
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzyme-catalyzed polyurethane adhesive degradation†
Angela
Romano
a,
Antonella
Rosato
a,
Laura
Sisti
 *a,
Giulio
Zanaroli
a,
Svajus Joseph
Asadauskas‡
b,
Paulina
Nemaniutė
b,
Dalia
Bražinskienė
b,
Asta
Grigucevičienė
b and
Grazia
Totaro
a
*a,
Giulio
Zanaroli
a,
Svajus Joseph
Asadauskas‡
b,
Paulina
Nemaniutė
b,
Dalia
Bražinskienė
b,
Asta
Grigucevičienė
b and
Grazia
Totaro
a
aDipartimento di Ingegneria Civile, Chimica, Ambientale e dei Materiali, Università di Bologna, Via Terracini 28, 40131, Bologna, Italy. E-mail: laura.sisti@unibo.it
bCenter for Physical Sciences and Technology, Department of Chemical Engineering and Technologies, Sauletekio Av. 3, Vilnius, LT-10257 Lithuania
First published on 11th September 2024
Abstract
Polyurethanes represent a class of highly versatile synthetic polymers, suitable for a wide range of applications. Their biological degradation is of great interest since it can allow the design of specific formulations by selecting suitable building blocks and it can contribute to the development of sustainable recycling processes. In the current study, a commercial hydrolytic enzyme (cutinase from Humicola insolens, HiC) was investigated for its ability to degrade various polyurethane adhesive formulations, by focusing first on macrodiols, then on specific polyurethanes. The aim was to identify solvent-based polyurethane formulations susceptible to enzymatic hydrolysis. First, a semi-quantitative assay, namely the emulsion turbidity test, was carried out on some macrodiols. Then, weight loss tests were carried out on specific solvent-based polyurethane formulations, and three promising formulations have shown 90, 60 and 40% degradation, after 96 h of incubation with HiC. A study of the enzymatic degradation mechanism of macrodiols and the most degradable polyurethanes was also carried out, through the characterization of the solid residues after the enzymatic degradation by infrared spectroscopy, calorimetric and thermogravimetric analysis, and the identification and/or quantification of the monomers released during the hydrolysis of macrodiols within the liquid fraction (by high-performance liquid chromatography). According to the results, a prevalent exo-type action mode for HiC against some macrodiols was found under the conditions tested, while, from a chemical point of view, the degradation seems to determine, on the polyurethane residues, a general increase in crosslinking.
1. Introduction
Thanks to the invention of the diisocyanate polyaddition technique by Otto Bayer and his collaborators in 1937, the polyurethane (PUR) industry was developed, by producing PUR from diisocyanate and polyester-based polyols. Since then, the PUR industry has significantly grown, becoming a global business sector, involving a market of billions of euros.1Currently, polyurethanes are one of the most common, versatile materials in the world, equivalent to poly(ethylene terephthalate) (PET) in terms of global plastic consumption (around 21 Mt in 2021),2 ranking 6th among global polymer production.2,3
Polyurethanes are typically block copolymers with soft and hard segments, based on polyols of low glass transition temperature and diisocyanates, respectively. While the block domain of soft segments offers high flexibility and resilience, those made of hard segments endow polyurethane with mechanical strength and improved thermal performance. When applicable, crosslinking is carried out with aromatic or aliphatic isocyanates. According to the chemical composition and content of the soft and hard segments, the material properties can be largely tuned, therefore they are suitable for many applications, involving biomedical, building and construction, automotive, textiles, etc.3 Based on the diverse range of sources and targeted applications, PUR can be classified into several classes: flexible, rigid, waterborne, thermoplastic, binders, coating, sealants, adhesives, and elastomers.4 By focusing on the adhesive field, polyurethanes are largely diffused in flexible packaging, because of low levels of migration-capable compounds. Food packaging indeed must meet strict requirements to avoid contamination of packaged items: additives (i.e. catalysts, flame retardants, antioxidants, plasticizers, solvents) included in the formulation to impart specific properties, or residues of polymerization, degradation compounds, and by-products from curing reaction or raw material production could migrate into food through the polymer films, but the most recent generation of PUR adhesives has a very low monomer concentration, therefore the risk of their migration is reduced.5 Solvent-based and solvent-free two-component system formulations, and, to a lesser extent, water-based PUR are usually employed to laminate multilayer packaging materials for food.
Within the circular economy concept, the end-of-life of polyurethanes must be assessed: currently, the most consolidated strategies, at the industrial level, are mechanical and chemical recycling. The first involves regrinding and compression moulding, while the latter occurs mainly through glycolysis.6 Both require high temperatures and pressure, and/or the use of hazardous chemicals (i.e. organic solvents, catalysts, etc.). Energy recovery is also reported as a large-scale end-of-life option for PUR, but extreme conditions are necessary, and toxic fumes can be released.7 On the other hand, recycling through biodegradation, which involves the use of microorganisms and enzymes, is still at the lab-scale but is highly promising since mild conditions are required (i.e. low temperatures, no hazardous chemicals).6 Therefore, in terms of environmental benefits and cost-effectiveness, recycling through biodegradation represents a field worthy of investigation.
In this context, the development of degradable polyurethane formulations is of great interest, and the identification and characterization of enzymes and microbes capable of breaking down PUR chains is the subject of an increasing number of studies. The literature indeed reports several studies related to the degradation of PUR, for example by laccase EC 1.10.3.2, esterase EC 3.1.1, Candida antarctica or cylindracea lipase EC 3.1.1.3, protease K EC 3.4.21.64, urease EC 3.5.1.5,8 and cutinase EC 3.1.1.74.3 Such a class of enzymes is of particular interest: it shows both lipase and esterase characteristics, and it is a serine hydrolase able to hydrolyze the insoluble cutin from plant cuticles, usually consisting of C16 or C18 hydroxy fatty acids. Several studies focus on cutinase from Humicola insolens (HiC),9,10 which was active on both ester and urethane bonds,11 causing degradation of different synthetic polymers, i.e. polyamides, polyesters.9,12 Moreover, its thermal stability can be easily increased by immobilization in inorganic layers, widening its applicability within the plastic industry.13,14
In view of these considerations, the commercial HiC cutinase has been chosen as the target enzyme for the degradation of a series of polyurethanes for solvent-based adhesive formulations. The rationale of the work focused first on a series of low molecular weight polyfunctional alcohols (also defined as polyols or macrodiols), then a series of specific PUR formulations was prepared and tested. A general scheme of PUR-based adhesives is shown in Fig. 1. The initial preliminary screening is based on semi-quantitative assays, namely an emulsion turbidity test. Subsequently, a quantitative weight loss test was performed on target PUR formulations to select the most degradable ones and to evaluate their degradation rates. Additionally, a hypothesis regarding the degradation mechanisms was developed based on the characterization of the solid residues following enzymatic degradation (using techniques such as infrared spectroscopy (FT-IR), calorimetric analysis (DSC)), as well as the identification and/or quantification of the monomers released during the hydrolysis of macrodiols within the liquid fraction through high-performance liquid chromatography (HPLC).
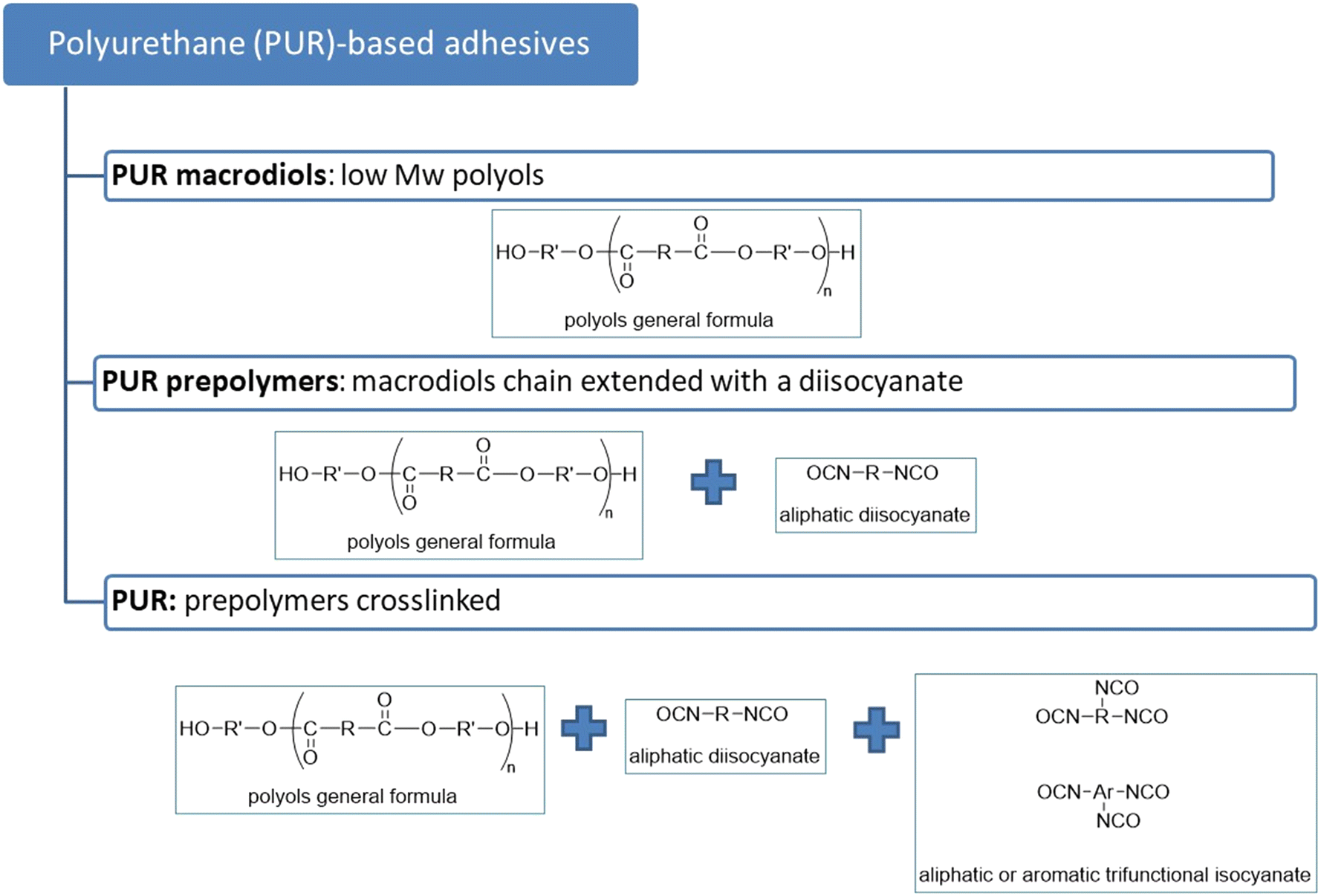 | ||
| Fig. 1 General scheme of PUR. | ||
2. Experimental
2.1 Materials
HiC is provided by ChiralVision (NZ 51032, 50 ml, 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 TBU ml−1). Seven macrodiols used in polyurethane formulations with the trade names PES1, PES2, PES3, PES4, PES5, PES6, and PES7 (PES = polyester) were provided by Covestro (Germany). These macrodiols, all of which are ester-based except for PES2, which is ether–ester based, were characterized by proton nuclear magnetic resonance (1H NMR), gel permeation chromatography (GPC), FT-IR, DSC and thermogravimetry (TGA). The codes and compositions are reported in Table 1. One difunctional isocyanate, namely 1,6-hexamethylene diisocyanate (HDI), and two trifunctional isocyanates, namely an adduct of 2,4-toluene diisocyanate (TDI) and trimethylol propane (TMP), trade name Desmodur L75 (coded L75), and a 1,6-hexamethylene diisocyanate trimer, trade name Desmodur N3300 (coded N3300), were supplied by Covestro (Germany). A commercial polyurethane sample NOVACOTE SF 716A + CA336, which is a two-component solvent free (SF) polyurethane adhesive coded NV, was provided by STTP Emballage SAS (France). Such a commercial adhesive system is used in a wide range of industrial applications, especially in flexible packaging, and consists of 65% diisocyanate (NCO component) Novacote SF 716 and 35% hardener (OH component) CA 336. Anhydrous ethyl acetate (C4H8O2, ≥99.8%) and dibutyl amine ((CH3CH2CH2CH2)2NH, ≥99.5%) were obtained from Sigma-Aldrich (now Merck, USA).
000 TBU ml−1). Seven macrodiols used in polyurethane formulations with the trade names PES1, PES2, PES3, PES4, PES5, PES6, and PES7 (PES = polyester) were provided by Covestro (Germany). These macrodiols, all of which are ester-based except for PES2, which is ether–ester based, were characterized by proton nuclear magnetic resonance (1H NMR), gel permeation chromatography (GPC), FT-IR, DSC and thermogravimetry (TGA). The codes and compositions are reported in Table 1. One difunctional isocyanate, namely 1,6-hexamethylene diisocyanate (HDI), and two trifunctional isocyanates, namely an adduct of 2,4-toluene diisocyanate (TDI) and trimethylol propane (TMP), trade name Desmodur L75 (coded L75), and a 1,6-hexamethylene diisocyanate trimer, trade name Desmodur N3300 (coded N3300), were supplied by Covestro (Germany). A commercial polyurethane sample NOVACOTE SF 716A + CA336, which is a two-component solvent free (SF) polyurethane adhesive coded NV, was provided by STTP Emballage SAS (France). Such a commercial adhesive system is used in a wide range of industrial applications, especially in flexible packaging, and consists of 65% diisocyanate (NCO component) Novacote SF 716 and 35% hardener (OH component) CA 336. Anhydrous ethyl acetate (C4H8O2, ≥99.8%) and dibutyl amine ((CH3CH2CH2CH2)2NH, ≥99.5%) were obtained from Sigma-Aldrich (now Merck, USA).
| Macrodiol | Full name | Compositiona |
|---|---|---|
| a Composition of copolymers from 1H NMR (see the ESI†). | ||
| PES1 | Poly(ethylene adipate) (PEA) | — |
| PES2 | Poly(diethylene adipate) (PDEA) | — |
| PES3 | Poly(hexylene-co-neopentyl adipate) | (PHA)0.7-(PNA)0.3 |
| PES4 | Poly(butylene adipate) (PBA) | — |
| PES5 | Poly(ethylene-co-propylene adipate) | (PEA)0.8-(PPA)0.2 |
| PES6 | Poly(hexylene adipate-co-isophthalate) | (PHA)0.7-(PHI)0.3 |
| PES7 | Poly(hexylene orthophthalate) (PHO) | — |
2.2 Preparation of PUR
For the synthesis of solvent based (SB) polyurethane, OH-terminated prepolymers, ethyl acetate and either an aromatic (L75) or aliphatic (N3300) crosslinker were used.First, the OH-terminated prepolymers were synthesized using standard laboratory glassware, by chain-extension of PES with HDI using molar ratios of 97:3 for the macrodiol and HDI, respectively. To initiate the synthesis, a predetermined amount of PES macrodiol was poured into a 3-neck flask, which was then placed in a thermostat equipped with a reflux condenser. Typically, a 100 mL flask was chosen when 10 g of macrodiol was used for the synthesis. Dry nitrogen was used to purge the flask, and the temperature was set at 80 °C ± 1 °C while stirring the reactants with a mechanical stirrer. 3 mol% of preheated HDI (with respect to PES) was then carefully added to the flask. Stirring was continued at the specified temperature, and small samples ranging from 0.1 to 0.9 mL were periodically taken to determine the amount of isocyanates as %NCO by back-titration with dibutyl amine (0.1 M). The reaction was considered complete when the titration indicated the absence of isocyanates.15 The flask was then removed from the thermostat to terminate the reaction. The synthesized OH-terminated prepolymers were stored in glass vials at room temperature (20 °C) until further testing/use.
The preheated prepolymer was brought to 35–50 °C and then metered into a clean dry vessel. To prepare the first PUR component, the prepolymer was diluted with ethyl acetate in a ratio of 1:1 (w/w) while stirring for 5 minutes. Ethyl acetate was used to reduce the viscosity of the prepolymer. An aromatic or aliphatic crosslinker was used as the second PUR component in a 1.4 mol excess. This ratio was chosen because NCO/OH ratios greater than 1.0 resulted in PUR adhesives with increased strength due to a more complete reaction between the isocyanate adducts and polyol.16 The total amount of the crosslinker was added within 10 to 50 seconds while stirring until a uniform solution was obtained. By manual blending with a spatula for 30 s at 20 °C, the mixture was immediately transferred into a 1 cm2 mold and the average thickness of the adhesives was 0.24 ± 0.05 mm. The samples in the mold were placed in an oven at 50 °C for 3 days to cure.
The synthesized PUR samples were characterized by FT-IR and DSC. The codes and compositions are reported in Table 2. The chemical structures of the main components are shown in Table 3. The samples were coded PXHL or PXHN: P = PES, H = HDI, L = L75, N = N3300, X denotes the PES used (2 for PES2, 3 for PES3 and so on).
| PUR adhesives | Macrodiol | Diisocyanate | OH/NCO (mol) | Curing agent | NCO/OH (mol) | Thickness (mm) |
|---|---|---|---|---|---|---|
| Chain extension | Crosslinking | |||||
| NV | — | — | — | — | — | 0.33 |
| P1HL | PES1 | HDI | 0.3 | L75 | 1.4 | 0.20 |
| P1HN | PES1 | HDI | 0.3 | N3300 | 1.4 | 0.17 |
| P2HL | PES2 | HDI | 0.3 | L75 | 1.4 | 0.20 |
| P2HN | PES2 | HDI | 0.3 | N3300 | 1.4 | 0.25 |
| P3HL | PES3 | HDI | 0.3 | L75 | 1.4 | 0.31 |
| P3HN | PES3 | HDI | 0.3 | N3300 | 1.4 | 0.23 |
| P6HL | PES6 | HDI | 0.3 | L75 | 1.4 | 0.23 |
| P6HN | PES6 | HDI | 0.3 | N3300 | 1.4 | 0.20 |
| Chemical structure | Name |
|---|---|
 Poly(ethylene adipate) (PEA) Poly(ethylene adipate) (PEA) |
PES1 |
 Poly(diethylene adipate) (PDEA) Poly(diethylene adipate) (PDEA) |
PES2 |
 Poly(hexylene-co-neopentyl adipate) Poly(hexylene-co-neopentyl adipate) |
PES3 |
 Poly(butylene adipate) Poly(butylene adipate) |
PES4 |
 Poly(ethylene-co-propylene adipate) Poly(ethylene-co-propylene adipate) |
PES5 |
 Poly(hexylene adipate-co-isophthalate) Poly(hexylene adipate-co-isophthalate) |
PES6 |
 Poly(hexylene orthophthalate) Poly(hexylene orthophthalate) |
PES7 |
| Diisocyanate | |
 1,6-Hexamethylene diisocyanate 1,6-Hexamethylene diisocyanate |
HDI |
| Curing agent | |
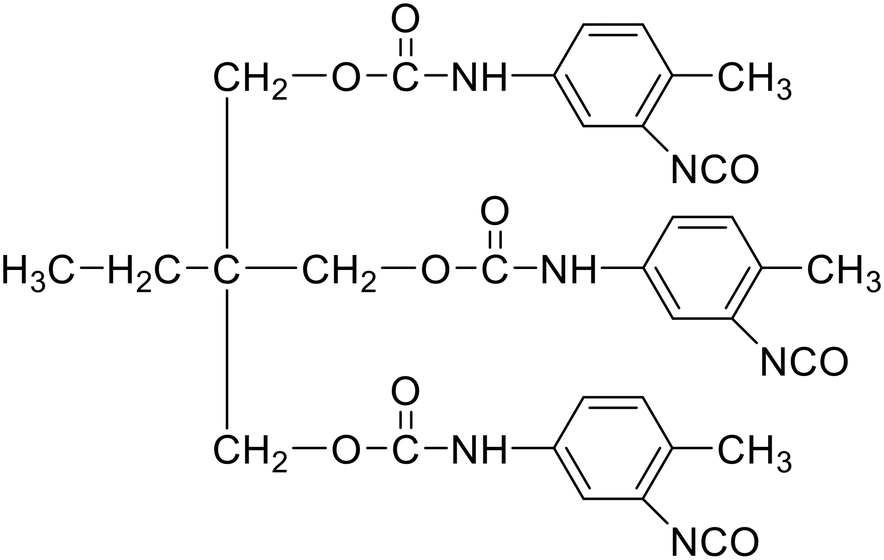
|
L75 |
 HDI trimer HDI trimer |
N3300 |
2.3 Measurements
1H NMR analysis was performed with a Varian Mercury 400 spectrometer and spectra report the chemical shifts in ppm downfield from tetramethylsilane (TMS); the samples were dissolved in CDCl3.GPC analysis was performed using a Knauer Azura device, equipped with a refractive index detector and a PL gel 5 μm Minimixed-C column (detection limit 500 g mol−1), in CHCl3 at ambient temperature. Number average molecular weight (Mn), weight average molecular weight (Mw) and polydispersity index (PD) were determined by comparison with a universal calibration curve with polystyrene standards.
ATR FT-IR analysis was conducted using a Perkin Elmer Spectrum One spectrometer. All spectra were recorded after 16 scans over the wavenumber range of 650–4000 cm−1.
DSC analysis was carried out under nitrogen flow, using a Perkin-Elmer DSC6. All the samples (around 10 mg) were subjected to a first heating step (to erase their thermal history), a cooling step, and a second heating step. The temperature program started from ambient temperature until melting (rate 20 °C min−1), followed by 2 min of isotherm at high temperature, cooling down to maximum −70 °C (rate 10 °C min−1), then second heating up to the melting (rate 10 °C min−1). The glass transition temperature (Tg), the cold crystallization temperature (TCC), the melting temperature (Tm), and their corresponding enthalpies (ΔHCC, ΔHm) were measured during the 2nd heating scan, while the crystallization temperature (TC) and its enthalpy (ΔHC) were recorded during the cooling scan. Tg was determined as the midpoint of the heat capacity increment related to the glass-to-rubber transition.
A Perkin Elmer TGA4000 apparatus was used for the TGA analysis. All the samples (around 15 mg) were heated from 50 to 900 °C (heating rate 10 °C min−1, N2 flow 40 mL min−1). From the intersections of the tangents of the initial and the inflection points, the onset degradation temperature (Tonset) was measured.
000 g mol−1) in 250 mL of phosphate buffer. A disperser was used to homogenize the emulsion, then the solvent was removed by heating the sample for 1 h at 50 °C. After that, a mixture containing 4 mL of PES emulsion (0.1% w/v) in buffer phosphate (0.1 M), plus 1 U mL−1 of enzyme, was incubated in a screw-cap glass vial (10 mL) with stirring in a thermostatic bath set at the optimal temperature and pH of cutinase (40 °C, pH 8). Free-enzyme controls were also prepared under the same conditions. The turbidity, derived from insoluble PES, was measured at 650 nm over time, and the biodegradation rate was calculated as the decrease of absorbance over time (OD650nm h−1) through linear fitting up to 60 min.
3. Results and discussion
3.1 Macrodiols' enzymatic degradation
Some macrodiols, whose codes, full names and formulas are specified in Tables 1 and 3, were characterized. They are low molecular weight polyols, all ester-based apart from PES2, which contains both ether and ester moieties. The samples from PES1 to PES5 present aliphatic structures while PES6 and PES7 are aromatic. For the copolymers (PES3, PES5, PES6), the ratio between the two moieties has been evaluated by 1H NMR analysis through comparison of the integration areas of opportune resonance peaks. Such values are reported in Table 1 while the spectra and the details are reported in the ESI† (Fig. S1–S3).Molecular and thermal properties of the macrodiols are reported in Table 4. GPC data show that, as expected, the samples have low Mw in the range 5000–11000 g mol−1. From DSC analysis, PES2, PES5 and PES7 are amorphous, with a glass transition temperature around −51, −50, and −21 °C, respectively. PES1, PES3, PES4 and PES6 are semicrystalline: they all melt below 50 °C, and they show a Tc at 7, 1.5, 30 and 8 °C, respectively. The curves are reported in ESI,† Fig. S4. TGA data show the high thermal stability of all the samples, since Tonset data are within the range 350–396 °C. The thermal degradation pathway of all the samples occurs in one main process, as visible from the thermogravimetric curves and their corresponding derivative profiles (ESI,† Fig. S5).
| GPC | TGA | DSC | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sample | M w (×10−3 g mol−1) | PD | T onset (°C) | Cooling scan | 2nd heating scan | |||||
| T c (°C) | ΔHc (J g−1) | T g (°C) | T cc (°C) | ΔHcc (J g−1) | T m (°C) | ΔHm (J g−1) | ||||
| nd: not detectable in the condition tested; /: thermal transition absent. | ||||||||||
| PES1 | 7.2 | 2.1 | 350 | 7 | 57 | −46 | / | / | 46 | 62 |
| PES2 | 11 | 1.9 | 384 | / | / | −51 | / | / | / | / |
| PES3 | 7.8 | 2.1 | 396 | 1.5 | 31 | nd | / | / | 19–28 | 28 |
| PES4 | 9.8 | 2.2 | 386 | 30 | 61 | nd | / | / | 45–53 | 62 |
| PES5 | 7.8 | 2.0 | 363 | / | / | −50 | 6.9 | 22 | 35 | 23 |
| PES6 | 9.5 | 1.7 | 389 | 8 | 40 | nd | / | / | 27–34 | 37 |
| PES7 | 5.1 | 1.9 | 367 | / | / | −21 | / | / | / | / |
IR curves are consistent with the chemical structures: CH2 stretching from the aliphatic chains is visible around 2930 cm−1 as well as an intense carbonyl band, around 1715 cm−1. The double bond C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C from the aromatic ring is located around 1580 cm−1 in the PES7 profile (Fig. S6†).
C from the aromatic ring is located around 1580 cm−1 in the PES7 profile (Fig. S6†).
In order to evaluate the potential degradability of PES in the presence of cutinase HiC, emulsion turbidity tests were carried out.17 The obtained data, reported in Table 5, are compared to free-enzyme controls, tested under the same conditions. The absorbance of PES emulsions incubated in the absence of enzyme (free-enzyme controls) did not significantly decrease. Conversely, the turbidity of the emulsions rapidly and extensively decreased in the presence of cutinase, indicating that almost all PES, apart from PES7, are biodegradable. The aromatic nature of PES7 and the steric hindrance of the structure are probably responsible for its low biodegradation. No complete resolution of the turbidity was achieved under the conditions tested.

|
The degradation mechanisms of PES were investigated through the identification and quantification, via HPLC-RID, of the monomers released from the corresponding emulsions at the end of incubation (i.e. ethylene glycol and adipic acid for PES1, diethylene glycol and adipic acid for PES2, neopentylglycol, 1,6-hexanediol and adipic acid for PES3, 1,4-butanediol and adipic acid for PES4, ethylene glycol, 1,2 propanediol and adipic acid for PES5, 1,6-hexanediol, i-phthalic acid and adipic acid for PES6, and 1,6-hexanediol and o-phthalic acid for PES7). In more detail, no monomers were detected in all PES controls (enzyme-free), as well as in all enzymatically treated PES7 emulsions (data not shown), according to the absence of degradation observed in these samples. Conversely, different monomers were detected in all the other polyol samples after treatment with cutinase (Fig. 2). By comparing the polyols, higher monomer concentrations were observed in PES3 (i.e. neopentylglycol, 1,6-hexanediol and adipic acid) and PES5 emulsions (i.e. 1,2 propanediol), while lower concentrations were found in PES4 (i.e. 1,4-butanediol and adipic acid), PES6 (i.e. 1,6-hexanediol and adipic acid), PES1 (i.e. adipic acid), and PES2 emulsions (i.e. diethylene glycol). These results suggest a prevalent exo-type action mode for cutinase on PES3 and PES5, and a mixed exo/endo action mode for PES1, PES2, PES4 and PES6, under the conditions tested. However, further analyses would be necessary to confirm the hypothesis, since dimers, trimers, oligomers and/or other degradation by-products could be present. It should be also underlined that higher concentrations of diols (with respect to the acids) were generally detected in all the samples, probably due to the preferential hydrolysis of the OH-end by cutinase12 and the fact that PES are OH-terminated macrodiols. In any case, HiC proved to be efficient in the degradation of all the polyols apart from PES7.
 | ||
| Fig. 2 Total monomer concentrations (mmol L−1) detected in PES1 (A), PES2 (B), PES3(C), PES4 (D), PES5 (E), and PES6 (F) emulsions in the presence of different enzymes and in the negative controls (enzyme-free) at the end of incubation. No monomers were detected in PES7 emulsion (data not shown). Red bars indicate the maximum achievable concentration (mmol L−1). | ||
3.2 Polyurethanes' enzymatic degradation
The preparation of solvent-based polyurethanes was carried out through the reaction of selected PES1, PES2, PES3, and PES6 with 3 mol% of HDI, and an aromatic (L75) or aliphatic (N3300) crosslinking agent.16 Ethyl acetate was chosen as the solvent since it has relatively low toxicity compared to some other solvents (i.e. acetone and methyl-ethyl ketone). Additionally, the fast evaporation rate of ethyl acetate helps in the drying and curing process of the adhesive. Formulations with PES4 and PES5 were excluded because under the conditions tested (curing at 50 °C), the reaction was significantly slower compared to the other macrodiols, and the final PUR formulations presented liquid-like properties. A commercial sample (code NV) was used as a reference. The codes and compositions are reported in Table 2, while Table 3 reports the chemical structures of the main components.From DSC analysis, all the samples were found amorphous, with a single and intense Tg within the range −48-15 °C (Table 6). The presence of hard and soft segments in polyurethanes typically results in a phase-separated morphology, but both phases are generally amorphous in highly crosslinked systems. The amorphous nature is predominant due to the constraints imposed by the crosslinks, which prevent long-range ordering.18 IR curves are consistent with the chemical structures (Fig. 3). In more detail, PUR present the stretching vibration of the N–H group at 3300 cm−1, while the asymmetric and symmetric stretching of CH2 is visible at 2935 cm−1 and 2850 cm−1, respectively. The CO stretching vibrations of ester and urethane (amide I band) are detectable at 1730 cm−1 and 1705 cm−1, respectively, while the C–O stretching vibration can be found within the region 1300–1100 cm−1. The amide II band, corresponding to the N–H bending vibration with C–N stretching vibration from the –C–NH group, is located at 1530 cm−1. The bands around 1597 cm−1 (CC from the aromatic ring) can be coupled with the absorbance around 814 cm−1 (C–H out of plane bending vibration in the 1,4-disubstituted aromatic ring).19
| Sample | DSC |
|---|---|
| 2nd heating scan | |
| T g (°C) | |
| NV | 15 |
| P1HL | −31 |
| P1HN | −39 |
| P2HL | −37 |
| P2HN | −44 |
| P3HL | −35 |
| P3HN | −48 |
| P6HL | −27 |
| P6HN | −39 |
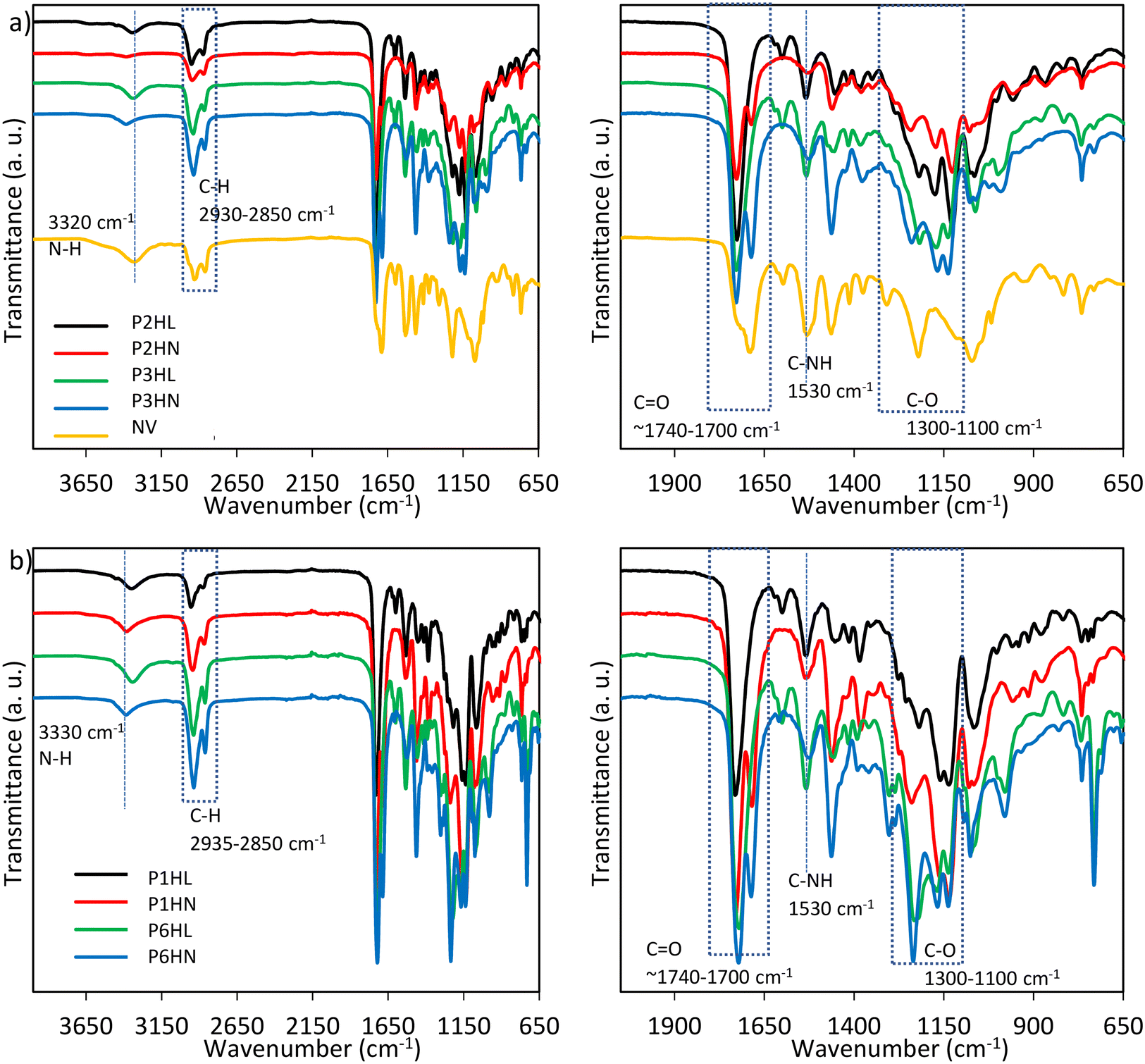 | ||
| Fig. 3 FT-IR spectra of PUR adhesives: a) P2HL, P2HN, P3HL, P3HN, NV; b) P1HL, P1HN, P6HL, P6HN; zoom region (1950–650 cm−1) on the right side. | ||
To evaluate the degradation rate of the formulated polyurethanes, a quantitative screening through a weight loss test was performed in the presence of HiC. Negligible weight loss (around 0.0–3.7%) occurred in the negative (enzyme-free) controls for all the PUR adhesives after 96 h of incubation in buffer, as well as with the reference (Table 7). Conversely, cutinase was particularly active on P2HL, P1HN and P2HN, leading to approximately 90, 60 and 40% of degradation, respectively, after 96 h of incubation. In particular, the degradation rate of cutinase on P2HL, P1HN and P2HN was 0.30 and 0.12 mg h−1 cm−2, respectively (Table 7). The weight loss (%) of PUR over time in the presence of cutinase is shown in Fig. 4. Compared to the literature, these are highly encouraging results since for example, Magnin et al.3 obtained a weight loss of 33% after 51 days in the presence of esterase, on a polycaprolactone polyol-based polyurethane, while Olivito et al.20 obtained a weight loss of 24% after 30 days in the presence of cholesterol esterase, on a polyether-polyurethane foam. Higher weight losses (i.e. 40% in the presence of Bacillus subtilis MZA-75 and Pseudomonas aeruginosa MZA-85, or 60.6% in the presence of Aspergillus flavus (ITCC 6051)) were reported by Liu et al.21 for polyester-PUR, but for longer times (i.e. 30 days) with respect to the conditions of the current study.
| PUR adhesive | Enzyme (activity) | % weight loss (96 h) | Degradation rate (mg h−1 cm−2) |
|---|---|---|---|
| NV | Negative control | 0.2 | — |
| Cutinase | 0.7 | 0.00 | |
| P1HL | Negative control | 2.1 | — |
| Cutinase | 14.4 | 0.02 | |
| P1HN | Negative control | 2.9 | — |
| Cutinase | 60.9 | 0.12 | |
| P2HL | Negative control | 2.2 | — |
| Cutinase | 87.1 | 0.30 | |
| P2HN | Negative control | 3.7 | — |
| Cutinase | 41.3 | 0.12 | |
| P3HL | Negative control | 2.1 | — |
| Cutinase | 5.0 | 0.01 | |
| P3HN | Negative control | 1.6 | — |
| Cutinase | 8.3 | 0.02 | |
| P6HL | Negative control | 0.3 | — |
| Cutinase | 2.8 | 0.01 | |
| P6HN | Negative control | 0.0 | — |
| Cutinase | 5.1 | 0.01 |
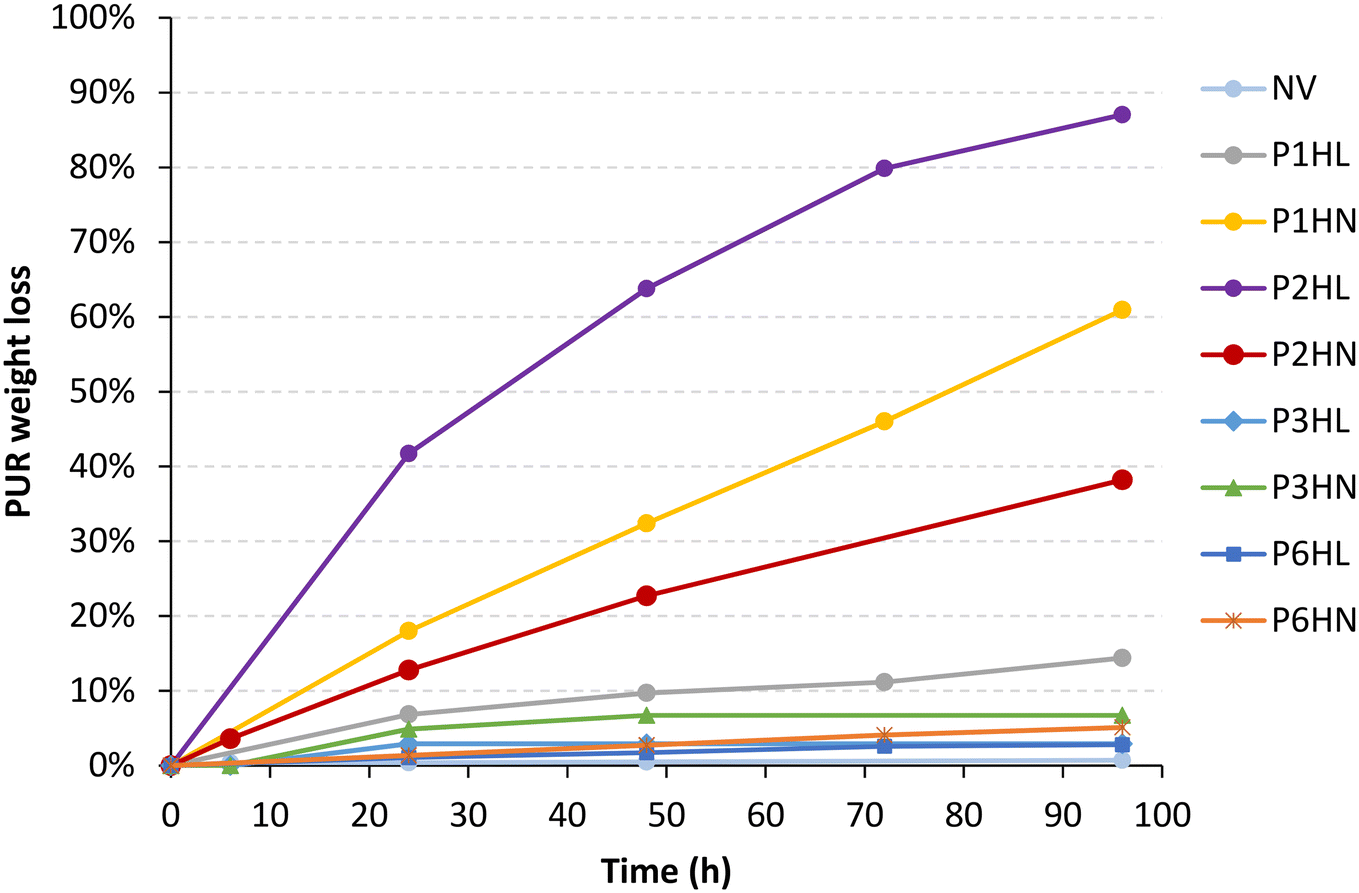 | ||
| Fig. 4 Weight loss (%) by cutinase HiC (100 U mL−1) of PUR adhesives, as a function of the incubation time (hours). | ||
Therefore, P2HL, P1HN and P2HN formulations were found very promising in terms of enzymatic degradation, in particular the one containing the poly(diethylene adipate) (PES2) chain extended with 1,6-hexamethylene diisocyanate and crosslinked with the aromatic trifunctional isocyanate (L75). The latter was found preferable with respect to the aliphatic N3300, possibly because its aromatic structure contains ester bonds, while N3300 does not. For this reason, PUR formulations in which the aromatic crosslinker has been used have a higher number of ester linkages and cutinase, particularly effective in catalyzing their cleavage, resulted in more efficient biodegradation of those PUR formulations. In addition, the aromatic crosslinker involved a faster crosslinking rate compared to the aliphatic one, 2 days against 5, respectively. The latter often requires high temperatures of 50–60 °C for effective crosslinking during PUR reticulation. Desmodur L75 is also much more widely used in industrial SB formulations than other isocyanate crosslinkers. In general, more flexible PUR chains may offer some advantages when it comes to degradation because they can be more accessible to environmental factors such as water, oxygen, and enzymes, which can facilitate the breakdown of the polymer.
