
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetically controlled synthesis of rotaxane geometric isomers†
Dillon R.
McCarthy‡
a,
Ke
Xu‡
 b,
Mica E.
Schenkelberg
ab,
Nils A. N.
Balegamire
ab,
Huiming
Liang
a,
Shea A.
Bellino
a,
Jianing
Li
ab and
Severin T.
Schneebeli
*ab
b,
Mica E.
Schenkelberg
ab,
Nils A. N.
Balegamire
ab,
Huiming
Liang
a,
Shea A.
Bellino
a,
Jianing
Li
ab and
Severin T.
Schneebeli
*ab
aDepartments of Chemistry, Pathology, and Materials Science Program, University of Vermont, Burlington, VT 05405, USA. E-mail: Schneebeli@purdue.edu
bDepartments of Industrial & Molecular Pharmaceutics, Chemistry, and Medicinal Chemistry & Molecular Pharmacology, Purdue University, West Lafayette, IN 47907, USA
First published on 1st February 2024
Abstract
Geometric isomerism in mechanically interlocked systems—which arises when the axle of a mechanically interlocked molecule is oriented, and the macrocyclic component is facially dissymmetric—can provide enhanced functionality for directional transport and polymerization catalysis. We now introduce a kinetically controlled strategy to control geometric isomerism in [2]rotaxanes. Our synthesis provides the major geometric isomer with high selectivity, broadening synthetic access to such interlocked structures. Starting from a readily accessible [2]rotaxane with a symmetrical axle, one of the two stoppers is activated selectively for stopper exchange by the substituents on the ring component. High selectivities are achieved in these reactions, based on coupling the selective formation reactions leading to the major products with inversely selective depletion reactions for the minor products. Specifically, in our reaction system, the desired (major) product forms faster in the first step, while the undesired (minor) product subsequently reacts away faster in the second step. Quantitative 1H NMR data, fit to a detailed kinetic model, demonstrates that this effect (which is conceptually closely related to minor enantiomer recycling and related processes) can significantly improve the intrinsic selectivity of the reactions. Our results serve as proof of principle for how multiple selective reaction steps can work together to enhance the stereoselectivity of synthetic processes forming complex mechanically interlocked molecules.
Complex interlocked molecules have become integral components for the development of next-generation supramolecular catalysts,1 molecular machines,2 and molecular motors.3 In particular, rotaxanes with either oriented tracks4 or facially dissymmetric macrocycles5 have shown promise for ribosome-inspired peptide synthesis6 and cargo transport.7 To impart additional degrees of spatial control and unidirectionality into these systems, it would be desirable to combine oriented axles with facially dissymmetric (i.e. rim-differentiated) macrocyclic components in a selective fashion, which leads to geometric isomers.8
We now report a through-space controlled9 aminolysis reaction, which can selectively form specific geometric isomers of [2]rotaxanes under kinetic control. Our approach starts with a readily accessible [2]rotaxane with a symmetric axle, which is then desymmetrized based on selective stopper exchange accelerated10 by the presence of nearby glyme functional groups. We have recently applied this concept in the context of interlocked molecules with through-space glyme-activating groups (Fig. 1a), which enabled the two reactive ends in rotaxanes to communicate with each other.10 However, in our initial system, the ring components of the rotaxanes were facially symmetrical. Therefore, our initial system did not address the complexity of forming specific rotaxane geometric isomers selectively, which has now been accomplished in this work. Furthermore, in our initial glyme-catalyzed rotaxane system (Fig. 1a), we had observed only modest selectivity for the glyme-activated reactions with the maximum selectivity for mono- vs. difunctionalization ∼8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at the beginning of the reaction.10 Overall, our glyme-activated directional stopper-exchange process represents an alternative way to accomplish kinetic selection of reaction barriers in interlocked molecules. Our results complement existing approaches to control/augment chemical reactivity through space across the mechanical bond.11 Related processes have also been implemented in chemically fueled molecular machines, where the position2a,12 or facial dissymmetry13 of the macrocycle determine the rate of addition and/or removal or a barrier.
:1 at the beginning of the reaction.10 Overall, our glyme-activated directional stopper-exchange process represents an alternative way to accomplish kinetic selection of reaction barriers in interlocked molecules. Our results complement existing approaches to control/augment chemical reactivity through space across the mechanical bond.11 Related processes have also been implemented in chemically fueled molecular machines, where the position2a,12 or facial dissymmetry13 of the macrocycle determine the rate of addition and/or removal or a barrier.
 | ||
| Fig. 1 (a) Through-space controlled aminolysis in a rotaxane system. (b) This work applies the concept to the selective synthesis of rotaxane geometric isomers, while also introducing a strategy to enhance the selectivity in such reactions by coupling (see also Scheme 1) a formation reaction selective for the major geometric isomer with a depletion reaction with inverse selectivity. | ||
Here, we now find that the selectivity for forming specific rotaxane geometric isomers increases exponentially during such glyme-activated reactions. After ∼300 hours, the d.r. for formation of the major geometric isomer increases to >40:1, which represents a remarkable improvement from the prior 8:1 ratio. Our reactions lead (Fig. 1b) to specific rotaxane geometric isomers with high selectivity. This improved selectivity was enabled by coupling (Scheme 1) two through-space controlled aminolysis reactions8f,10,14 with each other: the first reaction leads to the major geometric isomer with modest selectivity, while the second through-space controlled aminolysis reaction selectively depletes the minor product. Thereby, the overall selectivity of the coupled reaction system is enhanced significantly compared to the two individual reactions. While this concept is related to minor enantiomer recycling15 and related photo-deracemization processes,16 the minor isomer is not recycled in our system. Therefore, while the selectivity rapidly increases over time (which can lead to simpler purification), the increased selectivity arises at the cost of the overall yield in our system, which decreases over time.
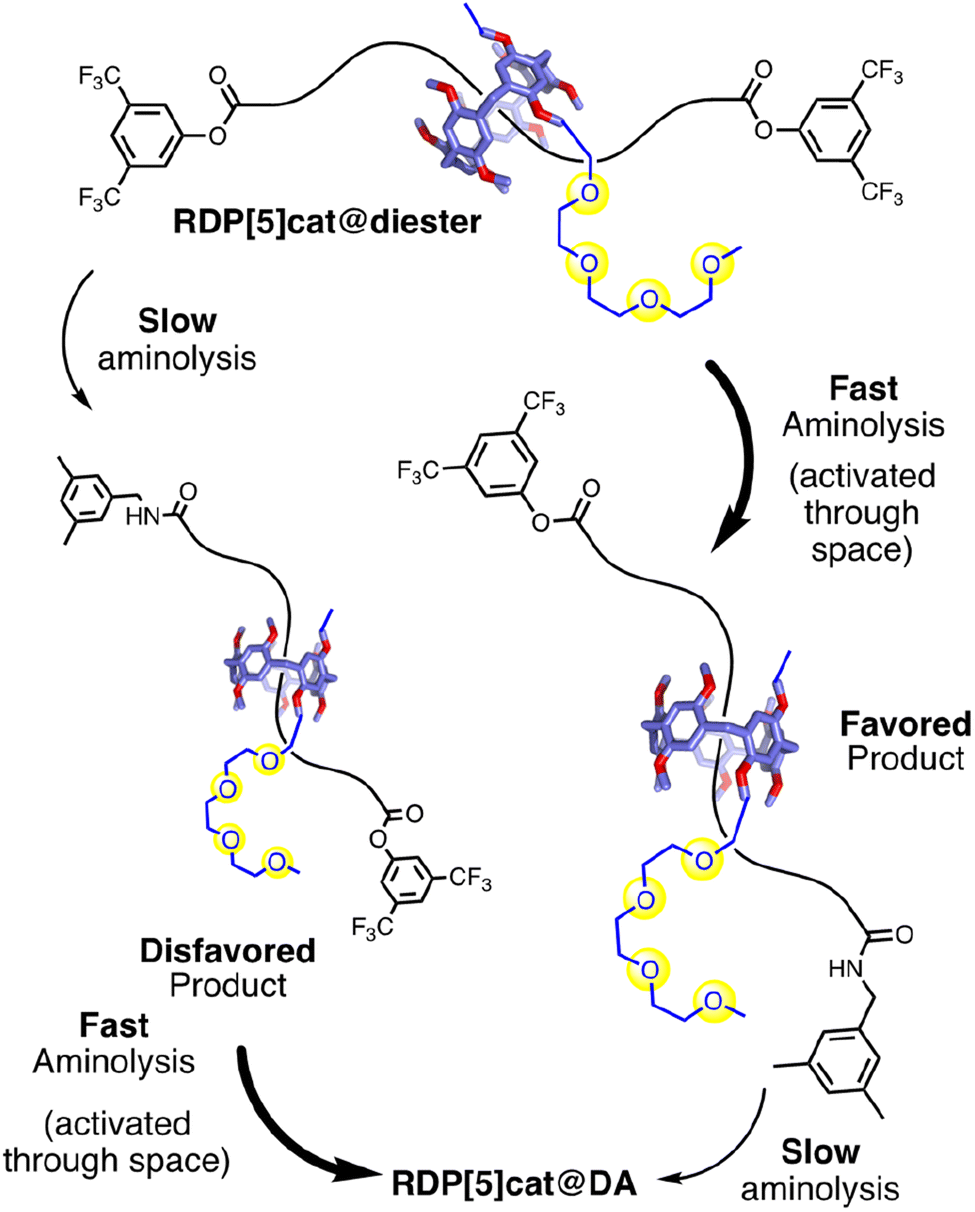 | ||
| Scheme 1 A matrix of fast and slow aminolysis reactions (all fast ones are through-space controlled by the glyme-activating groups) leads to kinetic control of geometric isomerism with >40:1 selectivity for the major geometric isomer. | ||
To establish proof-of-concept for our kinetically controlled synthesis, a rim-differentiated pillar[5]arene17 was chosen as the facially dissymmetric macrocycle given the ease of synthesis,18 excellent chemical stability and solubility,19 and the ability to control the directionality of the catalyst/activating group.17,20 The triglyme activating group (needed to selectively enhance the rate of stopper exchange as illustrated in Fig. 1b) is readily installed and is a known10,14a–d,21 organocatalyst for aminolysis reactions in relatively nonpolar organic solvents like chloroform. With these building blocks in hand, we synthesized RDP[5]cat@diester (Scheme 1) in 54% yield by threading10,22 the rim-differentiated pillar[5]arene RDP[5]cat (synthesis detailed in the ESI†) onto a hexadecanedioic acid dichloride axle in the presence of excess 3,5-bis(trifluoromethyl)phenol stopper and triethylamine. Next, we subjected RDP[5]cat@diester to aminolysis with 3,5-dimethylbenzylamine at 30 °C. We worked up the reaction early (after 60 hours), to ensure that we could isolate both the major and the minor geometric isomers of the mono-amide products as the NMR standards for the quantitative 1H NMR experiments (Fig. 3). As measured by 1H NMR spectroscopy (see Fig. 3c), the minor geometric isomer disappears almost completely at later time points.
