
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biphasic phase-transfer catalysis: epoxidation of vegetable oils by surface active ionic liquids in water†
Markus
Hegelmann
a,
Wilson F.
Bohórquez
b,
Johannes
Luibl
c,
Andreas
Jess
c,
Alvaro
Orjuela
 b and
Mirza
Cokoja
*a
b and
Mirza
Cokoja
*a
aSchool of Natural Sciences, Department of Chemistry and Catalysis Research Center, Technical University of Munich, Ernst-Otto-Fischer-Straße 1, D-85747 Garching bei München, Germany. E-mail: mirza.cokoja@tum.de
bDepartment of Chemical and Environmental Engineering, Universidad Nacional de Colombia, 111321 Bogotá D.C., Colombia
cFaculty of Engineering Science, Chair of Chemical Engineering, University of Bayreuth, Universitätsstraße 30, D-95447 Bayreuth, Germany
First published on 26th July 2024
Abstract
Vegetable oils (VOs) are an environmentally benign alternative and sustainable carbon feedstock for various industrially relevant compounds, e.g. epoxidized products (EVOs). The commercial production of EVOs is a heterogeneous liquid–liquid reaction with low reaction rates and a limited epoxide selectivity. Furthermore, the separation of the EVOs from the reaction mixture is very intricate, limiting large-scale applicability. In this work, we introduce surface-active imidazolium tungstate ionic liquids (SAILs) as sustainable catalysts for the epoxidation of VOs in water using hydrogen peroxide as a green oxidant. Micelle formation and substrate uptake into the aqueous phase depend on the nature of the cation of the SAIL catalyst, which was studied by dynamic light scattering (DLS), transmission electron microscopy (TEM) and cryo-TEM at various concentrations and temperatures. Recycling studies demonstrate that the catalyst remains in the aqueous phase and can be recovered completely. The absence of the catalyst and additive in the product phase is verified by inductively coupled plasma mass spectrometry (ICP-MS) and 31P-NMR spectroscopy.
Introduction
The limited supply and growing consumption of fossil carbon resources require sustainable alternatives. Vegetable oils (VOs) are abundant and renewable, and thus industrially relevant precursors for lubricants, plasticizers and polymer stabilizers, cosmetics, surfactants, coatings and resins.1 For instance, the epoxidation of waste lipids (e.g. used cooking oils, yellow/brown grease, rendering oils or oil mill effluents) offers pathways for environmentally friendly waste recycling towards sustainable value-added products.2,3 On an industrial scale, unsaturated VOs are converted to their epoxidation products (EVOs) via the Prilezhaev reaction with peracetic acid or performic acid, which are formed in situ by the acid-catalyzed oxidation of the corresponding acids with aqueous hydrogen peroxide (H2O2).2–4 However, this method suffers from intrinsic drawbacks, such as mass transport limitations between the organic substrate and the oxidant in the aqueous phase, difficult separation of the product/non-recyclable catalyst mixture, formation of corrosive carboxylic acids and low selectivities due to acid-catalyzed ring opening reactions.5 Most often, these systems require large amounts of acetic acid in addition to sulfuric acid to efficiently activate H2O2 compared to common catalysis systems.6 Epoxidation of VOs with molecular transition metal catalysts was studied as well. Methyltrioxorhenium, a well-known epoxidation catalyst, was used for VO epoxidation with good reaction rates under mild conditions.7 Yet, catalyst recycling has not been systematically studied.8 Other homogeneous approaches involve molybdenum complexes which, however, demand comparably rough conditions making catalyst recycling difficult.9 Polyoxometalates (POMs) are among the most prominent epoxidation catalysts and they have been used in biphasic epoxidation since the seminal studies by Venturello and Ishii,10 and hence, they were also used for the epoxidation of VOs via in situ transfer into the VO phase.11 Due to quasi-homogeneous conditions, product/catalyst separation has not been the focus.12,13 Biphasic approaches involving manganese complexes with sophisticated ligand systems show good reaction rates under moderate conditions.14 Several groups reported on heterogeneous organocatalysts consisting of strongly acidic ion exchange resins which in situ form peroxo species upon treatment with various peroxides.3,15–17 These systems often suffer from lower activities/selectivities due to diffusion limitations and acid-mediated ring opening of the epoxide moiety although the heterogeneous nature of the catalyst allows facile recycling. Other heterogeneous approaches are based on mesoporous silica with an active metal incorporated into the network structure, such as Ti or Mn.18,19 However, these catalysts exhibit drawbacks which are similar to those of heterogenized organocatalysts. Silica-supported peroxycarboxylic acids show activities comparable to classic Prilezhaev-type reactions for the biphasic epoxidation of unsaturated fatty acids and esters with good conversions and epoxide selectivities.20 The catalyst is recyclable; yet, the conversion and selectivity decreases with each catalysis run, pointing to deactivation mechanisms and/or catalyst leaching.In order to combine the benefits of high catalytic activity and catalyst recycling, biphasic systems offer a suitable platform by switching towards the aqueous phase as a reaction medium. Nevertheless, to the best of our knowledge, there are no reports on the biphasic liquid–liquid epoxidation of VOs with molecular transition metal catalysts operating in the aqueous phase. Such a reaction requires the use of surfactants, which have to meet the following conditions: (i) the catalyst is either bound to the surfactant, or is part of it, (ii) the catalyst/surfactant is exclusively present in the aqueous phase, (iii) the VO substrate can be solubilized in the aqueous phase by the surfactant, and (iv) the surfactant is inert in the oxidative environment and does not decompose the oxidant.12,13 Surface-active (mostly imidazolium-based) ionic liquids (SAILs) are well known to form micelles in aqueous media21 and solubilize organic molecules therein while not being soluble in the organic media themselves.22 However, reports on multitasking single component SAILs – acting as phase transfer agents and catalytically active species – in water are still rare.12,13,23 A schematic illustration of the principle of micellar catalysis with SAILs is shown in Scheme 1.
