
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxime derivatives of betulonic acid and platanic acid as novel cytotoxic or antiviral agents†
Lucie
Černá‡
a,
Uladzimir
Bildziukevich‡
c,
Lucie
Rárová‡
d,
Jana
Trylčová‡
e,
David
Šaman
e,
Jan
Weber
e,
Petra
Lovecká
a and
Zdeněk
Wimmer
 *bc
*bc
aDepartment of Biochemistry and Microbiology, University of Chemistry and Technology in Prague, Technická 5, 16628 Prague 6, Czech Republic
bDepartment of Chemistry of Natural Compounds, University of Chemistry and Technology in Prague, Technická 5, 16628 Prague 6, Czech Republic. E-mail: wimmerz@vscht.cz
cInstitute of Experimental Botany of the Czech Academy of Sciences, Isotope Laboratory, Vídeňská 1083, 14220 Prague 4, Czech Republic. E-mail: wimmer@biomed.cas.cz
dLaboratory of Growth Regulators, Institute of Experimental Botany of the Czech Academy of Sciences, Faculty of Science, Palacký University, Šlechtitelů 27, CZ-78371 Olomouc, Czech Republic
eInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo náměstí 2, 16610 Prague 6, Czech Republic
First published on 12th February 2024
Abstract
Less frequently studied plant triterpenoids betulonic acid and platanic acid were selected to design, synthesize and investigate their oxime derivatives as novel and potentially effective cytotoxic and/or antiviral agents. The target compounds were subjected to cytotoxicity screening tests in several human cancer cell lines. Several of them displayed cytotoxicity against T-lymphoblastic leukemia (CCRF-CEM), breast adenocarcinoma (MCF7), malignant melanoma (G-361) and cervical cancer (HeLa) cell lines. Betulonic acid oxime (3) displayed cytotoxicity against CCRF-CEM (IC50 = 18.9 ± 1.1 μM; SI = 2.4) and G-361 (IC50 = 21.3 ± 2.8 μM; SI = 2.2) cancer cell lines. The benzyl ester of platanic acid oxime (17) displayed an anti-HIV-1 effect (EC50 = 3.2 ± 0.43 μM; SI > 31) and no anti-HSV-1 effect (EC50 > 100 μM), while platanic acid oxime (15) showed lower effects, EC50 = 36 ± 4.0 μM (HIV-1) and EC50 = 48 ± 6.0 μM (HSV-1), respectively. Compound 17 showed high selectivity in antiviral effects, being effective in HIV-1 and inactive in HSV-1. A side product 18 showed antiviral activity with EC50 = 15 ± 1.3 μM (HIV-1) and EC50 = 12 ± 0.21 μM (HSV-1), while another side product 19 was cytotoxic to HeLa (IC50 = 24.5 ± 1.8 μM).
1. Introduction
Cancer and viral infections are among the most important human diseases affecting a large part of the human population.1,2 Therefore, a search for new agents capable of treating either of these threats has permanently been required. Plants have always served as sustainable sources of important natural products in drug discovery, and recent review articles have clearly shown the potential of plant products, namely plant triterpenoids, to be applied in cancer and viral infection treatments.1–5Viral infections are the main infectious diseases worldwide,6 representing more than two thirds of infectious diseases. Viruses affecting human bodies are divided into two categories, either viruses representing long-term parasites in the human body, or viruses representing long-term parasites in animals, which live in vicinity to humans. These viruses can also infect humans across species.6
Human immunodeficiency virus type 1 (HIV-1) features a common mechanism to catalyse membrane fusion as other enveloped viruses with class I fusion proteins. The envelope glycoproteins of HIV-1 play important roles in the early stage of HIV-1 entry. The surface glycoprotein subunit of HIV-1 engages in receptor binding, and the transmembrane subunit mediates the fusion of the virus with the target cell.6 Therefore, anti-HIV-1 agents can be designed by targeting different steps of the enveloped virus replication cycle (maturation inhibitors).7 Among the various life cycle events, the entry of enveloped viruses into host cells by fusion of the viral membrane with the cellular membrane is the first step.6
Herpes simplex virus (HSV) is a double-stranded DNA virus.8 It exists in two types of HSV strains: HSV-1 causing infection of the oral mucosa, and HSV-2 affecting the genital mucosa and causing neonatal infections.9 According to the latest report of the WHO, more than 65% of the world population under the age of 50 is infected with HSV-1, and the incidence of herpes simplex virus encephalitis (HSE) caused by HSV-1 infection is increasing every year.6 Moreover, HSV-1 infections are associated with neurodegenerative diseases, e.g., Alzheimer's disease.10–12 HSV-1 has also been identified for its association with certain types of cancers, e.g., cervical carcinoma or acute lymphocytic leukemia.13 At present, anti-HSV-1 drugs mainly belong to nucleoside drugs (e.g., acyclovir, ganciclovir, valacyclovir, etc.), capable of inhibiting viral replication by interfering with viral DNA polymerase. Based on the wide use of these drugs, drug-resistant strains of the virus appeared within a relatively short time period.12 In order to cope with the problem of drug resistance, the investigation has been focused on developing novel anti-HSV agents targeting different viral life cycle steps.14
