
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe solubility product controls the rate of calcite dissolution in pure water and seawater†
Minjun
Yang‡
 a,
Ling
Tan‡
a,
Christopher
Batchelor-McAuley
b and
Richard G.
Compton
*a
a,
Ling
Tan‡
a,
Christopher
Batchelor-McAuley
b and
Richard G.
Compton
*a
aPhysical and Theoretical Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford, UK. E-mail: Richard.Compton@chem.ox.ac.uk
bThe School of Chemistry, Trinity College Dublin, The University of Dublin. College Green, Dublin 2, Ireland
First published on 10th January 2024
Abstract
Quantification of calcite dissolution underpins climate and oceanographic modelling. We report the factors controlling the rate at which individual crystals of calcite dissolved. Clear, generic criteria based on the change of calcite particle dimensions measured microscopically with time are established to indicate if dissolution occurs under kinetic or thermodynamic control. The dissolution of calcite crystals into water is unambiguously revealed to be under thermodynamic control such that the rate at which the crystal dissolved is controlled by the rate of diffusion of ions from a saturated surface layer adjacent to the calcite surface. As such the dissolution rate is controlled by the true stoichiometric solubility product which is inferred from the microscopic measurement as a function of the concentration of NaCl. Comparison with accepted literature values shows that the role of ion pairing at high ionic strengths as in seawater, specifically that of CaCO3 and other ion pairs, exerts a significant influence since these equilibria control the amount of dissolved calcium and carbonate ions in the later of solution immediately adjacent to the solid.
Introduction
The surface of the world's oceans is oversaturated with respect to calcium carbonate (calcite) where levels of calcium and carbonate/bicarbonate ions partly reflect the exchange of carbon dioxide with the atmosphere,1 the weathering of rocks and the extent of biomineralization. The global importance of this oceanic carbonate equilibria is partially reflected by the fact that the Ocean absorbs roughly 30% of anthropogenic CO2.2 Despite the oversaturation of calcite in the surface of our ocean CaCO3(s) is not known to precipitate abiotically. However, some marine phytoplankton, notably “coccolithophores”, have evolved to encrust themselves with platelets of CaCO3.3–5 The formation of CaCO3 by these photosynthetic single-cells act as a ballast sinking the surface mineralized CO2 to the deep where it is stored for over millennia.6,7 Beyond a critical depth, however, the calcite saturation falls below unity (increase in pressure and lower temperature) causing the surface biomineralized CaCO3 particulates to be dissolved.1 A better understanding of this dynamic carbonate equilibria is thus crucial to modelling marine carbon cycle, understanding ocean health and how this might change with climate change.The saturation state of calcite depends on a knowledge of the true solubility product defined in terms of the activities of the ions. However, in the case of seawater it has been usual to work with the stoichiometric solubility constant defined by
| K0sp = [Ca2+][CO32−] | (1) |
| K0sp,apparent = ∑[Ca2+]∑[CO32−] | (2) |
The stoichiometric solubility product controls the maximum rate at which calcite can dissolve since this corresponds to the rate at which ions diffuse into bulk solution from the solid–liquid interface under conditions where the interfacial concentration is pinned at the maximum level consistent with the value of K0sp. Under other conditions, notably for ultra-small particles the rate can be slower and reflects a surface kinetic controlled dissolution.1 Other than particle size,12 the pH and salinity of seawater,13 and the adsorption of surface-active components in seawater, notably magnesium ions,14–16 are thought to impact the rate of calcite dissolution.
Typical seawater contains a total calcium (∑[Ca2+]) concentration of approximately 10 mM, a total carbonate ions (∑[CO32−]) of 0.2 mM and a total magnesium concentration at a relatively large excess of 55 mM.9,17 Under seawater conditions carbonate ion is known to form ion pairs18 most notably with Mg, Ca and Na cations,17,19 resulting in merely 8% of the total carbonate existing as free ions.17 It is evident that the ionic composition of seawater plays an integral role in the calcium carbonate chemistry not only at the heterogeneous calcite–water interface20,21 but also chemically in the solution phase. Note that more recent studies observed the recession/growth of pristine surface and kink sites of calcite surfaces using atomic force microscopes allows mechanistic insights into particle dissolution/crystallisation at the nanoscale level with or without calcite dissolution/growth inhibitors to be inferred.15,22–27 Most notably, the dissolution step velocities of obtuse 〈48![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 〉 and acute 〈
〉 and acute 〈![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 41〉 sites on a pristine (104) calcite surface were found to reduced by 97% and 18% with the addition of 0.8 mM Mg2+, respectively.15
41〉 sites on a pristine (104) calcite surface were found to reduced by 97% and 18% with the addition of 0.8 mM Mg2+, respectively.15
In contrast the work in this paper concerns the overall rate of macroscopic dissolution of micron-sized calcite crystals as entire single entities. This is different to atomic force microscope studies where the latter necessarily focuses on the evolving recess of surfaces, steps and kink sites on pristine crystalline facets. The rate of dissolution of the crystal (particle) as a holistic entity is perplexed by the intertwined effect of the surface kinetics and rate of mass-transport of solute to and from the crystal surfaces. In the limiting cases, the overall rate of particle dissolution can either be limited by the rate of surface processes (surface kinetics) or the rate of mass-transport of materials (thermodynamic dissolution). Note that the intertwined effects of surface kinetics and mass-transport are commonly seen in reaction kinetics at interfaces such as, but not limited to, crystal precipitation28 and dissolution,29 biosensors30 and electrochemistry.31,32
A discussion of the limiting cases is given in ESI Section 3† and below where it is shown that the variation of the particle dimensions with time shows generically different behavior for the two cases reflecting either a constant flux of dissolving solid (surface kinetic control) or dissolution controlled by the rate of diffusion of the dissolved solid away from the crystal as happens when the concentration local to the dissolving surface is pinned, for example by the solubility of the solid, in which case the solubility product controls the local concentration and hence indirectly the rate of dissolution. In the following we exploit the generic probe for fingerprinting and hence resolving the two kinetic limits of surface and thermodynamic control. In the thermodynamic limit it should be stressed that the physical process controlling the rate is that of diffusion of dissolved solid into the bulk of solution but that this reflects the prevailing concentration gradients adjacent to the solid and these in turn reflect the pinned concentration(s) at the surface as dictated by the solubility product. In this limit the rate at which dissolved solute diffuses into bulk is matched by an equal (net) rate of dissolution of the crystal so maintaining the saturated layer at the dissolving interface.