The glyme-activated stopper-exchange reaction with a first amine nucleophile (3,5-dimethylbenzylamine) led to a mixture of three aminolysis products, which included the two geometric isomers of the mono-substituted rotaxanes RDP[5]cat@MAfav and RDP[5]cat@MAdisfav, as well as the disubstituted rotaxane RDP[5]cat@DA in 96% combined yield (calculated based on recovered starting material). The excess amine in the reaction mixture posed a challenge during the workup as attempts to remove the solvent increased the amine concentration, which led to the complete substitution of the remaining active esters. Therefore, we developed a protocol (see ESI† for details) to remove the excess 3,5-dimethylbenzylamine reagent by simple filtration through an acid-chloride functionalized MP carboxylic acid resin before concentration and purification of the reaction mixture.
The structures of the reaction products with the 3,5-dimethylbenzylamine nucleophile (RDP[5]cat@MAfav, RDP[5]cat@MAdisfav, RDP[5]cat@DA) were confirmed with 1H NMR, 13C NMR, and 1H–1H ROESY NMR spectroscopy, as well as with high-resolution mass spectrometry (see the ESI†). Notably, the 1H–1H ROESY NMR spectrum of the major, monosubstituted rotaxane product RDP[5]cat@MAfav (Fig. 2a) shows a cross peak between the Het proton resonance (the –CH3 proton resonance of the ethyl group on the pillar[5]arene macrocycle, observed as a triplet at 1.41 ppm) and the Ho aromatic resonance at 7.64 ppm (which corresponds to the ortho-protons on the remaining active-ester stoppering unit). The presence of this cross-peak seems to indicate that the pillar[5]arene macrocycle possesses an energetically favorable co-conformation, in which the ring binds to the remaining active ester stopper.
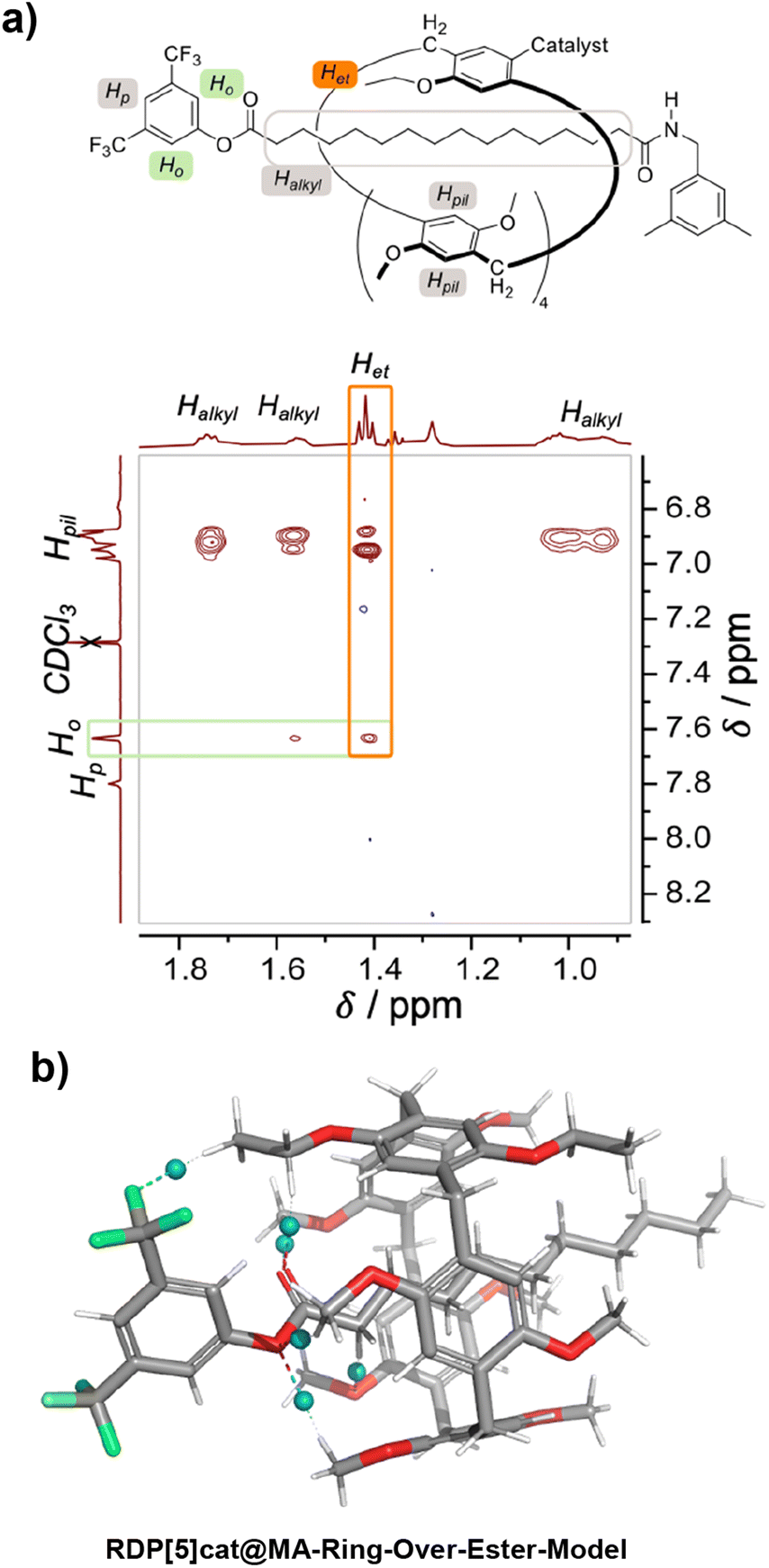 | ||
| Fig. 2 (a) Partial 1H–1H ROESY NMR (500 MHz, DCDl3) spectrum of the major geometric isomer (RDP[5]cat@MAfav) obtained from the aminolysis reaction of RDP[5]cat@diester with 3,5-dimethylbenzylamine. See the ESI† for additional characterization data as well as the full 1H–1H ROESY NMR spectrum. The 1H–1H ROESY NMR spectrum shown in the figure clearly shows that the ethyl group on the pillar[5]arene ring is located proximal to the remaining active ester present in the axle of the favored geometric rotaxane isomer. The key NOE cross-peak between Het and Ho—which leads us to this conclusion—is highlighted in orange. (b) Non-covalent interaction plots23 calculated at the B3LYP-MM/LACVP* level of theory with the NCI method implemented in Jaguar (version 8.8) as detailed in the ESI.† The NCI plots show the presence of attractive [C–H]⋯F interactions between the ethyl groups on the pillararene ring and one of the –CF3 functionalities of the 3,5-bis(trifluoromethyl)phenyl stopper. We hypothesize that these non-covalent interactions are primarily responsible for biasing the equilibrium distribution of the pillararene ring toward the side of the active-ester stopper, which results in the clear NOE cross-peak shown in panel (a). | ||
To investigate the origin of this attractive interaction between the ring and the active ester stopper, we optimized a DFT model (Fig. 2b) of the corresponding complex and calculated the noncovalent interactions from the DFT-optimized electron density with the NCI method.23,24 Based on our DFT results, there are attractive [C–H⋯O] and [C–H⋯F] interactions (illustrated as blue spheres in Fig. 2b), which seem to be playing a key role in stabilizing the co-conformation with the pillar[5]arene macrocycle residing next to the active-ester stopper.
