
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of acrylonitrile–butadiene–styrene copolymers through interface-initiated room-temperature polymerization†
Shijie
Wu
,
Yao
Fu
,
Soham
Das
,
Miles Pamueles
Duan
 and
Tan
Zhang
*
and
Tan
Zhang
*
Division of Natural and Applied Sciences, Duke Kunshan University, Kunshan, Jiangsu 215316, China. E-mail: tan.zhang@dukekunshan.edu.cn
First published on 7th May 2024
Abstract
Acrylonitrile–butadiene–styrene (ABS) copolymers were synthesized in emulsions and silica-containing emulsion gels at 20, 40, and 60 °C. The room-temperature polymerization was achieved by decomposing 2,2′-azobisisobutyronitrile (AIBN) at oil–water interfaces. The decomposition rate constants of the AIBN decomposed in bulk phases and at interfaces were measured for the first time. At room temperatures, the decomposition of AIBN primarily occurred at oil–water interfaces. At 60 °C, the decomposition of AIBN occurred both in bulk phases and at interfaces. In a dark environment without inert gas protection, the polymerizations in emulsions and emulsion gels were conducted at room temperatures successfully. The presence of fumed silica particles enhanced the interfacial initiation and the subsequent polymerization. In the presence of fumed silica, the polymerizations at 40 °C can be as fast as that conducted without silica at 60 °C. The molar mass of the ABS copolymers increased with decreased polymerization temperatures. The ABS copolymers with an ultra-high molar mass and narrow molar mass distribution were synthesized. The ABS copolymers with ultra-high molecular masses exhibit improved ductility and thermal properties without compromising Young's modulus and surface hardness. Interfacial initiation is an eco-friendly technique to produce high-performance polymer materials.
1. Introduction
Polymer materials are ubiquitous in daily life with their applications in packaging,1 coating,2 construction,3 medical,4 electronics,5 energy storage,6 and catalysis.7 Over 400 million tons of polymer materials were produced worldwide in 2021.8,9 Polymer materials have drawn great concerns because of their environmental and sustainable issues. Current concerns mainly focus on the degradation and the recycling of post-consumer polymer waste.10–12 Very few studies focus on the sustainability of polymer production. Many commonly used vinyl polymers are synthesized through free radical polymerizations. The initiation of free radical polymerization usually requires elevated temperatures (60 °C or above) for thermal initiators, such as 2,2′-azobisisobutyronitrile (AIBN), to decompose.13 Considering the volume of polymers produced each year, the energy consumed by high-temperature polymerization techniques is enormous.14,15 With the Paris Agreement, improving energy efficiency in industries is critical for reducing the emissions of greenhouse gases and improving their carbon footprint.16Conducting free radical polymerization at room temperature can be achieved through several approaches. The most straightforward approach is using a thermal initiator that can be decomposed at room temperature. Perfluorodiacyl peroxides, trialkyl boranes, and some azo compounds can produce radicals efficiently at room temperature, but these low-temperature initiators require strict regulation for storage and use.17,18 Redox initiators can be an effective initiation approach at room temperature. Still, their applications are limited due to the presence of salts and transitional metal compounds.15,16,19 The initiation using radiation, such as UV light and γ-ray, is limited by their penetration depth and production scale.20,21 Recently, oil–water interfaces have been found to accelerate some chemical reactions.22 Surfactant-stabilized oil–water interface processes chemical and physical properties that are different from bulk, such as interfacial tension,23 polarity or reactant gradient,24–26 steric effect,27,28 complex formation,29 and electrostatic activities.30 Some of these interfacial properties may enhance the reaction kinetics at oil–water interfaces.22,27,28,31,32 In a dark environment at room temperature, many commonly used thermal initiators, such as AIBN and potassium persulfate, can decompose at oil–water interfaces or through interfacial initiation at room temperature.13,23,32 In light of interfacial initiation, the oil–water interfaces in emulsions can be catalytic sites where thermal initiators can decompose at low temperatures. As a result, a low-temperature radical polymerization is achievable through interfacial initiation in emulsion systems to improve energy efficiency.
Acrylonitrile–butadiene–styrene (ABS) copolymer is an important engineering thermoplastic and is widely used in automotive, electronics, and membranes.33,34 The acrylonitrile/styrene segments make the ABS copolymer mechanically strong and resistant to chemicals while the butadiene segments adjust their toughness.33 The global production of ABS materials was 14 million tons in 2022 and was projected to increase by 37% by 2027.35 Emulsion polymerization is one of the favorite methods to obtain ABS copolymer materials.33 In light of interfacial initiation, the energy consumption of ABS copolymers and their composite materials can be reduced by decreasing the polymerization temperatures in emulsion systems.
Herein, we report the synthesis of ABS copolymer composite monoliths using AIBN as an initiator in emulsions or silica-containing emulsion gels through interfacial initiation. Polymer monoliths are important engineering materials used in separation, extraction, medical, and catalysis.36,37 The polymerizations were conducted at the normal decomposition temperature for AIBN (60 °C) or the lower-than-normal temperatures (20 and 40 °C). The kinetics for initiation and polymerization, the structural characterization, and the mechanical properties of the ABS monoliths synthesized at different temperatures were investigated.
2. Materials and methods
Styrene, acrylonitrile, and 1,3-butadiene were purchased from Aldrich, Adamas, and TCI, respectively. The stabilizers in the monomers were removed by passing through a basic alumina-packed column. Cetyltrimethylammonium bromide (CTAB) was obtained from Aldrich. 2,2′-Azobisisobutyronitrile (AIBN) (Adamas) was purified by recrystallization in methanol before use. Fumed silica Cab-O-Sil M5 with a diameter of 20 nm was provided by Cabot. Toluene, dodecane, 1-butanol, and deuterated chloroform were purchased from Adamas.Silica-containing emulsion gels were prepared using the following procedure: 24 ml monomers (acrylonitrile/1,3-butadiene/styrene 30/10/60 by volume), 1.2 g fumed silica, and 0.5 g AIBN were mixed first in a glass vial using a vortex mixer. 6 ml CTAB aqueous solution (0.5 M) was then added, and the mixtures were shaken for 1 min to form stable emulsion gels. The appearance of the silica-containing emulsion gels can be found in the literature.38,39 Emulsion samples were prepared using the same procedure without adding fumed silica. All the samples were capped and sealed with Teflon tape.
The rheological properties of the emulsion gels were tested using an Anton Paar MCR 302e rheometer with parallel plates. For each sample, the monomers were replaced by a high-boiling point organic solvent dodecane, and 0.5 mL materials were used for the testing. A steady shear was applied before each rheological test.