The distinction between surface and thermodynamic dissolution in the context of calcite dissolution has proved challenging with a diversity of views expressed.1 The terms “surface kinetics” and “thermodynamic dissolution” in the context of this work are discussed in ESI Section 3.† In our opinion this partly arises since the approaches used have been to monitor the net release/consumption of calcium and/or carbonate ions in the solution phase – an inherently indirect method. Driven by the need for unambiguous mechanistic distinction coupled with the fact that the dissolution/precipitation process is intimately linked to the evaluation of K0sp, in this paper we use a direct method for measuring the dissolution rate and show that the rate of change of the size of calcite particles with time inferred microscopically via recording the rate of recession of the particle interface gives the sought clear, unambiguous mechanistic distinction and allows the quantification of the real, stoichiometric K0sp of calcite prompting reconsideration of Mucci's approach. The method is extended to consider retarded dissolution in the presence of Mg2+ ions at concentrations similar to those encountered in seawater with a simple model used to explain all observations.19
Results and discussion
In this work we newly report the dissolution of calcite single particles in aqueous solutions at a constant ionic strength of 0.3 M with various concentrations of Mg2+, where the latter ranges from 0 mM to 0.1 M. We first, however, investigate the effect of ion pairing on the stoichiometric solubility constant of calcite in solutions absence of Mg2+ using data reported Fan et al.33 The experimental approach used by Fan et al. and the present work is summarised in Fig. 1A where optical microscopy (see ESI Section 2 Experimental details†) allows us to record the top-down projection area of the dissolving calcite particle as a function of time.4,34 We adapt from our prior work33 and show in ESI Section 3† that for a thermodynamic dissolution (or strictly dissolution with a constant saturated interface) the rate of change of the projection area (A) of the cubic particle with time (t) is constant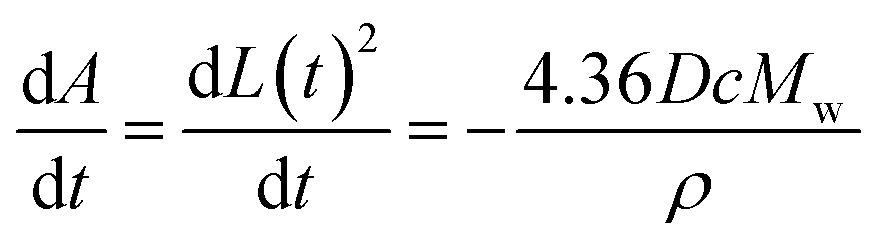 | (3) |
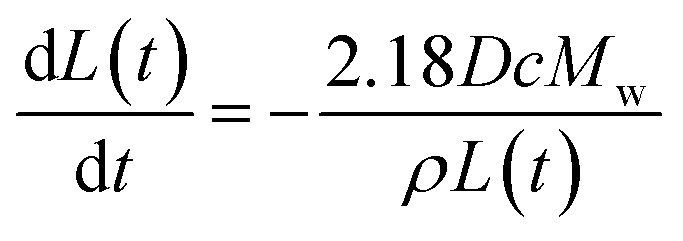 | (4) |
 | ||
Fig. 1 Single particle dissolution. (A) An illustration of the dissolution of a single calcite particle on a plate and the observed projection area of the particle as a function of time viewed top-down. (B) Optical images of the dissolution of a representative calcite particle. The top row shows raw images and the bottom row shows the same images after thresholding. (C) Projected areas, A, and (D) particle lengths, L, of three calcite particles are plotted as a function of time in a solution containing 0.7 M NaCl. The particle length was calculated from 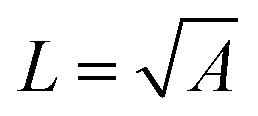 . The x-axis of each particle was shifted to depict the dissolution trend collectively as a function of particle size. Data obtained from the reanalysis of work reported by Fan et al.33 . The x-axis of each particle was shifted to depict the dissolution trend collectively as a function of particle size. Data obtained from the reanalysis of work reported by Fan et al.33 | ||
Note that, in the limiting case of thermodynamic dissolution, which is discussed in detail in more ESI Section 3,† the interfacial concentration of the solute is a constant pinned by the thermodynamic solubility. A revaluation of the right-hand side of eqn (3) and (4) reveals that, under thermodynamic dissolution, 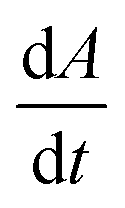 is a constant whereas
is a constant whereas 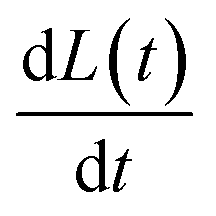 becomes increasingly more negative as the particle size reduces during dissolution. Note that the opposite is true if the dissolution is limited by the surface kinetics. The dissolution pattern as described by eqn (3) and (4) provides a generic mechanistic distinction for the dissolution of calcite single-particles analysed first in the absence of inhibitors and, second, with sea-level quantities of Mg2+.
becomes increasingly more negative as the particle size reduces during dissolution. Note that the opposite is true if the dissolution is limited by the surface kinetics. The dissolution pattern as described by eqn (3) and (4) provides a generic mechanistic distinction for the dissolution of calcite single-particles analysed first in the absence of inhibitors and, second, with sea-level quantities of Mg2+.
Single calcite particle dissolution in the absence of Mg2+
The dissolution of micron-sized, pristine calcite particles in aqueous solutions ranging from 0 mM to 0.7 M ionic strengths was reported by Fan et al.33 The top row of Fig. 1B shows raw optical images of a representative calcite particle dissolving in 0.7 M NaCl and the bottom row are the same images after image-thresholding. Note that the solution contains no magnesium ions. As can be seen in the image, the calcite particle which is initially 9.0 microns in length is approximately halved in size over a period of 30 minutes. Fig. 1C and D present the changes in the geometric size of three calcite cubes with time studied in isolation, where the projection area of the particle was obtained directly from image analysis (see Experimental in ESI†) and the particle length was calculated from the measured area (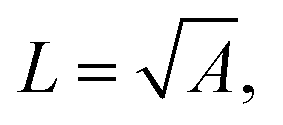 where A is area).
where A is area).
Fig. 1C shows that the rate of change in the projection area, A, displays a strong linear dependency on time (gradient = −2.1 × 10−14 m2 s−1, Parson's R = −0.99), which is not the case for particle length (Fig. 1D). ESI Section 4† shows more examples of transients analysed in ionic strengths ranging from 0 mM to 1 M NaCl(aq) with similar observations in respect of the variation of length and area with time. This strongly evidences calcite dissolution occurring at the thermodynamic limit; the rate of calcite dissolution and precipitation at the particle–solution interface is fast allowing local equilibrium and the overall rate of dissolution is dictated by how quickly the diffusion-limiting Ca2+ flux can diffuse down the concentration gradient away from the particle–solution interface into the bulk solution.
Fig. 2A shows the dissolution rate, 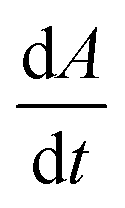 , of calcite single-particles measured experimentally in aqueous solutions containing exclusively NaCl(aq) reported by Fan et al.33 As can be seen, by increasing the overall ionic strength of the solution by 3 orders of magnitude, from 0.1 mM to 1 M, the rate of particle dissolution was seen to increase ca. three-fold from 1.6 × 10−14 m2 s−1 to 4.5 × 10−14 m2 s−1. At low ionic strengths, I < 10 mM, the data reported by Fan et al. agrees with the predicted thermodynamic rate of calcite dissolution using values reported by Mucci(6) but at higher ionic strengths, I > 10 mM, significant deviations were seen.33 Below we show this apparent paradox arises because of the neglect of ion pairing.
, of calcite single-particles measured experimentally in aqueous solutions containing exclusively NaCl(aq) reported by Fan et al.33 As can be seen, by increasing the overall ionic strength of the solution by 3 orders of magnitude, from 0.1 mM to 1 M, the rate of particle dissolution was seen to increase ca. three-fold from 1.6 × 10−14 m2 s−1 to 4.5 × 10−14 m2 s−1. At low ionic strengths, I < 10 mM, the data reported by Fan et al. agrees with the predicted thermodynamic rate of calcite dissolution using values reported by Mucci(6) but at higher ionic strengths, I > 10 mM, significant deviations were seen.33 Below we show this apparent paradox arises because of the neglect of ion pairing.