Finally, to confirm our kinetic model for the reaction with the 3,5-dimethylbenzylamine nucleophile, we conducted detailed kinetic studies with quantitative 1H NMR spectroscopy to investigate the selectivity of the reaction over time. For this purpose, RDP[5]cat@diester was reacted with an excess of 3,5-dimethylbenzylamine in CDCl3 at 30 °C in an NMR tube. Our reaction system is governed by four rate constants, k1, k1′, k2, and k2′ as defined in Fig. 3a. Reaction progression was monitored by 1H NMR in CDCl3 using 1,2,4,5-tetrabromobenzene (TBB) as the internal standard. The unique amide protons for all three rotaxane products were readily apparent (Fig. 3b), which allowed us to integrate them against the internal TBB standard to yield absolute concentrations. The resulting concentration–time plots (Fig. 3c) were fit to the kinetic model shown in Fig. 3a with Dynafit,25 providing the four rate constants k1, k2, k1′, and k2′. The kinetic model showed that the rate constant corresponding to the formation of the favored rotaxane RDP[5]cat@MAfav-1 (k1 = 0.55 ± 0.03) is about an order of magnitude larger than the corresponding rate constant for formation of the disfavored rotaxane RDP[5]cat@MAdisfav-1 (k2 = 0.08 ± 0.005). Moreover, both k1 and k1′ are also about an order of magnitude larger than either k2 or k2′, demonstrating the increased reactivity at the end of the rotaxane nearest to the catalyst.
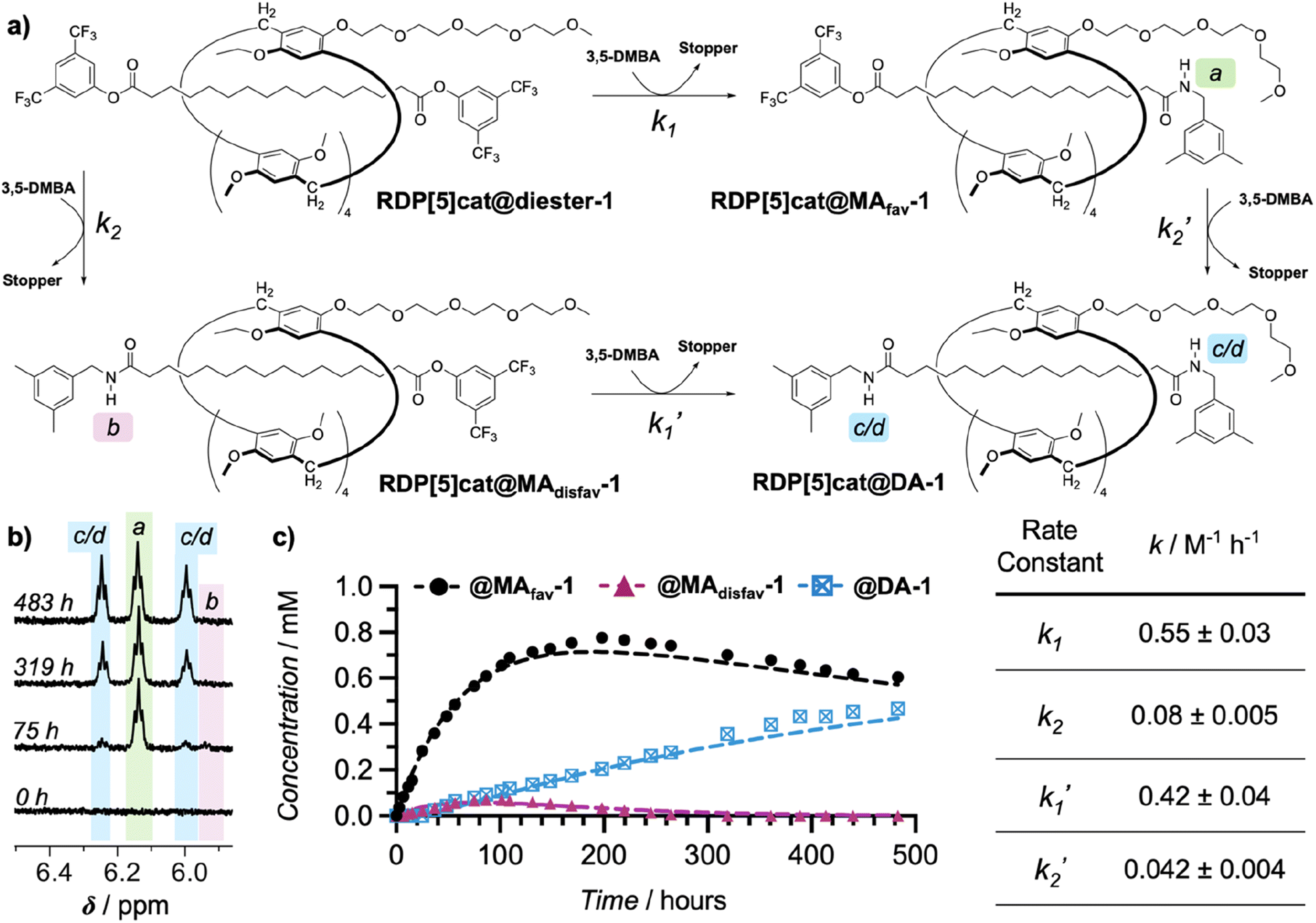 | ||
| Fig. 3 (a) Complete kinetic pathway for through-space controlled stopper exchange with 3,5-dimethylbenzylamine as the nucleophile. 3,5-DMBA = 3,5-dimethylbenzylamine; Stopper = 3,5-bis(trifluoromethyl)phenol. Rate constants k1 and k1′ denote substitution at the activated ester (proximal to the catalytic the side-chain), while k2 and k2′ denote substitution at the ester distal to the catalyst. (b) Four representative 1H NMR spectra (500 MHz, CDCl3, 300 K) recorded at different time points over the course of the kinetics experiment. The three sets of amide protons (1 NH each for both RDP[5]cat@MAfav and RDP[5]cat@MAdisfav, 2 NH for RDP[5]cat@DA) are highlighted. A complete stack of the entire kinetics spectrum is shown in Fig. S1 in the ESI.† (c) Concentrations of all three reaction products measured by quantitative 1H NMR spectroscopy with the TBB internal standard over the course of the reaction. The reaction was run at 30 °C as detailed in the ESI.† Kinetics fits are shown as dashed lines. The kinetic fits were obtained using the Dynafit software package as detailed in the ESI.† Derived rate constants with error bars (standard errors obtained from the Dynafit kinetic fits) are shown in the table on the right. | ||
Our Dynafit model, which was fit to the quantitative 1H NMR data shown in Fig. 3c, provides concentrations of 0.70 mM for RDP[5]cat@MAfav-1 and 0.02 mM for RDP[5]cat@MAdisfav-1 at ∼250 hours, which leads to a selectivity of approximately 31:1 d.r. at this reaction time point. After 300 hours, the selectivity for the formation of the major geometric isomer rises even further to about ∼45:1. This finding provides proof of principle for the enhanced selectivity enabled by our kinetically coupled reaction system.
With the kinetic model established for 3,5-dimethylbenzylamine as the nucleophile, we generalized (Fig. 4) our selective rotaxane synthesis to other amine nucleophiles, including 1-naphthalenemethanamine and 9-anthracenemethanamine.