The polymerizations were conducted in a dark environment to eliminate the possibility of UV exposure. The room-temperature polymerizations were carried out at laboratory temperature, 20 °C, and the high-temperature polymerizations (40 and 60 °C) were conducted in an oil bath. At different times, aliquots of the polymerizing samples were taken out and dried under vacuum to remove water and unreacted monomers. The conversions of monomers were calculated by subtracting the masses of fumed silica and CTAB from the masses of the dried samples.
Pure polymers were extracted from the polymerized monoliths by dissolving them in dimethylformamide at 60 °C. The undissolved portion (fumed silica and CTAB) was discarded. The extracted polymers were dried and redissolved in tetrahydrofuran to measure molar mass and dispersity. Gel permeation chromatography (GPC) measurement was carried out using Waters E2695 with a Heleos light-scattering detector and an Optilab rEX refractive index detector.
For the decomposition of AIBN, the compositions of the emulsion and emulsion gel samples are the same as the polymerizing sample, except the monomers were replaced by non-polymerizable toluene. At different times, portions of the samples were taken out and converted to microemulsions by adjusting the composition to their microemulsion range, i.e., toluene/CTAB/water/1-butanol 55/15/9/21 by mass.32 The absorption spectra for the microemulsions were characterized using an Agilent Cary 60 UV/visible spectrophotometer and the absorption at 344–346 nm was recorded for azo absorption.32 The UV spectra for the bulk samples (toluene/AIBN) were measured directly without converting to microemulsions.
The morphologies of the as-synthesized ABS materials were characterized by Hitachi Regulus 8100 scanning electron microscope (SEM). The Fourier transform infrared spectroscopy (FTIR) was recorded using a Bruker Invenio S spectrometer. The nuclear magnetic resonance (NMR) spectrum was tested using a Bruker Avance Core 400 MHz spectrometer with deuterated chloroform. The differential scanning calorimetry (DSC) thermograms were measured using a TA DSC 25 with a ramping rate of 10 °C min−1, and the thermograms from the second heating scan were recorded. The mechanical properties of the as-synthesized monoliths were tested using an Anton Paar UNHT nanoindentation tester with a maximum load of 10.00 mN, loading/unloading rate of 60.0 mM min−1, and a pause of 5.0 s. For each sample, at least 5 indents were performed, and the average values were reported.
3. Results and discussion
3.1 Initiation at interfaces
AIBN as a commonly used thermal initiator decomposes at 60 °C or above to generate two radicals and release one nitrogen. The decomposition of AIBN in toluene, emulsion, and emulsion gel samples was measured from the azo-absorption in UV spectra, and the decomposition rate constant (kd) was calculated based on the following equation:| At = A0e(−kdt) |
 | ||
| Fig. 1 log(At/A0) as a function of time for AIBN in toluene (bulk), emulsion, and emulsion gel samples at 20, 40, and 60 °C. | ||
Interestingly, the decomposition of AIBN at 20 °C was observed in emulsion systems. AIBN is insoluble in water and barely decomposes in toluene at this low temperature. The observed decomposition in the emulsions most likely originated from the surfactant-stabilized oil–water interfaces. Due to the heterogeneous environment and the complicated initiation process in emulsions,43,44 the decomposition of AIBN contributed from interfaces is difficult to measure directly. By assuming that the decomposition of AIBN in bulk phases and at interfaces are two independent processes, the decomposition rate constants of AIBN at interfaces were calculated using the following equation:
| kd,t = kd,b + kd,i |
| Temperature | Toluene (bulk) | Emulsion | Emulsion gel | |||||
|---|---|---|---|---|---|---|---|---|
| k d,b × 10−6 | R p | k d,t × 10−6 | k d,i × 10−6 | R p | k d,t × 10−6 | k d,i × 10−6 | R p | |
| 20 °C | ∼0 | N/A | 0.31 | 0.31 | 0.08 | 0.61 | 0.61 | 0.28 |
| 40 °C | 0.19 | N/A | 0.86 | 0.67 | 0.76 | 2.78 | 2.59 | 3.62 |
| 60 °C | 21.97 | N/A | 42.58 | 20.61 | 4.42 | 37.11 | 15.03 | 6.08 |
The reaction temperatures for some aqueous and organic phase reactions can be lowered at oil–water interfaces, consequently accelerating the reaction rates.22,26 In these cases, the catalytic effect was achieved through the accumulation of reactants at or near the oil–water interfaces or within micelles, resulting in a higher localized reactant concentration.25,26 The reactions involving multiple components therefore can be catalyzed through this mechanism or so-called micellar catalysis. AIBN decomposes through a homolytic cleavage of its own C–N bonds, which should not be sensitive to the concentration of AIBN.13 In fact, the decomposition temperature for AIBN was slightly lowered if they were diluted in an organic solution.40 Although AIBN initiators prefer to migrate to the oil–water interfaces in emulsions,23 the concentration of AIBN at the interfaces is unlikely to be the reason for the lowered decomposition temperature observed in this study.
In addition to the concentration effects, the interfacial energy of the oil–water interfaces and the complex formation may be the other possible mechanisms. The former is based on the fact that the interfacial energy of the micelles are 200 times greater than the thermal energy needed for AIBN to decompose.23 With the assistance of the additional interfacial energy, AIBN should readily decompose on the micelle surfaces at room temperature. The latter proposed the formation of a CTAB–AIBN complex roughly in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, which lowered the activation energy for AIBN decomposition.29 Both theories seem to explain the decomposition of AIBN in emulsions at 20 °C.
:1 molar ratio, which lowered the activation energy for AIBN decomposition.29 Both theories seem to explain the decomposition of AIBN in emulsions at 20 °C.
By increasing the temperature to 40 °C, the observed decomposition of AIBN in both bulk phases and at interfaces was enhanced. The higher the temperature, the more thermal energy was provided to the molecules in the emulsion systems. The slow decomposition of AIBN in toluene at 40 °C is agreed with the onset decomposition temperature of AIBN in the same conditions measured using calorimetry.40 If the complexes of CTAB–AIBN were formed at interfaces with a lowered activation energy, increasing temperature provides extra energy and the kinetics for AIBN decomposition at interfaces should be accelerated. Increasing thermal energy may not have a significant effect on the interfacial energy at oil–water interfaces, but the diffusion of molecules in emulsions increases with temperature.45 As temperature elevates, AIBN molecules are expected to migrate to the surfactant-stabilized oil–water interfaces faster, at which they decompose with the assistance of additional interfacial energy. With increased temperature, both interfacial energy and complexation mechanisms are expected to have a greater catalytic effect on the AIBN decomposition at interfaces than in bulk. Indeed, the increment of the kd,i in emulsions (3.6 × 10−7 s−1) from 20 to 40 °C is greater than that of the kd,b (1.9 × 10−7 s−1) in bulk phases. At 60 °C, as the thermal energy overcomes the activation energy for AIBN decomposition, AIBN can decompose in an emulsion system regardless of their locations. Therefore, the difference between kd,b and kd,i is minimized.