The calcite dissolution equilibrium is expressed in terms of free ions in solution and ion pairs, CaCO30(aq) and CaHCO3+(aq)
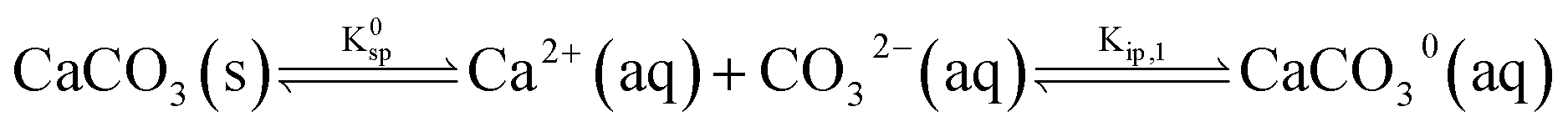 | (5) |
 | (6) |
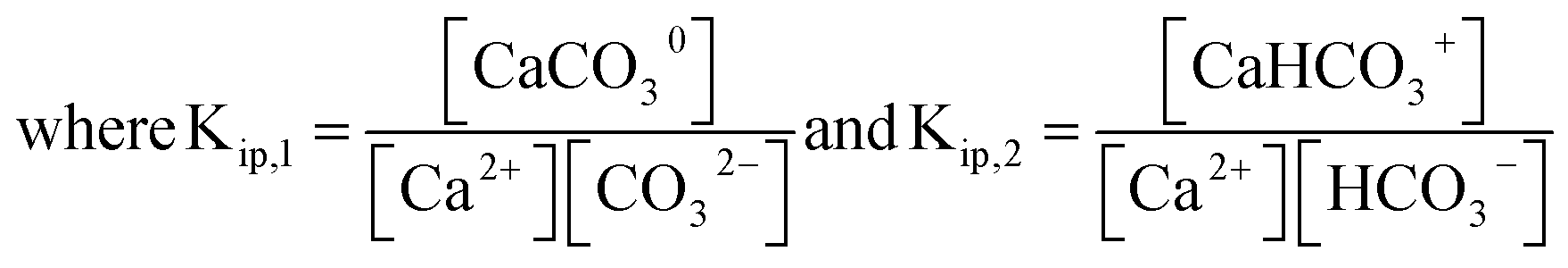 | (7) |
Noting that the stoichiometric ion pair association constants defined herein, Kip,1 and Kip,2, are in terms of concentration, not activities. As the ionic strength of an aqueous medium increases, the effect of the ion pairing effect between calcium and carbonate ions becomes non-negligible.19 Separate from the calcite dissolution equilibrium, calcium binds with hydroxide to form
 | (8) |
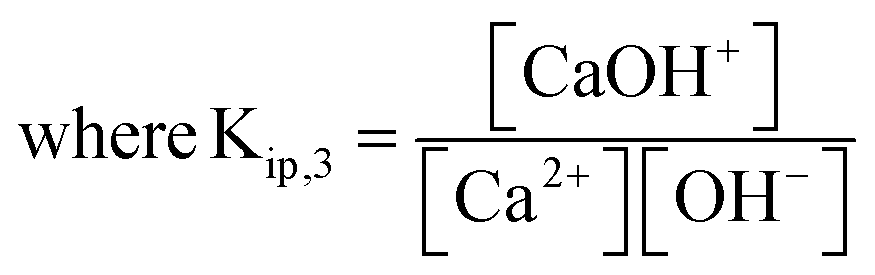
At 0.7 M ionic strength, the stoichiometric ion pair association constants, Kip,1 and Kip,2 has a reported value of 162 M−1 and 1.96 M−1, respectively.17 A Kip,3 value of 3.3 M−1 was calculated using the PHREEQC software36 by accounting for the activity coefficients of species involved at 0.7 M ionic strength via the Davies equation. In the next section we first calculate the effect of ion pairings in the calcite single-particle experiments before discussing the extent of ion pairings in Mucci's experiments.
The extent of ion pairing in calcite single-particle auto-dissolution in brine
It is evident from the data shown in Fig. 1C that the linearity of confirms that the rate of dissolution the calcite particles occurs under conditions where the interfacial concentration of calcite is constant under steady-state conditions and the value of which is controlled, largely, by the solubility product of calcite. On this basis, we first infer the concentration of calcium ions formed at the particle–solution interface using eqn S15† where we further assume that Ca2+ and the ion pair CaCO3 are equilibrated so that c in that equation represents the total Ca concentration defined by
confirms that the rate of dissolution the calcite particles occurs under conditions where the interfacial concentration of calcite is constant under steady-state conditions and the value of which is controlled, largely, by the solubility product of calcite. On this basis, we first infer the concentration of calcium ions formed at the particle–solution interface using eqn S15† where we further assume that Ca2+ and the ion pair CaCO3 are equilibrated so that c in that equation represents the total Ca concentration defined by| c = ∑[Ca2+]0 = [Ca2+]0 + [CaCO30]0 + [CaHCO3+]0 + [CaOH+]0 | (9) |
Using the experimentally determined ∑[Ca2+]0, the apparent stoichiometric solubility product is inferred via modelling (K0sp,apparent = ∑[Ca2+]∑[CO32−]) and ∑[CO32−] are obtained and shown in Table 1 by solving the CaCO3 and carbonate equilibrium for the given ionic strength and temperature recognising that the solid CaCO3 dissolution releases both calcium and carbonate ions in equal amounts.33 Separately, to investigate the effects of ion pairing, the CaCO3 and carbonate equilibrium, fully described in ESI section 5,† is solved simultaneously additionally with the above ion pairing equilibria (eqn (5), (6) and (8)) to infer the fraction of the total dissolved solute existing as free ions, Ca2+(aq), CO32−(aq), and ion pairs assuming that the ion-pairing equilibria are fast and not rate determining.
| Ionic strength (mM) | K0sp,apparent (M2) | Total concentrations (including ion pairing, M) | Concentrations of free ions and ion pairs (M) | |||||
|---|---|---|---|---|---|---|---|---|
| ∑[Ca2+]0 | ∑[CO32−]0 | [Ca2+]0 | [CO32−]0 | [CaCO30]0 | [CaHCO3+]0 | [CaOH+]0 | ||
| 0.1 | 3.9 × 10−9 | 1.3 × 10−4 | 2.9 × 10−5 | 1.2 × 10−4 | 2.9 × 10−5 | 5.7 × 10−7 | 2.6 × 10−8 | 1.0 × 10−5 |
| 160 | 2.6 × 10−8 | 2.2 × 10−4 | 1.2 × 10−4 | 2.0 × 10−4 | 1.2 × 10−4 | 3.7 × 10−6 | 4.0 × 10−8 | 1.6 × 10−5 |
| 300 | 3.0 × 10−8 | 2.3 × 10−4 | 1.3 × 10−4 | 2.1 × 10−4 | 1.3 × 10−4 | 4.4 × 10−6 | 4.2 × 10−8 | 1.6 × 10−5 |
| 684.5 | 3.4 × 10−8 | 2.4 × 10−4 | 1.4 × 10−4 | 2.2 × 10−4 | 1.4 × 10−4 | 4.9 × 10−6 | 4.3 × 10−8 | 1.7 × 10−5 |
| 1000 | 8.5 × 10−8 | 3.5 × 10−4 | 2.4 × 10−4 | 3.2 × 10−4 | 2.3 × 10−4 | 1.2 × 10−5 | 7.0 × 10−8 | 2.9 × 10−5 |
The right-hand side of Table 1 tabulates the concentrations of the ions that exist as free ions and as ion pairs where
| ∑[CO32−]0 = [CO32−]0 + [CaCO30]0 | (10) |
It is evident from Table 1 that the extent of ion pairing in the calcite single-particle dissolution experiment reported by Fan et al. is negligible. This is because the bulk solution contains only NaCl ions but no calcium other than that resulting from calcite dissolution. Fig. 2B plots the values of K0sp,apparent and K0sp values of calcite inferred from data reported by Fan et al. As can be seen, the effect of ion pairing can be essentially ignored in those experiments since K0sp,apparent ≈ K0sp. Next, we turn to investigate the extent of ion pairing in Mucci's ‘long-term’ calcite equilibrium experiments in natural seawater and we will discuss how this might affect the true stoichiometric solubility product of calcite, K0sp.