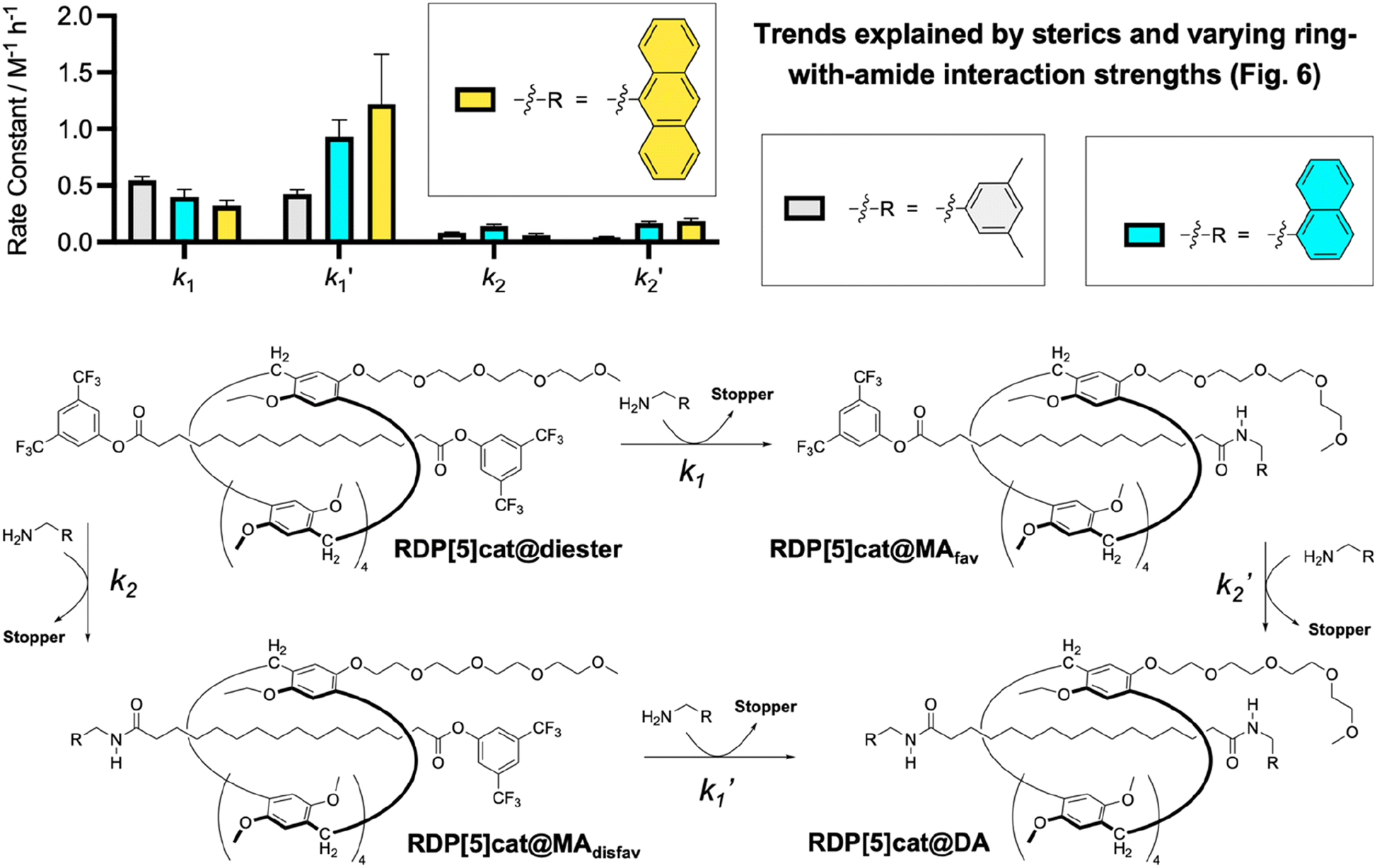 | ||
| Fig. 4 Comparison of aminolysis rate constants for RDP[5]cat@diester with different amine nucleophiles. All reactions were run at 30 °C as detailed in the ESI.† See Fig. S1–S5† for the kinetic fits and stacks of the time-dependent 1H NMR spectra, which were used to determine all the rate constants. The kinetic fits were obtained using the Dynafit software package as detailed in the ESI.† Numerical values for the derived rate constants with error bars (standard errors obtained from the Dynafit kinetic fits) are listed in Fig. 3c, S3b, and S5b.† | ||
Both systems performed qualitatively similar to the reaction system with the 3,5-dimethylbenzylamine, which confirms the generality of our kinetically controlled rotaxane geometric isomer synthesis. However, we also observed (Fig. 4 and 6) clear trends in the rate constants, based on (i) the sterics of the nucleophiles/amide stoppers and (ii) the sterics of the secondary (non-activating) face of the ring, which (when positioned over an active ester) seems to slow down the aminolysis reactions.
(i) Steric effects of the nucleophile/amide stopper on the aminolysis rates: First, the observed trend in k1 rate constants (Fig. 4) clearly shows that the k1 rate constants decrease with increasing steric bulk of the nucleophile, as one would expect for a classical acyl substitution mechanism.
At the same time, the rate constants k1′ increased significantly from R = 3,5-dimethylbenzyl, to R = 1-naphtyl, and R = 9-anthracenyl, which is contrary to the trend observed for k1. We hypothesize that this inverted trend is the result of reduced supramolecular interactions between the pillararene ring and the amide stoppers in the monofunctionalized rotaxane products RDP[5]cat@MAdisfav-2 (the naphthyl case) and RDP[5]cat@MAdisfav-3 (the anthracenyl case). This hypothesis was confirmed by DFT-calculated binding energies (Fig. 6) between the ring and the amide stoppers.
Based on the DFT results, we find that 3,5-dimethylbenzylamide stopper in RDP[5]cat@MAdisfav-1 binds the strongest with the pillararene ring, while the 9-anthracenemethanamide and the 1-naphthalenemethanamide stoppers showed a reduced affinity with the ring.
Once again, this trend is caused by the increasing steric bulk of the initial amine nucleophiles, which ultimately leads to bulkier amide stoppers in the anthracenyl/naphthyl cases for the monofunctionalized rotaxane products RDP[5]cat@MAdisfav. As shown by our DFT calculations (Fig. 6), the increased steric bulk of the 9-anthracenemethanamide and the 1-naphthalenemethanamide stoppers even forces one of the methoxyl groups out of conjugation with the aromatic units on the pillararene rings. As a result, the supramolecular interaction strength between the rings and the amide stoppers is significantly reduced in the anthracenyl/naphthyl cases, which favors the co-conformations with the glyme activating groups residing over the remaining active esters. Consequently, the k1′ rate constants with RDP[5]cat@MAdisfav-2 and RDP[5]cat@MAdisfav-3 are faster than with RDP[5]cat@MAdisfav-1.
Since the fastest k1′ results with 9-anthracenemethanamine as the nucleophile, the minor geometric rotaxane isomer (RDP[5]cat@MAdisfav) reacts away even faster in the anthracenyl case, which further increases the selectivity for the formation of the major geometric isomer (as shown in Fig. 5) with 9-anthracenemethanamine as the nucleophile. Overall, near exponential growth of the reaction selectivity over time is observed (Fig. 5) with all three amine nucleophiles, since—as more of the desired major product forms over time—the undesired product also keeps reacting away faster than the desired product, which leads to a continuously increasing selectivity of the reaction for the major geometric isomer.
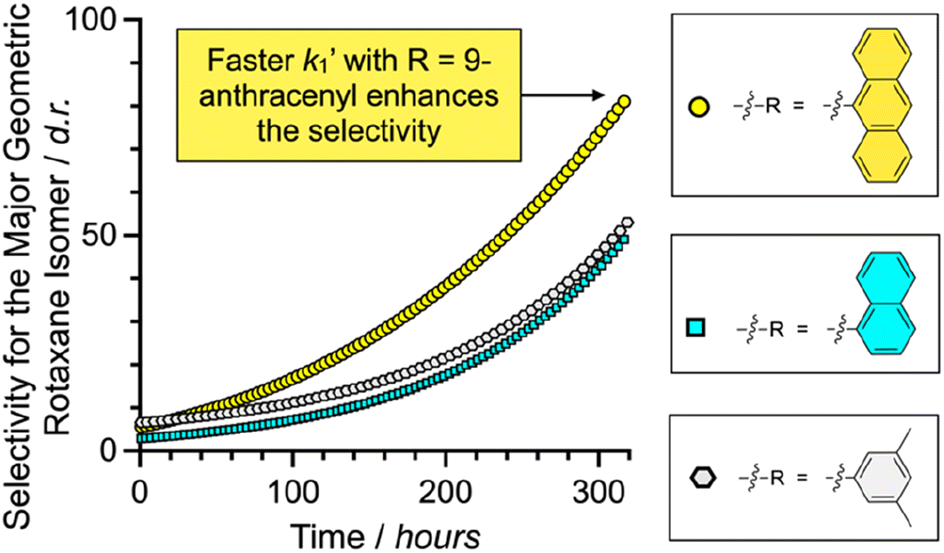 | ||
| Fig. 5 Plots of the diastereoselectivity (d.r. = [RDP[5]cat@MAfav]/[RDP[5]cat@MAdisfav]) for the major geometric rotaxane isomers formed over time for the aminolysis reactions shown in Fig. 4. The concentrations of the products were obtained from the kinetic fits to the quantitative 1H NMR data shown in Fig. 3, S3b, and S5b.† | ||
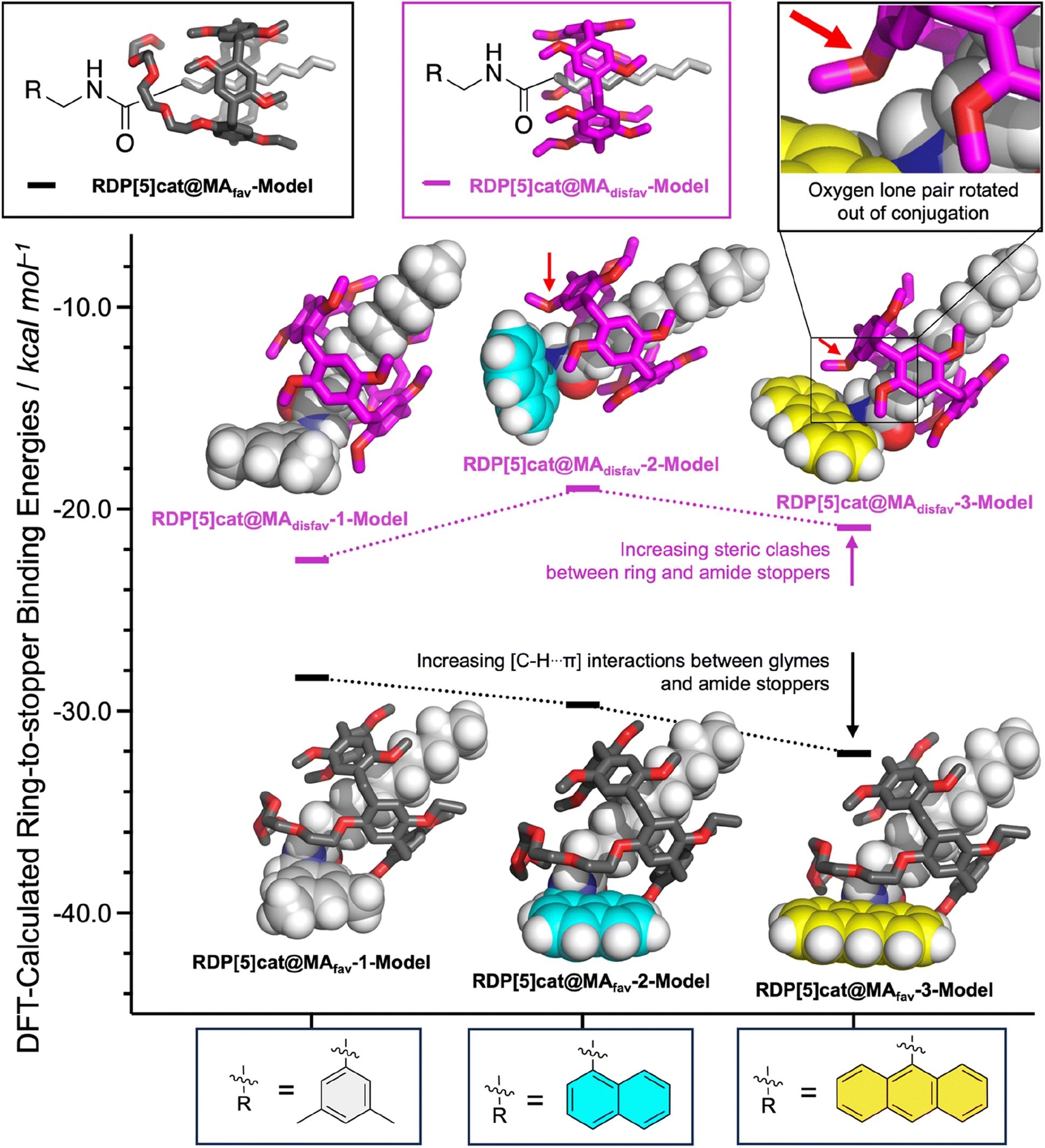 | ||
| Fig. 6 DFT-calculated binding energies (B3LYP-MM/aug-cc-pVDZ//B3LYP-MM/LACVP* level of theory) between the different faces of the RDP[5]cat ring and the varying amide stoppers for both geometric isomers. The model systems used to calculate the binding energies are shown in insets at the top left of the figure. In the model systems for the disfavored rotaxane products (RDP[5]cat@MAdisfav-1-Model, RDP[5]cat@MAdisfav-2-Model, and RDP[5]cat@MAdisfav-3-Model), the tetraglyme chains do not directly interact with the varying amide stoppers. Therefore, for the models of the disfavored rotaxane products, the tetraglyme chains on the ring were replaced with ethyl substituents to simplify the conformational space and enable a more accurate search of the conformational space at the DFT level with these smaller model systems. | ||
(ii) Steric effects of the ring on the aminolysis rates: While the face of the ring with the tetraglyme chains clearly speeds up the aminolysis reactions as discussed above, the aminolysis reactions slow down when the secondary face of the ring (i.e., the face without the glyme functions) is sitting over an active ester. Based on our computational model shown in Fig. 2b, we explain this slow-down effect by the simple steric bulk of the macrocycle, which partially blocks attack of the nucleophile when the secondary face of the ring is positioned over the active ester. Related inhibition effects of reactivity by the mechanical bond have been observed previously in the literature.26