Triterpenoids are important plant metabolites. Studies reported that triterpenoids have a wide range of pharmacological effects and important biological activities, especially anti-inflammatory, hypoglycemic, anti-tumour, and antiviral activities. Triterpenoids have been proven to be important in the defence mechanisms of plants against pathogenic infection. Recently, an increasing number of studies have shown that triterpenoids exhibit excellent antiviral activity. These compounds can prevent viral adsorption and invasion into host cells in the early stage and thereby inhibit the viral replication process at the entry step.6
In this partial investigation, attention has been focused on two less explored plant triterpenoids, betulonic acid15 and platanic acid.16
Betulonic acid, 3-oxo-lup-20(29)-en-28-oic acid (1; Scheme 1), belongs to the lupane family of plant triterpenoids and is found in different plant species.17,18 Mostly, the content of betulonic acid in the plant sources is low, and, therefore a semisynthetic oxidation procedure from betulin often takes place to obtain betulonic acid in a higher quantity.19 The solubility of betulonic acid in aqueous media is low, and so is its bioavailability. Despite this fact, betulonic acid shows important biological effects, such as antiviral, antimicrobial, anti-inflammatory, hepatoprotective, anticancer, etc.17 To enhance its bioavailability, and thus, to enhance the biological effects, structural modifications have taken place to prepare its amides and other nitrogen-group-bearing derivatives or compounds with an extended number of cycles, either heteroaromatic or heterocyclic ones.17 Betulonic acid derivatives bearing excessive ring(s) have been found to display ability to interfere with human corona virus 229E replication via the nsp15 endoribonuclease.20 Aryl pyrimidine amides of betulonic acid have been developed as anti-inflammatory agents,21 and piperazine-based amides exhibited cytotoxicity.22
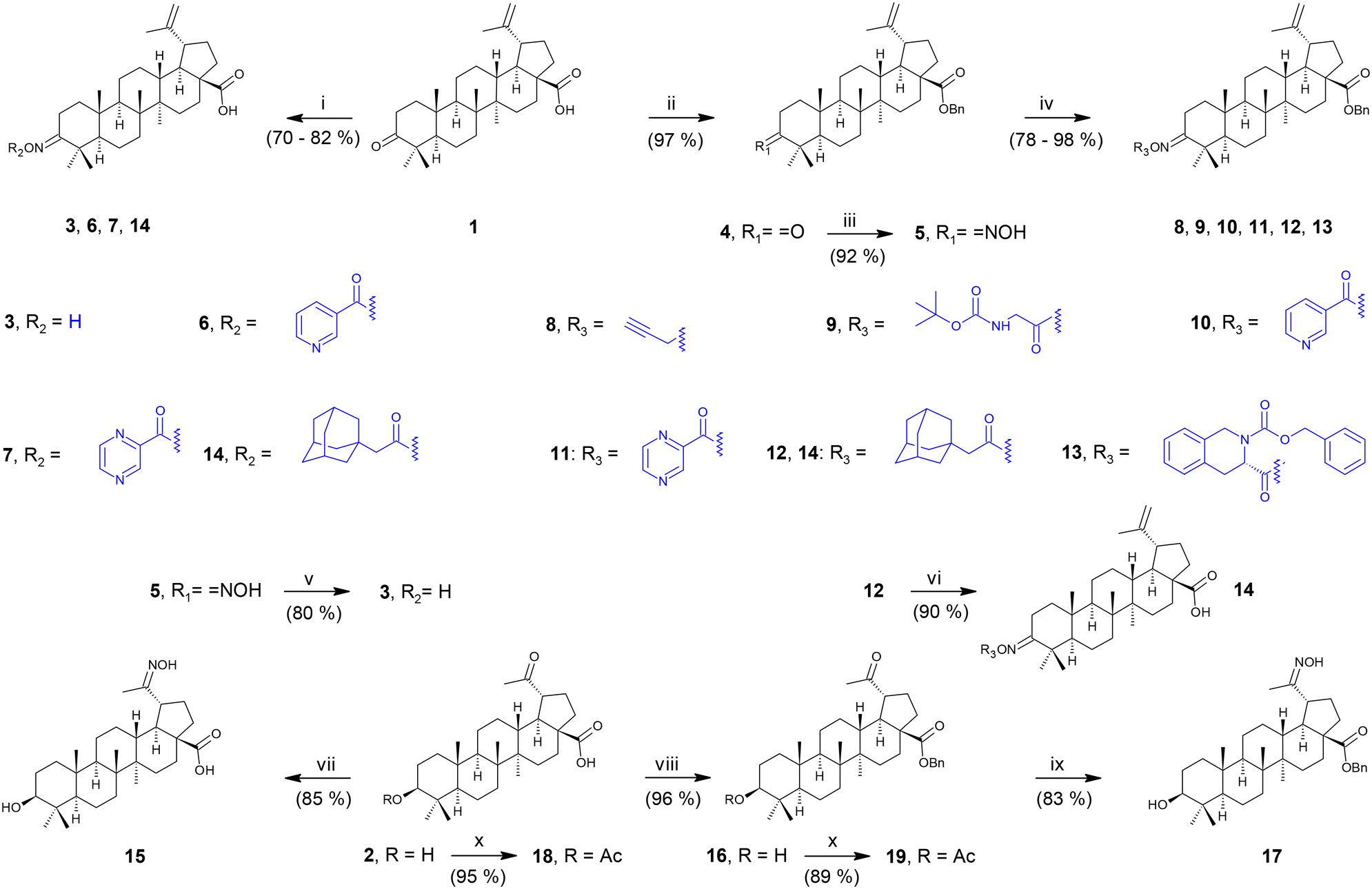 | ||
Scheme 1 Synthetic procedures. Reaction conditions: i: a) NH2OH·HCl, pyridine, 96% EtOH (to prepare 3), b) selected reagent, 50% T3P in ethyl acetate, pyridine (to prepare 6, 7 and 14); ii: BnBr, K2CO3, Et3N, DMF; iii: NH2OH·HCl, pyridine, 96% EtOH; iv: a) NaH, DMF, b) propargyl bromide, DMF (to prepare 8); or selected reagent, 50% T3P in ethyl acetate, pyridine (to prepare 9); or selected reagent, EDC·HCl, DMAP, DCM (to prepare 10–13); v: 10% Pd/C, 1,4-cyclohexadiene, EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF (1:1); vi: 10% Pd/C, 1,4-cyclohexadiene, EtOH:THF (1:1); vii: NH2OH·HCl, pyridine, 96% EtOH; viii: BnBr, K2CO3, Et3N, DMF; ix: NH2OH·HCl, pyridine, 96% EtOH; x: Ac2O, DMAP, THF, argon atmosphere. :THF (1:1); vi: 10% Pd/C, 1,4-cyclohexadiene, EtOH:THF (1:1); vii: NH2OH·HCl, pyridine, 96% EtOH; viii: BnBr, K2CO3, Et3N, DMF; ix: NH2OH·HCl, pyridine, 96% EtOH; x: Ac2O, DMAP, THF, argon atmosphere. | ||
Platanic acid, (3β)-3-hydroxy-20-oxo-30-norlupan-28-oic acid (2; Scheme 1), belongs to the norlupane family of pentacyclic plant triterpenoids. It is mostly extracted from sycamore trees, Platanus occidentalis L. and Platanus x hybrida Brot. (Platanaceae).23 It is also found in Melaleuca leucadendra L. (Myrtaceae)24 and in Leptospermum scoparium J. R. Forst. & G. Forst. (Myrtaceae).25 The solubility in aqueous media and bioavailability of platanic acid are also low. Nevertheless, it exhibits important anti-HIV activity26 and cytotoxicity.27,28 We have reviewed platanic acid, besides other triterpenoids, and its selected nitrogen-group-bearing derivatives recently.3 Nevertheless, before the review paper has been published, several new papers have appeared in the literature: pentacyclic triterpenoid sulphamates used for radiotherapy of breast cancer,29 nitrogen-group-bearing bicyclic compounds used as inhibitors of butyrylcholinesterase,30,31 pyridinylidene derivatives developed for targeting apoptosis,32 platanic acid amides33,34 and even more complex derivatives of platanic acid35 for investigating cytotoxicity.
The objectives of this investigation have been set, and the attention has been focused on: (a) synthesis and analysis of oximes, oxime-ethers and oxime-esters of betulonic acid and oximes of platanic acid; (b) determination and evaluation of the cytotoxicity and antiviral activity of the target compounds and selected intermediate compounds; (c) evaluation and comparison of the significance of the parent triterpenoids in the target compounds, regarding the type of their pharmacological effect.
2. Results and discussion
2.1. Synthetic protocol and structure elucidation
To prepare the oxime of betulonic acid directly, 1 was subjected to a reaction with NH2OH·HCl in 96% ethanol in the presence of pyridine as a base to obtain 3 (Scheme 1, path i). To synthesize three compounds of the variety of oxime esters (6, 7 and 14), 3 was allowed to react with the appropriate carboxylic acid, using 1-propanephosphonic acid anhydride (T3P) as the condensation agent, in pyridine (path i).36 To synthesize further the variety of oxime ether and oxime esters of betulonic acid, 1 was converted into its benzyl ester (4) to protect the carboxyl group (path ii). Subsequently, the C(3) keto group was converted into the oxime using again NH2OH·HCl in 96% ethanol in the presence of pyridine as a base yielding 5 (path iii). The Bn-protected oxime 5 was further etherified to obtain oxime ether 8 in a two-step procedure: oxime 5 was first converted into its sodium salt using sodium hydride in DMF, and the salt was then reacted with propargyl bromide affording the oxime ether 8 (path iv).36 To synthesize other target oxime ester derivatives of 1, i.e., compounds 9–13, oxime 5 was reacted with the appropriate carboxylic acids, using either T3P in pyridine as the condensation reagent (to prepare 9; path iv) or N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl) as the condensation reagent, in DCM with DMAP (to prepare 10–13; path iv).37 The benzyl protecting group in 5 was removed by transfer hydrogenation, using 10% Pd on charcoal as a catalyst and 1,4-cyclohexadiene as a hydrogen donor, in an ethanol/THF (1:1) mixture yielding 3 by the indirect way to compare the yields of 3 (path v). Last but not least, product 14 was prepared from 12 by the same transfer hydrogenation as described above (path vi), which was more effective than that mentioned above (path i).
The first step in the modification of platanic acid (2; Scheme 1) consisted of conversion of the keto group into the oxime group by NH2OH·HCl in ethanol in the presence of pyridine (15; path vii). The other way consisted of protection of the carboxyl group of 2 in the form of benzyl ester of platanic acid (16; path viii) to investigate the effect of the free carboxyl group vs. esterified carboxyl group in the synthesis of the oxime derivatives (15vs.17). The esterified platanic acid 16 gave a higher yield of the oxime derivative (15vs.17; path ix). Finally, a side product 18 was prepared from 2 using acetic anhydride and DMAP in THF under an argon atmosphere (path x).38 Using the same procedure (path x), 19 was prepared from 16. Compounds 18 and 19 were originally considered to become the intermediates in the synthesis of 17; however, the protection of the C(3)–OH group in 2 was finally not required. Nevertheless, 18 and 19 were subjected to biological screening tests, displaying their antiviral effects on HIV-1 and HSV-1 cell culture models. We also tried to synthesize a series of platanic acid derivatives to be analogous to 6–14; however, the platanic acid oxime ether and oxime ester derivatives were more difficult to prepare in comparison with the betulonic acid oxime derivatives. The reactions either gave very low yields of the required products or did not proceed at all, and, therefore, the synthetic effort to engage 2 was stopped.
To elucidate the configuration of the platanic acid oximes and their derivatives, we have studied the so far published results from the literature. The search has been based on the analysis of the 1H and 13C NMR spectra of compounds 15 and 17 (ESI,† Table S1). The chemical shift of the C(29) carbon atom of the (20E)-isomer of the earlier published oxime derivative was reported at δ = 11.5 ppm, while the same signal of the (20Z)-isomer was at δ = 16.1 ppm.39 These values of the corresponding chemical shift in the 13C NMR spectra, found for 15 and 17, are δ = 13.1 ppm and δ = 11.9 ppm, respectively. Similarly, much earlier published data of the 1H NMR chemical shifts of the H–C(19) hydrogen atom were found at δ = 3.44 ppm [(20E)-isomer of the earlier published compound] and δ = 4.63 ppm [(20Z)-isomer of the same compound].40 The corresponding chemical shifts of the H–C(19) hydrogen atom in compounds 15 and 17 were found at δ = 3.43 ppm and δ = 3.32 ppm, respectively. The search resulted in a finding that compounds 15 and 17 bear the (20E)-absolute configuration of the oxime group. Table S1† also shows chemical shifts of several additional signals.