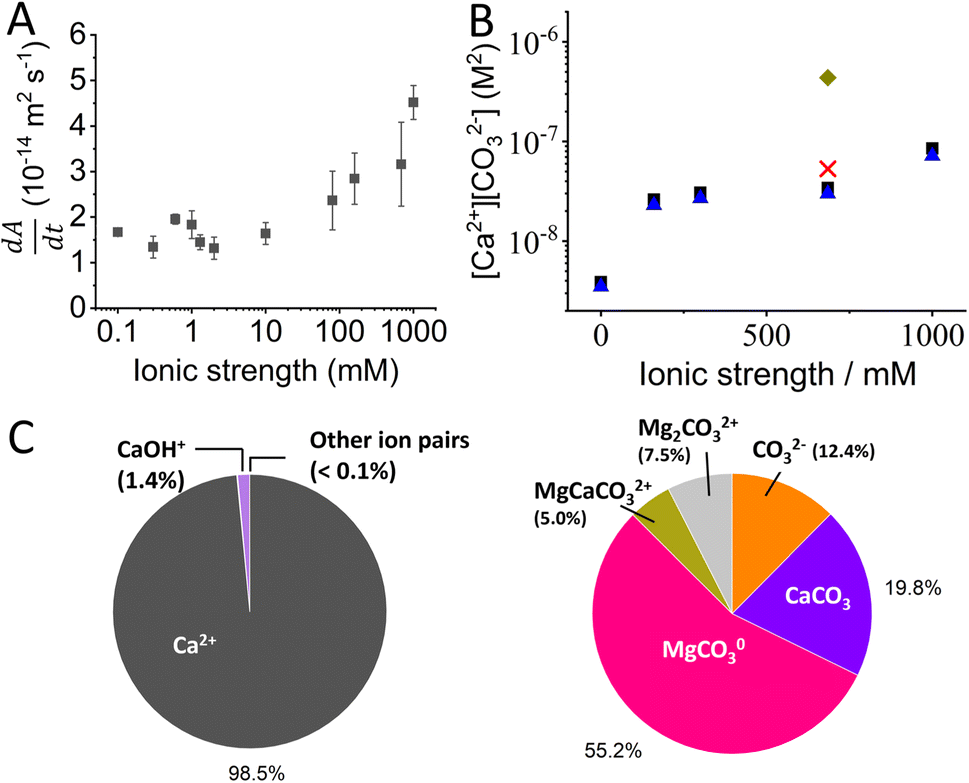 | ||
| Fig. 2 Dissolution of calcite single-particles. (A) Experimentally measured rate of change in the projection area of CaCO3 microparticles with time using aqueous solutions containing variable concentrations of NaCl. Data is obtained from ref. 33, an open-access article in ACS Measurement Science Au. (B) The stoichiometric solubility product of calcite as a function of ionic strength in NaCl solution and in seawater, with and without ion pair correction. Black squares shows the calcite solubility product in NaCl solutions using data reported by Fan et al.33 (no ion pair correction). Blue triangles shows the true calcite solubility product in NaCl after ion pair correction (reanalysed in this work). Green rhombus is the calcite solubility product reported by Mucci in seawater (without ion pair correction). Red cross is the true stoichiometric solubility product of calcite in seawater after ion pair correction (reanalysed in this work). (C) Pie charts showing the calculation of the percentage of the speciation of (i) Ca2+ and (ii) CO32− in natural seawater as inferred by the analysis (see text) of the data reported by Mucci. | ||
The extent of ion pairing in seawater
The chemical composition of natural seawater is complex. The ions present at sufficient concentrations which can ion-pair with (bi)carbonate anion are calcium and magnesium at concentrations of 10 and 55 mM, respectively.9 Similar to the calcite single-particle dissolution, we solve the calcium carbonate, carbonate and the corresponding ion pairing equilibria to infer the speciation of ions as either free ions or ion paired. The model for seawater is fully described in ESI Section 6† with the additional inclusion of magnesium (bi)carbonate and magnesium hydroxide ion pairings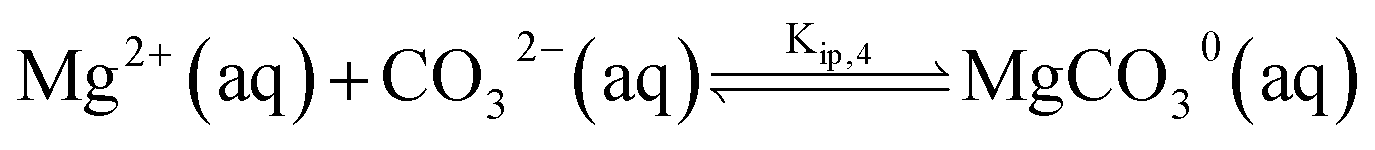 | (11) |
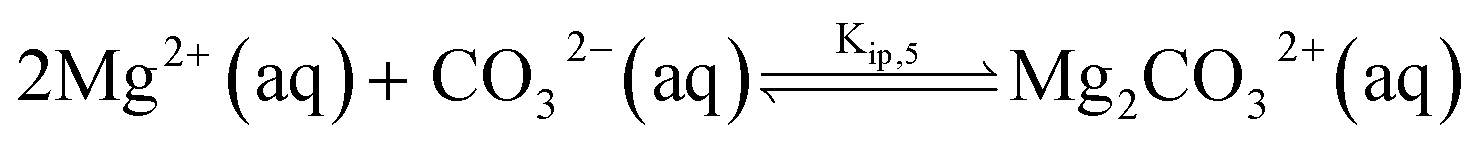 | (12) |
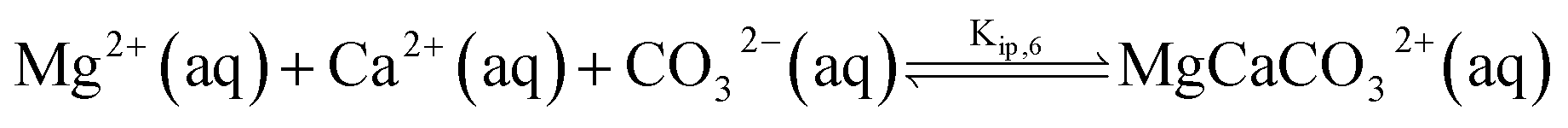 | (13) |
 | (14) |
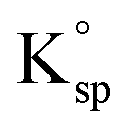 of calcite is 5.4 × 10−8 M2. This is approximately one order of magnitude lower than the K0sp,apparent value reported by Mucci, 4.4 × 10−7 M2. These two data points is shown on Fig. 2B as red cross and green rhombus. Shown in Fig. 2C is the fraction of the calcium and carbonate in Mucci's experiment existing as either free ions and ion pair after long-term equilibrium with CaCO3. It is evident that, as a direct result of ion pairs, CO32− free ion is approximately 1/10 the total carbonate concentration, ∑[CO32−]0, agreeing with those reported in the literature.17,37,38 In comparison, calcium is effectively unaffected due to the enhanced levels of calcium present before the dissolution of the calcite. The exact concentrations are tabulated in ESI Table 1.† We note that, the factor of ca. 10 decrease in the true stoichiometric K0sp, as compared to the apparent stoichiometric K0sp,apparent (eqn (2)) reported by Mucci, is a direct consequence of the presence of carbonate ion pairs. Importantly, once the role of ion pairing is quantified there is excellent agreement for the value of the true stoichiometric solubility product, K0sp (red cross and blue triangles) between the single calcite particle dissolution reported by Fan and those after ‘long-term’ equilibration by Mucci. Note to compare other ionic strengths one would need the value of Kip3, Kip4 and Kip5 at the corresponding ionic strengths, which is not available to the best of the authors knowledge.
of calcite is 5.4 × 10−8 M2. This is approximately one order of magnitude lower than the K0sp,apparent value reported by Mucci, 4.4 × 10−7 M2. These two data points is shown on Fig. 2B as red cross and green rhombus. Shown in Fig. 2C is the fraction of the calcium and carbonate in Mucci's experiment existing as either free ions and ion pair after long-term equilibrium with CaCO3. It is evident that, as a direct result of ion pairs, CO32− free ion is approximately 1/10 the total carbonate concentration, ∑[CO32−]0, agreeing with those reported in the literature.17,37,38 In comparison, calcium is effectively unaffected due to the enhanced levels of calcium present before the dissolution of the calcite. The exact concentrations are tabulated in ESI Table 1.† We note that, the factor of ca. 10 decrease in the true stoichiometric K0sp, as compared to the apparent stoichiometric K0sp,apparent (eqn (2)) reported by Mucci, is a direct consequence of the presence of carbonate ion pairs. Importantly, once the role of ion pairing is quantified there is excellent agreement for the value of the true stoichiometric solubility product, K0sp (red cross and blue triangles) between the single calcite particle dissolution reported by Fan and those after ‘long-term’ equilibration by Mucci. Note to compare other ionic strengths one would need the value of Kip3, Kip4 and Kip5 at the corresponding ionic strengths, which is not available to the best of the authors knowledge.