Concerning the non-degradable formulations, they are based on poly(hexylene-co-neopentyl adipate) (PES3), poly(hexylene adipate-co-isophthalate) (PES6), HDI, L75 or N3300. Despite the fact that pristine macrodiols PES3 and PES6 were more degradable than PES1 and PES2 (Table 5), their corresponding final formulations do not degrade significantly (weight loss below 10%, Fig. 4). The reasons of such behaviour could be ascribable to the crosslinking degree of the materials, possibly hindering the diffusion and adsorption (and consequently the activity) of cutinase within the soft segments. The enzyme adsorption indeed represents a major limitation in PUR hydrolysis.22
To investigate the possible mechanism of the enzymatic degradation, P2HL, P2HN and P1HN were analyzed by FT-IR and DSC after incubation with HiC. The mobility of polymeric chains is known to be restricted by crosslinking, which causes an increase of the Tg. This could be the result of increased chemical crosslinking in polyurethane chains by covalent bonding, increased physical crosslinking by hydrogen bonding, or both. Since polyurethanes have N–H and CO groups, it is possible for hydrogen bonds to form between hard and soft segments, and this can have a substantial impact on the physical properties of polyurethanes. Infrared spectra of the H-bonded N–H band show frequency shifts, as well as band width and intensity variations. H-bonded N–H groups (3300–3340 cm−1) are shifted to lower frequencies, whereas non-H-bonded N–H groups (3400–3460 cm−1) exhibit no shift in frequency. Consequently, by monitoring the frequency shift of N–H groups, variation of strength of H-bonding can be determined and provide indications of the presence of physical crosslinking.18 By observing such regions of interest in FT-IR spectra of the degraded PUR compared to the pristine adhesives, a shift to lower frequency appears in all the samples (Fig. 5).
 | ||
| Fig. 5 FT-IR spectra of degraded PUR adhesives: a) P2HL, b) P2HN, c) P1HN; zoom region (1950–650 cm−1) on the right side. | ||
In addition, in the case of P2HN and P1HN (Fig. 5b and c), by observing the bidentate band in the carbonyl region (1730–1650 cm−1), the relative intensity between the carbonyl stretching at 1726 cm−1 and the one at 1685 cm−1 changes in favor of the latter after enzymatic incubation, which is related to strongly hydrogen bonded carbonyl groups. The first band, on the other hand, is related to non-hydrogen bonded carbonyl groups.23 This means that the number of well-ordered and strongly hydrogen bonded carbonyl groups increases, probably due to the increase of OH/NH end-groups able to form hydrogen bonds. Moreover, in P2HN treated with cutinase, the band related to hydroxyl and amine moieties resulting from hydrolysis appears around 3400 cm−1.
In the case of P2HL (Fig. 5a), the monodentate carbonyl band after incubation with HiC shifts from 1725 to 1703 cm−1, confirming the formation of ordered H-bonded carbonyls. However, this can also indicate a cleavage of the ester bonds:24 the reduction of peaks at 1170 and 1125 cm−1 (C–O stretching) and the relative increase of the band at 1223 cm−1 (C–N stretching of urethane bonds) would confirm a change in the ester components indeed.8
An overall increase in crosslinking is, to some extent, confirmed by the thermal analysis. DSC data are reported in Table 8: a single and intense Tg is present in the second heating scan. The degraded P2HL showed an increase of Tg with respect to the pristine PUR, suggesting the formation of more crosslinked bonds that reduce the mobility of soft segments.25,26 This could be related to the increase of OH/NH end-groups that are able to form more hydrogen bonds, thus lowering the mobility of polymeric chains. In the case of P2HN and P1HN, no significant differences compared to pristine adhesives were detected. In addition, all the samples present small endothermic transitions during the first heating scans (not shown), induced by room temperature annealing, around 170 °C (P2HL) or around 60 °C (all the other samples), while P2HN also showed transition during the second heating scan around 160 °C. Such phenomena are reported to be related to the disruption of short-range order hard segments (around 55–80 °C) or the disruption of long-range order hard segments (around 100–180 °C).27
| DSC | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Enzyme | Units/mL | Time (h) | 1st heating scan | Cooling scan | 2nd heating scan | ||||||||
| T g1 (°C) | ΔCp1 (J g−1 °C−1) | T g2 (°C) | ΔCp2 (J g−1 °C−1) | T m (°C) | ΔHm (J g−1) | T c (°C) | T g1 (°C) | ΔCp1 (J g−1 °C−1) | T m (°C) | ΔHm (J g−1) | ||||
| nd: not detectable in the condition tested; /: thermal transition absent. | ||||||||||||||
| P2HL | / | / | / | −42 | 0.647 | 33 | 0.059 | 171 | 8.7 | / | −37 | 0.686 | / | / |
| Cutinase | 100 | 96 | −42 | 0.519 | / | / | 65 | 6.7 | / | −34 | 0.488 | / | / | |
| P2HN | / | / | / | −48 | 0.638 | / | / | 55 | 7.2 | / | −44 | 0.628 | 163 | 1.7 |
| Cutinase | 100 | 96 | nd | nd | / | / | nd | nd | / | −44 | 0.585 | / | / | |
| P1HN | / | / | / | −42 | 0.531 | / | / | 61 | 7.5 | / | −39 | 0.556 | / | / |
| Cutinase | 100 | 96 | −42 | 0.532 | / | / | 61 | 5.6 | / | −39 | 0.573 | / | / | |
In summary, from a chemical point of view, the degradation seems to determine, on the residues, a general increase in crosslinking, turning into more intense H-bonded N–H and CO groups (FT-IR), and, to some extent, to an increase of Tg (DSC).
4. Conclusions
The enzymatic degradation of some PUR adhesive formulations is described. The study has focused first on single components (i.e. macrodiols), then specific formulations were assessed. According to current findings, cutinase from Humicola insolens is active against almost all macrodiols selected, then some PUR formulations were prepared, and weight loss tests were carried out. Cutinase was particularly effective on P2HL, P1HN and P2HN, leading to approximately 90, 60 and 40% degradation, respectively, after 96 h of incubation. The degradation rate of cutinase was 0.30 mg h−1 cm−2 on P2HL, and 0.12 mg h−1 cm−2 on P1HN and P2HN. Thus, at least three promising formulations resulted from all the characterization carried out: P2HL, P2HN and P1HN. Such formulations, where the most promising one contains a poly(diethylene adipate) chain extended with 1,6-hexamethylene diisocyanate and crosslinked with an aromatic trifunctional isocyanate, could be used as adhesives in multilayer materials. The idea of the future work is to embed a protected cutinase (i.e. immobilized in a layered double hydroxide) within the formulation in order to obtain multilayer materials able to delaminate on-demand, as already done for other polymeric adhesives.13,14 Finally, it must be highlighted that the monomers/oligomers resulting from degradation could be recovered and chemically upcycled into new materials, contributing to a more sustainable plastic economy.Data availability
The data supporting this article have been shown in figures or tables, and they are included as part of the article and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study was carried out within the framework of the TERMINUS project, funded by the European Union under Horizon 2020. Call: H2020-NMBP-ST-IND-2018. Grant Agreement: 814400. This report reflects only the views of the authors. European Commission and Research Executive Agency are not responsible for any use that may be made of the information it contains, see §29.5 of H2020 General Model Grant Agreement for details. Covestro Deutschland AG (Germany) is gratefully acknowledged for the preparation of prepolymers. STTP Emballage SAS (France) is gratefully acknowledged for supplying the commercial PUR.References
- P. Alves, P. Ferreira and M. H. Gil, Biomedical polyurethanes-based materials, in Polyurethane: properties, structure and applications, Polymer science and technology, Nova Science Publishers, New York, 2012, pp. 25–50 Search PubMed.
- Plastics – the Facts 2022, https://plasticseurope.org/knowledge-hub/plastics-the-facts-2022/ (accessed on 2023/12/12).
- A. Magnin, E. Pollet, R. Perrin, C. Ullmann, C. Persillon, V. Phalip and L. Avérous, Enzymatic recycling of thermoplastic polyurethanes: Synergistic effect of an esterase and an amidase and recovery of building blocks, Waste Manage., 2019, 85, 141–150, DOI:10.1016/j.biotechadv.2019.107457.
