
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
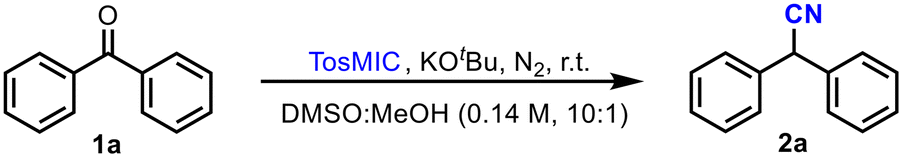
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cyanide-free synthesis of nitriles exploiting flow chemistry†
Niamh
Disney
 a,
Megan
Smyth
b,
Scott
Wharry
b,
Thomas S.
Moody
bc and
Marcus
Baumann
*a
a,
Megan
Smyth
b,
Scott
Wharry
b,
Thomas S.
Moody
bc and
Marcus
Baumann
*a
aSchool of Chemistry, University College Dublin, Science Centre South, Belfield, Dublin 4, Dublin, Ireland. E-mail: marcus.baumann@ucd.ie
bAlmac Sciences, Technology Department, Craigavon BT63 5QD, UK
cArran Chemical Company, Roscommon N37 DN24, Ireland
First published on 12th October 2023
Abstract
A continuous flow process for the synthesis of aryl nitriles in a cyanide-free manner is reported. Using a simple setup and mild conditions, TosMIC (p-tosylmethyl isocyanide) is used as a readily available precursor to convert ketones into nitriles via the van Leusen reaction. The resulting continuous process is fast (1.5 minutes residence time) and both the scalability (up to 8.8 g h−1) and reproducibility of this approach has been demonstrated for a variety of nitrile products. Through these studies, 4-tosyloxazole was isolated as a side-product that aids in understanding the mechanistic pathways of this transformation. Overall, this efficient flow method provides for a straightforward flow synthesis of nitrile-containing building blocks in high yields and with an improved safety profile.
Introduction
Nitriles are versatile structural motifs and ubiquitous in the pharmaceutical industry. Replacing a carbonyl group with a nitrile group in a drug molecule can lead to an improved absorption, distribution, metabolism, and excretion (ADME) profile making them a key asset in early-stage drug development.1 Nitriles are also convenient handles in API intermediates as they can be readily converted to other functional groups such as carboxylic acids, ketones, amines, amides, esters, and aldehydes.2 Typically, nitriles are synthesised from an alkyl or aryl halide and a metal cyanide (e.g., Sandmeyer cyanation,3 Rosenmund–von Braun reaction,4 and Kolbe nitrile synthesis) (Scheme 1, top)5 or alternatively via the hydrocyanation of alkenes or ammoxidation reactions.6–8 However, synthesising nitriles in this way can present challenges on a manufacturing scale due to the use and associated release of highly toxic cyanide waste products. | ||
| Scheme 1 Overview of different batch and flow-based cyanation processes. A: A large-scale continuous flow cyanation process towards Remdesivir. B: A cyanide-free cyanation in batch mode using an oxazole-based masked nitrile source. | ||
Various approaches have been taken to develop methodologies which circumvent this issue, including the use of masked nitrile transfer reagents,9,15 biocatalysis10,11 and continuous flow chemistry.12–15 To date, none have been as attractive and robust as the van Leusen reaction, which was first reported in 1977, and is frequently used in the total synthesis of medicinal compounds.16–19 The reaction relies on ketones reacting with TosMIC (p-tosylmethyl isocyanide),20 a nitrile transfer reagent, which prevents the inadvertent release of cyanide side products via a mechanistic sequence involving ring formation and subsequent ring-opening. To the best of our knowledge, no broad-scope optimisation work has been reported on the reaction since its first publication almost 50 years ago. As the van Leusen reaction is a safe way to synthesise nitriles, we aimed to improve the efficiency and increase the understanding of the reported conditions by transferring the reaction into a continuous flow process.