Heretofore, the results hint at the importance of considering, and the need to correct for ion-pairing since any changes in the chemical composition, in seawater or other media, will have an impact on the value of K0sp,apparent. Using the ion-pair corrected true stoichiometric 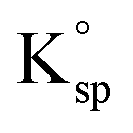 of calcite in seawater calculated in this study (5.4 × 10−8 M2) and assuming that the concentration of Ca2+ is in large excess (10 mM in surface waters) the concentration of CO32− free ions needed to reach saturation (Ω = 1) is 5.4 μM. In natural seawater the total carbonate concentration is reported to be in the range of 50–250 μmol kg−1,39,40 the carbonate free ions in seawater, however, is approximately 7.99% of the total concentration17,38 which gives rise to a range of 4–20 μmol kg−1. This is entirely consistent with the claim that the surface of our ocean is saturated/supersaturated with respect to calcite, however, we note the literature is often unclear if free or total ion concentrations are used in the calculation.40–45
of calcite in seawater calculated in this study (5.4 × 10−8 M2) and assuming that the concentration of Ca2+ is in large excess (10 mM in surface waters) the concentration of CO32− free ions needed to reach saturation (Ω = 1) is 5.4 μM. In natural seawater the total carbonate concentration is reported to be in the range of 50–250 μmol kg−1,39,40 the carbonate free ions in seawater, however, is approximately 7.99% of the total concentration17,38 which gives rise to a range of 4–20 μmol kg−1. This is entirely consistent with the claim that the surface of our ocean is saturated/supersaturated with respect to calcite, however, we note the literature is often unclear if free or total ion concentrations are used in the calculation.40–45
Further, we recognised that the true saturation of calcium carbonate can rigorously only be asserted relative to a true stoichiometric solubility product, in seawater or in a well-defined NaCl aqueous solution, or anything in between. Of the many components in seawater, however, one of considerable impact is Mg2+ cations where it is important to note that in a sample of solid CaCO3 particles equilibrated with seawater-type media, significant amounts of magnesite (MgCO3) have been detected even after just 7 days of exposure.46 Clearly assertions of ‘supersaturation’ require a precise knowledge of the composition of the solid phase controlling the equilibrium! Given the apparent role of Mg2+, in the following, we explore the role of this ion in the dissolution of CaCO3.
Single calcite particle dissolution – effect of Mg2+
Focus is next turned to investigating the effect of dissolved Mg2+ ions have on the dissolution of single-calcite particles. Dissolution experiments were conducted similar to those described above but with variable amounts of added Mg(NO3)2 whilst the overall ionic strength of the medium was maintained at a total value of 0.3 M by the contribution of Mg(NO3)2 and the addition of a suitable amount of KNO3. Shown in Fig. 3A and B are the projected area and the measured length (inferred from the area) of calcite particles in a 0.3 M ionic strength solution containing 10 mM of Mg2+. As can be seen, the projection area of the calcite particle decreases linearly with time, similar to that seen in the absence of Mg2+.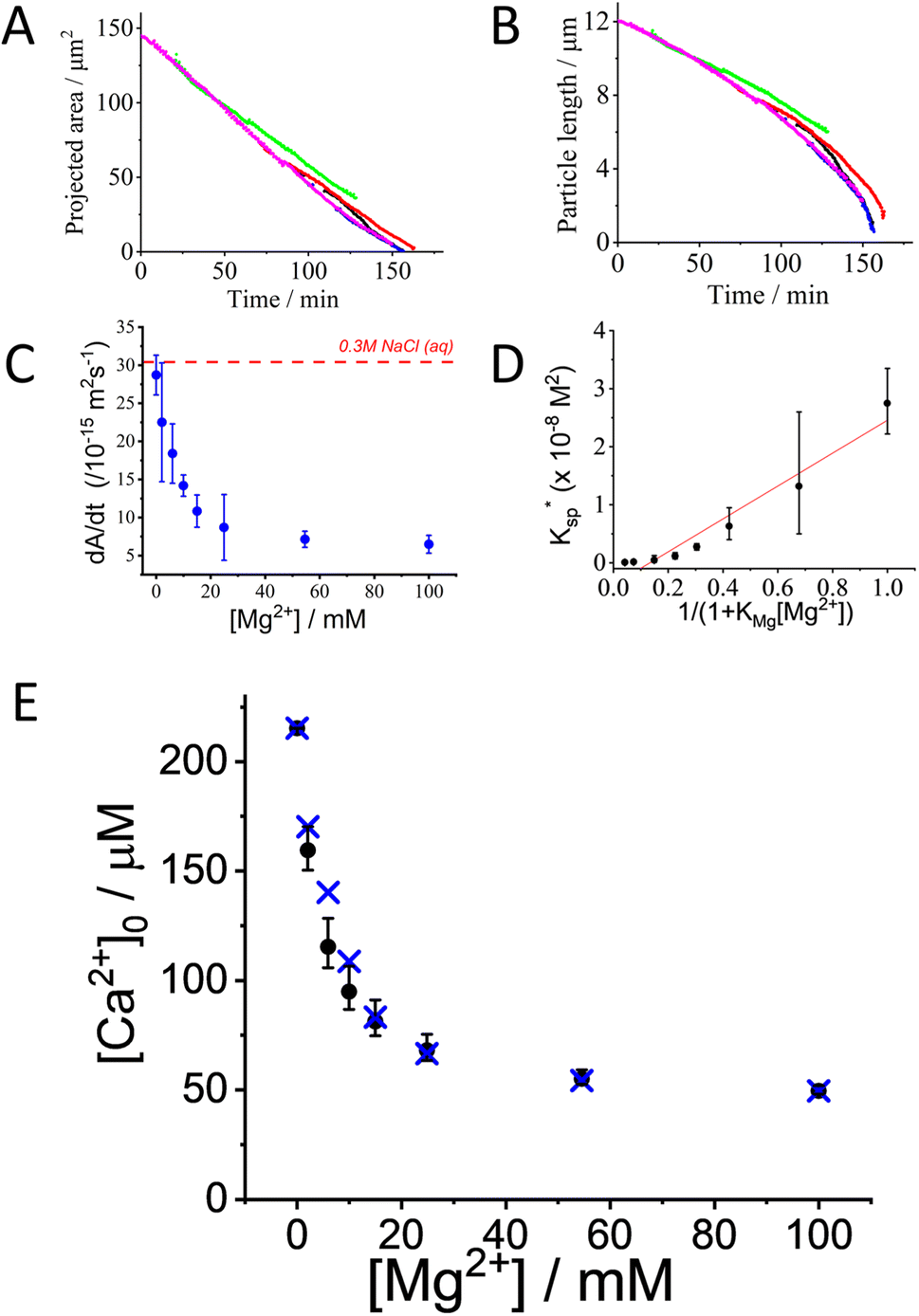 | ||
Fig. 3 Calcite dissolution in the presence of magnesium. (A) Projected area and (B) side length of 5 different dissolving calcite particles plotted against time. The calcite particles were exposed to 10 mM Mg(NO3)2·6H2O solution to which. KNO3 was added to make the total ionic strength of the solution equal to 0.3 M. Different colours relate to different particles. (C) The average rate of dissolution of calcite as a function of magnesium concentration. The overall ionic strength of the aqueous solution is 0.3 M with the addition of a suitable amounts of KNO3. The dashed red line corresponds to the value in the absence of magnesium. (D) Plot of apparent stoichiometric solubility product inferred from single particle dissolution measurements against 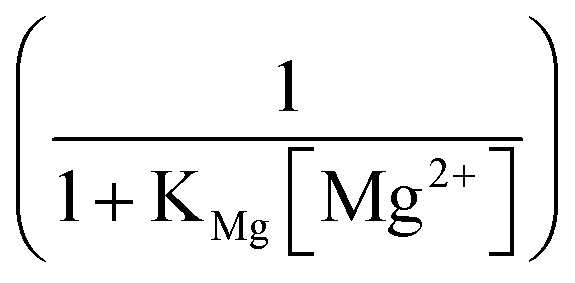 . The slope of the linear fit line is 2.83 (±0.27) × 10−8 mol2 dm−6, Pearson's r = 0.973. (E) The concentration of calcium free-ions at the surface inferred from single particle dissolution measurements plotted against the concentration of magnesium ions. The crosses represent the ion-pair corrected calcium ion concentration inferred from experimental data, and the dots represent the theoretical value accounting for the adsorption of magnesium ions calculated using KMg2+ (=230 ± 60 M−1) reported in ref. 14. . The slope of the linear fit line is 2.83 (±0.27) × 10−8 mol2 dm−6, Pearson's r = 0.973. (E) The concentration of calcium free-ions at the surface inferred from single particle dissolution measurements plotted against the concentration of magnesium ions. The crosses represent the ion-pair corrected calcium ion concentration inferred from experimental data, and the dots represent the theoretical value accounting for the adsorption of magnesium ions calculated using KMg2+ (=230 ± 60 M−1) reported in ref. 14. | ||
Fig. 3C shows the average rate of change in projection area,  , of calcite particles measured at different concentrations of Mg2+. The rate is reduced by approximately 6-fold with the addition of Mg2+ from 2.9 × 10−14 m2 s−1 to a limiting value of approximately 6.5 × 10−15 m2 s−1 at [Mg2+] of 25–100 mM. Interestingly, the fact that the rate does not decrease to zero but, rather, to a non-zero plateau suggests that the different calcite sites are affected differently by the magnesium adsorption as suggested elsewhere.15 An examination of the particle-size evolution of the calcite particles, the transients of which are shown in ESI Section 7,† reveals that