To further investigate the possible mechanisms, fumed silica was added to the emulsion systems. It is worth noting that the fumed silica particles used in this study are of high purity without any potential catalytic elements and impurities.46 On one hand, fumed silica surfaces are negatively charged which adsorb cationic surfactant CTAB readily in emulsions.47 By introducing fumed silica to a similar emulsion system, the amount of CTAB surfactants at oil–water interfaces was reduced by 50% for an emulsion formed with a nonpolar monomer and 10% for that with a polar monomer.38,39 The adsorbed CTAB surfactants on fumed silica surfaces form densely packed aggregates,47 which are unlikely available for complexation with AIBN limited by diffusion as illustrated in Fig. 2. Since the molar ratio of surfactant and AIBN used in this study is 1:1, smaller than the proposed complex ratio of 3:1, a decrease in the amount of CTAB surfactants at interfaces should reduce the probability of complex formation, consequently lowering the rate constants for the AIBN decomposition. This expectation is contradictory to the experimental results where the AIBN decomposition is more pronounced in the presence of fumed silica. The kd,i for the silica-containing emulsion gels are 2.0 times and 3.9 times greater than that without silica at 20 and 40 °C, respectively. The decomposition of AIBN in emulsion systems is unlikely due to the complexation with surfactants.
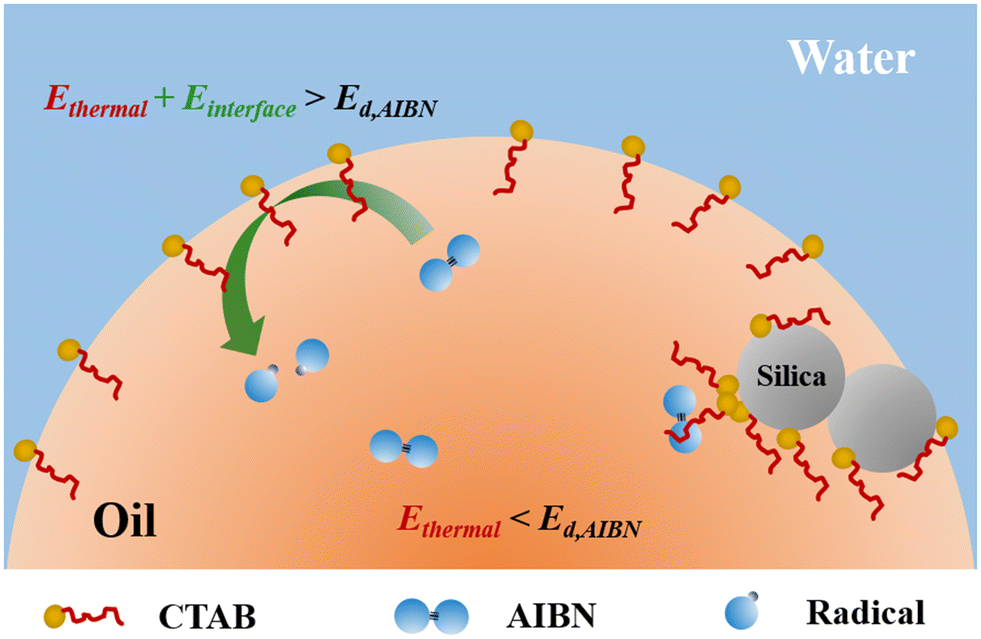 | ||
| Fig. 2 Proposed locations for AIBN decomposition in the silica-containing emulsion gels at 20 °C. | ||
On the other hand, fumed silica particles can form a three-dimensional network and bridge the dispersed oil phases in emulsions,39 leading to gelation in emulsions. It has been shown that high viscosity promotes the AIBN decomposition at interfaces.32,38,39 The enhanced decomposition of AIBN in emulsion gels is likely due to the gelation from fumed silica. In the emulsion gels at 60 °C, the decomposition of AIBN contributed from the interfaces was smaller than that in the bulk phases. The rheological properties of the emulsion gels provide a clue for this phenomenon. As seen in Fig. 3, the loss factors (tanδ), the ratios of the storage modulus and the loss modulus, for the emulsion gels are smaller than 1, indicating that these samples behaved elastically under shear.48 The tanδ of the emulsion gels at 20 and 40 °C are almost identical. The emulsion gels at 60 °C had a greater value of the tanδ, suggesting that the gel structures were weakened at 60 °C. As the temperature increases, the aggregate of fumed silica and the surfactant-stabilized oil–water interfaces in emulsions may change. The temperature-induced rearrangement of fumed silica particles leads to enhanced gelation with increased temperatures,34 which is contradictory to the observations at 60 °C in this study. At high temperatures, the desorption of surfactants became more pronounced and consequently destabilized oil–water interfaces in emulsion gels.48–50 As a result, the desorption of the surfactants from oil–water interfaces reduces the interfacial viscosity and the corresponding gel strength.51 It is reasonable to deduce that the destabilized oil–water interfaces reduce the interfacial energy, providing insufficient loci for AIBN to decompose. The interfacial initiation and the catalytic effect of silica became less pronounced as the interface destabilized at high temperatures.