 | ||
| Scheme 1 Concept of micellar epoxidation catalysis of olefins with SAIL catalysts. Upon addition of the olefin substrate (S) to the SAIL-containing aqueous phase, a biphasic system is formed (step 1). Upon heating and stirring, the substrate is solubilized in the aq. phase and converted to the epoxide product P (step 2). After reaction completion, the product phase (P) and the SAIL-containing aq. phase are fully separated (step 3). | ||
Surface-active imidazolium perrhenate SAILs were investigated as multitasking catalysts for the epoxidation of olefins in aqueous hydrogen peroxide.24 This work was expanded to cheaper and more abundant tungsten-based SAIL catalysts, which are significantly more active – especially with organophosphonic acid additives.25 Hence, in this study, the application of tungstate SAILs as catalysts for the epoxidation of VOs is presented, which is the first example of single-component surface active catalysts for VO epoxidation. The influence of cation variation on the substrate solubilization in the aq. phase and on the catalytic activity, respectively, is investigated. The catalytic system is subjected to consecutive catalysis runs to demonstrate facile product separation and catalyst recovery.
Results and discussion
Synthesis and characterization of tungstate-based SAILs
For the SAIL synthesis, imidazolium bromide precursors were synthesized according to published procedures (see the ESI†). The imidazolium tungstate SAILs [RR′MIm]2[WO4] (R = octyl [O], dodecyl [Do]; R′ = H, methyl [M]) were obtained from the respective imidazolium bromide precursors via an anion exchange procedure.25,26 The bromide precursor was eluted in a basic anion exchange resin and subsequently treated with tungstic acid to yield the product in high yields (>95%) and purities according to 1H- and 13C-NMR and elemental analysis (see the ESI†). The presence of tungstate was confirmed by 183W-NMR by a signal at −8.89 ppm for [DoMIm]2[WO4] (ESI,† Fig. S10). This shift is well in the range of [OMIm]2[WO4] at 6.02 ppm and the Na2WO4 reference signal at 0 ppm.25,27As the catalytic activity in multiphase systems is directly correlated to the presence of micelles, their concentration, size and shape, the critical micelle concentrations (CMCs) were determined by tensiometry at different temperatures (Table 1 and ESI,† Fig. S1–S3). Generally, increasing alkyl chain lengths from C8 to C12 significantly decrease the CMC by a factor of 18, which is in accordance with our previous studies.25 The additional methyl substituent at the C2-position of the imidazole of [DoR′MIm]2[WO4] (R′ = H, Me) does not notably influence the CMC. In contrast, increasing temperature leads to increasing CMC values, most likely due to greater solubilization of the respective IL in aqueous H2O2 at higher temperatures.
| IL | CMC [mmol L−1] | Mean size [diameter in nm] | ||
|---|---|---|---|---|
| 25 °C | 60 °C | Before | After | |
| [OMIM]2[WO4] | 46.2 | 55.4 | 3.0 (±0.44) | 4.0 (±0.86) |
| [DoMIM]2[WO4] | 2.6 | 5.5 | 2.0 (±0.35) | 3.1 (±0.72) |
| [DoMMIM]2[WO4] | 2.6 | 4.0 | 2.3 (±0.60) | 3.1 (±0.59) |
The micelle diameters were investigated by dynamic light scattering (DLS). At catalytic concentrations of the catalysts in aq. H2O2, the detected micelles are all in the same range of 2–3 nm (see Table 1 and the ESI,† Fig. S4). This phenomenon most likely results from the ensemble of dominating supramolecular interactions, such as the electrostatic repulsion of the polar head groups versus van der Waals (vdW) interactions of the hydrophobic alkyl chains. Therefore, in the case of [OMIm]2[WO4], the repulsion outweighs the vdW interaction leading to larger micelles, while for [DoMIm]2[WO4] the longer alkyl chain allows more vdW contacts and results in a denser packing of the supramolecular structure. Uptake of the organic substrate into the micellar aggregates is crucial for solubilizing the substrate in the aq. phase to overcome the diffusion and mass transfer limitations of the biphasic reaction medium. Therefore, all samples were additionally treated with high oleic palm oil prior to the measurement.