Over the past two decades, continuous flow technology has been gaining momentum as a manufacturing tool of the future. There are several advantages associated with performing chemical reactions in continuous flow mode, including better heat and mass transfer, improved reproducibility, and better scalability.21 This technology platform is particularly attractive to industry as it allows for the synthesis of compounds ‘on tap’ in a fast, safe, and efficient manner.22 A number of recent literature reports utilise flow technology for cyanation reactions, including in the derivatisation of amines.12,13 In 2020, Vieira et al. reported a large-scale continuous flow cyanation process towards Remdesivir, an antiviral drug used for the treatment of COVID-19 (Scheme 1A).14 A mechanistically insightful report by Sharma et al. detailed a cyanide-free cyanation for the derivatisation of sp2 and sp carbons via an oxazole-based masked nitrile source (Scheme 1B).15 However, a cyanide-free continuous flow synthesis of nitriles via the van Leusen reaction is unreported in the literature despite its significant advantages.
Results & discussion
Initial exploration in batch mode
Initial exploratory work on the reaction was carried out using traditional batch methods. Employing benzophenone as the model substrate and van Leusen's reported conditions as a baseline reaction, the following parameters were screened: reaction time, temperature, and reagent addition. The results of this screen are detailed in Table 1.|
|
||||
|---|---|---|---|---|
| Entry | TosMIC (equiv.) | KOtBu (equiv.) | Reaction time (h) | 1H-NMR yield 2aa |
| a Using 1,3,5-trimethoxybenzene as internal standard. b Addition after 60 min. c Gradient used: r.t. for 1 h, then 30 °C for 30 min, then 50 °C for 30 min, then 70 °C for 30 min, then 90 °C for 30 min. | ||||
| 1 | 1.3 | 3.0 | 2 | 63% |
| 2 | 1.3 + 1.0b | 1.0 + 2.0b | 4 | 91% |
| 3 | 1.3 + 1.0b | 1.0 + 2.0b | 3 | 63% |
| 4c | 1.1 + 1.0b | 1.0 + 2.0b | 4 | 93% |
Firstly, conditions similar to those reported by van Leusen were replicated on a smaller scale (1 mmol) and it was found that a shorter reaction time of 2 h gave 63% conversion to the nitrile product 2a (Table 1, entry 1) compared with a 69% yield reported by van Leusen for the same reaction after 17 h. Additional portions of TosMIC (1.3 equiv.) and KOtBu (2 equiv.) were added after 1 h which gave a significantly improved conversion of 91% after 4 h (Table 1, entry 2). Gradually increasing the reaction temperature by 20 °C every 30 min did not show any improvement on the reaction conversion (Table 1, entry 3). The most efficient conditions for the batch process are shown in entry 4 which give high conversion whilst limiting the excess of TosMIC reagent used. To understand the positive effect of adding further aliquots of TosMIC and base, a kinetic study was carried out in which the amount of each component was monitored by GC analysis over a period of 3 h (Scheme 2).
 | ||
| Scheme 2 Reaction monitoring via GC analysis. | ||
The rate of nitrile formation was initially high but then levelled off after 50 min. There was no TosMIC left in the reaction after 60 minutes despite an excess of 1.1 equiv. being used. A second addition of TosMIC and KOtBu at this point led to an increased amount of nitrile product 2a before leveling off at 90 min. Two new peaks were observed by GC analysis (side products 1 & 2 (Scheme 2)), possibly resulting from a degradation pathway, or a competing pathway involving TosMIC. At this stage it was hoped that a continuous flow setup would enable a quicker reaction due to better mass transfer. Additionally, with the knowledge of competing reactant decomposition in batch mode, we anticipated that spatiotemporal processing in flow mode might be advantageous as the propagating reaction mixture is separated from the feed streams containing base and TosMIC.