This inhibition effect is also clearly visible when comparing the k2 and k2′ rate constants (Fig. 3c) for the aminolysis reaction with 3,5-dimethylbenzylamine. In this case, k2′ is significantly slower than k2, since in the monoamide RDP[5]cat@MAfav-1 the ring spends a significant portion of time over the active ester (based on the NOESY NMR shown in Fig. 2a), thereby partially blocking access of the nucleophile to the active ester in this monoamide. In contrast, the ring is expected to be much more evenly distributed between the two active ester sites in the starting material RDP[5]cat@diester, which ultimately leads to k2 being significantly faster than k2′ with the 3,5-dimethylbenzylamine nucleophile.
At the same time, the k2′ rate constants also increased notably (Fig. 4) in the anthracenyl and naphthyl cases, compared to the case with R = 3,5-dimethylbenzyl. Again, we hypothesize that this effect is caused by the secondary face of the ring inhibiting nucleophilic attack, and by changing the balance of supramolecular interactions between the ring and the varying amide stoppers. In this case, the DFT calculations show that the tetraglyme groups interact27 more strongly with the amide stoppers when R = naphthyl/anthracenyl than with R = 3,5-dimethylbenzyl. Therefore, the stronger supramolecular interactions between the tetraglyme groups and the aromatic stoppers in the anthracenyl/naphthyl case favor the co-conformation with the ring residing over the side of the amide stopper in the case of RDP[5]cat@MAfav-2 and RDP[5]cat@MAfav-3, which frees up the active ester on the other end of the rotaxane for faster nucleophilic attack and leads to overall faster k2′ rate constants.
Conclusions
We developed a kinetically controlled strategy to selectively access specific geometric isomers of complex interlocked molecules through a coupled reaction system involving selective stopper exchange reactions. Our reaction system was able to achieve high selectivity by enhancing the intrinsic selectivity of the selective stopper exchange reactions based on coupled reactions of inverse selectivity. While the use of a glyme catalyst/activating group as a means of promoting stopper exchange in rotaxanes was previously reported by our group,10 this work expands the synthetic toolbox available to selectively access rotaxane geometric isomers. We are currently applying our synthetic strategy for the synthesis of new living polymerization catalysts and are also expanding our methodology to other macrocycles and catalysts/activating groups to access complex interlocked molecules in a more effective manner.Data availability
The datasets generated during this study are available from the authors upon reasonable request.Author contributions
S. T. S. guided the project, performed the DFT calculations together with D. R. M. and K. X., discussed the experimental results, and wrote the paper together with D. R. M. and K. X. D. R. M., K. X., N. B., H. L., and S. B. performed the synthesis and analysed the data. All the authors discussed the results and revised the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank M. Ivancic for NMR support and B. O'Rourke for high-resolution mass spectrometry. Synthetic material for this research was supported by the NSF (CAREER Award CHE-1848444/2317652 awarded to STS), while the kinetic analysis was supported by the U.S. Army Research Office (Grant 71015-CH-YIP awarded to STS). Part of the computational resources were supported by the NVIDIA GPU Grant Seed Program (awarded to STS) and by the NIH (MIRA award 1R35GM147579-01). Partial support was also provided by NSF's CTMC Program via an NSF CAREER Award (CHE-1945394/2410514) awarded to JL. H. L. was supported by a summer undergraduate research fellowship from Middlebury College.Notes and references
- (a) V. Blanco, A. Carlone, K. D. Hänni, D. A. Leigh and B. Lewandowski, A Rotaxane-Based Switchable Organocatalyst, Angew. Chem., Int. Ed., 2012, 51, 5166–5169 CrossRef CAS PubMed; (b) V. Blanco, D. A. Leigh, V. Marcos, J. A. Morales-Serna and A. L. Nussbaumer, A Switchable [2]Rotaxane Asymmetric Organocatalyst That Utilizes an Acyclic Chiral Secondary Amine, J. Am. Chem. Soc., 2014, 136, 4905–4908 CrossRef CAS PubMed; (c) Y. Cakmak, S. Erbas-Cakmak and D. A. Leigh, Asymmetric Catalysis with a Mechanically Point-Chiral Rotaxane, J. Am. Chem. Soc., 2016, 138, 1749–1751 CrossRef CAS PubMed; (d) K. Eichstaedt, J. Jaramillo-Garcia, D. A. Leigh, V. Marcos, S. Pisano and T. A. Singleton, Switching between Anion-Binding Catalysis and Aminocatalysis with a Rotaxane Dual-Function Catalyst, J. Am. Chem. Soc., 2017, 139, 9376–9381 CrossRef CAS PubMed; (e) A. W. Heard and S. M. Goldup, Synthesis of a Mechanically Planar Chiral Rotaxane Ligand for Enantioselective Catalysis, Chem, 2020, 6, 994–1006 CrossRef CAS PubMed; (f) C. Kwamen and J. Niemeyer, Functional Rotaxanes in Catalysis, Chem.–Eur. J., 2021, 27, 175–186 CrossRef CAS PubMed; (g) J. Y. C. Lim, N. Yuntawattana, P. D. Beer and C. K. Williams, Isoselective Lactide Ring Opening Polymerisation using [2]Rotaxane Catalysts, Angew. Chem., Int. Ed., 2019, 58, 6007–6011 CrossRef CAS PubMed; (h) A. Martinez-Cuezva, A. Saura-Sanmartin, M. Alajarin and J. Berna, Mechanically Interlocked Catalysts for Asymmetric Synthesis, ACS Catal., 2020, 10, 7719–7733 CrossRef CAS; (i) A. Martinez-Cuezva, A. Saura-Sanmartin, T. Nicolas-Garcia, C. Navarro, R.-A. Orenes, M. Alajarin and J. Berna, Photoswitchable interlocked thiodiglycolamide as a cocatalyst of a chalcogeno-Baylis–Hillman reaction, Chem. Sci., 2017, 8, 3775–3780 RSC.