Elucidating the configuration of the C(3) oxime group in the betulonic acid derivatives is much more difficult (ESI,† Table S2). The relevant signal seems to belong to the C(24) carbon atom in the 13C NMR spectra. The chemical shift of the C(24) carbon atom was found to be δ = 14.3 ppm.41 We have found these chemical shifts at δ = 14.3 ppm (3) and δ = 14.5 ppm (5), respectively. In the 1H NMR spectra, the H–C(24) and H–C(23) hydrogen atom chemical shifts were found to be δ = 0.83 ppm and δ = 1.06 ppm, respectively, for the (3E)-isomer.41 We have found these chemical shifts at δ = 0.86 ppm and δ = 0.78 ppm (3) and δ = 1.06 ppm and δ = 1.16 ppm (5), respectively. These findings also support the (3E)-configuration of 3 and 5. Table S2† also shows chemical shifts of several additional signals.
Similar values of the chemical shifts of the corresponding signals in the 1H and 13C NMR spectra can be found in the spectra of other compounds of the published series. However, we could not find relevant NMR data comparing (E)- and (Z)-isomers of the structurally identical substances in the literature. The available literature data either do not contain the determination of the stereochemistry or they establish it as (E)-configuration while stating the presence of the only stereoisomer. In addition, we have not identified the (Z)-isomers in the prepared series of compounds. It seems realistic that they appeared as minor components in the reaction mixtures that were separated from the major products during column chromatography purification.
2.2. Cytotoxicity
Only several compounds of this series showed cytotoxicity; however, the IC50 values are mostly low (Table 1). The oxime of betulonic acid (3) displayed cytotoxicity against T-lymphoblastic leukemia (CCRF-CEM), breast adenocarcinoma (MCF7), malignant melanoma (G-361) and cervical cancer (HeLa) cell lines comparable with those displayed by the parent betulonic acid (1) (Table 1). Compound 14 was partly successful in cytotoxicity screening tests showing IC50 values of 7.4 ± 0.3 μM (G-361; SI (selectivity index) = 2.1) and 8.3 ± 2.8 μM (HeLa; SI = 1.9), respectively. The other disadvantage of this series of compounds is the fact that the cytotoxicity of the compounds is almost equal to their toxicity to the reference cells, normal human fibroblasts, BJ. CDDP (cisplatin) was used as a positive reference compound. It should be noted that CDDP displayed cytotoxicity comparable with 1, 6 and 14 against the HeLa cancer cell line, and with 14 against the G-361 cancer cell line. Moreover, compounds 1, 6 and 14 showed a better ratio between cytotoxicity to the cancer cell lines and toxicity to the reference cells (Table 1).| Compounda | IC50 ± SE [μM], 72 h, DMSO | ||||
|---|---|---|---|---|---|
| CCRF-CEMb | MCF7c | G-361d | HeLae | BJf | |
| a Compounds 4, 5, 8, 9, 10, 12, 13, 15 and 18 showed no cytotoxicity against the cancer cell lines, and no toxicity to BJ cells (IC50 > 50 μM). b CCRF-CEM, human T-lymphoblastic leukemia. c MCF7, human breast adenocarcinoma. d G-361, human malignant melanoma. e HeLa, human cervical cancer. f BJ, normal human fibroblasts, reference cells. g SI (selectivity index) = IC50 (BJ)/IC50 (cancer cell line), for 3: SI = 2.4 (CCRF-CEM), SI = 2.0 (MCF7), SI = 2.2 (G-361), SI = 1.6 (HeLa). h SI = 2.1 (G-361), SI = 1.9 (HeLa). i cis-Diaminedichloridoplatinum(II), cisplatin, a pharmacologically used agent for treating cancers, a positive reference compound. | |||||
| 1 | 20.5 ± 1.6 | 31.1 ± 4.2 | 27.2 ± 10.5 | 11.7 ± 2.1 | >50 |
| 2 | >50 | >50 | >50 | >50 | >50 |
| 3 | 18.9 ± 1.1 | 23.3 ± 0.9 | 21.3 ± 2.8 | 27.8 ± 1.8 | 46.2 ± 1.1 |
| 6 | 44.0 ± 4.4 | 37.2 ± 2.5 | 29.8 ± 2.2 | 13.5 ± 2.6 | 47.8 ± 3.1 |
| 7 | 44.5 ± 3.5 | 23.8 ± 2.5 | 22.0 ± 0.4 | 27.7 ± 5.5 | >50 |
| 11 | 26.5 ± 2.5 | >50 | >50 | 25.6 ± 4.3 | 20.4 ± 0.8 |
| 14 | 16.2 ± 0.3 | 16.3 ± 0.3 | 7.4 ± 0.3 | 8.3 ± 2.8 | 15.6 ± 1.2 |
| 16 | >50 | 47.9 ± 0.2 | >50 | >50 | >50 |
| 17 | 33.3 ± 1.3 | 22.3 ± 3.4 | 27.1 ± 1.7 | >50 | >50 |
| 19 | 36.5 ± 4.5 | >50 | >50 | 24.5 ± 1.8 | >50 |
| CDDP | 0.8 ± 0.1 | 7.7 ± 1.7 | 4.5 ± 0.6 | 11.4 ± 3.8 | 6.9 ± 0.9 |
2.3. Antiviral activity
To determine the anti-HIV-1 and anti-HSV-1 activities of the prepared derivatives of 1 and 2, the ability of the compounds to inhibit the virus-induced cytopathic effect (CPE) in MT-4 and Vero cells, respectively, was measured. The most important result of the anti-HIV-1 tests was observed with 17 displaying a high anti-HIV-1 effect (EC50 = 3.2 ± 0.43 μM; SI > 31; Fig. 1 and Table 2), while being inactive in anti-HSV-1 tests (Fig. 2 and Table 2). Compound 18, a side product in this series of compounds, showed EC50 = 15 ± 1.3 μM (HIV-1) and EC50 = 12 ± 0.21 μM (HSV-1) (Table 2; Fig. 1 and 2). Inhibition of the virus-induced CPE in MT-4 and Vero cells by 17 and 18, as well as the viability of the MT-4 or Vero cells, respectively, is also shown in Fig. 1 and 2. Only the compounds showing no or very low cytotoxicity in the screening tests (Table 1) were subjected to the antiviral tests subsequently (Table 2; Fig. 1 and 2; ESI,† Fig. S1). Therefore, graphical dependence of inhibition of the virus-induced CPE in the MT-4 and Vero cells on the concentration of the less active compounds 4, 5, 8 and 15, the source triterpenoids (1 and 2), and the positive reference compounds (acyclovir or saquinavir) is summarized in ESI,† Fig. S1.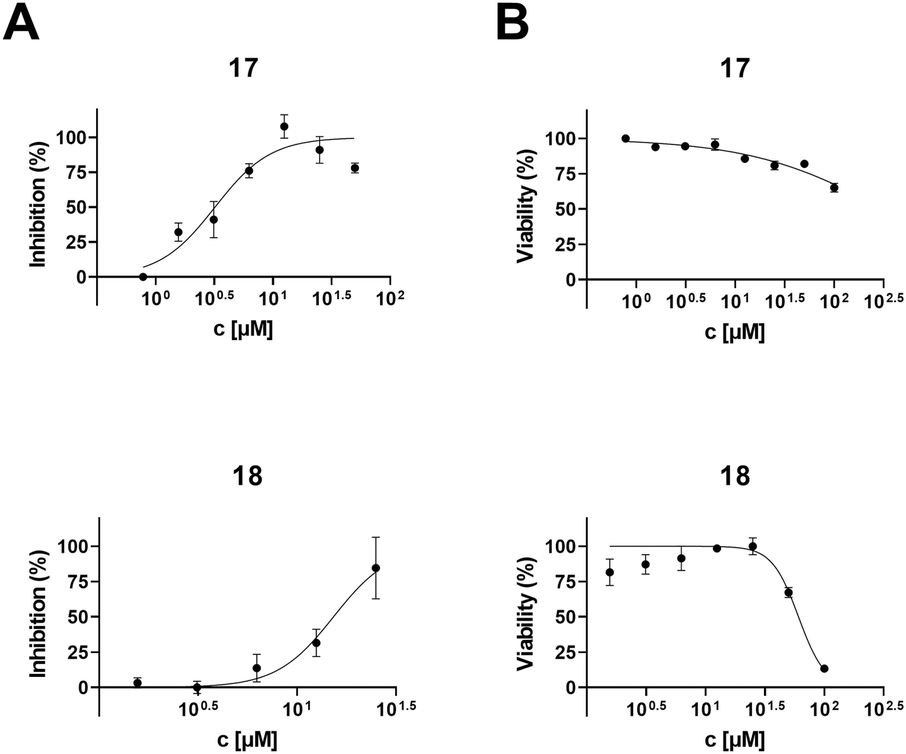 | ||
| Fig. 1 HIV-1 inhibition (A) and cytotoxicity (B) of 17 and 18 against MT-4 cells. | ||
| Compounda | Anti-HIV-1 activity and cytotoxicity against the MT-4 cells | Anti-HSV-1 activity and cytotoxicity against the Vero cells | ||||
|---|---|---|---|---|---|---|
| EC50 ± SE [μM] | CC50 ± SE [μM] | SIb | EC50 ± SE [μM] | CC50 ± SE [μM] | SIb | |
| a Compounds 4, 5 and 8 showed no antiviral activity (EC50 > 100 μM) and no cytotoxicity (CC50 > 100 μM). b SI (selectivity index) = CC50/EC50. c n.d. = not determined. | ||||||
| 1 | >71 | 71 ± 6.7 | <1 | 17 ± 1.1 | 92 ± 21 | 5.4 |
| 2 | 47 ± 5.0 | >100 | >2.1 | 75 ± 2.2 | >100 | >1.3 |
| 15 | 36 ± 4.0 | >100 | >2.8 | 48 ± 6.0 | >100 | >2.1 |
| 17 | 3.2 ± 0.43 | >100 | >31 | >100 | >100 | n.d.c |
| 18 | 15 ± 1.3 | 61 ± 4.5 | 4.1 | 12 ± 0.21 | ∼27 | 2.3 |
| Saquinavir | 0.0015 ± 0.0005 | >1 | >670 | n.d. | n.d. | n.d. |
| Acyclovir | n.d. | n.d. | n.d. | 9.4 ± 0.90 | >100 | >11 |
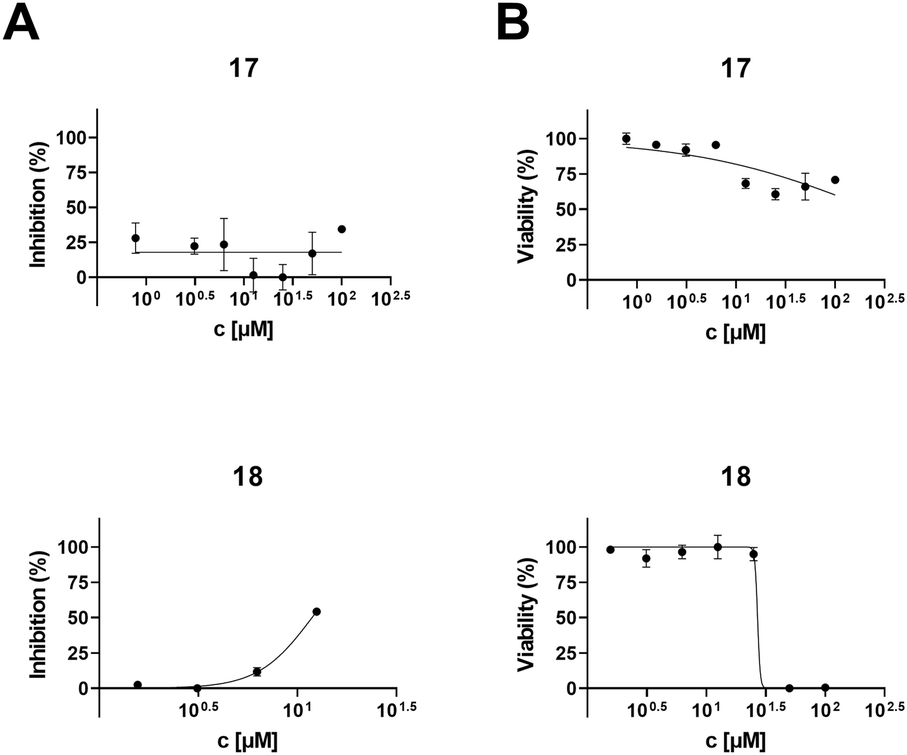 | ||
| Fig. 2 HSV-1 inhibition (A) and cytotoxicity (B) of 17 and 18 against Vero cells. | ||
2.4. A comparison of the oxime derivatives of betulonic acid and platanic acid in cytotoxicity and antiviral activity
A remarkable result was achieved with the oximes 3 and 17, in which the former compound (3) displayed cytotoxicity against CCRF-CEM, MCF7, G-361 and HeLa cancer cell lines (Table 1), while the latter compound (17) showed anti-HIV-1 activity displaying IC50 = 3.2 ± 0.43 μM (SI > 31) (Table 2 and Fig. 1) besides low cytotoxicity (Table 1). In fact, 17 is the benzyl ester of the oxime of platanic acid. When the benzyl group was removed to obtain 15, the anti-HIV-1 activity decreased 10 times, and the selectivity for the anti-HIV-1 effect was lost (Table 2 and Fig. 1). Compound 14 showed cytotoxicity against G-361 and HeLa cancer cell lines; however, it was also toxic to normal human fibroblasts (BJ). The side product 18 displayed no cytotoxicity; however, it showed activity both in HIV-1 and HSV-1 tests (EC50 = 15 ± 1.3 μM and 12 ± 0.21 μM, respectively; Table 2; Fig. 1 and 2). Another side product 19 displayed medium to low cytotoxicity against HeLa and CCRF-CEM cancer cell lines (Table 1).3. Conclusions
A series of more than 15 oxime derivatives of betulonic acid and platanic acid that include several oxime esters and an oxime ether were synthesized and subjected to screening tests for cytotoxicity and antiviral activity. It should be stressed here that oxime derivatives of betulonic acid and platanic acid have been studied earlier by other authors. However, with exception of the structures identical to compounds 3, 15 and 19 that have been prepared before,39,41–43 all other compounds of the prepared series of oxime derivatives of betulonic acid and platanic acid are new compounds. None of the synthesized compounds showed high cytotoxicity. However, a partly remarkable result was a finding that the betulonic acid oxime derivative 3 displayed medium cytotoxicity against CCRF-CEM, while the platanic acid oxime derivative 17 showed high anti-HIV-1 activity and was inactive against HSV-1. This finding corresponds to the cytotoxicity and antiviral activity of the parent triterpenoids 1 and 2 (cf.Tables 1 and 2). Compound 14 was cytotoxic against G-361 and HeLa cancer cell lines; however, it was also toxic to the reference BJ cells. The side product 18 showed medium antiviral activity against both HIV-1 and HSV-1, while another side product 19 displayed medium cytotoxicity against the HeLa cancer cell line. Based on these partial conclusions, the platanic acid oxime derivative 17 should be pointed out as the most promising and selective anti-HIV-1 compound of the studied series of the prepared oxime derivatives. It seems reasonable for future investigation of antiviral activity of a broader series of triterpenoid oxime derivatives to focus attention on less polar compounds, which were the results that revealed the advantage of ester-group bearing compounds over those bearing a free carboxyl group or a free C(3)–OH group in the studied series of compounds.4. Experimental
4.1. General
The NMR measurements were performed on a Bruker AVANCE II 600 MHz spectrometer equipped with a 5 mm TCI cryoprobe in a 5 mm tube in different solvents. The 1H NMR and the 13C NMR spectra were recorded at 600.13 MHz and 150.90 MHz (AVANCE II 600 MHz) in CDCl3 or CD3OD using tetramethylsilane (δ = 0.0 – CDCl3) or the signal of the solvent (δ = 3.31 or 49.50 for 1H/13C – CD3OD) as internal references. 1H NMR data are presented in the following order: chemical shift (δ) expressed in ppm, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet), number of protons, and coupling constants in Hertz. For unambiguous assignment of both 1H and 13C signals, 2D NMR, 1H, 13C gHSQC and gHMBC spectra were measured using standard parameter sets and pulse programs delivered by the producer of the spectrometer. Infrared spectra (IR) were measured with a Nicolet iS5 FT-IR spectrometer. Mass spectra (MS) were measured with a ZMD mass spectrometer (Waters, Eschborn, Germany) in a positive ESI mode (coin voltage, CV = 10 to 20 eV). TLC was carried out on silica gel plates (Merck 60F254) and the visualization was performed by both UV detection and spraying with a methanolic solution of phosphomolybdic acid (5%) followed by heating. For column chromatography, silica gel 60 (0.063–0.200 mm) from Merck was used. All chemicals and solvents of analytical grade were purchased from regular commercial sources and the solvents were purified by general methods before use. Triterpenoids were purchased from Dr. Jan Šarek – Betulinines (https://www.betulinines.com). Analytical data and the scanned NMR spectra of the prepared compounds are presented in the ESI,† Experimental part.:2) as the mobile phase, affording product 8 in 78% yield.
:0 to 99:1) as the mobile phase, affording product 10 in 98% yield.
:1; 4 mL) mixture, under an argon atmosphere, with stirring at r.t. for 24 h. Then the solvents were evaporated, and the product was obtained as yellow crystalline compound 3 in 80% yield.
4.2. Cytotoxicity screening tests
The cytotoxicity screening tests were performed according to the standard experimental procedure published earlier.44,45 Cell viability was measured after 72 h using resazurin (Sigma-Aldrich).4.3. Antiviral tests
The anti-HIV-1 and anti-HSV-1 activity and cytotoxicity against MT-4 cells or Vero cells, respectively, were determined as previously described.46,47 Briefly, two-fold dilutions of compounds were added to MT-4 and Vero cells in triplicate, incubated for 1 hour at 37 °C in 5% CO2 and infected with HIV-1 (laboratory strain NL4-3 at multiplicity of infection [MOI] of 0.004 IU per cell) and HSV-1 (strain HF at MOI of 0.005 IU per cell), respectively. Saquinavir and acyclovir were used as controls for HIV-1 and HSV-1, respectively. Following three (HSV-1) and five (HIV-1) days of incubation at 37 °C in 5% CO2, the cell viability was determined by the formazan-based cell viability XTT assay (Sigma-Aldrich). Briefly, 50 μL of a 50:1 mixture of an XTT labelling reagent (1 mg mL−1) and a PMS electron-coupling reagent (0.383 mg mL−1) was added to the wells and incubated for 4 hours at 37 °C in 5% CO2. The absorbance of the newly formed orange formazan solution was measured at 450 nm using an EnVision 2105 microplate reader (Perkin Elmer). Drug concentrations required to reduce viral CPE by 50% (EC50) were calculated using nonlinear regression from plots of percentage of cell viability vs. log10 drug concentration using GraphPad Prism v.9.5.1 (GraphPad Software). The cytotoxicity of the compounds was evaluated by incubating the same dilutions with uninfected MT-4 and Vero cells. After three days of incubation at 37 °C in 5% CO2, the cell viability was determined as above. The compound concentrations resulting in 50% reduction of absorbance (CC50) were calculated from plots of percentage of cell viability vs. log10 drug concentration as above.
Author contributions
Conceptualization: Z. W., J. W. and P. L.; methodology: L. C., U. B., L. R., J. T. and D. Š.; investigation: L. C., U. B., L. R., J. T. and D. Š.; resources: Z. W.; data curation: L. C., U. B., L. R., J. T. and D. Š.; writing – original draft preparation: Z. W., J. W., L. R. and P. L.; writing – review and editing: Z. W., J. W., L. R. and P. L.; project administration: Z. W.; funding acquisition: Z. W. and J. W. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Part of this investigation was funded by grant no. FV30300 (MPO). The antiviral experiments were supported by the project National Institute Virology and Bacteriology (Programme EXCELES, Project No. LX22NPO5103) – funded by the European Union – Next Generation EU. The authors thank Ms. Martina Wimmerová for her excellent technical assistance.References
- R. Dutt, V. Garg, N. Khatri and A. K. Madan, Anti-Cancer Agents Med. Chem., 2019, 19, 172–183 CrossRef CAS PubMed.
- P. Darshani, S. S. Sarma, A. K. Srivastava, R. Baishya and D. Kumar, Phytochem. Rev., 2022, 21, 1761–1842 CrossRef CAS PubMed.
- Z. Özdemir and Z. Wimmer, Phytochemistry, 2022, 203, 113340 CrossRef PubMed.
- Z. Özdemir, Nonappa and Z. Wimmer, ACS Appl. Nano Mater., 2022, 5, 16264–16277 CrossRef.
- U. Bildziukevich, M. Wimmerová and Z. Wimmer, Pharmaceuticals, 2023, 16, 386 CrossRef CAS PubMed.
- Y. Liu, L. Yang, H. Wang and Y. Xiong, Pharmaceuticals, 2022, 15, 1169 CrossRef CAS PubMed.
- O. Pornillos and B. K. Ganser-Pornillos, Curr. Opin. Virol., 2019, 36, 47–55 CrossRef CAS PubMed.
- X. Liu, D. Main, Y. Ma and B. He, J. Virol., 2018, 92, e00402–e00418 CAS.
- R. J. Whitley, Antiviral Res., 2006, 71, 141–148 CrossRef CAS PubMed.
- P. Pecak, B. Orzechowska, E. Chrobak and S. Boryczka, Eur. J. Med. Chem., 2021, 225, 113738 CrossRef CAS PubMed.
- J. Ye, Z. Wang, J. Jia, F. Li, Y. Wang, Y. Jiang, Y. Wang, Z. Ren and H. Pu, Acta Virol., 2021, 65, 254–263 CrossRef CAS PubMed.
- R. Ramu, P. S. Shirahatti, S. N. Swamy, F. Zameer, B. L. Dhananjaya and M. N. N. Prasad, PLoS One, 2016, 11, e0151135 CrossRef PubMed.
- A. Kolokotronis and S. Doumas, Clin. Microbiol. Infect., 2006, 12, 202–211 CrossRef CAS PubMed.
- D. M. Coen and P. A. Schaffer, Nat. Rev. Drug Discovery, 2003, 2, 278–288 CrossRef CAS PubMed.
- G. Nistor, C. Trandafirescu, A. Prodea, A. Milan, A. Cristea, R. Ghiulai, R. Racoviceanu, A. Mioc and M. Mioc, Molecules, 2022, 27, 6552 CrossRef CAS PubMed.
- N. Nakano, K. Fukuda, E. Tashiro, H. Ishikawa, W. Nagano, R. Kawamoto, A. Mori, M. Watanabe, R. Yamazaki, T. Nakane, M. Naito, I. Okamoto and S. Itoh, J. Biochem., 2022, 171, 631–640 CrossRef CAS PubMed.
- A. Lombrea, A. D. Scurtu, S. Avram, I. Z. Pavel, M. Turks, J. Luginina, U. Peipins, C. A. Dehelean, C. Soica and C. Danciu, Int. J. Mol. Sci., 2021, 22, 3676 CrossRef CAS PubMed.
- Z. Chu, Y. Luo, T. Ni, M. Zhu, X. Feng, Y. Liu and H. Wang, Molecules, 2022, 27, 1025 CrossRef CAS PubMed.
- S. A. Popov, L. P. Kozlova, L. M. Kornaukhova and A. V. Shpatov, Ind. Crops Prod., 2016, 92, 197–200 CrossRef CAS.
- A. Stevaert, B. Krasniqi, B. Van Loy, T. Nguyen, J. Thomas, J. Vandeput, D. Jochmans, V. Thiel, R. Dijkman, W. Dehaen, A. Voet and L. Naesens, J. Med. Chem., 2021, 64, 5632–5644 CrossRef CAS PubMed.
- M. D. Semenova, S. A. Popov, I. V. Sorokina, Y. V. Meshkova, D. S. Baev, T. G. Tolstikova and E. E. Shults, Steroids, 2022, 184, 109042 CrossRef CAS PubMed.
- G. V. Gyniyatullina and O. B. Kazakova, Chem. Nat. Compd., 2021, 57, 698–705 CrossRef.
- T. G. Halsall and R. T. Aplin, A pattern of development in the chemistry of pentacyclic triterpenes, in Fortschritte der Chemie Organischer Naturstoffe/Progress in the Chemistry of Organic Natural Products/Progrès dans la chimie des substances organiques naturelles, ed. L. Zechmeister, Springer Vienna, Vienna, 1964, pp. 153–202 Search PubMed.
- C. K. Lee, J. Nat. Prod., 1998, 61, 375–376 CrossRef CAS PubMed.
- R. Mayer, Arch. Pharm., 1996, 329, 447–450 CrossRef.
- T. Fujioka, Y. Kashiwada, R. E. Kilkuskie, L. M. Cosentino, L. M. Ballas, J. B. Jiang, W. P. Janzen, I.-S. Chen and K.-H. Lee, J. Nat. Prod., 1994, 57, 243–247 CrossRef CAS PubMed.
- A. A. El-Gamal, Nat. Prod. Res., 2008, 22, 191–197 CrossRef CAS PubMed.
- B. A. Ganaie, M. Shahid, A. Rashid, T. Ara, J. A. Banday, F. Malik and B. A. Bhat, Chem. Biodiversity, 2021, 18, e2100292 CrossRef CAS PubMed.
- M. Petrenko, A. Güttler, E. Pflüger, I. Serbian, M. Kahnt, Y. Eiselt, J. Kessler, A. Funtan, R. Paschke, R. Csuk, D. Vordermark and M. Bache, Eur. J. Med. Chem., 2021, 224, 113721 CrossRef CAS PubMed.
- N. Heise, S. Friedrich, V. Temml, D. Schuster, B. Siewert and R. Csuk, Eur. J. Med. Chem., 2022, 227, 113947 CrossRef CAS PubMed.
- N. V. Heise, D. Ströhl, T. Schmidt and R. Csuk, J. Mol. Struct., 2022, 1249, 131646 CrossRef CAS.
- O. Kazakova, C. Soica, M. Babaev, A. Petrova, E. Khusnutdinova, A. Poptsov, I. Macasoi, G. Draghici, S. Avram, L. Vlaia, A. Mioc, M. Mioc, C. Dehelean and A. Voicu, Int. J. Mol. Sci., 2021, 22, 10695 CrossRef CAS PubMed.
- S. Hoenke, M. A. Christoph, S. Friedrich, N. Heise, B. Brandes, H.-P. Deigner, A. Al-Harrasi and R. Csuk, Molecules, 2021, 26, 2102 CrossRef CAS PubMed.
- M. Kozubek, S. Hoenke, T. Schmidt, D. Ströhl and R. Csuk, Med. Chem. Res., 2022, 31, 1049–1059 CrossRef CAS.
- E. Khusnutdinova, Z. Galimova, A. Lobov, I. Baikova, O. Kazakova, H. N. T. Thu, N. Van Tuyen, Y. Gatilov, R. Csuk, I. Serbian and S. Hoenke, Nat. Prod. Res., 2022, 36, 5189–5198 CrossRef CAS PubMed.
- U. Bildziukevich, Z. Özdemir, D. Šaman, M. Vlk, M. Šlouf, L. Rárová and Z. Wimmer, Org. Biomol. Chem., 2022, 20, 8157–8163 RSC.
- B. K. Bettadaiah, S. C. S. Kumar, N. V. Kumar and P. Srinivas, Synthesis, 2014, 46, 1847–1852 CrossRef.
- U. Bildziukevich, M. Šlouf, L. Rárová, D. Šaman and Z. Wimmer, Soft Matter, 2023, 19, 7625–7634 RSC.
- L. Heller, M. Kahnt, A. Loesche, P. Grabandt, S. Schwarz, W. Brandt and R. Csuk, Eur. J. Med. Chem., 2017, 126, 652–668 CrossRef CAS PubMed.
- A. Vystrčil, V. Křeček, M. Buděšínský and J. Protiva, Collect. Czech. Chem. Commun., 1986, 51, 581–592 CrossRef.
- S. C. B. Gnoatto, A. Dassonville-Klimpt, S. A. Da Nascimento, P. Galera, K. Boumediene, G. Gosmann, P. Sonnet and S. Moslemi, Eur. J. Med. Chem., 2008, 43, 1865–1877 CrossRef CAS PubMed.
- E. F. da Silva, A. S. de Vargas, J. B. Willig, C. B. de Oliveira, A. R. Zimmer, D. A. Pilger, A. Buffon and S. C. B. Gnoatto, Chem.-Biol. Interact., 2021, 344, 109535 CrossRef CAS PubMed.
- O. B. Flekhter, E. I. Boreko, L. R. Nigmatullina, N. I. Pavlova, N. I. Medvedeva, S. N. Nikolaeva, E. V. Tretyakova, O. V. Savinova, L. A. Baltina, L. T. Karachurina, F. Z. Galin, F. S. Zarudii and G. A. Tolstikov, Pharm. Chem. J., 2004, 38, 148–152 CrossRef CAS.
- U. Bildziukevich, N. Vida, L. Rárová, M. Kolář, D. Šaman, L. Havlíček, P. Drašar and Z. Wimmer, Steroids, 2015, 100, 27–35 CrossRef CAS PubMed.
- U. Bildziukevich, M. Malík, Z. Özdemir, L. Rárová, L. Janovská, M. Šlouf, D. Šaman, J. Šarek, Nonappa and Z. Wimmer, J. Mater. Chem. B, 2020, 8, 484–491 RSC.
- J. Humpolíčková, J. Weber, J. Stárková, E. Mašínová, J. Günterová, I. Flaisigová, J. Konvalinka and T. Majerová, Sci. Rep., 2018, 8, 10438 CrossRef PubMed.
- J. Konč, M. Tichý, R. Pohl, J. Hodek, P. Džubák, M. Hajdúch and M. Hocek, Med. Chem. Commun., 2017, 8, 1856–1862 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Analytical data of the prepared compounds, scanned 1H and 13C NMR spectra, and additional data on the antiviral activity of several target compounds. See DOI: https://doi.org/10.1039/d4re00032c |
| ‡ These authors contributed equally, in different topics of this paper. |
| This journal is © The Royal Society of Chemistry 2024 |