Transfer to continuous flow
Initial efforts in transferring the batch-based van Leusen reaction into continuous flow continued with the use of KOtBu as the base. Table 2 details the results of optimising the reaction with a focus on investigating the stoichiometry of TosMIC as well as the residence time when using either normal PFA coils (perfluoroalkoxy, 10 mL volume) or modified coils containing static mixing elements (PFA, 22.4 mL, see ESI† for details).|
|
||||
|---|---|---|---|---|
| Entry | TosMIC (equiv.) | Flow rate (mL min−1) | t Res (min) | 1H-NMR yield 2aa |
| Reactions performed at 1 mmol scale.a Determined by 1H-NMR, using 1,3,5-trimethoxybenzene as an internal standard.b Standard PFA coil (10 mL).c Static mixer coil (22.4 mL).d Reaction performed at 50 °C.e Overall concentration 0.37 M. | ||||
| 1b | 1.2 | 0.5 | 15 | 21 |
| 2b | 1.2 | 0.5 | 20 | 31 |
| 3b | 1.2 | 0.5 | 40 | 23 |
| 4b | 1.4 | 0.5 | 20 | 12 |
| 5b | 1.05 | 0.5 | 20 | 19 |
| 6b,d | 1.2 | 0.5 | 20 | 18 |
| 7c | 1.2 | 0.56 | 20 | 33 |
| 8c | 1.2 | 1.12 | 20 | 46 |
| 9c,e | 1.2 | 1.12 | 20 | 52 |
| 10c,e | 1.2 | 1.68 | 20 | 50 |

Variation of the residence time indicated that 20 min was preferable to shorter and longer residence times (entries 1–3, Table 2). Following this, the amount of TosMIC was varied from 1.2 equivalents (entries 4 and 5), however, no improvement in reaction conversion was observed. Increasing the reaction temperature to 50 °C resulted in a lower reaction conversion, similar to what was observed in batch mode (entry 6). To study the effect of mixing on the reaction, the initially used standard 10 mL PFA coil was replaced with a bespoke static mixer coil (see ESI† for images). Although the diameter of this static mixer coil is wider, increased mixing is typically achieved via the screw-like internal structure. First, a 20 min residence time (0.56 mL min−1 flow rate) in the static mixer coil caused only a negligible improvement in conversion to 33% (entry 7). However, increasing the flow rate while maintaining a 20 min residence time caused the most significant increase in reaction conversion to 46% (entry 8). When this was coupled with an increase in the reaction concentration to 0.37 M (from 0.27 M) further improvement to 52% was noted (entry 9). Increasing the flow rate further and therefore reducing the residence time to 15 min resulted in a slight decrease in the reaction conversion (entry 10). From this study it was clear that mixing is a key parameter for this reaction, however, due to the poor solubility of KOtBu in DMSO, the concentration of the reaction mixture could not be increased and thus further improvements in product yield were not possible. Several other mixing strategies were tried (see ESI† for details), but none offered an improvement on the best results detailed above (entry 9). Given the low productivity of these optimised conditions and the limitations in scaling this initial flow process, several other metal alkoxide bases were tried in the reaction (Fig. 1).
 | ||
| Fig. 1 Evaluation of different alkoxide bases in batch. | ||
This comparative study identified NaOMe as well as NaOtBu as promising alternatives for this reaction. The reaction with NaOMe was stopped after just 1 h as previous experiments revealed that the GC %area of the nitrile product is highest at 0.5 h despite a second addition of reagents at 1 h. As NaOMe (commercial solution in MeOH, 25% w/v) is readily available at low cost, and has the best atom economy, the reaction was initially studied in continuous flow mode with this base. The optimised conditions however, led to low yields and a very poor substrate scope (see ESI† for details). Therefore, efforts turned to using commercially available solutions of NaOtBu (2 M in THF) and the following optimisation employed design of experiment (DOE) strategies (Table 3).