- J. O. Akindoyo, M. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, Polyurethane types, synthesis and applications–a review, RSC Adv., 2016, 6(115), 114453–114482, 10.1039/C6RA14525F.
- F. Isella, E. Canellas, O. Bosetti and C. Nerin, Migration of non intentionally added substances from adhesives by UPLC–Q-TOF/MS and the role of EVOH to avoid migration in multilayer packaging materials, J. Mass Spectrom., 2013, 48(4), 430–437, DOI:10.1002/jms.3165.
- G. Rossignolo, G. Malucelli and A. Lorenzetti, Recycling of polyurethanes: where we are and where we are going, Green Chem., 2024, 26, 1132–1152, 10.1039/d3gc02091f.
- A. Kemona and M. Piotrowska, Polyurethane Recycling and Disposal: Methods and Prospects, Polymer, 2020, 12(8), 1752, DOI:10.3390/polym12081752.
- A. Magnin, E. Pollet, V. Phalip and L. Avérous, Evaluation of biological degradation of polyurethanes, Biotechnol. Adv., 2020, 39, 107457, DOI:10.1016/j.biotechadv.2019.107457.
- V. Ferrario, A. Pellis, M. Cespugli, G. M. Guebitz and L. Gardossi, Nature inspired solutions for polymers: will cutinase enzymes make polyesters and polyamides greener?, Catalysts, 2016, 6(12), 205, DOI:10.3390/catal6120205.
- A. Romano, S. Varriale, C. Pezzella, G. Totaro, J. M. Andanson, V. Verney and L. Sisti, Natural deep eutectic solvents as thermostabilizer for Humicola insolens cutinase, New Biotechnol., 2023, 76, 118–126, DOI:10.1016/j.nbt.2023.05.00.
- F. Di Bisceglie, F. Quartinello, R. Vielnascher, G. M. Guebitz and A. Pellis, Cutinase-catalyzed polyester-polyurethane degradation: elucidation of the hydrolysis mechanism, Polymers, 2022, 14(3), 411, DOI:10.3390/polym14030411.
- A. Rosato, A. Romano, G. Totaro, A. Celli, F. Fava, G. Zanaroli and L. Sisti, Enzymatic degradation of the most common aliphatic bio-polyesters and evaluation of the mechanisms involved: an extended study, Polymers, 2022, 14(9), 1850, DOI:10.3390/polym14091850.
- A. Romano, A. Rosato, G. Totaro, G. Zanaroli, A. Celli and L. Sisti, Recycling by-design of plastic through formulation with thermally protected enzymes in layered double hydroxide structures, J. Cleaner Prod., 2023, 384, 135517, DOI:10.1016/j.jclepro.2022.135517.
- A. Romano, A. Rosato, S. Bianchi, G. Zanaroli, A. Celli, G. Totaro and L. Sisti, Triggering of Polymer-Degrading Enzymes from Layered Double Hydroxides for Recycling Strategies, Int. J. Mol. Sci., 2023, 24(1), 831, DOI:10.3390/ijms24010831.
- S. J. Asadauskas, P. Nemaniutė, D. Bražinskienė, O. Eicher-Lorka and V. Verney, Time–temperature superposition for kinetic mapping of solventless autocatalytic addition of diisocyanates and macrodiols, RSC Adv., 2023, 13(14), 9686–9696, 10.1039/D2RA08326D.
- A. Khoon Poh, L. Choy Sin, C. Sit Foon and C. Cheng Hock, Polyurethane wood adhesive from palm oil-based polyester polyol, J. Adhes. Sci. Technol., 2014, 28(11), 1020–1033, DOI:10.1080/01694243.2014.883772.
- P. Sriyapai, K. Chansiri and T. Sriyapai, Isolation and characterization of polyester-based plastics-degrading bacteria from compost soils, Microbiology, 2018, 87, 290–300, DOI:10.1134/S0026261718020157.
- H. C. Jung, S. J. Kang, W. N. Kim, Y. B. Lee, K. H. Choe, S. H. Hong and S. B. Kim, Properties of crosslinked polyurethanes synthesized from 4, 4′-diphenylmethane diisocyanate and polyester polyol, J. Appl. Polym. Sci., 2000, 78(3), 624–630, DOI:10.1002/1097-4628(20001017)78:3<624::AID-APP180>3.0.CO;2-O.
- D. Rosu, L. Rosu and C. N. Cascaval, IR-change and yellowing of polyurethane as a result of UV irradiation, Polym. Degrad. Stab., 2009, 94(4), 591–596, DOI:10.1016/j.polymdegradstab.2009.01.013.
- F. Olivito, P. Jagdale and G. Oza, Synthesis and Biodegradation Test of a New Polyether Polyurethane Foam Produced from PEG 400, L-Lysine Ethyl Ester Diisocyanate (L-LDI) and Bis-hydroxymethyl Furan (BHMF), Toxics, 2023, 11(8), 698, DOI:10.3390/toxics11080698.
- J. Liu, J. He, R. Xue, B. Xu, X. Qian, F. Xin, L. M. Blank, J. Zhou, R. Wei, W. Dong and M. Jiang, Biodegradation and up-cycling of polyurethanes: Progress, challenges, and prospects, Biotechnol. Adv., 2021, 48, 107730, DOI:10.1016/j.biotechadv.2021.107730.
- C. Gamerith, E. H. Acero, A. Pellis, A. Ortner, R. Vielnascher, D. Luschnig and G. M. Guebitz, Improving enzymatic polyurethane hydrolysis by tuning enzyme sorption, Polym. Degrad. Stab., 2016, 132, 69–77, DOI:10.1016/j.polymdegradstab.2016.02.025.
- J. Kucińska-Lipka, I. Gubanska and A. Skwarska, Microporous polyurethane thin layer as a promising scaffold for tissue engineering, Polymers, 2017, 9(7), 277, DOI:10.3390/polym9070277.
- J. Schmidt, R. Wei, T. Oeser, L. A. Dedavid e Silva, D. Breite, A. Schulze and W. Zimmermann, Degradation of polyester polyurethane by bacterial polyester hydrolases, Polymers, 2017, 9(2), 65, DOI:10.3390/polym9020065.
- B. Pilch-Pitera, Examination of the enzyme resistance of polyurethane powder coatings, J. Polym. Environ., 2013, 21, 215–223, DOI:10.1007/s10924-012-0519-1.
- T. L. Yu, T. L. Lin, Y. M. Tsai and W. J. Liu, Morphology of polyurethanes with triol monomer crosslinked on hard segments, J. Polym. Sci., Part B: Polym. Phys., 1999, 37(18), 2673–2681, DOI:10.1002/(SICI)1099-0488(19990915)37:18<2673::AID-POLB11>3.0.CO;2-N.
- Y. M. Tsai, T. L. Yu and Y. H. Tseng, Physical properties of crosslinked polyurethane, Polym. Int., 1998, 47(4), 445–450, DOI:10.1002/(SICI)1097-0126(199812)47:4<445::AID-PI82>3.0.CO;2-B.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00253a |
| ‡ This research article is dedicated to the memory of Dr. Svajus Joseph Asadauskas, friend and colleague, who prematurely left us in July 2023. His enthusiasm for science is a stimulus and an inspiration for his collaborators. |
| This journal is © The Royal Society of Chemistry 2024 |