
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSTEP: profiling cellular-specific targets and pathways of bioactive small molecules in tissues via integrating single-cell transcriptomics and chemoproteomics†
Jiayun
Chen‡
a,
Zheng
Chu‡
a,
Qian
Zhang‡
b,
Chen
Wang
a,
Piao
Luo
b,
Ying
Zhang
a,
Fei
Xia
 a,
Liwei
Gu
a,
Yin Kwan
Wong
c,
Qiaoli
Shi
a,
Chengchao
Xu
*a,
Huan
Tang
*a and
Jigang
Wang
*abcd
a,
Liwei
Gu
a,
Yin Kwan
Wong
c,
Qiaoli
Shi
a,
Chengchao
Xu
*a,
Huan
Tang
*a and
Jigang
Wang
*abcd
aState Key Laboratory for Quality Ensurance and Sustainable Use of Dao-di Herbs, Artemisinin Research Center and Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing 100700, China. E-mail: ccxu@icmm.ac.cn; htang@icmm.ac.cn; jgwang@icmm.ac.cn
bSchool of Traditional Chinese Medicine and School of Pharmaceutical Sciences, Southern Medical University, Guangzhou 510515, China
cDepartment of Nephrology, Shenzhen Key Laboratory of Kidney Diseases, Shenzhen Clinical Research Centre for Geriatrics, Shenzhen People's Hospital, The First Affiliated Hospital, Southern University of Science and Technology, Shenzhen 518020, China
dState Key Laboratory of Antiviral Drugs, School of Pharmacy, Henan University, Kaifeng 475004, China
First published on 5th March 2024
Abstract
Identifying the cellular targets of bioactive small molecules within tissues has been a major concern in drug discovery and chemical biology research. Compared to cell line models, tissues consist of multiple cell types and complicated microenvironments. Therefore, elucidating the distribution and heterogeneity of targets across various cells in tissues would enhance the mechanistic understanding of drug or toxin action in real-life scenarios. Here, we present a novel multi-omics integration pipeline called Single-cell TargEt Profiling (STEP) that enables the global profiling of protein targets in mammalian tissues with single-cell resolution. This pipeline integrates single-cell transcriptome datasets with tissue-level protein target profiling using chemoproteomics. Taking well-established classic drugs such as aspirin, aristolochic acid, and cisplatin as examples, we confirmed the specificity and precision of cellular drug-target profiles and their associated molecular pathways in tissues using the STEP analysis. Our findings provide more informative insights into the action modes of bioactive molecules compared to in vitro models. Collectively, STEP represents a novel strategy for profiling cellular-specific targets and functional processes with unprecedented resolution.
Introduction
Identifying the target engagement of active small molecules (ASMs) and studying their subsequently mediated biological processes in complex organisms have been critical issues in understanding the mechanism of action (MoA) of drugs.1–3 In recent years, chemoproteomics has emerged as a powerful method for identifying protein targets of ASMs by coupling biorthogonal chemistry with activity-based protein profiling (ABPP) strategies. Although remarkable advances have been made in in vitro models such as cell lines and recombinant proteins by chemoproteomics,4–6 our knowledge of the target identification and functional validation of ASMs across various cell types in tissues is still limited. The inherent cellular heterogeneity and its crucial role in impacting the targeting bioactivity of ASMs underscore the urgent need for an approach or pipeline that enables the analysis of ASM target engagement in tissues with cell specific resolution.7Currently, dissecting the actions of cellular ASMs in living animals via chemoproteomics remains a long-standing challenge due to the high cost of large-scale bioorthogonal probe production, lower sensitivity, and a poor signal-to-noise ratio (SNR).8,9 Pang et al. developed a method termed CATCH, which improves the SNR by making tissues transparent, thereby enabling the imaging of small-molecule drug–target cellular interactions in brain tissues in situ.10 Building upon this, we are motivated to explore a high-throughput, low-cost, and user-friendly analysis strategy for dissecting drug–target engagement at single-cell resolution in mammalian tissues.
With recent advancements in cutting-edge analytical technologies, single-cell RNA sequencing (scRNA-seq) provides unprecedented resolution in revealing the gene expression profile and functional state in individual cells.4 Even though the consistency between RNA and protein levels is not always guaranteed due to the limitation of current technologies and the dynamic nature of biological systems, the divergence of expression patterns among different cell types might persist at the transcriptome and protein levels.11 Inspired by the power and advantage of scRNA-seq in addressing cellular heterogeneity, we here developed a pipeline called Single-cell TargEt Profiling (STEP) for investigating cellular targets in mammalian tissues by integrating chemoproteomics with scRNA-seq approaches. Using this strategy, we successfully obtained the target engagement profiles of three well-known ASMs (aspirin, aristolochic acid, and cisplatin) at single-cell resolution in various tissues (brain, kidney, and tumor), demonstrating the feasibility, reliability, and versatility of this strategy. Furthermore, our findings regarding the distribution and enriched pathways of protein targets across various cellular populations within the tissues provide valuable insights into the pharmacological and toxicological effects of ASMs, which could greatly advance drug discovery and clinical translation.