|
|
|||
|---|---|---|---|
| Entry | TosMIC (equiv.) | NaOtBu (equiv.) | 1H-NMR yielda |
| a Determined by 1H-NMR using 1,3,5-trimethoxybenzene as internal standard. Full DOE results available in ESI.† | |||
| 1 | 1.0 | 2.0 | 37 |
| 2 | 2.5 | 3.5 | 56 |
| 3 | 3.0 | 4.0 | 61 |
| 4 | 3.5 | 4.0 | 61 |
| 5 | 1.75 | 5.0 | 62 |
| 6 | 2.0 | 6.0 | 65 |
| 7 | 3.25 | 5.0 | 66 |
| 8 | 2.5 | 6.5 | 72 |
| 9 | 2.0 | 4.0 | 72 |
| 10 | 2.5 | 5.0 | 73 |
| 11 | 3.0 | 6.0 | 75 |

Two DOE studies (central composite design) were carried out. The first with lower limit 1, upper limit 3 equiv. of TosMIC and lower limit 2, upper limit 6 equiv. of NaOtBu (entries 1–6). A consecutive DOE study followed with more refined limits: lower limit 2, upper limit 3 equiv. of TosMIC and lower limit 4, upper limit 6 equiv. of NaOtBu (entries 7–11). As shown in Table 3, the highest yielding results were found when using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of TosMIC:NaOtBu (entries 9–11). The stoichiometry from entry 10 was used for further optimisation studies, however, the stoichiometry detailed in entry 9 would be more appropriate for an industrial-scale process due to the potential cost savings of using less reagents in the face of a slightly reduced yield (1%). Further optimisation was carried out on the residence time, solvent, and amount of MeOH used in the reaction (see ESI† for results). Fig. 2 details the optimised conditions for the reaction along with the substrate scope that was explored. For this study the reaction output was collected under steady state conditions. A range of acetophenones were first subjected to the optimised conditions. Both electron-donating and electron-withdrawing groups on the aryl ring were well tolerated, with compound 2e being synthesised in 96% yield. Compound 2f which has a fluorine in the 2-position of the ring was the lowest yielding of this study but still resulted in 60% yield. The phenyl group was replaced with heteroaromatic systems including a thiophene and furan ring which gave 88% and 78% yield respectively (2i, 2j). The cyclohexyl phenyl ketone was relatively low yielding (47%) compared with the other substrates (2k). A piperonyl system was well tolerated in the reaction and gave 76% yield of product. Lastly, selected α-tetralone substrates gave the corresponding nitriles in excellent yields (2m, 2n) as did propiophenone (2o).
:2 ratio of TosMIC:NaOtBu (entries 9–11). The stoichiometry from entry 10 was used for further optimisation studies, however, the stoichiometry detailed in entry 9 would be more appropriate for an industrial-scale process due to the potential cost savings of using less reagents in the face of a slightly reduced yield (1%). Further optimisation was carried out on the residence time, solvent, and amount of MeOH used in the reaction (see ESI† for results). Fig. 2 details the optimised conditions for the reaction along with the substrate scope that was explored. For this study the reaction output was collected under steady state conditions. A range of acetophenones were first subjected to the optimised conditions. Both electron-donating and electron-withdrawing groups on the aryl ring were well tolerated, with compound 2e being synthesised in 96% yield. Compound 2f which has a fluorine in the 2-position of the ring was the lowest yielding of this study but still resulted in 60% yield. The phenyl group was replaced with heteroaromatic systems including a thiophene and furan ring which gave 88% and 78% yield respectively (2i, 2j). The cyclohexyl phenyl ketone was relatively low yielding (47%) compared with the other substrates (2k). A piperonyl system was well tolerated in the reaction and gave 76% yield of product. Lastly, selected α-tetralone substrates gave the corresponding nitriles in excellent yields (2m, 2n) as did propiophenone (2o).
 | ||
| Fig. 2 Optimised continuous flow setup and exploration of substrate scope. A list of unsuccessful substrates are included in the ESI.†[a]Determined by 1H-NMR using 1,3,5-trimethoxybenzene as an internal standard. All nitriles were collected under steady state conditions. | ||
When the model benzophenone reaction was then scaled to 30 mmol the yield of the reaction was reproduced from the earlier small-scale tests producing 3.8 g of the desired nitrile within 26 minutes, which is equivalent to a throughput of 8.8 g h−1 (Scheme 3).