In all cases, the micelles grew larger upon addition of the substrate (Table 1), suggesting an incorporation of the substrate inside the micelles. In the case of [OMIm]2[WO4], the volume of the spherical micelles increased from 14 nm3 to 33 nm3 after treatment with palm oil, and for [DoMRIm]2[WO4] (R = H or Me) from about 5 nm3 to 16 nm3. In consideration of the CMC values at a catalytic concentration of 178 mmol L−1, around 1/4 of the SAIL [OMIm]2[WO4] is dissolved in aq. H2O2, while 3/4 of the SAIL forms micelles. In contrast, 99% of [DoMIm]2[WO4] molecules aggregate to micelles. This suggests that a SAIL catalyst with a larger hydrophobic moiety solubilizes more substrate in the aqueous phase as more micelles are formed, which leads to a micelle volume increase by a factor of 3.5 upon substrate contact, despite their small size.
As previously mentioned, the catalyst concentration likely affects the macroscopic parameters of the formed micelles. Then, the SAIL [DoMIm]2[WO4] was investigated at low concentrations of 2.7 and 26 mmol L−1 in aq. H2O2 at 20 °C. Meanwhile, at a catalytic concentration of 178 mmol L−1, the IL forms small micelles with a diameter of 2 nm; DLS detects large micelle aggregates of 130 nm at 26 mmol L−1 and even larger ones at 2.7 mmol L−1 (460 nm) as presented in Fig. 1a. These results underline the impact of concentration on the supramolecular aggregates. Additionally, TEM measurements of the respective samples were carried out (Fig. 1b–d). At a concentration slightly above the CMC, mostly large aggregates (>400 nm) are observed (Fig. 1b), which is in accordance with the DLS data. However, these aggregates are presumably formed from small primary micelles, but not detected by DLS, due to strongly differing scattering intensities (ESI,† Fig. S5). At 26 mmol L−1, the mean size of the spherical aggregates of small micelles decreases, in accordance with DLS, while also areas predominated by primary particles and network-like structure formation are detected (Fig. 1c and ESI,† Fig. S6). The TEM measurement at catalytic concentrations further supports the DLS data as mostly small micelles are present (Fig. 1d and ESI,† Fig. S7).
 | ||
| Fig. 1 (a) DLS measurements of [DoMIM]2[WO4] at different concentrations: 2.7 mmol L−1 (red), 26 mmol L−1 (blue) and 178 mmol L−1 (purple) in 50 wt% aq. H2O2 at 20 °C. (b)–(d) TEM pictures of [DoMIM]2[WO4] in 50 wt% aq. H2O2 at concentrations of 2.7 mmol L−1, 26 mmol L−1 and 178 mmol L−1, respectively. | ||
In order to imitate catalytic conditions more precisely, analogous measurements were performed using cryo-TEM. For this, the sample solutions were heated to 50 °C prior to drop casting, blotting and shock-frosting the sample, resulting in a rod-like structure, which swells upon substrate treatment (ESI,† Fig. S8). This most likely results from the strong electronic repulsion of the head groups at high concentrations, while hydrophobic interactions may be promoted by formation of linear structures.
Analogous measurements of the [OMIm]2[WO4] SAIL show a homogeneous distribution of spherical micelles with an average diameter of 15 nm (ESI,† Fig. S9) which is in accordance with our prior report.25 The increased size compared to the DLS data most likely results from the measurement procedure, as the copper grid is blotted in the case of cryo-TEM measurements which flattens the spherical micelle and then leads to an increase of the surface according to the previous volume calculations.
To further investigate the substrate uptake by the micellar system of [DoMIm]2[WO4], catalytic amounts of the SAIL (0.25 mmol) were dissolved in 1.4 mL of a 50 wt% solution of hydrogen peroxide in water (25 mmol) and stirred with an excess of palm oil. The precise amount of solubilized substrate was determined by quantitative 1H-NMR of an aliquot of the aqueous phase after full phase separation (ESI,† Fig. S11). Subsequently, at room temperature and a catalytic concentration of [DoMIm]2[WO4] (178 mmol L−1), 0.024 (±0.0012) mmol of palm oil is preserved in the aqueous phase, corresponding to a catalyst![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :substrate ratio of 1:10.
:substrate ratio of 1:10.
Two-phase epoxidation of vegetable oils catalyzed by SAILs
In a first case study, palm oil was chosen as the model substrate for the biphasic epoxidation with IL catalysts (Scheme 2). Firstly, SAILs with different cations were tested at a concentration of 2.5 mol% and a constant palm oil:H2O2 ratio of 1:40:100 at 60 °C. As vegetable oils contain several double bonds within triglyceride molecules with different chemical environments (i.e. variable combination of fatty acids), the respective amount of substrate is calculated according to the mean amount of double bonds per molecule. For instance, each experiment was performed with 4 mmol high oleic palm oil, which in average contains 2.5 double bonds per molecule, resulting in a ratio of 10 mmol double bonds vs. 25 mmol H2O2. Additionally, 5 mol% phenylphosphonic acid (PPA) was used as an additive, due to a significant activity boost as previously reported.25 This results from the formation of the catalytically active tungstate/PPA adduct instead of the kinetically hindered formation of the tetra(κ-peroxo)tungstate complex upon H2O2 contact. It needs to be noted here that in all experiments, the conversion and selectivity were determined by means of titration techniques (see the ESI† for details). Since both the substrates and their epoxidation products consist of complex organic matrices and are virtually non-volatile, gas or liquid chromatographic quantifications are not viable as samples have to be extracted from their matrices prior to analysis.28
 | ||
| Scheme 2 Catalytic two-phase epoxidation of vegetable oils with imidazolium tungstate SAILs as catalysts and aq. H2O2 as the oxidant. | ||
In comparison, spectroscopic methods such as NMR and FTIR are generally tedious. Analysis via1H-qNMR by integration of the respective olefin and epoxide signals for common organic substrates is not possible. However, the resonances of the educt and product are clearly shifted (ESI,† Fig. S12–S14) and specific NMR analysis procedures for (E)VOs are reported in the literature.29 Similarly, quantitative analysis via FTIR remains difficult, due to marginal differences between VOs and EVOs, also among different derivatives with each other (see the ESI,† Fig. S15–S17). Nonetheless, analysis by combination of FTIR with additional techniques such as gas chromatography is possible.29 Here, the substrate conversion was determined by iodometric titration using iodine monochloride to react with the unreacted C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of the substrate. The selectivity towards epoxides is determined via acid–base titration by ring opening of the epoxides with an excess of hydrochloric acid (for the detailed procedure see the ESI†). Note that the conversions obtained by this method correspond to the amount of the epoxidized CC bonds versus the total amount of all CC bonds in the substrate before the reaction starts. A precise determination of how many CC bonds per substrate molecule were converted is not possible.
C bonds of the substrate. The selectivity towards epoxides is determined via acid–base titration by ring opening of the epoxides with an excess of hydrochloric acid (for the detailed procedure see the ESI†). Note that the conversions obtained by this method correspond to the amount of the epoxidized CC bonds versus the total amount of all CC bonds in the substrate before the reaction starts. A precise determination of how many CC bonds per substrate molecule were converted is not possible.
The increasing alkyl chain length from C8 < C12 leads to a significant activity boost (Fig. 2). The most notable difference within [RMIm]2[WO4] is the low activity of R = octyl (40% conversion after 6 h), which is most presumably resulting from a weaker catalyst/substrate interaction than in the case of R = dodecyl (98% conversion after 6 h). Compared to common olefinic substrates, vegetable oils possess significantly larger molecule sizes with long hydrophobic alkyl chains that offer the possibility for vdW contacts, which are formed more extensively with dodecyl substituents. As previously mentioned, the dissolved amount of catalyst does not engage in vdW contacts, which are formed more extensively with dodecyl substituents. Also, as stated above, the dissolved amount of catalyst not taking part in micelle formation is 10 times higher for [OMIm]2[WO4] than for [DoMIm]2[WO4] at 60 °C. Increasing the hydrophobic content of the catalyst with a methyl substituent at the C2 position did not show a notable impact compared to C2 = H (95% vs. 92% conversion after 5 h). The selectivity decreased with the reaction time in all cases as the oxirane moiety is prone to the ring opening, and formation of diols, in the acidic environment of the aqueous phase owing to H2O2 and PPA. Nevertheless, the selectivity is independent of the hydrophobicity as all catalysts display selectivities of >70% (ESI,† Fig. S18).
 | ||
| Fig. 2 Conversion of high oleic palm oil in the epoxidation catalysis using imidazolium tungstate SAILs with varying alkyl chain lengths at 60 °C. Reaction conditions: 500 rpm, 3.8 mL palm oil (4 mmol, 100 equiv. double bonds), 1.4 mL H2O2 (50 wt% in water, 25 mmol, 250 equiv.), 2.5 mol% SAIL and 5 mol% PPA. | ||
In the following, the influence of the reaction temperature was studied using [DoMMIm]2[WO4] as the catalyst while maintaining analogue catalysts:PPA:palm oil:H2O2 ratios. Upon reducing the temperature to 50 °C, the conversion significantly declines to 55% after 5 h reaction time compared to 95% at 60 °C (Fig. 3). The activation energy value, based on the initial conversion rates, is 43.4 ± 3.3 kJ mol−1 (ESI,† Fig. S22), which is somewhat higher compared to the epoxidation of cyclooctene with tungstate ILs (32 kJ mol−1).25 However, the activation energy is notably lower compared to the activation energy for the Prilezhaev-type epoxidation of corn oil (74 kJ mol−1),30 indicating that mass transfer limitations do not play an important role. At 80 °C, the conv. increases to 98% after 2 h; however, the selectivity decreases to 68%, while at 60 °C, the selectivity is 82% at similar conversions (ESI,† Fig. S19). Hence, all following experiments were performed at 60 °C. Note that the activity towards to epoxidation of vegetable oils is somewhat lower compared to simple organic substrates, such as cyclooctene,25 which can be ascribed to the difficult accessibility of double bonds in the complex organic matrices.