- (a) S. Borsley, D. A. Leigh and B. M. W. Roberts, A Doubly Kinetically-Gated Information Ratchet Autonomously Driven by Carbodiimide Hydration, J. Am. Chem. Soc., 2021, 143, 4414–4420 CrossRef CAS PubMed; (b) S. F. M. v. Dongen, S. Cantekin, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, Functional interlocked systems, Chem. Soc. Rev., 2013, 43, 99–122 RSC; (c) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Artificial Molecular Machines, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed; (d) J. Groppi, L. Casimiro, M. Canton, S. Corra, M. Jafari-Nasab, G. Tabacchi, L. Cavallo, M. Baroncini, S. Silvi, E. Fois and A. Credi, Precision Molecular Threading/Dethreading, Angew. Chem., Int. Ed., 2020, 132, 14935–14944 CrossRef; (e) E. R. Kay and D. A. Leigh, Rise of the Molecular Machines, Angew. Chem., Int. Ed., 2015, 54, 10080–10088 CrossRef CAS PubMed; (f) J. E. M. Lewis, M. Galli and S. M. Goldup, Properties and emerging applications of mechanically interlocked ligands, Chem. Commun., 2016, 53, 298–312 RSC; (g) S. Mena-Hernando and E. M. Pérez, Mechanically interlocked materials. Rotaxanes and catenanes beyond the small molecule, Chem. Soc. Rev., 2019, 48, 5016–5032 RSC; (h) E. A. Neal and S. M. Goldup, Chemical consequences of mechanical bonding in catenanes and rotaxanes: isomerism, modification, catalysis and molecular machines for synthesis, Chem. Commun., 2014, 50, 5128–5142 RSC; (i) D. Sluysmans, P. Lussis, C.-A. Fustin, A. Bertocco, D. A. Leigh and A.-S. Duwez, Real-Time Fluctuations in Single-Molecule Rotaxane Experiments Reveal an Intermediate Weak Binding State during Shuttling, J. Am. Chem. Soc., 2021, 143, 2348–2352 CrossRef CAS PubMed; (j) C. A. Stanier, S. J. Alderman, T. D. W. Claridge and H. L. Anderson, Unidirectional Photoinduced Shuttling in a Rotaxane with a Symmetric Stilbene Dumbbell, Angew. Chem., Int. Ed., 2002, 41, 1769–1772 CrossRef; (k) T. Takata, Switchable Polymer Materials Controlled by Rotaxane Macromolecular Switches, ACS Cent. Sci., 2020, 6, 129–143 CrossRef CAS PubMed; (l) P. Wu, B. Dharmadhikari, P. Patra and X. Xiong, Rotaxane nanomachines in future molecular electronics, Nanoscale Adv., 2022, 4, 3418–3461 RSC; (m) B. Yao, H. Sun, L. Yang, S. Wang and X. Liu, Recent Progress in Light-Driven Molecular Shuttles, Front. Chem., 2022, 9, 832735 CrossRef PubMed; (n) H.-Y. Zhou, Q.-S. Zong, Y. Han and C.-F. Chen, Recent advances in higher order rotaxane architectures, Chem. Commun., 2020, 56, 9916–9936 RSC.
- (a) M. Baroncini, S. Silvi and A. Credi, Photo- and Redox-Driven Artificial Molecular Motors, Chem. Rev., 2020, 120, 200–268 CrossRef CAS PubMed; (b) Y. Feng, M. Ovalle, J. S. W. Seale, C. K. Lee, D. J. Kim, R. D. Astumian and J. F. Stoddart, Molecular Pumps and Motors, J. Am. Chem. Soc., 2021, 143, 5569–5591 CrossRef CAS PubMed.
- (a) R. J. Bordoli and S. M. Goldup, An Efficient Approach to Mechanically Planar Chiral Rotaxanes, J. Am. Chem. Soc., 2014, 136, 4817–4820 CrossRef CAS PubMed; (b) C. Casati, P. Franchi, R. Pievo, E. Mezzina and M. Lucarini, Unraveling Unidirectional Threading of α-Cyclodextrin in a [2]Rotaxane through Spin Labeling Approach, J. Am. Chem. Soc., 2012, 134, 19108–19117 CrossRef CAS PubMed; (c) F. d'Orchymont and J. P. Holland, Supramolecular Rotaxane-Based Multi-Modal Probes for Cancer Biomarker Imaging, Angew. Chem., Int. Ed., 2022, 61, e202204072 CrossRef PubMed; (d) E. M. G. Jamieson, F. Modicom and S. M. Goldup, Chirality in rotaxanes and catenanes, Chem. Soc. Rev., 2018, 47, 5266–5311 RSC; (e) M. A. Jinks, A. de Juan, M. Denis, C. J. Fletcher, M. Galli, E. M. G. Jamieson, F. Modicom, Z. Zhang and S. M. Goldup, Stereoselective Synthesis of Mechanically Planar Chiral Rotaxanes, Angew. Chem., Int. Ed., 2018, 57, 14806–14810 CrossRef CAS PubMed; (f) Y. Makita, N. Kihara and T. Takata, Synthesis and kinetic resolution of directional isomers of [2]rotaxanes bearing a lariat crown ether wheel, Supramol. Chem., 2021, 33, 1–7 CrossRef CAS; (g) J. R. J. Maynard and S. M. Goldup, Strategies for the Synthesis of Enantiopure Mechanically Chiral Molecules, Chem, 2020, 6, 1914–1932 CrossRef CAS; (h) K. Nakazono and T. Takata, Mechanical Chirality of Rotaxanes: Synthesis and Function, Symmetry, 2020, 12, 144 CrossRef CAS; (i) N. Pairault and J. Niemeyer, Chiral Mechanically Interlocked Molecules – Applications of Rotaxanes, Catenanes and Molecular Knots in Stereoselective Chemosensing and Catalysis, Synlett, 2018, 29, 689–698 CrossRef CAS.
- (a) M. Bazzoni, L. Andreoni, S. Silvi, A. Credi, G. Cera, A. Secchi and A. Arduini, Selective access to constitutionally identical, orientationally isomeric calix[6]arene-based [3]rotaxanes by an active template approach, Chem. Sci., 2021, 12, 6419–6428 RSC; (b) P. La Manna, C. Talotta, C. Gaeta, A. Soriente, M. De Rosa and P. Neri, Threading of an Inherently Directional Calixarene Wheel with Oriented Ammonium Axles, J. Org. Chem., 2017, 82, 8973–8983 CrossRef CAS PubMed.
- (a) G. De Bo, M. A. Y. Gall, M. O. Kitching, S. Kuschel, D. A. Leigh, D. J. Tetlow and J. W. Ward, Sequence-Specific β-Peptide Synthesis by a Rotaxane-Based Molecular Machine, J. Am. Chem. Soc., 2017, 139, 10875–10879 CrossRef CAS PubMed; (b) J. Echavarren, M. A. Y. Gall, A. Haertsch, D. A. Leigh, J. T. J. Spence, D. J. Tetlow and C. Tian, Sequence-Selective Decapeptide Synthesis by the Parallel Operation of Two Artificial Molecular Machines, J. Am. Chem. Soc., 2021, 143, 5158–5165 CrossRef CAS PubMed.
- J. Berná, D. A. Leigh, M. Lubomska, S. M. Mendoza, E. M. Pérez, P. Rudolf, G. Teobaldi and F. Zerbetto, Macroscopic transport by synthetic molecular machines, Nat. Mater., 2005, 4, 704–710 CrossRef PubMed.
- (a) T. Oshikiri, Y. Takashima, H. Yamaguchi and A. Harada, Kinetic Control of Threading of Cyclodextrins onto Axle Molecules, J. Am. Chem. Soc., 2005, 127, 12186–12187 CrossRef CAS PubMed; (b) M. Quiroga, M. Parajó, P. Rodríguez-Dafonte and L. García-Río, Kinetic Study of [2]Pseudorotaxane Formation with an Asymmetrical Thread, Langmuir, 2016, 32, 6367–6375 CrossRef CAS PubMed; (c) Q.-C. Wang, X. Ma, D.-H. Qu and H. Tian, Unidirectional Threading Synthesis of Isomer-Free [2]Rotaxanes, Chem.–Eur. J., 2006, 12, 1088–1096 CrossRef CAS PubMed; (d) M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Development of Pseudorotaxanes and Rotaxanes: From Synthesis to Stimuli-Responsive Motions to Applications, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS PubMed; (e) T. Oshikiri, Y. Takashima, H. Yamaguchi and A. Harada, Face-Selective [2]- and [3]Rotaxanes: Kinetic Control of the Threading Direction of Cyclodextrins, Chem.–Eur. J., 2007, 13, 7091–7098 CrossRef CAS PubMed; (f) P. Waelès, M. Gauthier and F. Coutrot, Study of [2]- and [3]Rotaxanes Obtained by Post-Synthetic Aminolysis of a Kinetically Stable Carbonate-Containing Pseudorotaxane, Eur. J. Org. Chem., 2022, 2022, e202101385 CrossRef; (g) H.-X. Wang, Z. Meng, J.-F. Xiang, Y.-X. Xia, Y. Sun, S.-Z. Hu, H. Chen, J. Yao and C.-F. Chen, Guest-dependent directional complexation based on triptycene derived oxacalixarene: formation of oriented rotaxanes, Chem. Sci., 2016, 7, 469–474 RSC.