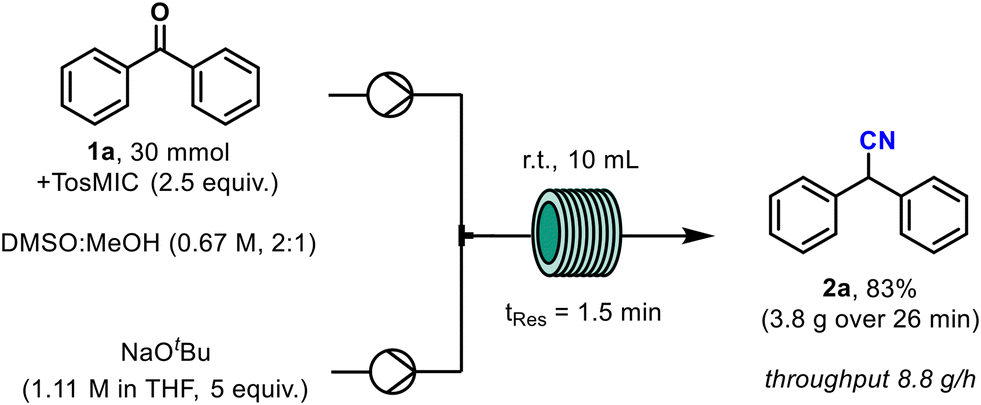 | ||
| Scheme 3 Reaction scalability demonstrated on 30 mmol scale. | ||
In relation to the competing reactivity of TosMIC, under continuous flow conditions only one distinct side product was observed and subsequently isolated. Based on NMR analysis this material was initially thought to be 4-tosylimidazole (5) which is believed to result from dimerisation of TosMIC followed by loss of the tosyl-CH2 group under basic conditions.23,24 However, an older report by Bull suggests that by using a ratio of 1:2 TosMIC:base, the formation of the dimer (4) can be prevented due to an absence of neutral TosMIC (3).25 This might explain why the highest yielding reactions from the DOE study (Table 3) were found to be along an axis which represents a 1:2 ratio of TosMIC to base. As our scale-up studies generated sufficient quantities of this side-product we were able to exploit single crystal X-ray diffraction analysis which revealed the structure of this compound to be 4-tosyloxazole (9) instead. Earlier reports by both van Leusen26 and Schöllkopf27 propose a mechanistic rationale for the formation of 9, however, its significance in the overall mechanism appears to have been overlooked due the absence of definitive structural data (Scheme 4).
 | ||
| Scheme 4 Proposed pathways for the formation of 4-tosylimidazole (5) and 4-tosyloxazole (9) from competing reaction pathways involving TosMIC. This route is secondary to that proposed by van Leusen for the formation of the nitrile product 2.16 | ||
In the mechanistic proposal shown below, intermediate 6 results from initial condensation between ketone and TosMIC followed by hydration of the isonitrile group (Scheme 4). This species (6) then abstracts a proton from MeOH and subsequently undergoes transfer of its formyl group to a deprotonated molecule of TosMIC. This leads to the formation of the desired nitrile product (2) as well as formyl species 8 that cyclises to give the aromatic 4-tosyloxazole 9. Crucially, this proposal accounts for the high yields observed in the reaction despite the formation of significant amounts of 4-tosyloxazole (9) which forms due to additional TosMIC reagent being available. Bull25 and van Leusen26 suggest that minimising the amount of MeOH used in the reaction may limit the formation of 9, however, our experiments did not corroborate this hypothesis.
Conclusions
In conclusion, we have developed a fast and straightforward continuous flow process for the formation of aryl nitriles from ketones and TosMIC in the presence of alkoxide bases. Screening different bases ultimately identified NaOtBu (2 M in THF) as the best choice in terms of solubility and product yield. The overall process is attractive as it does not require the handling of cyanide or any related reagents. Furthermore, the scalability of the flow method and its high throughput (8.8 g h−1) make this continuous van Leusen reaction an appealing strategy for future industrial applications. Systematic variation of the reaction parameters thereby yielded additional mechanistic insights. Importantly, the isolation and characterisation of a tosyloxazole side-product was accomplished whose formation accounts for the competitive consumption of the TosMIC reagent under the reaction conditions. This study is expected to generate renewed interest in the underutilised van Leusen nitrile synthesis and its use in accessing nitrile-containing organic building blocks via a safe and readily scalable process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was co-funded by Almac Group Ltd. and SSPC through provision of a PhD studentship (to ND, 12/RC/2275_P2 and 20/FFP-P/8712). We thank the technical staff in the School of Chemistry at UCD who supported this work: Dr Yannick Ortin, Hans Eckhardt, Dr Jimmy Muldoon, and Dr Julia Bruno-Colmenarez. We are grateful to Duncan Guthrie (Vapourtec) for provision of the static mixer coils used in this study.Notes and references
- F. Fleming, L. Yao, P. Ravikumar, P. Funk and B. Shook, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS.
- (a) A. Rakshit, H. N. Dhara, A. K. Sahoo and B. K. Patel, Chem. – Asian J., 2022, 17, e202200792 CrossRef CAS; (b) F. Schaefer, Nitrile Reactivity, John Wiley & Sons Ltd., 1970, pp. 239–305 Search PubMed.
- I. P. Beletskaya, A. Sigeev, A. Peregudov and P. Petrovskii, J. Organomet. Chem., 2004, 689, 3810–3812 CrossRef CAS.
- A. Pradal and G. Evano, Chem. Commun., 2014, 50, 11907–11910 RSC.
- Z. Wang, Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, 1st edn, 2010, pp. 1661–1663 Search PubMed.
- P. van Leusen, Homogeneous Catalysis, Springer, 2004, pp. 229–237 Search PubMed.
- R. Grasselli and M. Tenhover, Handbook of Heterogeneous Catalysis, Wiley-VCH, 2008, pp. 3489–3517 Search PubMed.
- (a) R. V. Jagadeesh, H. Junge and M. Beller, Nat. Commun., 2014, 5, 4123 CrossRef CAS; (b) A. Martin and B. Lücke, Catal. Today, 2000, 57, 61–70 CrossRef CAS.
- A. Schuppe, G. Borrajo-Calleja and S. Buchwald, J. Am. Chem. Soc., 2019, 141, 18668–18672 CrossRef CAS.
- Z. Chen, F. Mao, H. Zheng, Q. Xiao, Z. Ding, A. Wang and X. Pei, Enzyme Microb. Technol., 2021, 150, 109883 CrossRef CAS.
- A. Hinzmann, T. Betke, Y. Asano and H. Gröger, Chem. – Eur. J., 2021, 27, 5313–5321 CrossRef CAS.
- K. Tadele, S. Verma, M. N. Nadagouda, M. A. Gonzalez and R. S. Varma, Sci. Rep., 2017, 7, 16311 CrossRef PubMed.
- D. B. Ushakov, K. Gilmore, D. Kopetzki, D. T. Mcquade and P. H. Seeberger, Angew. Chem., Int. Ed., 2014, 53, 557–561 CrossRef CAS PubMed.
- T. Vieira, A. Stevens, A. Chtchemelinine, D. Gao, P. Badalov and L. Heumann, Org. Process Res. Dev., 2020, 24, 2113–2121 CrossRef CAS PubMed.
- B. Sharma, A. Nikam, S. Lahore, G. Ahn and D. Kim, Eur. J. Chem., 2021, 28, e202103777 CrossRef PubMed.
- O. Oldenziel, D. van Leusen and A. M. van Leusen, J. Org. Chem., 1977, 42, 3114–3117 CrossRef CAS.
- D. A. Siler, J. D. Mighion, E. J. Sorensen, I. Memory, D. C. Dittmer and H. H. Wasserman, Angew. Chem., Int. Ed., 2014, 53, 5332–5335 CrossRef CAS PubMed.
- D. S. Müller, N. L. Untiedt, A. P. Dieskau, G. L. Lackner and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 660–663 CrossRef PubMed.
- H. D. Hao and D. Trauner, J. Am. Chem. Soc., 2017, 139, 4117–4122 CrossRef CAS.
- D. van Leusen and A. M. van Leusen, Synthetic Uses of Tosylmethyl Isocyanide (TosMIC), Organic Reactions, John Wiley & Sons Ltd., 2001, vol. 57, pp. 417–466 Search PubMed.
- (a) L. Buglioni, R. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS PubMed; (b) A. Bonner, A. Loftus, A. Padgham and M. Baumann, Org. Biomol. Chem., 2021, 86, 14199–14206 CrossRef PubMed; (c) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117(18), 11796–11893 CrossRef CAS; (d) M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC; (e) J. Britton and C. L. Raston, Chem. Soc. Rev., 2017, 46, 1250–1271 RSC; (f) L. Capaldo, Z. Wen and T. Noël, Chem. Sci., 2023, 14, 4230–4247 RSC; (g) D. Dallinger, B. Gutmann and C. O. Kappe, Acc. Chem. Res., 2020, 53, 1330–1341 CrossRef CAS PubMed; (h) C. R. Sagandira, S. Nqeketo, K. Mhlana, T. Sonti, S. Gaqa and P. Watts, React. Chem. Eng., 2022, 7, 214–244 RSC.
- (a) M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2020, 24, 1802–1813 CrossRef CAS; (b) S. V. Ley, Y. Chen, A. Robinson, B. Otter, E. Godineau and C. Battilocchio, Org. Process Res. Dev., 2021, 25, 713–720 CrossRef CAS; (c) M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Eur. J. Org. Chem., 2020, 48, 7398–7406 CrossRef; (d) M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2015, 11, 1194–1219 CrossRef CAS PubMed; (e) A. Laybourn, K. Robertson and A. G. Slater, J. Am. Chem. Soc., 2023, 145, 4355–4365 CrossRef CAS PubMed; (f) G. Gambacorta, J. S. Sharley and I. R. Baxendale, Beilstein J. Org. Chem., 2021, 17, 1181–1312 CrossRef CAS; (g) S. Ötvös and C. O. Kappe, Green Chem., 2021, 23, 6117–6138 RSC.
- C. Zhao, M.-Z. Gu, Y.-Y. Chen, X.-W. Hu, Y.-B. Xu, X.-M. Lin, X.-N. Liu, L. Chen, G.-S. Chen and Y.-L. Liu, Org. Biomol. Chem., 2022, 20, 8623–8627 RSC.
- C. Necardo, A. I. Alfano, E. Del Grosso, S. Pelliccia, U. Galli, E. Novellino, F. Meneghetti, M. Giustiniano and G. C. Tron, J. Org. Chem., 2019, 84, 16299–16307 CrossRef CAS.
- J. R. Bull and A. Tuinman, Tetrahedron, 1975, 31, 2151–2155 CrossRef CAS.
- A. M. van Leusen, B. Hoogenboom and H. Siderius, Tetrahedron Lett., 1972, 13, 2369–2372 CrossRef.
- U. Schöllkopf and R. Schröder, Angew. Chem., Int. Ed. Engl., 1971, 10, 333 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00458a |
| This journal is © The Royal Society of Chemistry 2024 |