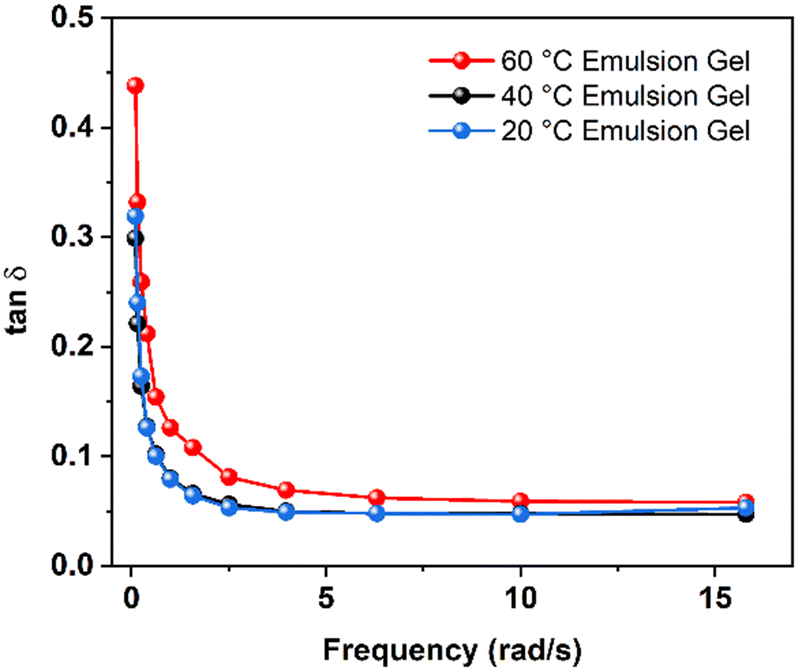 | ||
| Fig. 3 Loss factor tanδ as a function of frequency for the emulsion gels at 20, 40, and 60 °C. | ||
In addition, different surfactants should create different electrostatic environments and diffusion resistance near the oil–water interfaces,52 which affects the redox reactions and complex formation. The decomposition of thermal initiators in emulsions was found to be independent of the surfactant type as well as the initiator type.13 Therefore, it is not surfactant or their complexes but the nature of oil–water interfaces, such as interfacial energy, contributes to the catalytic decomposition of AIBN at low temperatures. An emulsion system with stable interfaces is critical to interfacial initiation. Further study is needed to determine the detailed mechanism of the catalyzed decomposition of AIBN at oil–water interfaces.
3.2 Polymerization at low temperatures
The radicals generated through interfacial initiation are sufficient to initiate free radical polymerizations at low temperatures. The emulsion and emulsion gel samples were polymerized successfully in a dark environment without inert gas protection at 20, 40, or 60 °C. High monomer–polymer conversions were reached for these samples (Fig. 4a and b). At the early stage of free radical polymerization, the monomer–polymer conversion follows a linear relationship.29 The average rates of polymerization in emulsions and emulsion gels were determined from the slope of the following equationwhere Δ[M] is the concentration change of the monomer in the period Δt. As the rate of free radical polymerization is affected by many factors, such as viscosity,29,46,53 initiation efficiency,44,54 and solvent/impurities,39,44,55 the Rp contributed from interfacial initiation cannot be calculated simply. Nonetheless, the rates of polymerization for the emulsion and emulsion gel samples are consistent with the total decomposition rate constants of AIBN. The Rp for emulsions and emulsion gels increased with temperature and the polymerization in emulsion gels is faster than that in emulsions. It is known that high viscosity usually slows polymerization by increasing the radical termination rate. In this study, as the decomposition of AIBN in emulsion gels is much faster than that in emulsion, considering a relatively small initiation efficiency (<0.4) for a slow-diffusion system,54 more active radicals are still present in the emulsion gels than in the emulsions at the same temperature. High radical flux initiates more radical chains in a polymerization system, therefore the monomers are consumed more rapidly. The polymerizations conducted at 20 °C are relatively slow, which took 2–3 days to reach high conversions. The rates of polymerization in emulsion gels at 40 °C are comparable with that in emulsions at 60 °C, about 3–4 hours to reach 90% conversion. By utilization of interfacial initiation, radical polymerizations can be conducted at low temperatures in emulsion gels with a comparable polymerization rate as these conducted at 60 °C.

 | ||
| Fig. 4 The monomer–polymer conversion (% by mass) in (a) emulsion and (b) emulsion gel samples at different temperatures. (c) The appearance of the as-synthesized monoliths from the emulsion (left) and the emulsion gels (right) at 20 °C. | ||
Due to the exothermic nature of chain propagation,53,56 the polymerizations in emulsions and emulsion gels at 60 °C are more intense than their low-temperature counterparts. The surface temperature of the glass vials increases rapidly during polymerization, especially for large-scale production (20 g or above). The heat generated from the chain propagation decomposed more AIBN molecules in the systems, which accelerated the overall polymerization and increased the viscosity dramatically. It is also known as the Trommsdorff effect.53 As a result, the nitrogen molecules released by AIBN decomposition became more difficult to escape and consequently were trapped in the as-synthesized ABS monoliths. The monoliths polymerized at 60 °C have many voids (Fig. S1†), and their measured densities, 0.67–0.74 g cm−3, are much smaller than the density of pure ABS (1.04 g cm−3).57,58 These voids originated from the fast decomposition of AIBN triggered by the Trommsdorff effect.
In comparison, the initiation and the subsequent polymerization conducted at 20 and 40 °C are steadier. The low-temperature environment dissipates heat generated during polymerization more efficiently. The surface temperatures for the glass vials remain almost the same during the polymerization. The monoliths polymerized in the emulsions and emulsion gels at low temperatures, as shown in Fig. 4c, are uniform in appearance. The densities of these monoliths synthesized at low temperatures are found to be 1.02–1.03 g cm−3 which are close to that of neat ABS materials. As the Trommsdorff effect was minimized at low temperatures, the nitrogen gas byproducts were easier to diffuse out of the systems. In addition to energy efficiency, conducting polymerization at low temperatures also improved the safety of the polymerizations and obtained products with a more uniform appearance. Compared with high-temperature polymerizations, conducting polymerization at low temperatures may not require cooling devices, and the production is as efficient as those conducted at 60 °C.