- (a) M. D. Struble, M. G. Holl, G. Coombs, M. A. Siegler and T. Lectka, Synthesis of a Tight Intramolecular OH···Olefin Interaction, Probed by IR, 1H NMR, and Quantum Chemistry, J. Org. Chem., 2015, 80, 4803–4807 CrossRef CAS PubMed; (b) M. G. Holl, M. D. Struble, P. Singal, M. A. Siegler and T. Lectka, Positioning a Carbon-Fluorine Bond over the π Cloud of an Aromatic Ring: A Different Type of Arene Activation, Angew. Chem., Int. Ed., 2016, 55, 8266–8269 CrossRef CAS PubMed; (c) D. D. Bume, C. R. Pitts, F. Ghorbani, S. A. Harry, J. N. Capilato, M. A. Siegler and T. Lectka, Ketones as directing groups in photocatalytic sp3 C-H fluorination, Chem. Sci., 2017, 8, 6918–6923 RSC; (d) L. Guan, M. G. Holl, C. R. Pitts, M. D. Struble, M. A. Siegler and T. Lectka, Through-Space Activation Can Override Substituent Effects in Electrophilic Aromatic Substitution, J. Am. Chem. Soc., 2017, 139, 14913–14916 CrossRef CAS PubMed; (e) K. E. Murphy, J. L. Bocanegra, X. Liu, H. K. Chau, P. C. Lee, J. Li and S. T. Schneebeli, Precise through-space control of an abiotic electrophilic aromatic substitution reaction, Nat. Commun., 2017, 8, 14840 CrossRef PubMed; (f) C. R. Pitts, M. G. Holl and T. Lectka, Spectroscopic Characterization of a [C-F-C]+ Fluoronium Ion in Solution, Angew. Chem., Int. Ed., 2018, 57, 1924–1927 CrossRef CAS PubMed; (g) J. P. Campbell, S. C. Rajappan, T. J. Jaynes, M. Sharafi, Y. T. Ma, J. Li and S. T. Schneebeli, Enantioselective Electrophilic Aromatic Nitration: A Chiral Auxiliary Approach, Angew. Chem., Int. Ed., 2019, 58, 1035–1040 CrossRef CAS PubMed; (h) M. Kazim, H. Foy, M. A. Siegler, T. Dudding and T. Lectka, Discovery and Mechanistic Study of a Totally Organic C(aryl)-C(alkyl)Oxygen Insertion Reaction, J. Org. Chem., 2019, 84, 14349–14353 CrossRef CAS PubMed; (i) M. Kazim, M. A. Siegler and T. Lectka, A Case of Serendipity: Synthesis, Characterization, and Unique Chemistry of a Stable, Ring-Unsubstituted Aliphatic p-Quinone Methide, Org. Lett., 2019, 21, 2326–2329 CrossRef CAS PubMed; (j) M. G. Holl, C. R. Pitts and T. Lectka, Quest for a Symmetric [C-F-C]+ Fluoronium Ion in Solution: A Winding Path to Ultimate Success, Acc. Chem. Res., 2020, 53, 265–275 CrossRef CAS PubMed; (k) M. Kazim, L. Guan, A. Chopra, R. Sun, M. A. Siegler and T. Lectka, Switching a HO···π Interaction to a Nonconventional OH···π Hydrogen Bond: A Completed Crystallographic Puzzle, J. Org. Chem., 2020, 85, 9801–9807 CrossRef CAS PubMed; (l) S. T. Schneebeli, M. Sharafi, J. P. Campbell, K. E. Murphy and R. Osadchey Brown, Chiral Auxiliaries for Stereoselective Electrophilic Aromatic Substitutions, Synlett, 2020, 32, 229–234 CrossRef; (m) K. F. Hoffmann, A. Wiesner, C. Mueller, S. Steinhauer, H. Beckers, M. Kazim, C. R. Pitts, T. Lectka and S. Riedel, Structural proof of a [C-F-C]+ fluoronium cation, Nat. Commun., 2021, 12, 5275 CrossRef CAS PubMed; (n) S. A. Harry, M. R. Xiang, E. Holt, A. Zhu, F. Ghorbani, D. Patel and T. Lectka, Hydroxy-directed fluorination of remote unactivated C(sp3)-H bonds: a new age of diastereoselective radical fluorination, Chem. Sci., 2022, 13, 7007–7013 RSC; (o) M. Kazim, Z. Feng, S. Vemulapalli, M. A. Siegler, A. Chopra, P. Minh Nguyen, M. Gargiulo Holl, L. Guan, T. Dudding, D. J. Tantillo and T. Lectka, Through-Space, Lone-Pair Promoted Aromatic Substitution: A Relay Mechanism Can Beat Out Direct Activation, Chem.–Eur. J., 2023, e202301550, DOI:10.1002/chem.202301550; (p) M. Kazim, M. Wang, S. Vemulapalli, P. M. Nguyen, Y. Wang, M. A. Siegler, T. Dudding and T. Lectka, How Do Face-to-Face Stacked Aromatic Rings Activate Each Other to Electrophilic Aromatic Substitution?, Org. Lett., 2023, 25, 4318–4322 CrossRef CAS PubMed; (q) H. J. Davis and R. J. Phipps, Harnessing non-covalent interactions to exert control over regioselectivity and site-selectivity in catalytic reactions, Chem. Sci., 2017, 8, 864–877 RSC; (r) W. A. Golding, R. Pearce-Higgins and R. J. Phipps, Site-Selective Cross-Coupling of Remote Chlorides Enabled by Electrostatically Directed Palladium Catalysis, J. Am. Chem. Soc., 2018, 140, 13570–13574 CrossRef CAS PubMed; (s) W. A. Golding and R. J. Phipps, Electrostatically-directed Pd-catalysis in combination with C-H activation: site-selective coupling of remote chlorides with fluoroarenes and fluoroheteroarenes, Chem. Sci., 2020, 11, 3022–3027 RSC; (t) Y.-H. Li, Y. Ouyang, N. Chekshin and J.-Q. Yu, PdII-Catalyzed γ-C(sp3)-H (Hetero)arylation of Ketones Enabled by Transient Directing Groups, ACS Catal., 2022, 12, 10581–10586 CrossRef CAS PubMed; (u) Y.-Q. Chen, S. Singh, Y. Wu, Z. Wang, W. Hao, P. Verma, J. X. Qiao, R. B. Sunoj and J.-Q. Yu, Pd-Catalyzed γ-C(sp3)-H Fluorination of Free Amines, J. Am. Chem. Soc., 2020, 142, 9966–9974 CrossRef CAS PubMed; (v) K. Hong, H. Park and J.-Q. Yu, Methylene C(sp3)-H Arylation of Aliphatic Ketones Using a Transient Directing Group, ACS Catal., 2017, 7, 6938–6941 CrossRef CAS PubMed; (w) Y. Wu, Y.-Q. Chen, T. Liu, M. D. Eastgate and J.-Q. Yu, Pd-Catalyzed γ-C(sp3)-H Arylation of Free Amines Using a Transient Directing Group, J. Am. Chem. Soc., 2016, 138, 14554–14557 CrossRef CAS PubMed; (x) G. Xia, J. Weng, L. Liu, P. Verma, Z. Li and J.-Q. Yu, Reversing conventional site-selectivity in C(sp3)-H bond activation, Nat. Chem., 2019, 11, 571–577 CrossRef CAS PubMed; (y) L.-J. Xiao, K. Hong, F. Luo, L. Hu, W. R. Ewing, K.-S. Yeung and J.-Q. Yu, PdII-Catalyzed Enantioselective C(sp3)-H Arylation of Cyclobutyl Ketones Using a Chiral Transient Directing Group, Angew. Chem., Int. Ed., 2020, 59, 9594–9600 CrossRef CAS PubMed; (z) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Functionalization of C(sp3)-H bonds using a transient directing group, Science, 2016, 351, 252–256 CrossRef CAS PubMed; (a a) R.-Y. Zhu, L.-Y. Liu and J.-Q. Yu, Highly Versatile β-C(sp3)-H Iodination of Ketones Using a Practical Auxiliary, J. Am. Chem. Soc., 2017, 139, 12394–12397 CrossRef CAS PubMed.
- S. C. Rajappan, D. R. McCarthy, J. P. Campbell, J. B. Ferrell, M. Sharafi, O. Ambrozaite, J. Li and S. T. Schneebeli, Selective Monofunctionalization Enabled by Reaction-History-Dependent Communication in Catalytic Rotaxanes, Angew. Chem., Int. Ed., 2020, 59, 16668–16674 CrossRef CAS PubMed.