Results and discussion
Workflow of STEP for identifying the cellular targets of ASMs in tissues
As shown in Fig. 1, the workflow of STEP includes four main modules. Firstly, to figure out which tissues or organs were the primary targets or deposition sites of specific ASMs, the distribution and bioactivity of ASMs should be determined by pharmacokinetic studies including the distribution and bioactivity of ASMs in vivo. Of note, chemoproteomics can also provide an alternative approach to locate the most targeted tissue by the chemical labeling effect. Secondly, the selected tissues were then subjected to scRNA-seq and chemoproteomics analysis, obtaining the transcriptomics dataset of various cell types and targeted protein profiling of ASMs in the selected tissues. Furthermore, the expression matrix of targeted proteins was extracted from the scRNA-seq dataset in corresponding tissues. Finally, the divergence in the gene expression level of protein targets in tissues indicates the heterogeneity of ASMs across various cell types, thereby determining the cellular targets and related biological pathways of ASMs in tissues at single-cell resolution.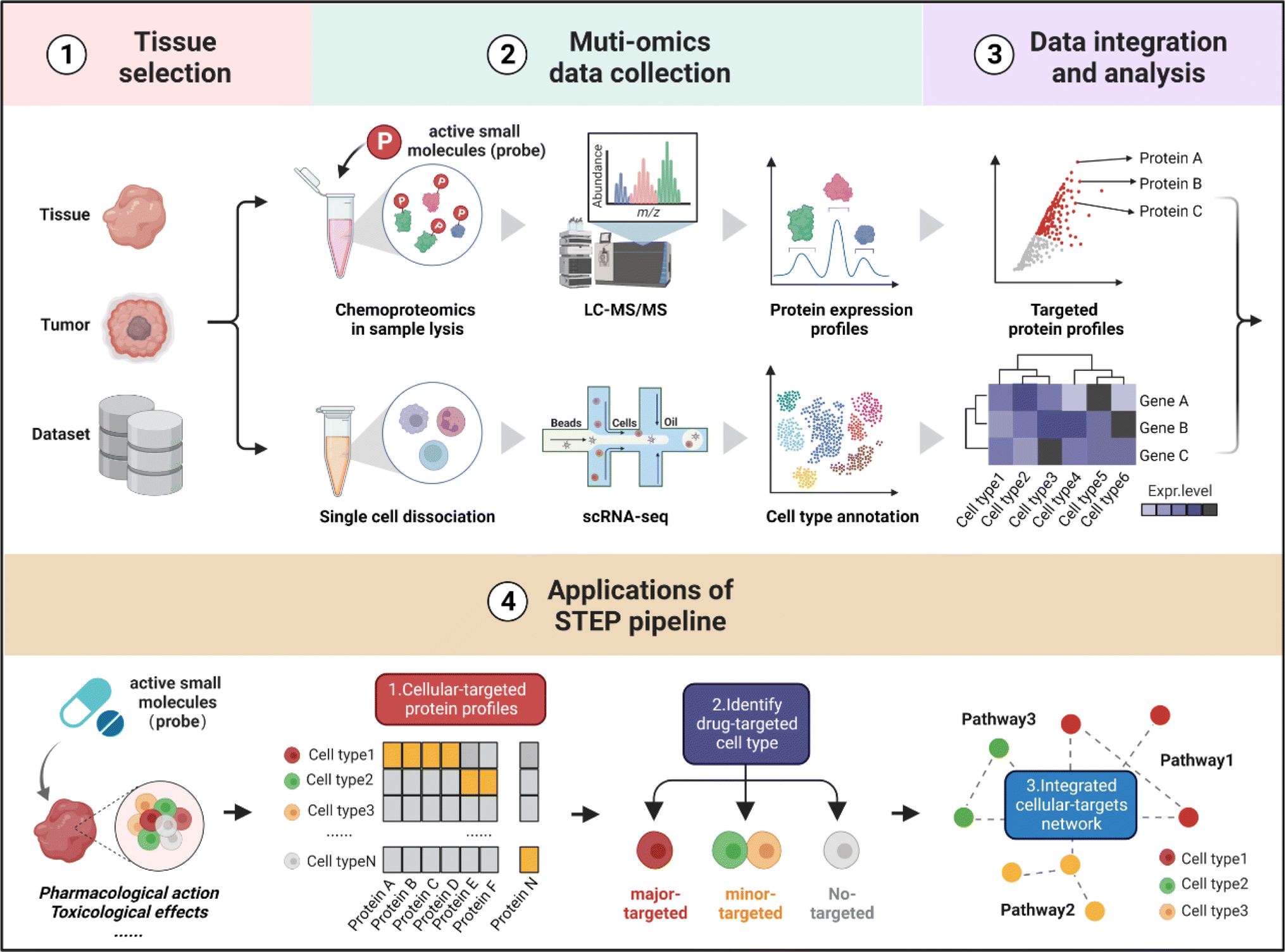 | ||
| Fig. 1 Schematic illustration of cellular target identification of ASMs in tissues by STEP. The workflow of STEP consists of four main modules: ① tissue selection, ② multi-omics data acquisition, ③ data integration and analysis, and ④ cellular target identification and functional analysis, including cellular-targeted protein profiling, drug-targeted cell type identification, and integrated cellular-target networks. | ||
In the following analysis, by taking well-established covalent drugs such as aspirin, aristolochic acid, and cisplatin as examples, we will demonstrate the precision, predictability and informative nature of our strategy. According to the features of ASMs to be explored, this pipeline as well as the datasets can be extended to analyze the pharmacological or toxicological effects of other drugs on their targeted tissues. Furthermore, this pipeline offers a guideline to select suitable cell lines as in vitro models by comparing the distribution of protein targets of ASMs for studying the bioactivities of drugs in specific organs in an unbiased manner.
STEP identification of the cellular-specific targets of the aspirin probe in brain tissues
Aspirin (ASP), a well-known drug that is widely used worldwide, has broad-spectrum bioactivities including antipyretic, analgesic, antithrombotic, and anti-inflammatory.12,13 Referring to our previous study, we have previously developed a chemical ASP probe and used it to identify protein targets in vitro.14 To further investigate the main targeted tissue where ASP functions, ten types of mouse tissues including liver, kidney, and brain were lysed and further labeled by using the biorthogonal ASP probe. As shown in Fig. S1,† brain tissue showed the greatest labelling intensity among these tissues, indicating that proteins in the brain had the most affinity to aspirin at the tissue level, in line with the clinical role of aspirin in treating cerebrovascular diseases.15 Therefore, we selected the brain as the subject of further analysis. Besides, the labeling intensity of the ASP probe in brain lysates gradually increased in a concentration dependent manner (Fig. S2a†). Moreover, the fluorescence intensity of proteins labelled by the ASP probe could be effectively competed away by pretreatment with unfunctionalized ASP (Fig. S2b†), indicating that the proteins captured by the ASP probe were indeed the targets of ASP.As shown in Fig. 2a and S3,† we applied the STEP pipeline to generate the protein targets engaged by the ASP probe in lysed brain tissues, as well as the scRNA-seq dataset of healthy mouse brains to profile the cellular drug targets at single-cell resolution. As shown in Fig. 2b, we identified 725 targeted proteins in total (probe group vs. DMSO group, FDR < 0.05 & fold change > 2). We then integrated the chemoproteomics results and scRNA-seq dataset to evaluate the expression patterns of the cellular targeted proteins (Fig. 2c and d) and further assigned these protein targets to each cell type based on the expression abundance (Fig. 2e). We found that neurons (neuron) and choroid plexus epithelial cells (CPC) had higher expression levels for most targeted proteins than other cell types such as microglia (micro). These results revealed that the heterogeneous distribution of aspirin protein targets could be observed not only at the tissue level, but also at the cellular level within the same tissue.
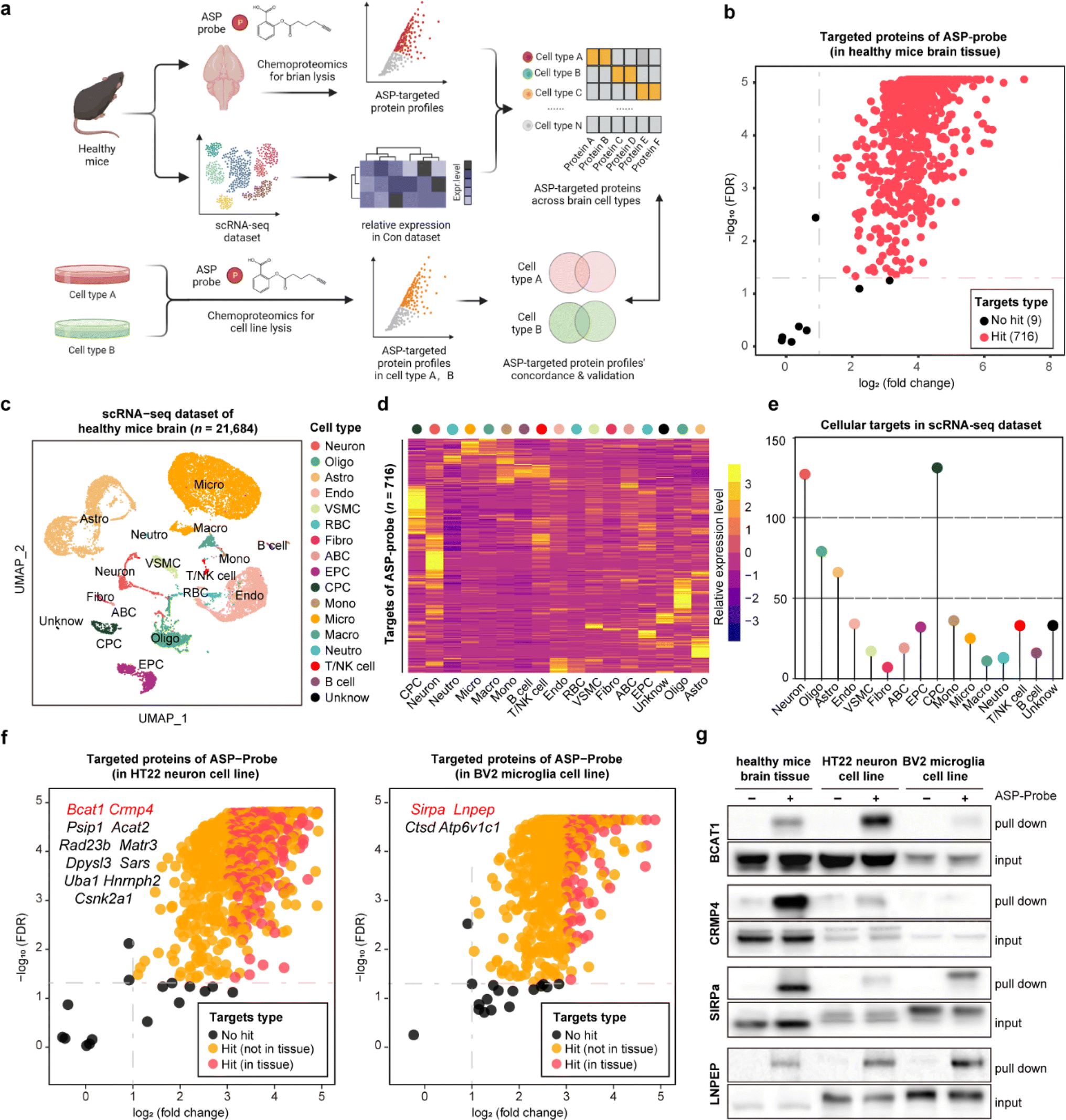 | ||
Fig. 2 The profiling of cellular protein targets of aspirin in brain demonstrating the precision and specificity of the STEP pipeline. (a) Schematic of cellular target profiling of aspirin (ASP) in mouse brains via STEP. (b) Volcano plot depicting the distribution of the targeted proteins captured by the ASP probe in mouse brains. (c) UMAP (uniform manifold approximation and projection) visualization showing 17 cell types based on 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 684 single cell transcriptomes. Neuron, oligo: oligodendrocyte, astro: astrocyte, endo: endothelial cells, mural cell, RBC: red blood cell, fibro: fibroblast, ABC: arachnoid barrier cells, EPC: ependymocytes, CPC: choroid plexus epithelial cells, mono: monocyte, micro: microglia, macro: macrophage, neutro: neutrophils and unknown. (d) The heatmap showing the relative expression level of ASP probe targeted proteins across 17 cell types in the scRNA-seq dataset. (e) Lollipop chart depicting the numbers of ASP probe targeted proteins across 17 cell types in the scRNA-seq dataset. (f) Volcano plot depicting the distribution of the targeted proteins captured by the ASP probe in the murine neuron cell line (left) and microglia cell line (right). (g) Pull-down western blotting experiments verifying the cellular targets of BCAT1, CRMP4, SIRPa, and LNPEP proteins in healthy brain tissue, the neuron cell line, and the microglia cell line. 684 single cell transcriptomes. Neuron, oligo: oligodendrocyte, astro: astrocyte, endo: endothelial cells, mural cell, RBC: red blood cell, fibro: fibroblast, ABC: arachnoid barrier cells, EPC: ependymocytes, CPC: choroid plexus epithelial cells, mono: monocyte, micro: microglia, macro: macrophage, neutro: neutrophils and unknown. (d) The heatmap showing the relative expression level of ASP probe targeted proteins across 17 cell types in the scRNA-seq dataset. (e) Lollipop chart depicting the numbers of ASP probe targeted proteins across 17 cell types in the scRNA-seq dataset. (f) Volcano plot depicting the distribution of the targeted proteins captured by the ASP probe in the murine neuron cell line (left) and microglia cell line (right). (g) Pull-down western blotting experiments verifying the cellular targets of BCAT1, CRMP4, SIRPa, and LNPEP proteins in healthy brain tissue, the neuron cell line, and the microglia cell line. | ||
To further confirm these findings, we performed the same chemoproteomics experiment in HT22 (murine neuron) and BV2 (murine microglia) cell lines. Comparing the labeling effects of the ASP probe in the brain tissue, HT22, and BV2 cells, we found the labeling intensity and specificity of these samples to be distinct from each other (Fig. S4†), indicating that the heterogeneity of protein targets of ASP was embodied in not only tissues but also in different cell types. There were 680 and 566 ASP probe targeted-proteins identified in HT22 neurons and BV2 cells, respectively (probe group vs. DMSO group) (Fig. 2f). In particular, we identified 39 proteins such as branched chain amino acid transaminase 1 (BCAT1) and collapsin response mediator protein 4 (CRMP4) overlapping with brain tissue in neuron, as well as 17 proteins such as signal regulatory protein alpha (SIRPa) and leucyl and cystinyl aminopeptidase (LNPEP) overlapping with brain tissue in micro.
To verify the heterogeneity of protein target expression and engagement in the two cell lines, we performed pull-down assays by using the ASP probe for the four proteins mentioned above, and the results matched well with the findings in tissues by STEP (Fig. 2g). Meanwhile, each identified target protein pulled down by the ASP probe could be similarly competed away by pre-incubating unmodified ASP in brain tissues and the respective cell line (Fig. S5†). Furthermore, the in situ labeling effect of the ASP probe in living HT-22 and BV2 cells showed that the ASP probe could penetrate the membrane into the cytoplasm and effectively label the identified cellular protein target in both cell lines (Fig. S6†). These results collectively demonstrate the high specificity and confidence of STEP in identifying cellular drug–target profiles in the tissues, providing more precise information on the target distribution and engagement across various cellular types.
It is noted that the most recognized aspirin target proteins, prostaglandin G/H synthases 1 and 2 (PTGS1 and PTGS2), were not included in our target list. PTGS1/2 are expressed in different tissues at different levels; however, the expressed levels of the recognized targets in brain tissue were relatively limited. Consistent with our former results PTGS1/2 are more likely to be pulled down in tissues with high levels of expression (Fig. S7†). Accordingly, the absence of prostaglandin-related proteins in our aspirin-brain dataset might primarily be ascribed to their low expression either in brain tissue or the relevant cell lines. Meanwhile, several studies have indicated that aspirin exerts pleiotropic therapeutic effects by engaging diverse proteins. For instance, aspirin predominantly diminishes the synthesis of PGE2 by inhibiting the activity of PTGS1/2 during anti-inflammatory application.16 However, in the context of anti-colorectal cancer, aspirin demonstrates tumor-suppressive properties through multiple pathways, including effecting transcriptional regulation through the acetylation of histones.14,17–19 Therefore, the target profiling of the drug and the specific mechanism of action might be diverse depending on tissue types and disease conditions. Our identification of aspirin target proteins in brain tissue would enhance the understanding of aspirin's versatility in treating neurological diseases.
STEP reveals the selective deactivation of pathways in proximal tubules in AAN tissues
Aristolochic acids (AAs), a class of carcinogenic and mutagenic natural products derived from Aristolochia and Asarum plants, can cause aristolochic acid nephropathy (AAN). Our previous scRNA-seq studies have demonstrated that aristolochic acid I (AAI, a major type of AA) could induce cell type-specific responses in AAN renal tissue compared with healthy controls,20 and identified target protein profiles of AAI in renal tissue by chemoproteomics technology.21 Here, we employed STEP to integrate these existing datasets including the AAI targeted protein lists, bulk RNA-seq, and scRNA-seq datasets to profile the cellular protein targets and mediate biological pathways engaged by the AAI probe. Next, we performed differentially expressed gene (DEG) analysis of each cell type between the AAN group and the healthy group, and further conducted gene set enrichment analysis (GSEA) to determine whether these pathways in various cell types were differentially activated after AAI treatment, thereby identifying the AAI-mediated nephrotoxicity mechanism (Fig. 3a).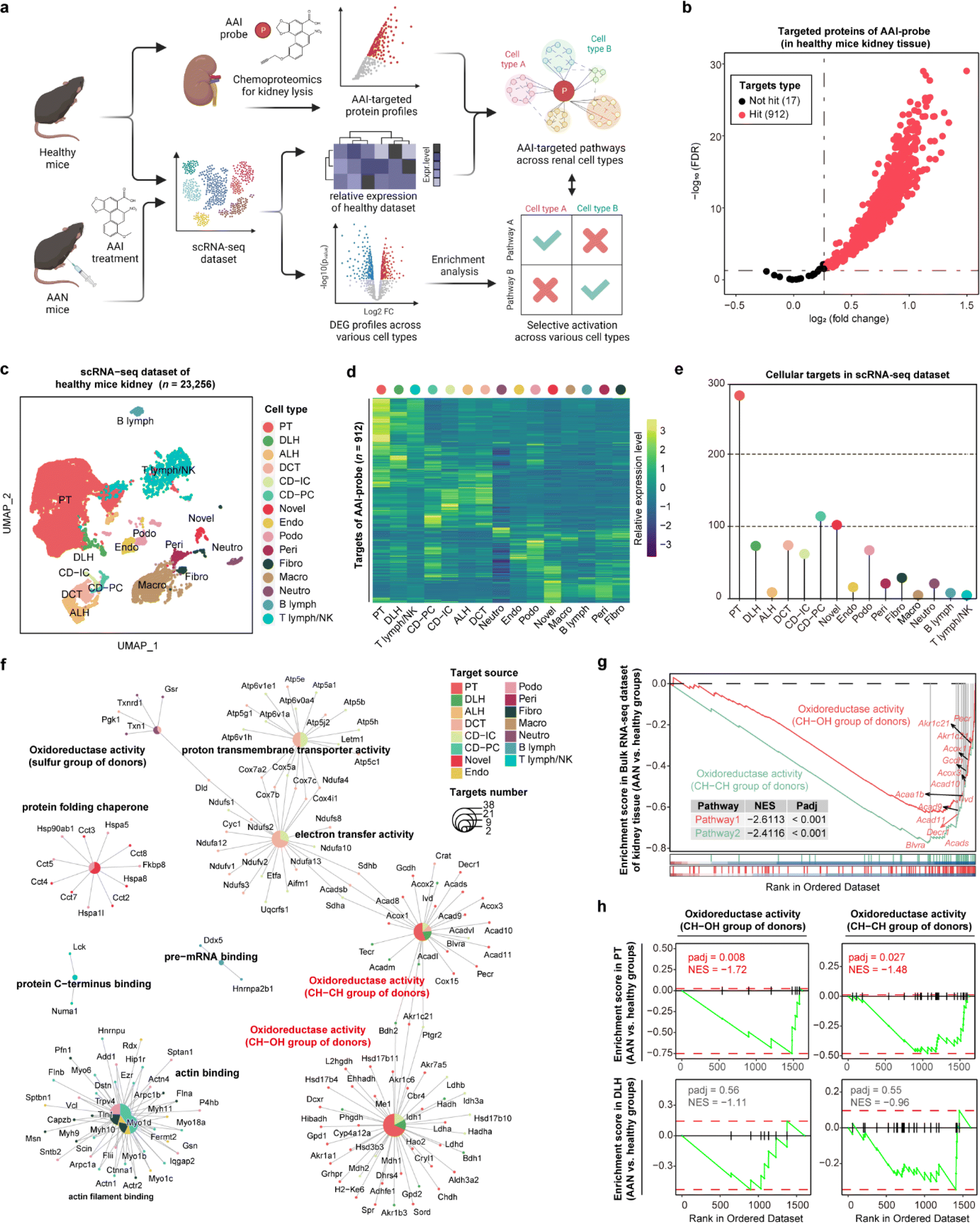 | ||
| Fig. 3 Aristolochic acid selectively activating biological pathways in proximal tubular cells via profiling cellular targets in the kidney. (a) Schematic of cellular target profiling of aristolochic acid (AAI) in mouse kidneys via STEP. (b) Volcano plot depicting the distribution of the targeted proteins captured by the AAI probe in healthy mouse kidney lysates. (c) UMAP visualization showing 15 distinct cellular types of 23256 cells in the scRNA-seq dataset for healthy mouse kidneys. PT, proximal tubule; DLH, descending loop of Henle; ALH, ascending loop of Henle; DCT, distal convoluted tubule; CD-IC, collecting duct intercalated cell; CD-PC, collecting duct principal cell; endo, endothelial; podo, podocyte; peri, pericytes and vascular smooth muscle cells; fibro, fibroblast; neutro, neutrophil; B lymph, B lymphocyte; T lymph, T lymphocyte; NK, NK cell. (d) The heatmap showing the relative expression level of AAI probe targeted proteins across 15 cell types in the mouse kidney. (e) Lollipop chart depicting the numbers of AAI probe targeted proteins across 15 cell types in the murine kidney. (f) Cnetplot depicting the biological pathways enriched by targeted proteins across various cell types in the scRNA-seq dataset. (g) GSEA results revealing the enrichment level of two oxidoreductase activity pathways (AAN vs. healthy groups) in the bulk RNA-seq dataset. (h) GSEA results revealing the enrichment level of two oxidoreductase activity pathways (AAN vs. healthy groups) in the scRNA-seq dataset for both PT cells (top panel) and DLH cells (bottom panel). | ||
In the gel-labeling assay, the AAI probe successfully labeled the protein targets in the range of physiological concentrations, and pretreatment of unfunctionalized AAI could markedly diminish the label intensity of the AAI probe on the targets, demonstrating that the proteins captured by the AAI probe represented the AAI targets well (Fig. S8†). Furthermore, chemoproteomics identified 912 AAI targeted proteins in total (probe group vs. DMSO group, FDR < 0.05 & fold change > 1.2) (Fig. 3b). We then integrated the chemoproteomics results and scRNA-seq dataset to evaluate the cellular targeted-protein expression pattern (Fig. 3c and d) and further acquired the protein target distribution at singe-cell resolution (Fig. 3e). These results revealed that proximal tubular (PT) cells have the largest number of targeted proteins compared to other cell types, in agreement with a previous report that PT cells are the primary target of AAI in the kidney.22 Gene ontology (GO) enrichment results indicated that AAI differentially affected the biological pathways involving protein targets among various cell types. In terms of enriched pathways, AAI-mediated pathways in PT cells, descending loop of Henle (DLH) cells, and other renal epithelial cells were mainly involved in CH–CH group of donors, CH–OH group of donors, and electron transfer activity (Fig. 3f), which coincides well with the fact that AAI induces apoptosis in renal cells by disturbing metabolic processes and mitochondrial respiration.21
Next, we wanted to determine the selective activation or deactivation of these pathways mentioned above across different cell types in the AAN and healthy groups. GSEA results of the bulk RNA-seq dataset indicated that the two oxidoreductase activity pathways in kidney tissue were significantly down-regulated after AAI treatment (Fig. 3g). To find out which renal cell types were responsible for the dysfunction of the two pathways, the DEG profiles of various cells in the scRNA-seq dataset were further subjected to GSEA. Our results revealed that these two pathways were significantly down-regulated in PT cells rather than DLH cells, indicating that AAI could selectively deactivate oxidoreductase activities in PT cells (Fig. 3h). These STEP results revealed the selective regulation of ASM-mediated pathways in a specific cell type, indicating the role of STEP as a powerful tool in identifying biological regulation among various cells in different tissues.
STEP provides novel insights into anti-tumor actions of cisplatin in xenograft tumors
As one of the most commonly used drugs in cancer chemotherapy and combination therapy, cisplatin (cis) is generally understood to kill tumor cells via acting on the purine and pyrimidine bases of DNA. Nevertheless, the intracellular platinum binding to DNA has been estimated as only about 1–10%23,24 because other intracellular nucleophilic substances such as RNA, cysteine residues on proteins and thiol-containing ligands can also engage with cisplatin.25,26 Several studies have recently indicated that cisplatin can also suppress tumor cells by directly interacting with proteins, supporting that proteins are important functional targets of cisplatin, in the same vein as their canonical nucleic acid targets.27,28 Therefore, we developed a cis-based activity probe to globally profile the protein targets in both the 4T1 murine breast tumor cell line and the corresponding cell-line-derived tumor xenograft (CDTX) model by chemoproteomics. The targeting efficiency and selectivity of the cis probe were confirmed by the labeling and competing experiments in the 4T1 cells and CDTX lysates (Fig. S9†). Meanwhile, a scRNA-seq dataset of CDTX tissue was also collected to dissect the drug–target engagement across various cell types via the STEP pipeline (Fig. 4a and S10†). There were 15 and 52 protein targets identified in the 4T1 cell line and CDTX tissue, respectively (probe group vs. DMSO group, FDR < 0.05 & fold change > 1.2) (Fig. 4b and c). As for the scRNA-seq dataset of CDTX tissue, we acquired 9452 cells including tumor cells (54.6%), macrophages (31.2%), T lymphocytes (4.08%), neutrophils (8.94%) and fibroblasts (1.26%), which comprehensively depicted the cellular composition of the tumor microenvironment (Fig. 4d). As expected, tumor cells were the cell subset in which most protein targets were enriched, and other cell subsets also had greater-than-expected numbers of distributed targets (Fig. 4e). As shown in Fig. 4f, there were 12 overlapping protein targets between 4T1 cells and CDTX tissue. Most of the protein targets in the 4T1 cell line (12/15) were also identified in CDTX tissue, while CDTX tissue had 40 exclusive targets. As for the cell specificity of target distribution in the tissue, tumor cells (in CDTX tissue) had 14 exclusive targets and 6 targets shared with 4T1 cells, and some targets were only identified in immune cells such as neutrophils and T lymphocytes.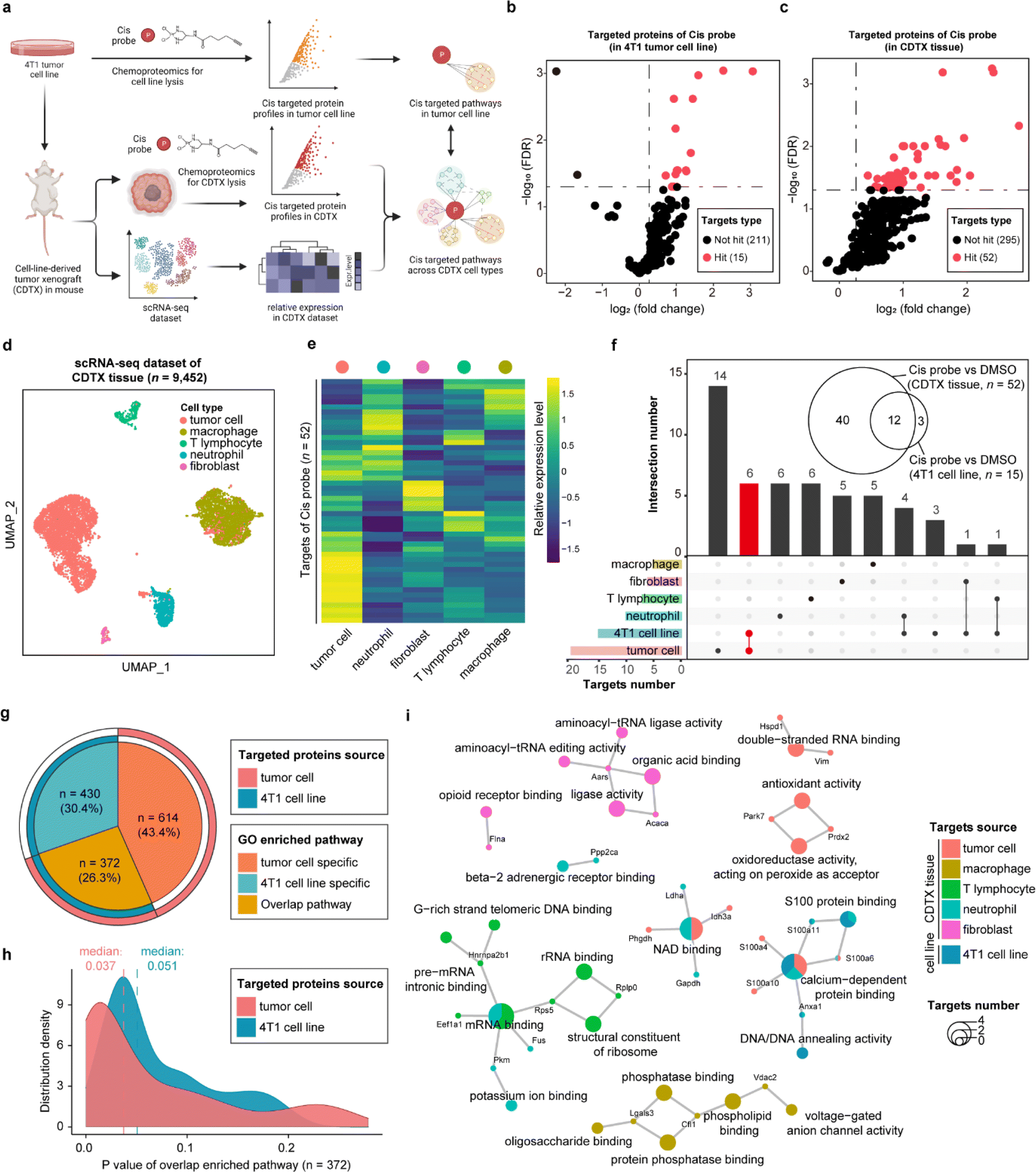 | ||
| Fig. 4 STEP providing more informative knowledge of cisplatin's anti-tumor mechanism by comparing target engagements in CDTX tissue and the tumor cell line. (a) Schematic of cellular target profiling of cisplatin (cis) in mouse kidneys via STEP. (b) Volcano plot depicting the distribution of the targeted proteins captured by the cis probe in 4T1 tumor line lysates. (c) Volcano plot depicting the distribution of the targeted proteins captured by the cis probe in mice CDTX tissue lysates. (d) UMAP visualization showing 5 distinct cellular identities of mouse tumor tissue in the scRNA-seq dataset. (e) The heatmap showing the relative expression level of cis probe targeted protein profiles across 5 cell types in the scRNA-seq dataset. (f) The UpSet plot showing the overlapping of targeted proteins captured by the cis probe among various cell types in CDTX tissue and the 4T1 tumor cell line. (g) The circle plot depicting the type (overlapping or specific) and the relative proportion of GO enriched pathways (biological process) involving the protein targets. (h) The density plot showing the distribution of overlapped GO enriched pathways' P value. (i) Cnetplot depicting the enriched biological pathways involving the targeted proteins across various cell types in CDTX tissue and the 4T1 cell line. | ||
To further compare the pathways mediated by drug–targets in tumor cells (in CDTX tissue) and 4T1 cells, GO enrichment analysis found that tumor cells enriched an additional 614 (accounting for 43.4%) exclusive pathways, indicating that the MOA of cis in CDTX tissue is much more complex than those in simplified cell lines (Fig. 4g). As for the overlapped pathways (n = 372, accounting for 26.3%), we found that the median P value of enriched pathways based on tumor cells (median = 0.037) was lower than the P value based on 4T1 cells, demonstrating that the statistical significance of the enrichment results was improved via enlarging the targets' scope with STEP analysis in CDTX tissue (Fig. 4h). Integrated analysis of GO enrichment results and targets profile revealed that xenograft tumor cells and 4T1 cells shared the pathways of NAD binding and calcium-dependent protein binding, while specific pathways such as double-strand RNA binding and oxidoreductase activity were exclusively enriched for targeted proteins in tumor cells. Interestingly, the pathways of mRNA binding and G-rich strand telomeric DNA binding involving the targeted proteins were shared in neutrophils and T lymphocytes (Fig. 4i). Compared with target profiling in the 4T1 cell line by chemoproteomics, the application of STEP in CDTX tissues not only brought out more informative knowledge in the understanding of the anti-tumor mechanism of cis within the tumor microenvironment, but also provided novel insights into the side-effects (e.g. granulocytopenia) by engaging cis targeted proteins in the immune cells.
The co-existing non-protein-based targets and protein targets of cisplatin indicated the versatility of its antitumor mechanism. In this study, an activity-based protein profiling strategy was employed to globally profile the distribution of proteins. This approach offers a novel perspective on understanding the anti-cancer activity of cisplatin. However, the precise binding mechanisms of the identified protein targets and their subsequent contribution to cis's therapeutic efficacy remain to be validated and studied. While the nucleic acid targets were not identified in the chemical proteomics experiments, our analytical framework should be flexible and applicable to integrate the nucleic acid targets with the scRNA-seq datasets in principle. For example, enriching and identifying cisplatin's nucleic acid targets in tissues would enable the profiling of their distribution and associated biological pathways across various cells by integrating the single-cell RNA sequencing dataset through a similar analytical pipeline. It is important to acknowledge certain limitations in our experimental design.
In this study, the use of homogenized lysates, as a common practice in chemical proteomics studies for enhanced labeling efficiency and maximal protein identification, inadvertently neglects considerations of permeability conditions and trafficking pathways of the probes. This limitation may influence the labeling behavior of functionalized probes in tissue lysates, potentially deviating from their behavior in living systems. To address this, future investigations could be extended to organoids or living animals, providing a more comprehensive understanding of drug–protein interactions under physiologically relevant conditions.
Conclusion
In summary, we developed a STEP pipeline to indentify the targets of ASMs in tissues at single-cell resolution by integrating scRNA-seq and chemoproteomics datasets. This proof of concept has been successfully verified by three classic drugs, with the identification of cellular drug–protein engagement. More importantly, the unprecedented resolution of their targets and biological processes in mammalian tissues provides novel insights for understanding the pharmacological and toxicological mechanisms of ASMs, which have been neglected or difficult to detect in cell models. We believe STEP is a powerful and reliable strategy to profile the cellular-specific targets, which would fill up the gaps in the knowledge of molecular mechanisms between in vivo and in vitro systems.Data availability
All transcriptomics and proteomics raw datasets supporting our findings in this study are available from the corresponding authors upon reasonable request.Author contributions
J. G. W., H. T., and C. C. X. supervised the project and conceived the idea. J. Y. C. designed the pipeline and conducted the main data analysis. Z. C. and Q. Z. performed the main experiments and validation. C. W. and P. L. performed the animal experiments. Y. Z. and F. X. performed probe synthesis. L. W. G. and Q. L. S. conducted some in vitro experiments and data analysis. J. Y. C. and H. T. drafted the original manuscript. J. G. W., H. T., C. C. X., and Y. K. W. revised the article. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the Scientific and Technological Innovation Project of the China Academy of Chinese Medical Sciences (CI2023D003 and CI2023E002); the National Key Research and Development Program of China (2020YFA0908000 and 2022YFC2303603); the Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (No. ZYYCXTD-C-202002); the Science and Technology Foundation of Shenzhen (JCYJ20210324115800001); National Natural Science Foundation of China (32201177, 82141001, and 82074098); the CACMS Innovation Fund (CI2021A05101 and CI2021A05104); the Science and Technology Foundation of Shenzhen (Shenzhen Clinical Medical Research Center for Geriatric Diseases); the Shenzhen Medical Research Fund (B2302051); the Fundamental Research Funds for the Central public welfare research institutes (ZZ14-YQ-061, ZZ16-ND-10-24, and ZZ14-FL-002); the Supportive Plan for Top Innovative PhD Students at the China Academy of Chinese Medical Sciences. All authors gave approval to the final version for publication.References
- M. Schenone, V. Dančík, B. K. Wagner and P. A. Clemons, Nat. Chem. Biol., 2013, 9, 232–240 CrossRef CAS PubMed.
- S. Ziegler, V. Pries, C. Hedberg and H. Waldmann, Angew Chem. Int. Ed. Engl., 2013, 52, 2744–2792 CrossRef CAS.
- X. Chen, Y. Wang, N. Ma, J. Tian, Y. Shao, B. Zhu, Y. K. Wong, Z. Liang, C. Zou and J. Wang, Signal Transduction Targeted Ther., 2020, 5, 72 CrossRef PubMed.
- Y. Zhu, Z. Ouyang, H. Du, M. Wang, J. Wang, H. Sun, L. Kong, Q. Xu, H. Ma and Y. Sun, Acta Pharm. Sin. B, 2022, 12, 4011–4039 CrossRef CAS PubMed.
- W. C. Chan, S. Sharifzadeh, S. J. Buhrlage and J. A. Marto, Chem. Soc. Rev., 2021, 50, 8361–8381 RSC.
- J. N. Spradlin, E. Zhang and D. K. Nomura, Acc. Chem. Res., 2021, 54, 1801–1813 CrossRef CAS.
- M. Labib and S. O. Kelley, Nat. Rev. Chem, 2020, 4, 143–158 CrossRef PubMed.
- V. Marx, Nat. Methods, 2019, 16, 809–812 CrossRef CAS PubMed.
- E. Levy and N. Slavov, Essays Biochem., 2018, 62, 595–605 CrossRef PubMed.
- Z. Pang, M. A. Schafroth, D. Ogasawara, Y. Wang, V. Nudell, N. K. Lal, D. Yang, K. Wang, D. M. Herbst, J. Ha, C. Guijas, J. L. Blankman, B. F. Cravatt and L. Ye, Cell, 2022, 185, 1793–1805 CrossRef CAS PubMed.
- H. Specht, E. Emmott, A. A. Petelski, R. G. Huffman, D. H. Perlman, M. Serra, P. Kharchenko, A. Koller and N. Slavov, Genome Biol., 2021, 22, 50 CrossRef CAS PubMed.
- J. R. Vane and R. M. Botting, Thromb. Res., 2003, 110, 255–258 CrossRef CAS.
- D. A. Drew, Y. Cao and A. T. Chan, Nat. Rev. Cancer, 2016, 16, 173–186 CrossRef CAS PubMed.
- J. Wang, C. J. Zhang, J. Zhang, Y. He, Y. M. Lee, S. Chen, T. K. Lim, S. Ng, H. M. Shen and Q. Lin, Sci. Rep., 2015, 5, 7896 CrossRef CAS PubMed.
- H. Hua, H. Zhang, Q. Kong, J. Wang and Y. Jiang, Med. Res. Rev., 2019, 39, 114–145 CrossRef CAS PubMed.
- N. R. Bin, S. L. Prescott, N. Horio, Y. Wang, I. M. Chiu and S. D. Liberles, Nature, 2023, 615, 660–667 CrossRef CAS PubMed.
- L. A. Bateman, B. W. Zaro, S. M. Miller and M. R. Pratt, J. Am. Chem. Soc., 2013, 135, 14568–14573 CrossRef CAS PubMed.
- S. Pathi, I. Jutooru, G. Chadalapaka, V. Nair, S. O. Lee and S. Safe, PLoS One, 2012, 7, e48208 CrossRef CAS PubMed.
- F. V. Din, A. Valanciute, V. P. Houde, D. Zibrova, K. A. Green, K. Sakamoto, D. R. Alessi and M. G. Dunlop, Gastroenterology, 2012, 142, 1504–1515 CrossRef CAS.
- J. Chen, P. Luo, C. Wang, C. Yang, Y. Bai, X. He, Q. Zhang, J. Zhang, J. Yang, S. Wang and J. Wang, JCI Insight, 2022, 7, e157360 CrossRef PubMed.
- Q. Zhang, P. Luo, J. Chen, C. Yang, F. Xia, J. Zhang, H. Tang, D. Liu, L. Gu, Q. Shi, X. He, T. Yang and J. Wang, Int. J. Biol. Sci., 2022, 18, 2003–2017 CrossRef CAS PubMed.
- X. W. Li, S. Yokota, D. Wang, X. Wang, Y. Shoyama and S. Q. Cai, Am. J. Chin. Med., 2014, 42, 1453–1469 CrossRef CAS PubMed.
- R. C. DeConti, B. R. Toftness, R. C. Lange and W. A. Creasey, Cancer Res., 1973, 33, 1310–1315 CAS.
- M. Groessl, O. Zava and P. J. Dyson, Metallomics, 2011, 3, 591–599 CrossRef CAS.
- B. Michalke, J. Trace Elem. Med. Biol., 2010, 24, 69–77 CrossRef CAS PubMed.
- R. N. Bose, S. K. Ghosh and S. Moghaddas, J. Inorg. Biochem., 1997, 65, 199–205 CrossRef CAS PubMed.
- T. Zimmermann, M. Zeizinger and J. V. Burda, J. Inorg. Biochem., 2005, 99, 2184–2196 CrossRef CAS.
- R. C. Todd and S. J. Lippard, Metallomics, 2009, 1, 280–291 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04826h |
| ‡ Jiayun Chen, Zheng Chu, and Qian Zhang contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2024 |