, of calcite particles measured at different concentrations of Mg2+. The rate is reduced by approximately 6-fold with the addition of Mg2+ from 2.9 × 10−14 m2 s−1 to a limiting value of approximately 6.5 × 10−15 m2 s−1 at [Mg2+] of 25–100 mM. Interestingly, the fact that the rate does not decrease to zero but, rather, to a non-zero plateau suggests that the different calcite sites are affected differently by the magnesium adsorption as suggested elsewhere.15 An examination of the particle-size evolution of the calcite particles, the transients of which are shown in ESI Section 7,† reveals that  remains linear with time whilst
remains linear with time whilst 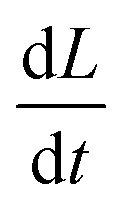 becomes increasingly non-linear and more steep over the dissolution duration (ca. ∼100 minutes). This indicates that, despite the presence of Mg2+ showing an unambiguous sign of rate reduction, the dissolution remains to proceed at the limit corresponding to a fixed surface concentration rather than a constant surface flux. This is a strong indication, as discussed above, that the rate-determining step is the diffusion of ions away from the crystal. The overall rate of the dissolution of the crystal is thus controlled by the constant composition in the adjacent layer.
becomes increasingly non-linear and more steep over the dissolution duration (ca. ∼100 minutes). This indicates that, despite the presence of Mg2+ showing an unambiguous sign of rate reduction, the dissolution remains to proceed at the limit corresponding to a fixed surface concentration rather than a constant surface flux. This is a strong indication, as discussed above, that the rate-determining step is the diffusion of ions away from the crystal. The overall rate of the dissolution of the crystal is thus controlled by the constant composition in the adjacent layer.
The dotted line in Fig. 3C shows the dissolution rate of calcite particles, in the form of 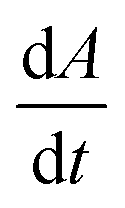 , reported separately by Fan et al. in 0.3 M NaCl solutions. However, increasing the concentration of magnesium ions from 0 to 53 mM is reported to increase the apparent solubility product of calcite (∑[Ca2+]∑[CO32−]) by ca. a factor of 1.5.47 This is due to the formation of magnesium (bi)carbonate ion pairs which encourages calcite dissolution. Therefore, the effect of magnesium adsorption has on slowing the rate of calcite dissolution may be slightly larger than that appears when referenced against the rate at 0.3 M NaCl solution (absent of Mg2+) as shown in Fig. 3C. Note that the variation of the error bars shown in Fig. 3C is likely due to an intertwined effect of the slight changes of particle geometry from particle to particle along with a slow drift in the optical focus away of the particle plane as the particle shrinks. A periodic readjustment of optical focus is usually required during the experiment.
, reported separately by Fan et al. in 0.3 M NaCl solutions. However, increasing the concentration of magnesium ions from 0 to 53 mM is reported to increase the apparent solubility product of calcite (∑[Ca2+]∑[CO32−]) by ca. a factor of 1.5.47 This is due to the formation of magnesium (bi)carbonate ion pairs which encourages calcite dissolution. Therefore, the effect of magnesium adsorption has on slowing the rate of calcite dissolution may be slightly larger than that appears when referenced against the rate at 0.3 M NaCl solution (absent of Mg2+) as shown in Fig. 3C. Note that the variation of the error bars shown in Fig. 3C is likely due to an intertwined effect of the slight changes of particle geometry from particle to particle along with a slow drift in the optical focus away of the particle plane as the particle shrinks. A periodic readjustment of optical focus is usually required during the experiment.
Hereto, it is clear that the auto dissolution of micron sized calcite particles, in 0.3 M ionic strength aqueous solutions both in the absence of, and in the presence of, Mg2+, occurs under conditions where the concentration of the dissolved material at the solid surface is constant, at least for most of the duration of dissolution. Accordingly, the question arises as to what is the interfacial equilibrium concentration of Ca2+ on the CaCO3(s) surface in the presence of Mg2+? And why might this be fixed? In the absence of Mg2+ we interpreted this above as indicating that the surface concentration is pinned by the solubility product of pure calcite.
Noting literature reports of significant Mg2+ adsorption on calcite surfaces and that this is approximately Langmuirian14 we suggest that the net interfacial dissolution rate can be described by the rate equation
| flux (mol cm−2 s−1) = kf(1 − θMg) − kb[Ca2+][CO32−] | (15) |
At steady-state, eqn (15) can be rearranged to give an effective stoichiometric solubility product of calcite as a function of magnesium coverage
 | (16) |
Note that  is different to the stoichiometric solubility constant, K0sp, defined in eqn (1) since a coverage of magnesium on calcite surfaces gives rise to a lower effective stoichiometric solubility product of calcite.
is different to the stoichiometric solubility constant, K0sp, defined in eqn (1) since a coverage of magnesium on calcite surfaces gives rise to a lower effective stoichiometric solubility product of calcite.
