-Cohn, E. F. V. Scriven, H. Suschitzky and P. T. Gallagher, Angew. Chem., Int. Ed., 1979, 18, 900–917 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2263048, 2263051 and 2263052. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05367a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |