
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into molecular accessibility in catalyst and sorbent materials using NMR porosity measurements†
J. A.
Ward-Williams
 a,
C. M.
Guédon
b,
M. D.
Mantle
a,
A. J.
Sederman
a and
L. F.
Gladden
*a
a,
C. M.
Guédon
b,
M. D.
Mantle
a,
A. J.
Sederman
a and
L. F.
Gladden
*a
aDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Philippa Fawcett Drive, Cambridge CB3 0AS, UK. E-mail: lfg1@cam.ac.uk
bShell Global Solutions International B.V., Amsterdam, Netherlands
First published on 26th April 2024
Abstract
NMR measurements of porosity are demonstrated for a range of catalyst and sorbent materials. The NMR method allows porosity measurements to be carried out directly with the molecules of interest for a given reaction or separation process, thereby providing valuable information regarding accessibility of that molecular species to the pore network during process operation.
The porosity, ε, of a solid material is an important characterization parameter that can be used to understand both the mechanical properties of a material1 and the transport properties of fluids confined within it.2,3 As such, porosity is a key parameter to control when optimising the properties of a wide range of heterogeneous systems. Many techniques already exist to measure porosity, but the most common methods still possess significant drawbacks. For example, the high pressures used for mercury porosimetry are well-known to damage pore structures,4 it cannot detect pores that are smaller than a few nm, the pore shielding effect can lead to inaccurate pore size distributions,5 and it is a destructive technique due to the inability to fully remove mercury from the sample after analysis.6 Helium pycnometry must be combined with an additional bulk or pore volume measurement to calculate porosity and can become erroneous for materials which adsorb helium, such as molecular sieves measured at high pressures.7 Furthermore, both methods rely on the use of indirect probe molecules to measure the accessible porosity.6 The use of indirect probe molecules may give misleading results if the probe is able to access different parts of the pore network compared to the actual fluids of interest. For example, in cases where the pore size is comparable to the size of the probe molecule even subtle changes in the structure can lead to large differences in the adsorption capacity, as was shown for the four-fold greater adsorption of toluene in EU type zeolites compared to the adsorption of 1,3,5-trimethyl benzene.8 The size selectivity of microporous materials such as zeolites makes it challenging to interpret porosity measurements that rely on indirect probe molecules such as He, N2, Ar, or Hg. NMR offers an opportunity to overcome this limitation as it can be implemented with any probe molecule that contains a nuclear spin with non-zero nuclear spin quantum number. The vast majority of existing NMR porosity methods use water as the probe molecule. CPMG measurements have been used extensively to measure total porosity and pore size distributions in macroporous media, including rock cores9–11 and cements.12 Intensity differences in MRI images between intra- and extra-particle liquid have been used to provide spatially resolved porosities,13 which agreed with gravimetric measurements to within 2% for rock core systems, and NMR cryoporometry measurements14 have been applied to measure the nanoporous pore size distribution and total porosity of titania films15 and silica foams.16 Typically, NMR porosity measurements require an independent measurement or estimate of the bulk volume of the porous material to yield a porosity value. However, in recent years, techniques based on three separate NMR measurements of intra-pellet fluid, intra- and extra-pellet fluid, and bulk fluid have been applied to measure the porosity of irregular shaped porous media without the need for independent measurements of porosity.17,18 Dick et al. have coupled such an approach with a CPMG porosity measurement for studying the porosity of rock core cuttings17 and Linck et al. used 1H spectroscopy to measure the porosity of silica spheres.18 For the latter, good agreement between NMR porosity measurements and gas sorption porosity measurements was observed for the large pore silicas, but much poorer agreement was observed for water in the smallest pore size silicas, with NMR underestimating the porosity by up to 9%. Further consideration of the factors contributing to the quantitative nature of the NMR measurement are required to ensure a robust implementation of these NMR methods for catalyst and sorbent materials, and to allow deeper physicochemical understanding of liquid accessibility.
Herein we present a simple and robust NMR porosity measurement that allows the apparent porosity experienced by any molecule containing NMR-active nuclei within a porous material to be directly measured. It is therefore possible to measure the porosity experienced by the reactants, products and sorbates of interest, with the scope to apply this method at the temperatures and pressures relevant to the chemical process under investigation. In the present work, porosity measurements are made using 1H nucleus observation. The method is applied to a range of porous oxides and zeolites initially using water as the probe molecule. When water is used as the probe molecule it is not necessary to dry the sorbent prior to analysis, making the NMR porosity method a rapid and simple characterisation tool. The method is then repeated with a range of different probe molecules to demonstrate the differences in apparent porosity that can be observed for different molecules, these insights could not be obtained unambiguously from conventional porosity measurements.
The principles of the measurement method are now given. The porosity of a material can be defined as:
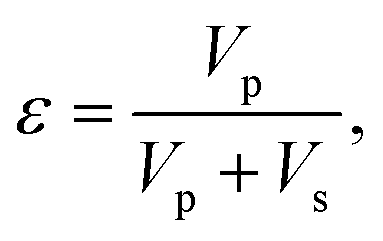 | (1) |
| Si = KSNi, | (2) |
 | (3) |
It follows that, when the size of the pores far exceeds the size of the probe molecules, the accessible and inaccessible volumes are well approximated by Vp and Vs respectively. Under these conditions and the assumption of a constant molar volume upon adsorption, eqn (1) and (3) can be combined to give:
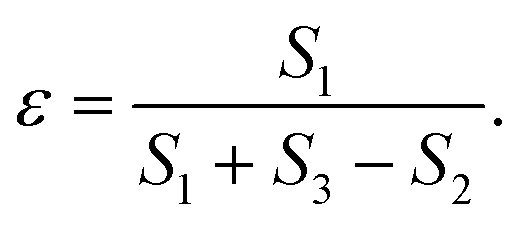 | (4) |
NMR experiments were carried out using a Bruker AV400 NMR spectrometer operating at a 1H frequency of 400.23 MHz, equipped with a Diff30 probe and 5 mm 1H/13C coil. Fig. 1 shows an excellent agreement between the NMR porosity measurement and conventional measurements when applied to a range of titania, alumina, and type A zeolite pellets. Here water was used as the probe and Fig. 1 shows that the NMR porosities were consistent to within ±2% of the conventional measurements. By choosing water as the probe molecule it is possible to remove sample drying protocols and the measurements here were taken in less than 15 min per sample. The method is, in theory, extendable to powder samples noting that due care must be taken to remove extra-particle liquid.
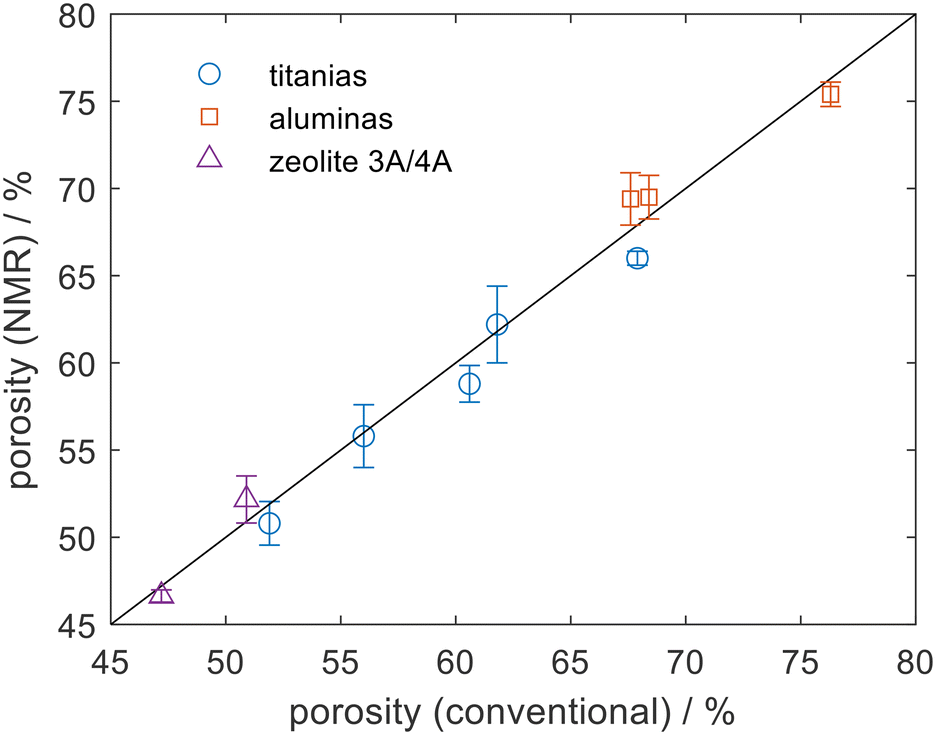 | ||
| Fig. 1 A comparison between the NMR porosity measurement and mercury porosimetry measurements applied to a range of titanias and aluminas. Skeletal densities of zeolites 3A and 4A were measured with helium pycnometry to ensure that the zeolite microporosity was captured. The solid line is a line of y = x. Errors bars represent the standard error of 3 measurements. | ||
The conventional porosity measurements were calculated as:22
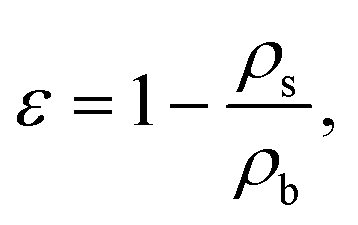 | (5) |
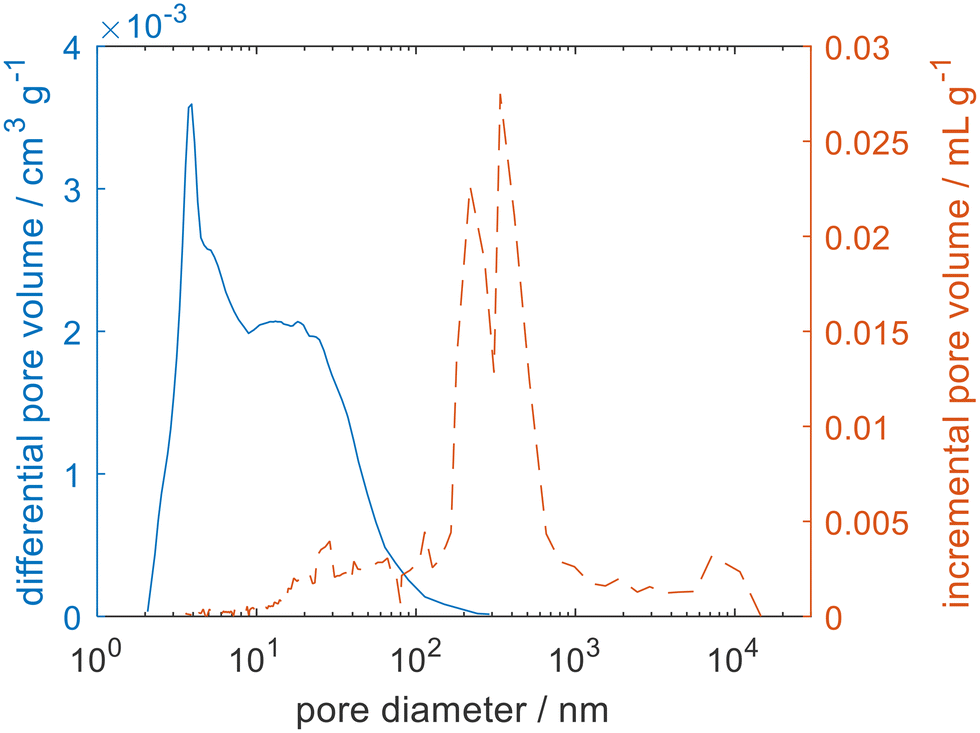 | ||
| Fig. 2 The pore size distribution measured for zeolite 4A using argon physisorption (blue solid line) and mercury porosimetry (orange dashed line). | ||
To ensure that the total porosity was being detected both mercury porosimetry and helium pycnometry were carried out for the zeolite 3A and 4A samples. The complex pore size distributions can be attributed to a hierarchical pore structure. For zeolite 4A, the zeolite crystals have a nominal pore opening of 4 Å. Surrounding the zeolite crystals is a porous network spanning several orders of magnitude of pore size, associated with the intercrystal space and the binder used to pelletise the zeolite beads, typical of such materials.24 Helium pycnometry gave a much larger porosity for the zeolites (εHP = 50.9%), when compared to mercury porosimetry (εMP = 32.9%). As mercury porosimetry is unable to access the microporosity, the large difference between the two measurements is consistent with a microporosity of 18%. Such a significant difference between εHP and εMP has previously been observed for similar materials that show a broad distribution of pore sizes and significant microporosity such as mudrocks25 and cements.26 It follows that the observed porosity will be a function of the size of the chosen probe molecule and that it is not always trivial to relate εHP and εMP to the porosity experienced by the actual sorbate–sorbent pairs of interest.