- (a) A. Martinez-Cuezva, D. Bautista, M. Alajarin and J. Berna, Enantioselective Formation of 2-Azetidinones by Ring-Assisted Cyclization of Interlocked N-(alpha-Methyl)benzyl Fumaramides, Angew. Chem., Int. Ed., 2018, 57, 6563–6567 CrossRef CAS PubMed; (b) M. Calles, J. Puigcerver, D. A. Alonso, M. Alajarin, A. Martinez-Cuezva and J. Berna, Enhancing the selectivity of prolinamide organocatalysts using the mechanical bond in [2]rotaxanes, Chem. Sci., 2020, 11, 3629–3635 RSC; (c) N. Pairault, H. Zhu, D. Jansen, A. Huber, C. G. Daniliuc, S. Grimme and J. Niemeyer, Heterobifunctional Rotaxanes for Asymmetric Catalysis, Angew. Chem., Int. Ed., 2020, 59, 5102–5107 CrossRef CAS PubMed; (d) C. Lopez-Leonardo, A. Saura-Sanmartin, M. Marin-Luna, M. Alajarin, A. Martinez-Cuezva and J. Berna, Ring-to-Thread Chirality Transfer in [2]Rotaxanes for the Synthesis of Enantioenriched Lactams, Angew. Chem., Int. Ed., 2022, 61, e202209904 CrossRef CAS PubMed.
- S. Amano, S. D. P. Fielden and D. A. Leigh, A catalysis-driven artificial molecular pump, Nature, 2021, 594, 529–534 CrossRef CAS PubMed.
- E. Liu, S. Cherraben, L. Boulo, C. Troufflard, B. Hasenknopf, G. Vives and M. Sollogoub, A molecular information ratchet using a cone-shaped macrocycle, Chem, 2023, 9, 1147–1163 CAS.
- (a) N. Basilio, L. García-Río, J. R. Leis, J. C. Mejuto and M. Pérez-Lorenzo, Novel catalytic effects in ester aminolysis in chlorobenzene, Chem. Commun., 2005, 3817–3819, 10.1039/B504629G; (b) N. Basilio, L. García-Río, J. C. Mejuto and M. Pérez-Lorenzo, A New Reaction Pathway in the Ester Aminolysis Catalyzed by Glymes and Crown Ethers, J. Org. Chem., 2006, 71, 4280–4285 CrossRef CAS PubMed; (c) R. D. Gandour, D. A. Walker, A. Nayak and G. R. Newkome, Catalysis of ester aminolysis by macrocyclic ionophores, J. Am. Chem. Soc., 1978, 100, 3608–3609 CrossRef CAS; (d) J. C. Hogan and R. D. Gandour, The remarkable catalytic power of glymes in ester aminolysis carried out in nonpolar media, J. Am. Chem. Soc., 1980, 102, 2865–2866 CrossRef CAS; (e) W. P. Jencks and J. Carriuolo, General Base Catalysis of the Aminolysis of Phenyl Acetate1, J. Am. Chem. Soc., 1960, 82, 675–681 CrossRef CAS.
- (a) R. Hertzberg, G. Monreal Santiago and C. Moberg, Synthesis of the β3-Adrenergic Receptor Agonist Solabegron and Analogous N-(2-Ethylamino)-β-amino Alcohols from O-Acylated Cyanohydrins - Expanding the Scope of Minor Enantiomer Recycling, J. Org. Chem., 2015, 80, 2937–2941 CrossRef CAS PubMed; (b) C. Moberg, Minor enantiomer recycling - a strategy to improve enantioselectivity, Pure Appl. Chem., 2016, 88, 309–316 CrossRef CAS; (c) Y.-Q. Wen, R. Hertzberg, I. Gonzalez and C. Moberg, Minor Enantiomer Recycling: Application to Enantioselective Syntheses of Beta Blockers, Chem.–Eur. J., 2014, 20, 3806–3812 CrossRef CAS PubMed; (d) E. Wingstrand, A. Laurell, L. Fransson, K. Hult and C. Moberg, Minor Enantiomer Recycling: Metal Catalyst, Organocatalyst and Biocatalyst Working in Concert, Chem.–Eur. J., 2009, 15, 12107 CrossRef CAS PubMed; (e) C. Moberg, Recycling in Asymmetric Catalysis, Acc. Chem. Res., 2016, 49, 2736–2745 CrossRef CAS PubMed.
- N. Y. Shin, J. M. Ryss, X. Zhang, S. J. Miller and R. R. Knowles, Light-driven deracemization enabled by excited-state electron transfer, Science, 2019, 366, 364–369 CrossRef CAS PubMed.
- P. Demay-Drouhard, K. Du, K. Samanta, X. Wan, W. Yang, R. Srinivasan, A. C. H. Sue and H. Zuilhof, Functionalization at Will of Rim-Differentiated Pillar[5]arenes, Org. Lett., 2019, 21, 3976–3980 CrossRef CAS PubMed.
- T. Ogoshi, Synthesis of novel pillar-shaped cavitands “Pillar[5]arenes” and their application for supramolecular materials, J. Inclusion Phenom. Macrocyclic Chem., 2012, 72, 247–262 CrossRef CAS.
- (a) P. J. Cragg and K. Sharma, Pillar[5]arenes: fascinating cyclophanes with a bright future, Chem. Soc. Rev., 2012, 41, 597–607 RSC; (b) K. Yang, S. Chao, F. Zhang, Y. Pei and Z. Pei, Recent advances in the development of rotaxanes and pseudorotaxanes based on pillar[n]arenes: from construction to application, Chem. Commun., 2019, 55, 13198–13210 RSC.
- T. Ogoshi, K. Demachi, K. Kitajima and T.-a. Yamagishi, Monofunctionalized pillar[5]arenes: synthesis and supramolecular structure, Chem. Commun., 2011, 47, 7164–7166 RSC.
- (a) S. D. P. Fielden, D. A. Leigh, C. T. McTernan, B. Pérez-Saavedra and I. J. Vitorica-Yrezabal, Spontaneous Assembly of Rotaxanes from a Primary Amine, Crown Ether and Electrophile, J. Am. Chem. Soc., 2018, 140, 6049–6052 CrossRef CAS PubMed; (b) M. Sharafi, K. T. McKay, M. Ivancic, D. R. McCarthy, N. Dudkina, K. E. Murphy, S. C. Rajappan, J. P. Campbell, Y. Shen, A. R. Badireddy, J. Li and S. T. Schneebeli, Size-Selective Catalytic Polymer Acylation with a Molecular Tetrahedron, Chem, 2020, 6, 1469–1494 CrossRef CAS PubMed.
- I. Nierengarten, E. Meichsner, M. Holler, P. Pieper, R. Deschenaux, B. Delavaux-Nicot and J.-F. Nierengarten, Preparation of Pillar[5]arene-Based [2]Rotaxanes by a Stopper-Exchange Strategy, Chem.–Eur. J., 2018, 24, 169–177 CrossRef CAS PubMed.
- Jaguar, version 8.8, 2015 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, Revealing noncovalent interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- P. Kuzmič, Program DYNAFIT for the Analysis of Enzyme Kinetic Data: Application to HIV Proteinase, Anal. Biochem., 1996, 237, 260–273 CrossRef PubMed.
- (a) A. M. Albrecht-Gary, Z. Saad, C. O. Dietrich-Buchecker and J. P. Sauvage, Interlocked macrocyclic ligands: a kinetic catenand effect in copper(I) complexes, J. Am. Chem. Soc., 1985, 107, 3205 CrossRef CAS; (b) A. H. Parham, B. Windisch and F. Vögtle, Chemical Reactions in the Axle of Rotaxanes – Steric Hindrance by the Wheel, Eur. J. Org. Chem., 1999, 1999, 1233–1238 CrossRef; (c) T. Oku, Y. Furusho and T. Takata, Rotaxane-stabilized thiophosphonium salt from disulfide and phosphine, Org. Lett., 2003, 5, 4923–4925 CrossRef CAS PubMed; (d) E. A. Neal and S. M. Goldup, Chemical consequences of mechanical bonding in catenanes and rotaxanes: isomerism, modification, catalysis and molecular machines for synthesis, Chem. Commun., 2014, 50, 5128–5142 RSC; (e) F. C. Hsueh, C. Y. Tsai, C. C. Lai, Y. H. Liu, S. M. Peng and S. H. Chiu, N-Heterocyclic Carbene Copper(I) Rotaxanes Mediate Sequential Click Ligations with All Reagents Premixed, Angew. Chem., Int. Ed., 2020, 59, 11278–11282 CrossRef CAS PubMed; (f) B. Leforestier, M. R. Gyton and A. B. Chaplin, Oxidative Addition of a Mechanically Entrapped C(sp)-C(sp) Bond to a Rhodium(I) Pincer Complex, Angew. Chem., Int. Ed., 2020, 59, 23500–23504 CrossRef CAS PubMed.
- S. Zhang, W. Li, J. Luan, A. Srivastava, V. Carnevale, M. L. Klein, J. Sun, D. Wang, S. P. Teora, S. J. Rijpkema, J. D. Meeldijk and D. A. Wilson, Adaptive insertion of a hydrophobic anchor into a poly(ethylene glycol) host for programmable surface functionalization, Nat. Chem., 2023, 15, 240–247 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04412b |
| ‡ Equal authorship contribution. |
| This journal is © The Royal Society of Chemistry 2024 |