3.3 Characterization and properties
The weight-average molar mass (Mw) and the dispersity (Đ) for the extracted ABS copolymers are listed in Table 2. Compared with bulk or solution polymerization, chain propagation in emulsion systems takes place more separately in these surfactant-stabilized particles, which produces polymers with higher molecular masses.43 At 60 °C, an Mw of 74 and 96 kg mol−1 were obtained for the ABS copolymers synthesized in emulsions and emulsion gels, respectively. High temperatures facilitated the diffusion of radicals and propagating chains, which facilitated intermolecular chain transfer or bimolecular termination at the high conversion (>90%) stage.43,59 It agrees with the relatively broad molar mass distribution obtained for 60 °C. It is difficult to synthesize polymers with an ultra-high molar mass and narrow molar mass distribution through free radical polymerization at high temperatures. Additionally, the molar mass distribution for the ABS copolymers at 60 °C is bimodal (Fig. 5), which is an indicator of the presence of the Trommsdorff effect.53| Temperature | Emulsion | Emulsion gel | ||
|---|---|---|---|---|
| M w | Đ | M w | Đ | |
| 20 °C | 1256 | 1.7 | 1416 | 1.6 |
| 40 °C | 274 | 1.9 | 693 | 2.1 |
| 60 °C | 74 | 4.1 | 96 | 2.8 |
 | ||
| Fig. 5 GPC traces for the extracted ABS copolymers synthesized in the emulsions (E) and the emulsion gels (EG) at 20, 40, and 60 °C. | ||
The Mw of the ABS copolymers increases as the polymerization temperature decreases. The molecular masses obtained from the ABS copolymers synthesized at low temperatures are very large, up to 1416 kg mol−1. At low temperatures, the initiation of AIBN was relatively slow, and a small number of propagating chains were initiated. The monomers available for each propagating chain to react with are more abundant than those with high radical flux. In addition, the low temperature facilitates exothermic chain propagation and minimizes chain diffusions. The polymer chains grow rapidly and reach an ultra-high molar mass before termination occurs. Surprisingly, the molar mass distribution became narrower as the polymerization temperature decreased. As the polymer chain grows very long at low temperatures, the active chain ends are easily trapped within the polymer coils and minimize the probability of encountering another propagating chain ends or other active transfer species.60 As a result, the bimolecular termination and chain transfer at low temperatures were limited even at a high conversion stage. As the polymerization temperature decreases, the distribution becomes unimodal, suggesting a mild and steady polymerization. The ABS copolymers with an ultra-high molar mass and narrow molecular distribution were synthesized at low temperatures.
The chemical compositions of the extracted ABS copolymers were analyzed using FTIR and NMR. The spectra can be found in the ESI.† The spectra for the ABS copolymers synthesized in emulsions and emulsion gels at different temperatures are very similar to each other, and also in line with those in literature.61–64 The chemical compositions of the as-synthesized ABS copolymers were determined from the NMR spectra.63 The as-synthesized ABS copolymers consist of acrylonitrile/butadiene/styrene segments 38/6/56 by mass, which is close to the original compositions for the monomers in the emulsion or emulsion gel systems. It suggests that the polymerization temperature has little effect on the chemical structures and compositions of the ABS copolymers synthesized in this study.
The glass transitions for the extracted ABS copolymers are plotted in Fig. 6. The ABS copolymers synthesized at 60 °C have glass transition temperatures (Tg) at 96–105 °C which is consistent with the ABS copolymers with similar molecular masses.65,66 The Tg of the ABS copolymers increased slowly as the polymerization temperature decreased. As the chemical compositions for the as-synthesized ABS copolymers are similar, the increase in the Tg is likely due to their molecular masses. According to the Fox–Flory equation, the Tg of a polymer depends on the free volume created by the mobile chain ends.67 The free-volume effect minimizes in the polymers with an ultra-high molecular mass. The Tgs saturate at a higher temperature, 115 °C, for the ABS copolymers with a molar mass greater than 1200 kg mol−1 (synthesized at 20 °C).
 | ||
| Fig. 6 DSC thermograms for the extracted ABS copolymers synthesized in the emulsions (E) and the emulsion gels (EG) at 20, 40, and 60 °C. | ||
The morphologies of the as-synthesized monoliths are shown in Fig. 7. The ABS materials synthesized at 20 °C are spherical particles with a diameter of 231 ± 65 (emulsions) and 190 ± 36 nm (emulsion gels). Similar spherical morphologies were obtained for these synthesized at 40 °C with a smaller size, e.g., 118 ± 14 and 91 ± 11 nm from the polymerization of emulsions and emulsion gels, respectively. The differences in the particle sizes originated from the molecular masses of the incorporated ABS copolymers. The ABS copolymers synthesized at lower temperatures have a longer polymer chain which occupied a larger volume and consequently produced larger particles. At 60 °C, the polymerization of the emulsions produced spherical particles with a diameter of 123 ± 49 nm while that of the emulsion gels formed porous structures without a well-defined particle shape. This morphology is consistent with the rheological results where the oil–water interfaces in emulsion gels were destabilized at 60 °C. The voids or pores observed in the ABS monoliths synthesized from emulsion gels at 60 °C likely originated from the trapped nitrogen gas during polymerization.
 | ||
| Fig. 7 Scanning electron micrographs of the ABS monolith synthesized from emulsions (E) or emulsion gels (EG) at 20, 40 or, 60 °C. The scale bar represents 1 μm. | ||
The mechanical properties of the as-synthesized monoliths were tested using nanoindentation. The presence of silica particles created heterogeneous surfaces in the monoliths, which complicates the surface mechanical analysis. To avoid the effects of silica, only these monoliths synthesized from emulsions were tested. As the chemical compositions and microstructures for these monoliths from emulsions are very similar, the major difference among these monoliths is the molar mass of the ABS copolymers, which varied from 74 kg mol−1 (synthesized at 60 °C) to 1256 kg mol−1 (synthesized at 20 °C). Theoretically, the presence of small molecules (surfactants) and the interfaces/voids between polymer particles in the monoliths should alter, most likely deteriorate, the overall mechanical strength of the materials. Therefore, the mechanical properties of the as-synthesized monoliths are not equal to that of the synthesized ABS copolymers. The tests on these monoliths are still meaningful as they provide a first insight into the mechanical properties of these ABS copolymer materials synthesized at low temperatures.
As seen in Fig. 8a, the Young's moduli for the monoliths synthesized at different temperatures are very close to each other, which are 4.3–4.5 GPa. The same trend was also observed for Vickers hardness and elastic deformation work (Fig. 8b and c). The ultra-high molecular masses of the ABS copolymers have little effect on these mechanical properties. The measured plastic deformation work as well as the total deformation work (elastic work + plastic work) are greater for these monoliths synthesized at lower temperatures. The greater total deformation work suggested that the monoliths are capable of absorbing more energies upon deformation, which is an indicator of fracture toughness.68 The brittleness of the ABS monoliths was estimated using the ratio of elastic work and plastic work.68,69 The ratios calculated for the ABS monoliths synthesized at 20, 40, and 60 °C are 0.37, 0.43, and 0.50, respectively. It suggested that the ABS monoliths synthesized at lower temperatures are less brittle or more ductile.68
 | ||
| Fig. 8 Mechanical properties of the ABS monolith synthesized from emulsions (a) modulus; (b) Vickers hardness, and (c) deformation work. | ||
Traditionally, ductile ABS materials were synthesized by incorporating more butadiene segments into the copolymers, which deteriorates the modulus and hardness of the materials.70,71 As the chemical compositions of the as-synthesized ABS copolymers are almost identical, the increment in ductility is unlikely due to the butadiene segments. Similar to other polymers with ultra-high molecular masses, long polymer chains can entangle and tie each other to a greater extent, which can transfer energy to the polymer backbone more efficiently.72 As more energy was absorbed upon deformation, the ABS monoliths with higher molecular masses were more ductile. The interface-initiated room-temperature polymerizations provide an alternative approach to enhance the ductility of ABS materials without compromising their modulus and surface hardness. The narrower molar mass distribution for the ABS copolymers synthesized at low temperatures is also desirable for high-performance applications because the mechanical properties are more predictable and reliable.73
4. Conclusion
ABS copolymers can be synthesized at low temperatures (20 or 40 °C) efficiently. The decomposition of AIBN contributed from the oil–water interfaces was measured for the first time. At low temperatures, the decomposition of AIBN at interfaces or interfacial initiation dominates the initiation in emulsion systems. Interfacial initiation depends on the temperature and the stability of the interfaces. The polymerization conducted through interfacial initiation meets the requirements for green chemistry in the following aspects.16 Energy efficiency: the lowered polymerization temperature saved tremendous energy for polymer production. It reduces greenhouse gas and pollutant emissions and improves the carbon footprint of polymer industries. Waste prevention and atom economy: the monomer conversions for interface-initiated room-temperature emulsion polymerizations are greater than 90%. They are the same as these emulsion polymerizations conducted at high temperatures.74 Compared with traditional techniques, room-temperature emulsion polymerization leaves no additional chemical waste to treat or clean up afterward. Accident prevention: the low-temperature approach can be conducted safely in a mild and steady condition. Additional cooling devices may be unnecessary if polymerization is conducted at low temperatures. It makes the emulsion polymerization process safer and simpler.In addition to being eco-friendly, low-temperature polymerization also produced ABS with ultra-high molecular masses and narrow molar mass distribution. Compared with the ABS materials obtained at 60 °C, those from the low-temperature polymerization showed higher Tg, and better ductility without compromising their modulus and surface hardness. Conducting low-temperature polymerization through interfacial initiation is an efficient eco-friendly technique to produce high-performance polymer materials.
Associated content
The picture for the monoliths synthesized at 60 °C, the FTIR spectra, and the NMR spectra were available in the ESI.†Data availability
Data will be made available on request.Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support of the Kunshan Municipal Government Research Fund (23KKSGR026). The authors also thank Cabot Corporation for providing the fumed silica used in this study. S. D. thank the support from Duke Kunshan University through the Summer Research Scholar (SRS) program.References
- H. D. Huang, P. G. Ren, G. J. Zhong, A. Olah, Z. M. Li, E. Baer and L. Zhu, Promising strategies and new opportunities for high barrier polymer packaging films, Prog. Polym. Sci., 2023, 144, 101722 CrossRef CAS.
- Q. H. Liu and M. W. Urban, Stimulus-responsive macromolecules in polymeric coatings, Polym. Rev., 2023, 63, 289 CrossRef CAS.
- Z. Ait-Touchente, M. Khellaf, G. Raffin, N. Lebaz and A. Elaissari, Recent advances in polyvinyl chloride (PVC) recycling, Polym. Adv. Technol., 2023, 6228 Search PubMed.
- H. Wang, J. Mills, B. R. Sun and H. G. Cui, Therapeutic supramolecular polymers: Designs and applications, Prog. Polym. Sci., 2024, 148, 101769 CrossRef CAS PubMed.
- Z. Y. Rao, A. Thukral, P. Y. Yang, Y. T. Lu, H. Shim, W. J. Wu, A. Karim and C. J. Yu, All-polymer based stretchable rubbery electronics and sensors, Adv. Funct. Mater., 2022, 32, 2111232 CrossRef CAS.
- L. T. Yang and N. Huang, Covalent organic frameworks for applications in lithium batteries, J. Polym. Sci., 2022, 60, 2225 CrossRef CAS.
- L. Zhou, K. He, N. Liu and Z. Q. Wu, Recent advances in asymmetric organocatalysis based on helical polymers, Polym. Chem., 2022, 13, 3967 RSC.
- Annual production of plastics worldwide from 1950 to 2021, https://www.statista.com/statistics/282732/global-production-of-plastics-since-1950/ (accessed January 2024).
- Synthetic rubber production worldwide from 2000 to 2022, https://www.statista.com/statistics/280536/global-natural-rubber-production/ (accessed January 2024).
- I. Olazabal, N. Goujon, D. Mantione, M. Alvarez-Tirado, C. Jehanno, D. Mecerreyes and H. Sardon, From plastic waste to new materials for energy storage, Polym. Chem., 2022, 13, 4222 RSC.
- A. Kulkarni, G. Quintens and L. M. Pitet, Trends in polyester upcycling for diversifying a problematic waste stream, Macromolecules, 2023, 56, 1747 CrossRef CAS.
- M. Zunita, H. P. Winoto, M. F. K. Fauzan and R. Haikal, Recent advances in plastics waste degradation using ionic liquid-based process, Polym. Degrad. Stab., 2023, 211, 110320 CrossRef CAS.
- T. Zhang, G. Xu and F. D. Blum, Eco-friendly room-temperature polymerization in emulsions and beyond, Polym. Rev., 2023, 63, 852 CrossRef CAS.
- Improving Energy Efficiency at U.S. Plastics Manufacturing Plants, U.S. Department of Energy, Washington DC, 2005 Search PubMed.
- M. Goikoetxea, R. Heijungs, M. J. Barandiaran and J. M. Asua, Energy efficient emulsion polymerization strategies, Macromol. React. Eng., 2008, 2, 90 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 2000 Search PubMed.
- M. Yamashina, Y. Sei, M. Akita and M. Yoshizawa, Safe storage of radical initiators within a polyaromatic nanocapsule, Nat. Commun., 2014, 5, 4662 CrossRef CAS PubMed.
- A. Székely and M. Klussmann, Molecular radical chain initiators for ambient- to low-temperature applications, Chem. – Asian J., 2019, 14, 105 CrossRef PubMed.
- N. Kohut-Svelko, R. Pirri, J. M. Asua and J. R. Leiza, Redox initiator systems for emulsion polymerization of acrylates, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2917 CrossRef CAS.
- I. Saenz-Dominguez, I. Tena, M. Sarrionandia, J. Torre and J. Aurrekoetxea, An analytical model of through-thickness photopolymerisation of composites: Ultraviolet light transmission and curing kinetics, Composites, Part B, 2020, 191, 107963 CrossRef CAS.
- L. Barner, J. F. Quinn, C. Barner-Kowollik, P. Vana and T. P. Davis, Reversible addition-fragmentation chain transfer polymerization initiated with γ-radiation at ambient temperature:: an overview, Eur. Polym. J., 2003, 39, 449 CrossRef CAS.
- J. Liu, Z. Yang, Z. Yan, S. Duan, X. Chen, D. Cui, D. Cao, T. Kuang, X. Ma and W. Wang, Chemical micromotors move faster at oil-water interfaces, J. Am. Chem. Soc., 2024, 146, 4221 CrossRef CAS PubMed.
- K. Tauer and N. Öz, Interfacial energy promotes radical heterophase polymerization, Macromolecules, 2004, 37, 5880 CrossRef CAS.
- M. Zhang, L. P. Fan, Y. F. Liu, S. Q. Huang and J. W. Li, Effects of proteins on emulsion stability: The role of proteins at the oil-water interface, Food Chem., 2022, 397, 133726 CrossRef PubMed.
- F. Migliorini, F. Dei, M. Calamante, S. Maramai and E. Petricci, Micellar catalysis for sustainable hydroformylation, ChemCatChem, 2021, 13, 2794 CrossRef CAS.
- E. Borrego, A. Caballero and P. J. Pérez, Micellar catalysis as a tool for C-H bond functionalization toward C-C bond formation, Organometallics, 2022, 41, 3084 CrossRef CAS PubMed.
- L. J. Zhang, S. Q. Yin, W. J. Guan and C. Lu, Steric barrier-steered reaction sites in micellar catalysis, Adv. Opt. Mater., 2023, 11, 2202705 CrossRef CAS.
- Q. Peng, R. Y. Wang, Z. L. Zhao, S. H. Lin, Y. Liu, D. Y. Dong, Z. Wang, Y. M. He, Y. Z. Zhu and J. Jin, et al., Extreme Li-Mg selectivity via precise ion size differentiation of polyamide membrane, Nat. Commun., 2024, 15, 2505 CrossRef CAS PubMed.
- G. Xu and F. D. Blum, Surfactant-enhanced free radical polymerization of styrene in emulsion gels, Polymer, 2008, 49, 3233 CrossRef CAS.
- M. Cerbelaud, A. Aimable and A. Videcoq, Role of electrostatic interactions in oil-in-water emulsions stabilized by heteroaggregation: An experimental and simulation study, Langmuir, 2018, 34, 15795 CrossRef CAS PubMed.
- T. Shen, S. D. Zhou, J. C. Ruan, X. Z. Chen, X. M. Liu, X. Ge and C. Qian, Recent advances on micellar catalysis in water, Adv. Colloid Interface Sci., 2021, 287, 644 CrossRef PubMed.
- G. Xu, R. R. Nambiar and F. D. Blum, Room-temperature decomposition of 2,2′-azobis(isobutyronitrile) in emulsion gels with and without silica, J. Colloid Interface Sci., 2006, 302, 658 CrossRef CAS PubMed.
- F. S. Kamelian, E. Saljoughi, P. S. Nasirabadi and S. M. Mousavi, Modifications and research potentials of acrylonitrile/butadiene/styrene (ABS) membranes: A review, Polym. Compos., 2018, 39, 2835 CrossRef CAS.
- H. Zhang, F. Ding, T. L. Liu, L. Y. Liu and Y. Q. Li, Additivity of the mechanical properties for acrylonitrile-butadiene-styrene resins, J. Appl. Polym. Sci., 2022, 139, 51923 CrossRef CAS.
- Production capacity of acrylonitrile butadiene styrene worldwide from 2022 to 2027, https://www.statista.com/statistics/856670/acrylonitrile-butadiene-styrene-global-production-capacity/ (accessed January 2024).
- I. Nischang and T. J. Causon, Porous polymer monoliths: From their fundamental structure to analytical engineering applications, TrAC, Trends Anal. Chem., 2016, 75, 108 CrossRef CAS.
- Y. H. Xie and M. A. Hillmyer, Nanostructured polymer monoliths for biomedical delivery aTrac-Trends in Analytical Chemistrypplications, ACS Appl. Bio Mater., 2020, 3, 3236 CrossRef CAS PubMed.
- T. Zhang, G. Xu, O. Regev and F. D. Blum, Low-temperature polymerization of methyl methacrylate emulsion gels through surfactant catalysis, J. Colloid Interface Sci., 2016, 461, 128 CrossRef CAS PubMed.
- T. Zhang, G. Xu, Z. F. Li, O. Regev, M. Maddumaarachchi and F. D. Blum, PS/CTAB/silica composites from room temperature polymerization of high internal phase emulsion gels, J. Colloid Interface Sci., 2015, 451, 161 CrossRef CAS PubMed.
- X. R. Li, X. L. Wang and H. Koseki, Study on thermal decomposition characteristics of AIBN, J. Hazard. Mater., 2008, 159, 13 CrossRef CAS PubMed.
- C. X. Zhang, G. B. Lu, L. P. Chen, W. H. Chen, M. J. Peng and J. Y. Lv, Two decoupling methods for non-isothermal DSC results of AIBN decomposition, J. Hazard. Mater., 2015, 285, 61 CrossRef CAS PubMed.
- N. Charton, A. Feldermann, A. Theis, M. H. Stenzel, T. P. Davis and C. Barner-Kowollik, Initiator efficiency of 2,2′-azobis(isobutyronitrile) in bulk dodecyl acrylate free-radical polymerizations over a wide conversion and molecular weight range, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5170 CrossRef CAS.
- P. A. Lovell and F. J. Schork, Fundamentals of emulsion polymerization, Biomacromolecules, 2020, 21, 4396 CrossRef CAS PubMed.
- S. C. Thickett and R. G. Gilbert, Emulsion polymerization: State of the art in kinetics and mechanisms, Polymer, 2007, 48, 6965 CrossRef CAS.
- B. Bera, R. Khazal and K. Schroën, Coalescence dynamics in oil-in-water emulsions at elevated temperatures, Sci. Rep., 2021, 11, 10990 CrossRef CAS PubMed.
- M. P. Duan, Z. R. Zhou and T. Zhang, Synthesis of Polymers with Narrow Molecular Mass Distribution through Interface-Initiated Room-Temperature Polymerization in Emulsion Gels, Polymers, 2023, 15, 4081 CrossRef CAS PubMed.
- T. Zhang, G. Xu, J. Puckette and F. D. Blum, Effect of silica on the structure of cetyltrimethylammonium bromide, J. Phys. Chem. C, 2012, 116, 11626 CrossRef CAS.