The major advantage of the NMR porosity method is that it can perform chemically-specific measurements and therefore can be used to directly measure the porosity of the sorbate–sorbent system of interest. It can also be used to measure porosity at relevant conditions of temperature and pressure. Fig. 3 shows porosity measurements for 3 materials: an alumina (Al-I) and zeolites 3A and 4A using 5 different probe molecules. Each material was dried (see Fig. S4 in ESI†) and the porosity measurements were repeated with each different probe molecule. Fig. 3 shows the porosity for each material as a function of the critical diameter of the probe molecule. For these experiments the critical diameter was defined as the second smallest molecular dimension of each molecule from ZINDO calculations,27,28 which would be the limiting dimension for accessing a circular pore opening of the zeolite crystals. The porosities measured by NMR were compared to helium pycnometry and mercury porosimetry methods, which are denoted as having critical diameters of 2.6 and 37 Å respectively. For helium pycnometry this value relates to the kinetic diameter of helium gas29 and for mercury porosimetry it corresponds to the pore size accessed at the highest pressure of the mercury porosimetry experiment used in these analyses. For the mesoporous alumina, Al-I, the pore size distribution was monomodal with an average pore size of 12 nm. The pores were far larger than the critical diameter of all probe molecules and no variation in porosity was noted as the probe molecule was varied. Excellent agreement was observed between the NMR method and both the helium pycnometry and mercury porosimetry methods for Al-I, which indicated that all molecules were able to access the full porosity. In contrast, the zeolite materials showed a significant decrease in porosity as the size of the probe molecule increased, with a step change from a high porosity region to a low porosity region at 4.2 Å. The change in effective porosity is consistent with a change in accessibility of the probe molecule. When the size of the probe molecule was smaller than the aperture size of the zeolite pores the probe molecule could access both the intracrystal zeolite porosity and the binder porosity, leading to a high value of porosity (48% and 52% for 3A and 4A respectively). When the size of the probe molecule exceeded that of the zeolite aperture then the probe molecule was excluded from the microporosity, and only the porosity of the intercrystalline space was measured (31% and 35% for 3A and 4A respectively). These arguments agree with the helium pycnometry and mercury porosimetry data.