The average surface coverage of magnesium, θMg2+, is given by the Langmuirian adsorption isotherm48,49
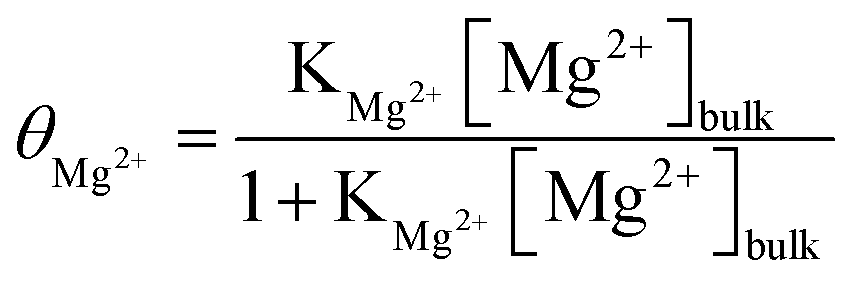 | (17) |
Substituting eqn (17) into eqn (16) shows
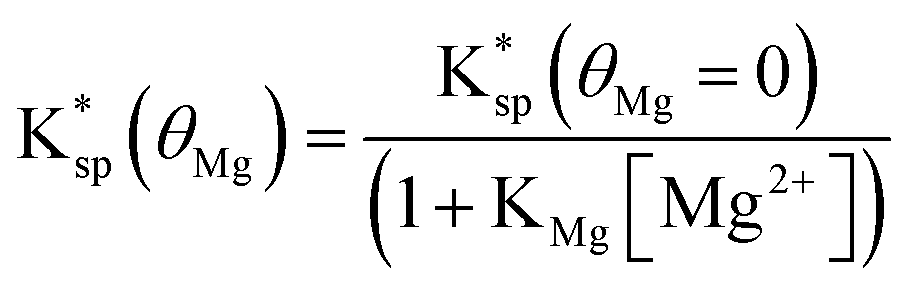 | (18) |
Fig. 3D shows a plot of  versus (1 + KMg[Mg2+])−1, where KMg is the adsorption constant of magnesium on Iceland Spar (230 ± 60 M−1) reported by Compton and Brown14 and
versus (1 + KMg[Mg2+])−1, where KMg is the adsorption constant of magnesium on Iceland Spar (230 ± 60 M−1) reported by Compton and Brown14 and 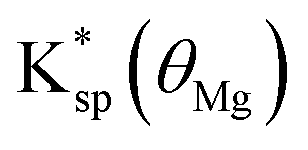 was inferred from the experimental measurements of
was inferred from the experimental measurements of  and by solving the CaCO3 and carbonate equilibrium shown in ESI Section 7.†
and by solving the CaCO3 and carbonate equilibrium shown in ESI Section 7.†
As can be seen in Fig. 3D, the experimentally inferred  as a function of magnesium concentration is in good agreement with the simple model using a magnesium adsorption constant derived from independent measurements.14 At high magnesium concentrations where
as a function of magnesium concentration is in good agreement with the simple model using a magnesium adsorption constant derived from independent measurements.14 At high magnesium concentrations where 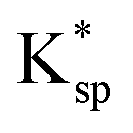 is close to zero the experimental data can be seen to deviate from the simple model. This is likely due to the presence of multiple sites available for adsorption on calcite surfaces as suggested by AFM studies.15 Finally, we note that the idea of a ‘partially blocked’ surface leading to a reduced average surface concentration is well established in the electrochemical literature (as summarised in ESI Section 9†,50–52) where the scale of the heterogeneity is small compared to diffusional lengths leads to a uniform, average surface concentration reflecting a balance of independent kinetic contributions. We accordingly view the calcite surface as a ‘partially blocked’ surface where the total macroscopic surface concentration is a sum of contributions by the ‘blocked’ and ‘unblocked’ zones:
is close to zero the experimental data can be seen to deviate from the simple model. This is likely due to the presence of multiple sites available for adsorption on calcite surfaces as suggested by AFM studies.15 Finally, we note that the idea of a ‘partially blocked’ surface leading to a reduced average surface concentration is well established in the electrochemical literature (as summarised in ESI Section 9†,50–52) where the scale of the heterogeneity is small compared to diffusional lengths leads to a uniform, average surface concentration reflecting a balance of independent kinetic contributions. We accordingly view the calcite surface as a ‘partially blocked’ surface where the total macroscopic surface concentration is a sum of contributions by the ‘blocked’ and ‘unblocked’ zones:
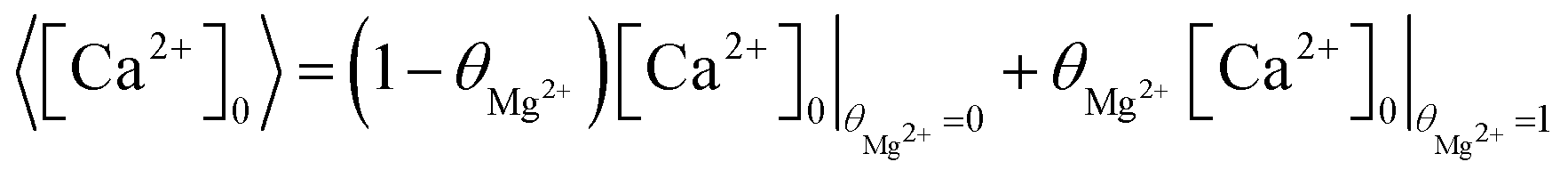 | (19) |
Conclusions
Microscopically monitored single particle dissolution experiments provide clear, generic mechanistic distinction between dissolution under constant flux or constant surface concentration conditions. In the case of calcite dissolving in aqueous solutions containing different amounts of NaCl at levels at and below those corresponding to the ionic strength of seawater the rate of dissolution is controlled by the solubility of calcite with the latter influenced, especially in elevated levels of Mg2+ and Ca2+ ions, by the formation of various ion pairs. The presence of dissolved Mg2+ reduces the rate of dissolution which takes place with a steady-state surface concentration reflecting a balance between dissolution and precipitation but with the latter taking place with incorporation of Mg ions into the solid lattice so that the calcium ion concentration at the surface is reduced from that seen in the absence of Mg. A phenomenological theory to quantify this effect has been developed.Data availability
All data are available in the main text or the ESI.† The codes used to solve the set of equilibria outlined in this manuscript can be requested via email to the corresponding author.Author contributions
Conceptualization: MY, RGC, methodology: MY, CBM, investigation: LT, software: CBM, visualization: MY, RGC, data analysis: LT, MY, RGC, funding acquisition: RGC, project administration: RGC, supervision: RGC, writing – original draft: LT, MY, writing – review & editing: MY, RGC, CBM.Conflicts of interest
Authors declare that they have no competing interests.Acknowledgements
Oxford Martin School Programme on Monitoring Ocean Ecosystems (MY, RGC).Notes and references
- C. Batchelor-McAuley, M. Yang, R. E. Rickaby and R. G. Compton, Chem.–Eur. J., 2022, 28, e202202290 CrossRef CAS PubMed.
- N. Gruber, D. Clement, B. R. Carter, R. A. Feely, S. Van Heuven, M. Hoppema, M. Ishii, R. M. Key, A. Kozyr and S. K. Lauvset, Science, 2019, 363, 1193–1199 CrossRef CAS.
- F. M. Monteiro, L. T. Bach, C. Brownlee, P. Bown, R. E. Rickaby, A. J. Poulton, T. Tyrrell, L. Beaufort, S. Dutkiewicz and S. Gibbs, Sci. Adv., 2016, 2, e1501822 CrossRef PubMed.
- M. Yang, C. Batchelor-McAuley, S. Barton, R. E. Rickaby, H. A. Bouman and R. G. Compton, Angew. Chem., 2021, 133, 21167–21174 CrossRef.