 | ||
| Fig. 3 Conversion of high oleic palm oil in the epoxidation catalysis with [DoMMIM]2[WO4] at different temperatures. Reaction conditions: 500 rpm, 3.8 mL palm oil (4 mmol, 100 equiv. double bonds), 1.4 mL H2O2 (50 wt% in water, 25 mmol, 250 equiv.), 2.5 mol% SAIL and 5 mol% PPA. | ||
To examine the effect of the catalyst and PPA loading alongside their respective ratio on the catalytic activity and selectivity of the reaction, experiments with catalyst loadings varying from 1 to 10 mol% and IL:PPA ratios between 2:1 and 1:5 were performed (Fig. 4 and ESI,† Fig. S20). At the lowest catalyst concentration of 1 mol%, conversions of 53% and 67% are obtained after 8 h with 2 mol% and 5 mol% PPA (Fig. 4, dark purple vs. purple), respectively, while the selectivity for the latter is slightly lower (81% vs. 76%). Increasing the catalyst loading to 5 mol% at 5 mol% PPA leads to almost full conversion (98%) after 4 h with a good selectivity of 86%. Increasing the PPA amount to 10 mol% results in somewhat higher activity at the start of the reaction; however, the selectivity declines significantly to 64% after 4 h with similar conversions. This suggests that the catalytically active SAIL–PPA adduct is almost quantitatively formed with stoichiometric amounts of PPA, while excessive amounts promote the ring opening reaction of the oxirane moiety due to the acidic nature of PPA. Interestingly, by increasing the SAIL amount to 10 mol% while maintaining a lower PPA loading of 5 mol% (2:1 ratio), the reaction rate is increased (95% conv. after 2 h) but the selectivity at 76% is lower compared to using half of the catalyst loading (Fig. 4, red and ESI,† Fig. S20). When the SAIL:PPA ratio is changed to 1:2, which corresponds to catalyst and PPA loadings of 10 and 20 mol% (Fig. 4, pink), respectively, the initial reaction rate is enhanced (77% vs. 52% conv. after 1 h, pink vs. red), while an almost quantitative conversion of 96% is also obtained after 2 h reaction time. However, the selectivity strongly decreases at excessive PPA loadings, especially in the case of a catalyst loading of 10 mol% and 20 mol% PPA (40% after 2 h reaction time). This further underlines the sufficiency of lower adduct (stoichiometric amounts) addition to achieve reasonable reaction rates without the promotion of side reactions.
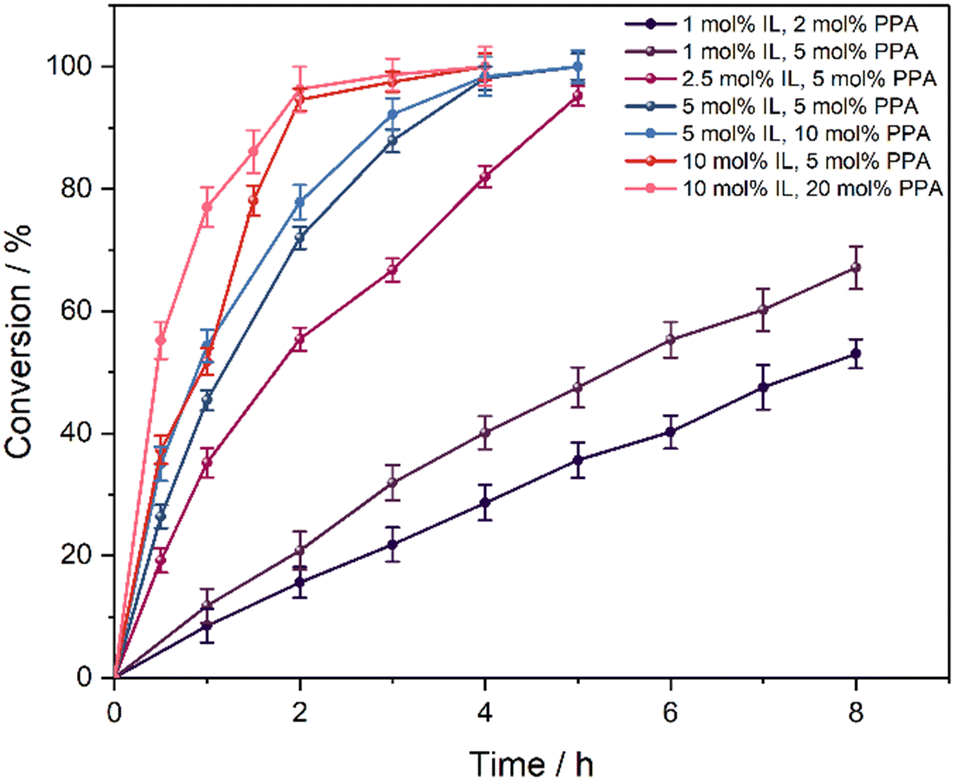 | ||
| Fig. 4 Conversion of high oleic palm oil in the epoxidation catalysis with different [DoMMIM]2[WO4] loadings and varying SAIL:PPA ratios at 60 °C. Reaction conditions: 500 rpm 3.8 mL palm oil (4 mmol, 100 equiv. double bonds), 1.4 mL H2O2 (50 wt% in water, 25 mmol, 250 equiv.). | ||
The applicability of the catalytic system for VOs with differing structural motifs, such as varying triglyceride backbones with different amounts and chemical environments of the olefinic bond, was tested with soybean and corn oil as additional substrates. Each vegetable oil has different amounts of CC double bonds (DBs) on average (high oleic palm oil: 2.5 DB, soybean oil: 4.5 DB, corn oil: 3.5 DB). The different chemical compositions of the respective vegetable oils are summarized in Table 2. Therefore, the amount of each VO for a standard catalytic experiment was calculated to be equivalent to 10 mmol of double bonds. The observed reaction rates for all three VOs are similar, as shown in Fig. 5. In contrast, the selectivity slightly decreases in the order high oleic palm oil > soybean oil > corn oil (ESI,† Fig. S21). The obtained profiles possibly result from differing accessibilities of the respective double bonds and subsequently the retention time of the substrate in the aqueous phase, as the oxirane moiety is prone to ring-opening and diol formation in an acidic environment. The micellar SAIL system working in water is more efficient than the Prilezhaev-based reaction, in which the catalysis takes place at the interface between the substrate phase and water. Also, it is superior to most homogeneous metal complex catalysts (see Table 3).6,7,9,14 Non-recyclable catalytic POM systems based on phase-transfer catalysis in the organic substrate phase display higher activities using methyl oleate as a model substrate, which is a pure, processed derivative of vegetable oils.11 As an alternative to heterogeneous solid–liquid systems,18,19 the SAIL catalysts show notably higher activities and selectivities under similar conditions, as presented in Table 3.
 | ||
| Fig. 5 Conversions of different vegetable oils with 5 mol% [DoMMIM]2[WO4] and 5 mol% PPA at 60 °C. Reaction conditions: 500 rpm, 3.8 mL palm oil (4 mmol, 100 equiv. double bonds), 1.4 mL H2O2 (50 wt% in water, 25 mmol, 250 equiv. per double bond). | ||
| Substrate | Catalyst | Oxidant | T [°C] | t [h] | Solvent | Conv. [%] | Sel. [%] | Recycling reported? | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Prilezshaev-type epoxidations, involving peracetic acids that are consumed (converted to the corresponding carboxylic acids) and not regenerated during the stoichiometric reaction. Sulfuric acid activates the peracids and/or stabilizes intermediates, but does not act as a classic catalyst, making catalyst recycling not necessary in these cases. b An epoxide yield of 78% was reported. | |||||||||
| Grape seed oil | Sulfuric acid | CH3COOH/H2O2 | 65 | 6 | None | 93 | 69 | Noa | 6 |
| Corn oil | Sulfuric acid | CH3COOH/H2O2 | 75 | n.a. | None | n.a. | n.a. | Noa | 30 |
| Soybean oil | CH3ReO3 | H2O2 | 25 | 2 | DCM | 100 | 95 | No | 7 |
| Soybean oil | [MoO2(acac)2] | TBHP | 110 | 24 | Toluene | 83 | 49 | No | 9 |
| Sunflower oil | rac-[Mn(OTf)2(BPBP)] | CH3COOH/H2O2 | 25 | 2 | None | 99 | 90 | No | 14 |
| Sunflower oil | Amberlyst 39 | CH3COOH/H2O2 | 75 | 4 | None | 90 | 96 | Yes | 16 |
| Castor oilb | Amberlite IR-120 | CH3COOH/H2O2 | 50 | 10 | Benzene | n.a. | n.a. | No | 17 |
| Soybean oil | Ti-HMS | TBHP | 60 | 6 | Ethyl acetate | 41 | 43 | No | 19 |
| Soybean oil | Mn-HMS | TBHP | 60 | 6 | Ethyl acetate | 43 | 51 | No | 19 |
| Soybean oil | [DoMIm]2[WO4] | H2O2 | 60 | 4 | None | 98 | 81 | Yes | This work |
Catalyst recycling
For a truly sustainable reaction protocol to convert VOs into EVOs, several requirements need to be fulfilled. For instance, using hydrogen peroxide as an oxidant is environmentally benign because it yields water as a by-product, meeting the criteria of green chemistry. Another important premise is a feasible product/catalyst separation without the need of additional product purification, and hence, the possibility to recycle the catalyst (if possible, on a long-term scale). To demonstrate this, the recyclability of the tungstate-SAILs was studied for five subsequent runs as shown in Fig. 6. The reaction was performed with [DoMIM]2[WO4] instead of [DoMMIM]2[WO4] as the catalyst using soybean oil as the substrate, due to the lower viscosity compared to high oleic palm oil (this also applies to the respective epoxidation products), and hence to promote a faster phase separation. For the recycling experiment, 5 mol% catalyst loading with a PPA ratio of 1:1 is used. The scale of the reaction was doubled and performed in a round bottom flask (see the ESI† for the detailed procedure).