- X. J. Wu, Y. Wang, W. Yang, B. H. Xie, M. B. Yang and W. Dan, A rheological study on temperature dependent microstructural changes of fumed silica gels in dodecane, Soft Matter, 2012, 8, 10457 RSC.
- N. R. Cameron and D. C. Sherrington, Biopolymers, liquid crystalline polymers, phase emulsion, Springer, 1996 Search PubMed.
- M. A. Kabong, W. W. Focke, E. L. Du Toit, H. Rolfes and S. Ramjee, Breakdown mechanisms of oil-in-water emulsions stabilised with Pluronic F127 and co-surfactants, Colloids Surf., A, 2020, 585, 124101 CrossRef CAS.
- T. F. Tadros, Emulsion Formation and Stability, Wiley, 2013 Search PubMed.
- R. Bradbury and M. Nagao, Effect of charge on the mechanical properties of surfactant bilayers, Soft Matter, 2016, 12, 9383 RSC.
- Y. Suzuki, Y. Shinagawa, E. Kato, R. Mishima, K. Fukao and A. Matsumoto, Polymerization-induced vitrification and kinetic heterogenization at the onset of the Trommsdorff effect, Macromolecules, 2021, 54, 3293 CrossRef CAS.
- M. Buback, B. Huckestein, F. D. Kuchta, G. T. Russell and E. Schmid, Initiator efficiency in 2,2′-azoisobutyronitrile-initiated free-radical polymerizations of styrene, Macromol. Chem. Phys., 1994, 195, 2117 CrossRef CAS.
- T. Zhang and F. D. Blum, Cationic surfactant blocks radical-inhibiting sites on silica, J. Colloid Interface Sci., 2017, 504, 111 CrossRef CAS PubMed.
- M. L. Coote and T. P. Davis, The mechanism of the propagation step in free-radical copolymerisation, Prog. Polym. Sci., 1999, 24, 1217 CrossRef CAS.
- F. Castles, D. Isakov, A. Lui, Q. Lei, C. E. J. Dancer, Y. Wang, J. M. Janurudin, S. C. Speller, C. R. M. Grovenor and P. S. Grant, Microwave dielectric characterisation of 3D-printed BaTiO3/ABS polymer composites, Sci. Rep., 2016, 6, 22714 CrossRef CAS PubMed.
- A. Mokhtari, N. Tala-Ighil and Y. A. Masmoudi, Nanoindentation to determine Young's modulus for thermoplastic polymers, J. Mater. Eng. Perform., 2022, 31, 2715 CrossRef CAS.
- A. Rudin and P. Choi, The elements of polymer science & Engineering, Academic Press, 2013 Search PubMed.
- Q. M. Jiang, D. Y. Xia, C. Liu, Q. L. Jiang, J. H. Li, B. Han, B. B. Jiang, W. Y. Huang, X. Q. Xue and H. J. Yang, et al., Does bimolecular termination dominate in benzoyl peroxide initiated styrene free-radical polymerization?, Polymer, 2020, 189, 122184 CrossRef CAS.
- A. K. Du, Q. Zhou, Z. B. Wen, J. W. Yang, J. M. N. van Kasteren and Y. Z. Wang, Denitrogenation of acrylonitrile-butadiene-styrene copolymers using polyethylene glycol/hydroxides, Polym. Degrad. Stab., 2011, 96, 870 CrossRef CAS.
- H. Kuleyin, R. Gümrük and S. Çaliskan, The effect of ABS fraction on the fatigue behavior of PMMA/ABS polymer blends, Mater. Today Commun., 2022, 33, 104139 CrossRef CAS.
- S. Kim, H. Y. Yu, C. H. Jeong, E. Choi and S. Ahn, Composition analysis of grafted ABS and its blend copolymers using a combination of 1H NMR spectroscopy and nitrogen element analysis, Bull. Korean Chem. Soc., 2021, 42, 1251 CrossRef CAS.
- M. I. Mohammed, D. Wilson, E. Gomez-Kervin, B. Tang and J. F. Wang, Investigation of closed-loop manufacturing with acrylonitrile butadiene styrene over multiple generations using additive manufacturing, ACS Sustainable Chem. Eng., 2019, 7, 13955 CrossRef CAS.
- K. M. M. Billah, F. A. R. Lorenzana, N. L. Martinez, R. B. Wicker and D. Espalin, Thermomechanical characterization of short carbon fiber and short glass fiber-reinforced ABS used in large format additive manufacturing, Addit. Manuf., 2020, 35, 101299 CAS.
- L. W. Jang, C. M. Kang and D. C. Lee, A new hybrid nanocomposite prepared by emulsion copolymerization of ABS in the presence of clay, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 719 CrossRef CAS.
- V. N. Novikov and E. A. Rössler, Correlation between glass transition temperature and molecular mass in non-polymeric and polymer glass formers, Polymer, 2013, 54, 6987 CrossRef CAS.
- J. Konnerth, A. Jäger, J. Eberhardsteiner, U. Müller and W. Gindl, Elastic properties of adhesive polymers.: II.: Polymer films and bond lines by means of nanoindentation, J. Appl. Polym. Sci., 2006, 102, 1234 CrossRef CAS.
- G. Wypych, Atlas of Material Damage, ChemTec Publishing, 2012 Search PubMed.
- H. Lee and D. Cho, Effects of A, B, and S components on fiber length distribution, mechanical, and impact properties of carbon fiber/ABS composites produced by different processing methods, J. Appl. Polym. Sci., 2021, 138, 50674 CrossRef CAS.
- Z. Y. Tan, X. F. Xu, S. L. Sun, C. Zhou, Y. H. Ao, H. X. Zhang and Y. Han, Influence of rubber content in ABS in wide range on the mechanical properties and morphology of PC/ABS blends with different composition, Polym. Eng. Sci., 2006, 46, 1476 CrossRef CAS.
- K. Patel, S. H. Chikkali and S. Sivaram, Ultrahigh molecular weight polyethylene: Catalysis, structure, properties, processing and applications, Prog. Polym. Sci., 2020, 109, 101290 CrossRef CAS.
- A. Godwin, M. Hartenstein, A. H. E. Müller and S. Brocchini, Narrow molecular weight distribution precursors for polymer-drug conjugates, Angew. Chem., Int. Ed., 2001, 40, 594 CrossRef CAS PubMed.
- J. M. Asua, Emulsion polymerization: From fundamental mechanisms to process developments, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1025 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00152d |
| This journal is © The Royal Society of Chemistry 2024 |