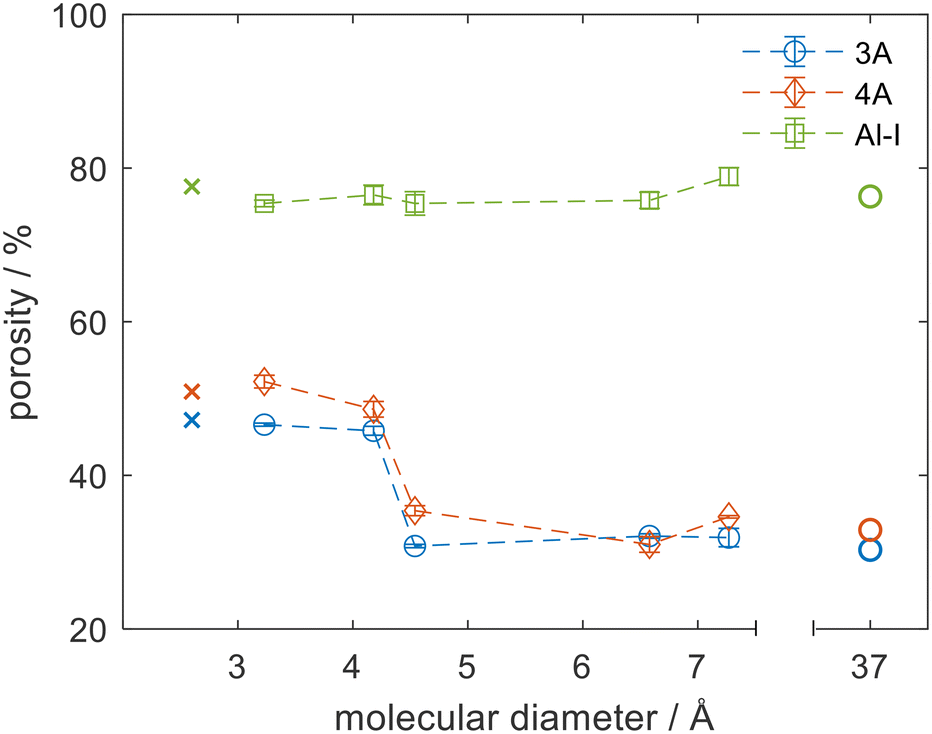 | ||
| Fig. 3 The porosity experienced by different probe molecules in alumina and zeolites 3A and 4A. The probe molecules are (left to right on the dashed line) water, methanol, n-decane, cyclohexane, and o-xylene. Helium pycnometry (x) and mercury porosimetry (o) results are shown for comparison. | ||
The sharp transition in porosity was expected for well-controlled zeolitic materials such as the 3A and 4A molecular sieves. The position of the transition, however, was not simple to predict a priori. Methanol (critical diameter of 4.2 Å) was shown to fully access the microporosity of zeolites 3A and 4A despite being slightly larger than the nominal pore size of the latter and much larger than the former. This result is consistent with previous drying (i.e. water removal) studies of primary alcohols, which suggested that competitive adsorption within the 3A crystals led to poorer drying of methanol by 3A zeolites compared to the drying of larger alcohols and esters, which could not access the zeolite microporosity.30 The data shown in Fig. 3 support the argument that as both water and methanol can enter the zeolite cages, competitive adsorption between water and methanol plays an important role in the drying process rather than a pure size exclusion of the methanol as may have been expected based on the zeolite pore sizes and molecular diameters. Such physicochemical insights are only possible when the species of interest are used as probe molecules themselves at relevant process conditions, as complex effects such as chemical interaction and structural flexing are not accurately detected with indirect probe molecules. Small changes in the system can result in significant changes in behaviour. For example, previous work has shown that when methanol adsorption on zeolite 3A is carried out in the gas phase rather than the liquid phase, poor methanol adsorption is observed.31,32 Conflicting literature results coupled with discrepancies in definitions of molecular diameters, differences in zeolite compositions,33 breathing phenomena34 and interactions between adsorbates and cations leading to trapdoor effects35 all complicate the process of predicting molecular accessibility computationally or from indirect measurements, and highlight the need for molecule-specific measurements of molecular accessibility.
In summary, a simple NMR methodology for measuring porosity has been outlined, and it has been demonstrated that by ensuring RF homogeneity of the magnetic field in which the NMR measurement is made and by quantitatively accounting for 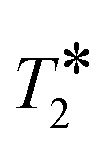 relaxation, agreement between magnetic resonance measurements of porosity and conventional measurements was reduced to <2% whilst also having the advantage of allowing rapid molecule-specific porosity values at any conditions of temperature. The chemical-specific nature of the measurement has been demonstrated in a case study in which the porosity of zeolites 3A and 4A was measured as a function of probe molecule. A significant decrease in porosity was observed between methanol and n-decane, corresponding to the latter being unable to access the microporosity of the zeolite crystals. Methanol was shown to access the micropores of zeolite 3A despite being significantly larger than the nominal pore opening. This result provides further evidence that methanol drying with zeolites 3A is not driven purely by size exclusion as is often assumed. For microporous systems a direct measurement of the porosity each molecule experiences is key to understanding the physicochemical process that occur within them, rather than relying on porosity measurements using indirect probes and calculations of molecular diameters. These measurements have been made on a 400 MHz spectrometer and using 1H nucleus observation. However, the measurements can be made using low field magnet hardware, time domain NMR methods, and nuclei other than 1H, as long as there is sufficient signal-to-noise. Therefore, the approach offers the potential for widespread use in research and analytical laboratories.
relaxation, agreement between magnetic resonance measurements of porosity and conventional measurements was reduced to <2% whilst also having the advantage of allowing rapid molecule-specific porosity values at any conditions of temperature. The chemical-specific nature of the measurement has been demonstrated in a case study in which the porosity of zeolites 3A and 4A was measured as a function of probe molecule. A significant decrease in porosity was observed between methanol and n-decane, corresponding to the latter being unable to access the microporosity of the zeolite crystals. Methanol was shown to access the micropores of zeolite 3A despite being significantly larger than the nominal pore opening. This result provides further evidence that methanol drying with zeolites 3A is not driven purely by size exclusion as is often assumed. For microporous systems a direct measurement of the porosity each molecule experiences is key to understanding the physicochemical process that occur within them, rather than relying on porosity measurements using indirect probes and calculations of molecular diameters. These measurements have been made on a 400 MHz spectrometer and using 1H nucleus observation. However, the measurements can be made using low field magnet hardware, time domain NMR methods, and nuclei other than 1H, as long as there is sufficient signal-to-noise. Therefore, the approach offers the potential for widespread use in research and analytical laboratories.
Author contributions
JWW: conceptualization, investigation, methodology, writing – original draft. CMG: conceptualization, investigation, resources, funding acquisition. MDM: methodology, supervision, writing – review and editing. AJS: methodology, supervision. LFG: conceptualization, funding acquisition, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Shell Global Solutions International B.V. for funding and J. W.-W. would like to thank the IChemE Andrew Fellowship for additional funding.Notes and references
- M. Zakeri, A. Samimi, M. Shafiee Afarani and A. Salehirad, Part. Sci. Technol., 2018, 36, 96–103 CrossRef CAS.
- A. Koponen, M. Kataja and J. Timonen, Phys. Rev. E, 1997, 56, 3319–3325 CrossRef CAS.
- P. Sudarsanam, E. Peeters, E. V. Makshina, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2019, 48, 2366–2421 RSC.
- X. Wang, Y. Peng, J. Wang and Q. Zeng, Materials, 2019, 12, 2220 CrossRef CAS PubMed.
- S. P. Rigby and L. F. Gladden, Chem. Eng. Sci., 2000, 55, 5599–5612 CrossRef CAS.
- L. Espinal, in Characterization of Materials, American Cancer Society, 2012, pp. 1–10 Search PubMed.
- A. Arami-Niya, T. E. Rufford, G. Birkett and Z. Zhu, Microporous Mesoporous Mater., 2017, 244, 218–225 CrossRef CAS.
- Z. Guo, W. Hao, J. Ma and R. Li, J. Chem. Res., 2020, 45, 187–193 CrossRef.
- P. Zhao, B. He, B. Zhang and J. Liu, Pet. Sci., 2022, 19, 509–517 CrossRef.
- M. Appel, Petrophysics – The SPWLA Journal of Formation Evaluation and Reservoir Description, 2004, vol. 45 Search PubMed.
- J. R. Hook, Petrophysics – The SPWLA Journal of Formation Evaluation and Reservoir Description 2003, vol. 44 Search PubMed.
- A. C. A. Muller, K. L. Scrivener, A. M. Gajewicz and P. J. McDonald, J. Phys. Chem. C, 2013, 117, 403–412 CrossRef CAS.
- F. Marica, Q. Chen, A. Hamilton, C. Hall, T. Al and B. J. Balcom, J. Magn. Reson., 2006, 178, 136–141 CrossRef CAS PubMed.
- J. Mitchell, J. B. W. Webber and J. H. Strange, Phys. Rep., 2008, 461, 1–36 CrossRef CAS.
- D. Vargas-Florencia, T. Edvinsson, A. Hagfeldt and I. Furó, J. Phys. Chem. C, 2007, 111, 7605–7611 CrossRef CAS.
- D. Vargas-Florencia, I. Furó and R. W. Corkery, Langmuir, 2008, 24, 4827–4832 CrossRef CAS.
- M. J. Dick, D. Veselinovic, T. Kenney and D. Green, Validation of Porosity of NMR Cuttings. Paper presented at the SPWLA 63rd Annual Logging Symposium, Stavanger, Norway, June 2022 Search PubMed.
- L. G. Linck, S. A. Maldonado Ochoa, M. Ceolín, H. Corti, G. A. Monti, F. V. Chávez and R. H. Acosta, Microporous Mesoporous Mater., 2020, 305, 110351 CrossRef CAS.
- S. K. Bharti and R. Roy, TrAC, Trends Anal. Chem., 2012, 35, 5–26 CrossRef CAS.
- Y. Yao, D. Liu, Y. Che, D. Tang, S. Tang and W. Huang, Fuel, 2010, 89, 1371–1380 CrossRef CAS.
- J. Lai, G. Wang, Z. Fan, J. Chen, S. Wang, Z. Zhou and X. Fan, Energy Fuels, 2016, 30, 10200–10214 CrossRef CAS.
- L. E. Flint and A. L. Flint, Methods of Soil Analysis, 2002, pp. 241–254 Search PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- L. Gueudré, M. Milina, S. Mitchell and J. Pérez-Ramírez, Adv. Funct. Mater., 2014, 24, 209–219 CrossRef.
- B. Andreas, S. Kevin, K. Niko, C. Ab, P. Vitaliy, F. Artem, L. Leon, A.-H. Alexandra and B. Pieter, Spec. Publ. - Geol. Soc. London, 2017, 454, 15–38 CrossRef.
- T. Tracz and T. Zdeb, Materials, 2019, 12, 192 CrossRef CAS PubMed.
- C. E. Webster, R. S. Drago, M. C. Zerner and V. Gaines, J. Am. Chem. Soc., 1998, 120, 5509–5516 CrossRef CAS.
- B. Chen, Y. Ji, M. Xue, F. R. Fronczek, E. J. Hurtado, J. U. Mondal, C. Liang and S. Dai, Inorg. Chem., 2008, 47, 5543–5545 CrossRef CAS.
- D. W. Breck and D. W. Breck, Zeolite molecular sieves: structure, chemistry, and use, John Wiley & Sons, 1973 Search PubMed.
- C. Pahl, C. Pasel, M. Luckas and D. Bathen, J. Chem. Eng. Data, 2012, 57, 2465–2471 CrossRef CAS.
- G. Pascual-Muñoz, R. Calero-Berrocal, M. Larriba, V. I. Águeda and J. A. Delgado, Microporous Mesoporous Mater., 2023, 360, 112669 CrossRef.
- P. Azhagapillai, M. Khaleel, F. Zoghieb, G. Luckachan, L. Jacob and D. Reinalda, ACS Omega, 2022, 7, 6463–6471 CrossRef CAS PubMed.
- R. Lin, A. Ladshaw, Y. Nan, J. Liu, S. Yiacoumi, C. Tsouris, D. W. DePaoli and L. L. Tavlarides, Ind. Eng. Chem. Res., 2015, 54, 10442–10448 CrossRef CAS.
- H. Shi, A. N. Migues and S. M. Auerbach, Green Chem., 2014, 16, 875–884 RSC.
- J. Shang, G. Li, R. Singh, Q. Gu, K. M. Nairn, T. J. Bastow, N. Medhekar, C. M. Doherty, A. J. Hill, J. Z. Liu and P. A. Webley, J. Am. Chem. Soc., 2012, 134, 19246–19253 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00190g |
| This journal is © The Royal Society of Chemistry 2024 |