 | ||
| Fig. 6 Conversion and selectivities of soybean oil in the epoxidation catalysis with 5 mol% [DoMIM]2[WO4] and 5 mol% PPA. Reaction conditions: 60 °C, 4 h, 500 rpm, 2.3 mL soybean oil (2.22 mmol, 100 equiv. double bonds), 1.4 mL H2O2 (50 wt% in water, 25 mmol, 250 equiv.). | ||
For each run, the reaction was stopped after 4 h, the two-phase reaction mixture was allowed to stand, and after 3 h, the organic phase was removed to ensure complete phase separation. The residual aqueous phase was washed with n-hexane three times and the solvent was removed under reduced pressure. The following runs were started by addition of fresh H2O2 and soybean oil and heating to reaction temperature. Throughout the five catalysis cycles, no notable decline in activity (conversion of soybean oil) or selectivity (to the respective epoxides) was observed, suggesting that neither the catalyst nor the additive is lost during the process. This also suggests that catalyst deactivation mechanisms, which are a common problem for molecular catalysts aside from the intriguing catalyst/product separation, are absent. Note that kinetic experiments for each recycling run – instead of reactions resulting in a single data point – and the subsequent determination of the respective reaction rates would underline these results.34 To ensure that the product phase is free of catalyst, a sample of epoxidized soybean oil was investigated via inductively coupled plasma mass spectrometry (ICP-MS) for traces of tungsten (see the ESI† for details). No metal was detected by this method, ensuring that the tungsten content is below the detection limit of 0.16 μg L−1. Additionally, the organic layer does not contain the additive PPA as revealed by 31P-NMR analysis, since no signal is detected during the measurement. These results underline that the product/catalyst separation of the biphasic system after the reaction is possible by simple mechanical methods.
Conclusion
This work shows the potential of multifunctional, surface-active catalysts in the industrially relevant epoxidation of vegetable oils by applying a two-phase reaction system which allows for carrying out the catalysis in water and for recycling the catalyst for reuse. Imidazolium tungstate SAILs are efficient epoxidation catalysts under moderate reaction conditions. The surfactant function of the SAILs induces a phase transfer of the hydrophobic substrates into water, where the catalytic function of the SAILs steps into action. Furthermore, the recyclability of the catalyst and additive, and the facile product separation by mechanical methods without the need for purification was demonstrated according to the principles of green chemistry. The model system showed no loss in activity or selectivity over five consecutive runs. The separated product did not contain any traces of catalyst or additive, as verified by 31P-NMR spectroscopy and ICP-MS. These results, together with the straightforward and scalable synthesis from cheap starting materials, are promising factors towards the implementation of IL-based epoxidation in continuous reactors, which are to date still limited to mostly heterogeneous composite materials and unfunctionalized substrates.The IL motif provides the possibility to catalyze a substrate with complex, functionalized organic molecules, which is still challenging for industrial applications. Furthermore, this platform offers a wide range of possible applications, due to the simplicity of cation/anion permutations and tuning towards the desired micellization, solubility, phase behavior and reactivities besides the epoxidation of olefins. This concept opens pathways for sustainable multitasking catalysts not limited to specific applications in the laboratory scope.
Experimental
A detailed description of the analytical methods, synthetic procedures, catalyst characterization and the catalytic setups are given in the ESI.†Data availability
The data supporting this article have been included as part of the ESI.† In case any further data are necessary, do not hesitate to contact us.Author contributions
M. H.: investigation (lead), data curation (lead), visualization (lead), formal analysis (lead), and writing – original draft; W. F. B.: investigation, data curation, and formal analysis; J. L.: investigation and formal analysis; A. O.: conceptualization, methodology, and writing – review & editing; A. J.: conceptualization, methodology, and writing – review & editing; M. C.: conceptualization, resources, funding acquisition, methodology, project administration, supervision, validation, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) for funding of the project ‘Surface-active ionic liquids as catalysts of olefin epoxidation: Reaction control by switchable phase behavior’ (project No. 326649877, grant No. Co 1543/1-2 and Je 257/24-2). W. F. B. thanks the Bavarian University Center for Latin America (BAYLAT) for funding. The authors further thank Dr. Carsten Peters for support with cryo-TEM measurements, and Christine Benning for ICP-MS measurements, as well as the staff of the Central Analytics Facility of the TUM Catalysis Research Center.References
- L. Maisonneuve, T. Lebarbé, E. Grau and H. Cramail, Polym. Chem., 2013, 4, 5472–5517 RSC.
- L. M. Ramírez, J. G. Cadavid, A. Orjuela, M. F. Gutiérrez and W. F. Bohórquez, Chem. Eng. Process., 2022, 176, 108963 CrossRef.
- M. Kurańska, H. Beneš, A. Prociak, O. Trhlíková, Z. Walterová and W. Stochlińska, J. Cleaner Prod., 2019, 236, 117615 CrossRef.
- E. Santacesaria, A. Renken, V. Russo, R. Turco, R. Tesser and M. Di Serio, Ind. Eng. Chem. Res., 2012, 51, 8760–8767 CrossRef CAS.
- S. M. Danov, O. A. Kazantsev, A. L. Esipovich, A. S. Belousov, A. E. Rogozhin and E. A. Kanakov, Catal. Sci. Technol., 2017, 7, 3659–3675 RSC.
- J. C. de Haro, I. Izarra, J. F. Rodríguez, Á. Pérez and M. Carmona, J. Cleaner Prod., 2016, 138, 70–76 CrossRef CAS.
- A. E. Gerbase, J. R. Gregório, M. Martinelli, M. C. Brasil and A. N. F. Mendes, J. Am. Oil Chem. Soc., 2002, 79, 179–181 CrossRef CAS.
- S. Verma, A. Joshi, S. R. De and J. L. Jat, New J. Chem., 2022, 46, 2005–2027 RSC.
- M. Farias, M. Martinelli and D. P. Bottega, Appl. Catal., A, 2010, 384, 213–219 CrossRef CAS.
- (a) C. Venturello, E. Alneri and M. Ricci, J. Org. Chem., 1983, 48, 3831–3833 CrossRef CAS; (b) Y. Ishii, K. Yamawaki, T. Ura, H. Yamada, T. Yoshida and M. Ogawa, J. Org. Chem., 1988, 53, 3587 CrossRef CAS.
- (a) M. Jabbour, I. Ben Talouba, L. Balland and N. Mouhab, J. Therm. Anal. Calorim., 2022, 147, 13447–13457 CrossRef CAS; (b) M. Musik, E. Janus, R. Pelech and L. Salaciński, Catalysts, 2021, 11, 1058 CrossRef CAS.
- F. Schmidt and M. Cokoja, Green Chem., 2021, 23, 708–722 RSC.
- M. Hegelmann, F. Schmidt and M. Cokoja, Encycl. Inorg. Bioinorg. Chem., 2024, 1–21 Search PubMed.
- J. Chen, M. de Liedekerke Beaufort, L. Gyurik, J. Dorresteijn, M. Otte and R. J. M. Klein Gebbink, Green Chem., 2019, 21, 2436–2447 RSC.
- L. A. Rios, D. A. Echeverri and A. Franco, Appl. Catal., A, 2011, 394, 132–137 CrossRef CAS.
- O. Gómez-de-Miranda-Jiménez-de-Aberasturi and J. Perez-Arce, Can. J. Chem. Eng., 2019, 97, 1785–1791 CrossRef.
- M. R. Janković, S. V. Sinadinović-Fišer and O. M. Govedarica, Ind. Eng. Chem. Res., 2014, 53, 9357–9364 CrossRef.
- L. A. Rios, P. Weckes, H. Schuster and W. Hoeldrich, J. Catal., 2005, 232, 19–26 CrossRef CAS.
- X. Ye, P. Jiang, P. Zhang, Y. Dong, C. Jia, X. Zhang and H. Xu, Catal. Lett., 2010, 137, 88–93 CrossRef CAS.
- M.-Y. Yao, Y.-B. Huang, X. Niu and H. Pan, ACS Sustainable Chem. Eng., 2016, 4, 3840–3849 CrossRef CAS.
- C. Jungnickel, J. Łuczak, J. Ranke, J. F. Fernández, A. Müller and J. Thöming, Colloids Surf., A, 2008, 316, 278–284 CrossRef CAS.
- S. Dorbritz, W. Ruth and U. Kragl, Adv. Synth. Catal., 2005, 347, 1273–1279 CrossRef CAS.
- T. Shen, S. Zhou, J. Ruan, X. Chen, X. Liu, X. Ge and C. Qian, Adv. Colloid Interface Sci., 2021, 287, 102299 CrossRef CAS PubMed.
- I. I. E. Markovits, W. A. Eger, S. Yue, M. Cokoja, C. J. Münchmeyer, B. Zhang, M.-D. Zhou, A. Genest, J. Mink, S.-L. Zang, N. Rösch and F. E. Kühn, Chem. – Eur. J., 2013, 19, 5972–5979 CrossRef CAS PubMed.
- (a) F. Schmidt, B. Zehner, W. Korth, A. Jess and M. Cokoja, Catal. Sci. Technol., 2020, 10, 4448–4457 RSC; (b) J. Schäffer, M. Alber, W. Korth, M. Cokoja and A. Jess, ChemistrySelect, 2017, 2, 11891–11898 CrossRef.
- F. Schmidt, B. Zehner, M. Kaposi, M. Drees, J. Mink, W. Korth, A. Jess and M. Cokoja, Green Chem., 2021, 23, 1965–1971 RSC.
- V. Nardello, J. Marko, G. Vermeersch and J. M. Aubry, Inorg. Chem., 1998, 37, 5418–5423 CrossRef CAS PubMed.
- E. O. Aluyor, C. E. Ozigagu, O. I. Oboh and P. Aluyor, Sci. Res. Essays, 2009, 4, 191–197 Search PubMed.
- P. T. Wai, P. Jiang, Y. Shen, P. Zhang, Q. Gu and Y. Leng, RSC Adv., 2019, 9, 38119–38136 RSC.
- C. Cai, H. Dai, R. Chen, C. Su, X. Xu, S. Zhang and L. Yang, Eur. J. Lipid Sci. Technol., 2008, 110, 341–346 CrossRef CAS.
- R. Zamiri, A. Zakaria, H. A. Ahangar, A. R. Sadrolhosseini and M. A. Mahdi, Int. J. Mol. Sci., 2010, 11, 4764–4770 CrossRef CAS PubMed.
- T. E. Clemente and E. B. Cahoon, Plant Physiol., 2009, 151, 1030–1040 CrossRef CAS PubMed.
- W. Carillo, C. Carpio, D. Morales, E. Vilcacundo, M. Álvarez and M. Silva, Asian J. Pharm. Clin. Res., 2017, 10, 150–153 Search PubMed.
- (a) C. W. Jones, Top. Catal., 2010, 53, 942–952 CrossRef CAS; (b) S. L. Scott, ACS Catal., 2018, 8, 8597–8599 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00215f |
| This journal is © The Royal Society of Chemistry 2024 |