- M. Yang, C. Batchelor-McAuley, S. Barton, R. E. Rickaby, H. A. Bouman and R. G. Compton, Environ. Sci.: Adv., 2022, 1, 156–163 CAS.
- R. A. Armstrong, C. Lee, J. I. Hedges, S. Honjo and S. G. Wakeham, Deep Sea Res., Part II, 2001, 49, 219–236 CrossRef.
- C. Klaas and D. E. Archer, Global Biogeochem. Cycles, 2002, 16(4), 63 CrossRef.
- F. J. Millero, Chem. Rev., 2007, 107, 308–341 CrossRef CAS.
- F. J. Millero, R. Feistel, D. G. Wright and T. J. McDougall, Deep Sea Res., Part I, 2008, 55, 50–72 CrossRef.
- A. Mucci, Am. J. Sci., 1983, 283, 780–799 CrossRef CAS.
- J. W. Morse, A. Mucci and F. J. Millero, Geochim. Cosmochim. Acta, 1980, 44, 85–94 CrossRef CAS.
- R. G. Compton, K. L. Pritchard and P. R. Unwin, Freshwater Biol., 1989, 22, 285–288 CrossRef CAS.
- J. W. Morse, R. S. Arvidson and A. Lüttge, Chem. Rev., 2007, 107, 342–381 CrossRef CAS PubMed.
- R. G. Compton and C. A. Brown, J. Colloid Interface Sci., 1994, 165, 445–449 CrossRef CAS.
- R. S. Arvidson, M. Collier, K. J. Davis, M. D. Vinson, J. E. Amonette and A. Luttge, Geochim. Cosmochim. Acta, 2006, 70, 583–594 CrossRef CAS.
- E. Ruiz-Agudo, C. Putnis, C. Jiménez-López and C. Rodriguez-Navarro, Geochim. Cosmochim. Acta, 2009, 73, 3201–3217 CrossRef CAS.
- R. Pytkowicz and J. Hawley, Limnol. Oceanogr., 1974, 19, 223–234 CrossRef.
- Y. Marcus and G. Hefter, Chem. Rev., 2006, 106, 4585–4621 CrossRef CAS PubMed.
- L. N. Plummer and E. Busenberg, Geochim. Cosmochim. Acta, 1982, 46, 1011–1040 CrossRef CAS.
- Y. Li, H. Zeng and H. Zhang, Materials Genome Engineering Advances, 2023, e4 CrossRef.
- M. Xu and S. R. Higgins, Geochim. Cosmochim. Acta, 2011, 75, 719–733 CrossRef CAS.
- M. D. Vinson, R. S. Arvidson and A. Luttge, J. Cryst. Growth, 2007, 307, 116–125 CrossRef CAS.
- J. V. Mills, H. A. Barnhart, D. J. DePaolo and L. N. Lammers, Geochim. Cosmochim. Acta, 2022, 334, 338–367 CrossRef CAS.
- L. Wang and C. V. Putnis, Acc. Chem. Res., 2020, 53, 1196–1205 CrossRef CAS PubMed.
- K. Miyata, K. Takeuchi, Y. Kawagoe, P. Spijker, J. Tracey, A. S. Foster and T. Fukuma, J. Phys. Chem. Lett., 2021, 12, 8039–8045 CrossRef CAS PubMed.
- C. Rodriguez-Navarro, A. Burgos Cara, K. Elert, C. V. Putnis and E. Ruiz-Agudo, Cryst. Growth Des., 2016, 16, 1850–1860 CrossRef CAS.
- E. Ruiz-Agudo, C. V. Putnis, L. Wang and A. Putnis, Geochim. Cosmochim. Acta, 2011, 75, 3803–3814 CrossRef CAS.
- S. Wachi and A. G. Jones, Chem. Eng. Sci., 1991, 46, 1027–1033 CrossRef CAS.
- X. Fan, C. Batchelor-McAuley, M. Yang and R. G. Compton, ACS Meas. Sci. Au, 2022, 2, 422–429 CrossRef CAS PubMed.
- T. M. Squires, R. J. Messinger and S. R. Manalis, Nat. Biotechnol., 2008, 26, 417–426 CrossRef CAS PubMed.
- R. G. Compton and C. E. Banks, Understanding Voltammetry, World Scientific, 3rd edn, 2018 Search PubMed.
- M. Yang and R. G. Compton, J. Electroanal. Chem., 2019, 836, 68–76 CrossRef CAS.
- X. Fan, C. Batchelor-McAuley, M. Yang and R. G. Compton, ACS Meas. Sci. Au, 2022, 2, 422–429 CrossRef CAS PubMed.
- T. Morton-Collings, M. Yang, C. Batchelor-McAuley, S. Barton, R. E. M. Rickaby, H. A. Bouman and R. G. Compton, Environ. Sci.: Adv., 2023, 2, 645–651 CAS.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC press, 2016 Search PubMed.
- D. L. Parkhurst and C. Appelo, U.S. Geol. Surv. Tech. Methods, 2013, 6, 497 Search PubMed.
- J. F. Adkins, J. D. Naviaux, A. V. Subhas, S. Dong and W. M. Berelson, Annu. Rev. Mar. Sci., 2021, 13, 57–80 CrossRef.
- T. R. Martz, H. W. Jannasch and K. S. Johnson, Mar. Chem., 2009, 115, 145–154 CrossRef CAS.
- R. A. Easley, M. C. Patsavas, R. H. Byrne, X. Liu, R. A. Feely and J. T. Mathis, Environ. Sci. Technol., 2013, 47, 1468–1477 CAS.
- T. Tyrrell and R. E. Zeebe, Geochim. Cosmochim. Acta, 2004, 68, 3521–3530 CrossRef CAS.
- R. A. Feely, C. L. Sabine, K. Lee, W. Berelson, J. Kleypas, V. J. Fabry and F. J. Millero, Science, 2004, 305, 362–366 CrossRef CAS PubMed.
- M. Gehlen, R. Gangstø, B. Schneider, L. Bopp, O. Aumont and C. Éthé, Biogeosciences, 2007, 4, 505–519 CrossRef CAS.
- K. Azetsu-Scott, A. Clarke, K. Falkner, J. Hamilton, E. P. Jones, C. Lee, B. Petrie, S. Prinsenberg, M. Starr and P. Yeats, J. Geophys. Res. Oceans, 2010, 115, C11021 CrossRef.
- J. D. Naviaux, A. V. Subhas, S. Dong, N. E. Rollins, X. Liu, R. H. Byrne, W. M. Berelson and J. F. Adkins, Mar. Chem., 2019, 215, 103684 CrossRef CAS.
- S. N. Chung, K. Lee, R. Feely, C. Sabine, F. Millero, R. Wanninkhof, J. Bullister, R. Key and T. H. Peng, Global Biogeochem. Cycles, 2003, 17(4), 1093 CrossRef.
- J. W. Morse, A. Mucci, L. M. Walter and M. S. Kaminsky, Science, 1979, 205, 904–905 CrossRef CAS PubMed.
- A. Mucci and J. W. Morse, Geochim. Cosmochim. Acta, 1984, 48, 815–822 CrossRef CAS.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS.
- H. Swenson and N. P. Stadie, Langmuir, 2019, 35, 5409–5426 CrossRef CAS PubMed.
- B. A. Brookes, T. J. Davies, A. C. Fisher, R. G. Evans, S. J. Wilkins, K. Yunus, J. D. Wadhawan and R. G. Compton, J. Phys. Chem. B, 2003, 107, 1616–1627 CrossRef CAS.
- T. J. Davies, C. E. Banks and R. G. Compton, J. Solid State Electrochem., 2005, 9, 797–808 CrossRef CAS.
- T. J. Davies, R. R. Moore, C. E. Banks and R. G. Compton, J. Electroanal. Chem., 2004, 574, 123–152 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04063a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |