
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyrolysis of biogas for carbon capture and carbon dioxide-free production of hydrogen†
Ahmet
Çelik
 ,
Iadh
Ben Othman
,
Heinz
Müller
,
Patrick
Lott
* and
Olaf
Deutschmann
,
Iadh
Ben Othman
,
Heinz
Müller
,
Patrick
Lott
* and
Olaf
Deutschmann
Institute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), Engesserstr. 20, 76131 Karlsruhe, Germany. E-mail: patrick.lott@kit.edu
First published on 18th September 2023
Abstract
Methane pyrolysis is considered an auspicious approach for large-scale hydrogen production and simultaneous carbon capture, hereby contributing to a decarbonization of the chemical industry. While commonly pure methane or natural gas serve as a feedstock, the usage of biogas may allow exploitation of the pyrolysis process as a carbon sink. In this context, the present study reports on biogas pyrolysis in a high-temperature reactor at temperatures between 1000 °C and 1600 °C, residence times between 1 s and 7 s, and molar CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2 ratios in the biogas between 1:1 and 4:1. Among these conditions, high residence times, a high CH4 content, and the introduction of a carbonaceous fixed bed in the reactor benefit high educt conversion, H2 selectivity, and solid carbon yield. A carbon fixation of up to 95% was achieved during reference measurements with pure CH4 feeds, whereas a carbon yield of 75% was found for biogas feeds. The analysis of the reaction product distribution uncovered a consumption of CO2via dry reforming, water gas shift, and Boudouard reactions, resulting in a maximum H2:CO ratio of 3:1 in the effluent gas stream. Herewith, the study underscores that optimized reactor operation parameters allow for maximizing CH4 and CO2 conversion as well as for achieving H2:CO ratios that are viable for further industrial applications, along with an efficient deposition of solid carbon.
:CO2 ratios in the biogas between 1:1 and 4:1. Among these conditions, high residence times, a high CH4 content, and the introduction of a carbonaceous fixed bed in the reactor benefit high educt conversion, H2 selectivity, and solid carbon yield. A carbon fixation of up to 95% was achieved during reference measurements with pure CH4 feeds, whereas a carbon yield of 75% was found for biogas feeds. The analysis of the reaction product distribution uncovered a consumption of CO2via dry reforming, water gas shift, and Boudouard reactions, resulting in a maximum H2:CO ratio of 3:1 in the effluent gas stream. Herewith, the study underscores that optimized reactor operation parameters allow for maximizing CH4 and CO2 conversion as well as for achieving H2:CO ratios that are viable for further industrial applications, along with an efficient deposition of solid carbon.
Introduction
The goals defined in the Paris agreement to limit global warming are linked to a drastic reduction in greenhouse gas emissions1 and require the establishment of a sustainable energy system. In this context, hydrogen (H2) is considered as one of the most important and promising energy carriers for the decarbonization of key technologies and hereby allows achievement of the climate targets.2–6 Hence, large-scale sustainable H2 production processes are key on the way towards a modern hydrogen economy. In this regard, the pyrolysis of methane (CH4) is a H2 production route that requires significantly less energy than water electrolysis and, compared to state-of-the-art steam reforming, does not exhibit any direct carbon dioxide (CO2) emissions.3,7–10 CH4 pyrolysis is an endothermic, thermal decomposition process during which gaseous H2 and solid carbon are formed from CH4 according to the global reaction eqn (1):11–13| CH4 → 2H2 + C ΔRH° = 75 kJ mol−1 | (1) |
The high stability of the CH4 molecule results in a highly endothermic nature of the pyrolysis reaction.26 Thus, temperatures between 500 °C and 1000 °C are needed to achieve technically relevant methane conversion rates and hydrogen yields even if catalytic systems, for example based on iron or nickel, are used.27–30 The thermocatalytic pyrolysis of methane requires temperatures well above 1000 °C to activate the CH4 molecule without a catalyst.31 Despite the higher energy demand, thermocatalytic methane decomposition offers several advantages compared to catalytic processes relying on catalysts such as iron or nickel.29,32 In particular, catalyst coking and impurities in the reactant stream that for instance may act as catalyst poison are essentially irrelevant. Hereby, longer and more stable operating times are achieved, and the resulting solid carbon can be extracted without any metallic impurities originating from a catalyst that may impede further usage.32,33 CH4 pyrolysis over carbon particles, which accelerate heterogeneous deposition reactions and provide additional surface area for particle growth, allows the above-mentioned advantages to be mostly maintained while lowering the temperature needed for a successful CH4 decomposition.32,34–39
To date, fossil natural gas is the main source of CH4 and therefore represents the main feedstock for methane pyrolysis processes. Although the pyrolytic conversion of natural gas extracts carbon from the gaseous energy carrier, the wide-spread usage of renewable methane sources rather than exploiting fossil sources would be much more elegant and desirable in the long term. Biogas obtained from the fermentation of biomass, for instance, is a promising alternative feedstock, but contains also up to almost 50% CO2.40 Under consideration of the harsh reaction conditions applied during the thermocatalytic pyrolysis of methane, additional reactions such as the dry reforming of methane (eqn (2)), the reverse water-gas shift (RWGS) reaction (eqn (3)), or the Boudouard reaction (eqn (4)) come into play.
| CH4 + CO2 ⇌ 2CO + 2H2 ΔRH° = 247 kJ mol−1 | (2) |
| H2 + CO2 ⇌ CO + H2O ΔRH° = 41 kJ mol−1 | (3) |
| CO2 + C ⇌ 2CO ΔRH° = 172 kJ mol−1 | (4) |
:CO ratio of the synthesis gas in the product stream are of particular interest.
Currently, pyrolysis processes enjoy great interest in academia and industry, especially in the context of chemical recycling and upcycling of carbonaceous materials and for energy generation. The potential impact on the environment is enormous: for example, waste from old fishing nets, wind turbine blades or conventional plastic waste can be converted to energy and high-value materials.41–43 Herein, biomass or biowaste has also been considered as a feedstock.44,45 However, even though some studies on the production of syngas or hydrogen from biomass or biogas have been conducted in the past,6,46–51 the usage of biogas under reaction conditions of thermocatalytic pyrolysis of methane remains mostly unexplored. When the current political tensions in the energy market and fluctuating availability of resources are taken into account, flexible operation of methane pyrolysis with varying feed gas streams becomes a valuable asset to reliably meet the increasing demand for H2 at all times. Most importantly, the use of biogas offers the potential for a negative carbon balance that actively reduces the greenhouse gas content in earth's atmosphere. In consideration of the overall biochemical process chain, namely CO2 capture in biomass, fermentation of biomass to form biogas, and high-temperature pyrolysis to extract carbon and to form H2, the carbon accrual during biogas pyrolysis can serve as a CO2 sink.
By exposing CO2-containing biogas as an alternative, sustainable CH4 feedstock to thermocatalytic pyrolysis conditions in a lab-scale high-temperature reactor, this work systematically investigates the influence of temperature, residence time, H2 dilution, and CH4:CO2 ratio in the biogas feed on CH4 and CO2 conversion as well as on H2 and solid carbon yield. By comparing empty reactor tube experiments with results obtained when the reactor was loaded with a carbonaceous fixed bed, our study identifies advantageous operating parameters. Hereby, our work provides guidance for possible reactor configurations and operation conditions that can be used to design industrially viable full-scale processes.
Experimental
All experiments were conducted in an in-house developed high-temperature setup that was already described in previous publications.14,52 The setup comprises a gas supply system, a reactor in plug-flow geometry, and an analysis and exhaust gas section as schematically depicted in Fig. 1a. By using mass flow controllers, a feed gas containing either pure methane or synthetic biogas, which is mixed from CH4 and CO2, was quantitatively fed and diluted with varying amounts of H2. Subsequently, the reaction gas stream entered an electrically heated Al2O3-based ceramic reactor tube (DEGUSSIT AL23 by Friatec/Aliaxis) with an inner diameter of 20 mm. To ensure efficient insulation and safe reactor operation even in the case of over-pressure in the reactor tube, the reactor was located in a stainless-steel vessel. For accurate and continuous temperature measurement, a platinum-based thermocouple was used, which was positioned directly on the outer wall of the ceramic tube in the center of the heated reactor zone (Fig. 1a). Since solid particles form during the reaction, a trap was positioned downstream of the reactor for separation, and an additional particle filter was installed in order to remove fine soot particles. Finally, the effluent product gases are quantitatively analyzed in a Hiden Analytical HPR-20 R&D mass spectrometer. Details regarding data evaluation can be found in the ESI.†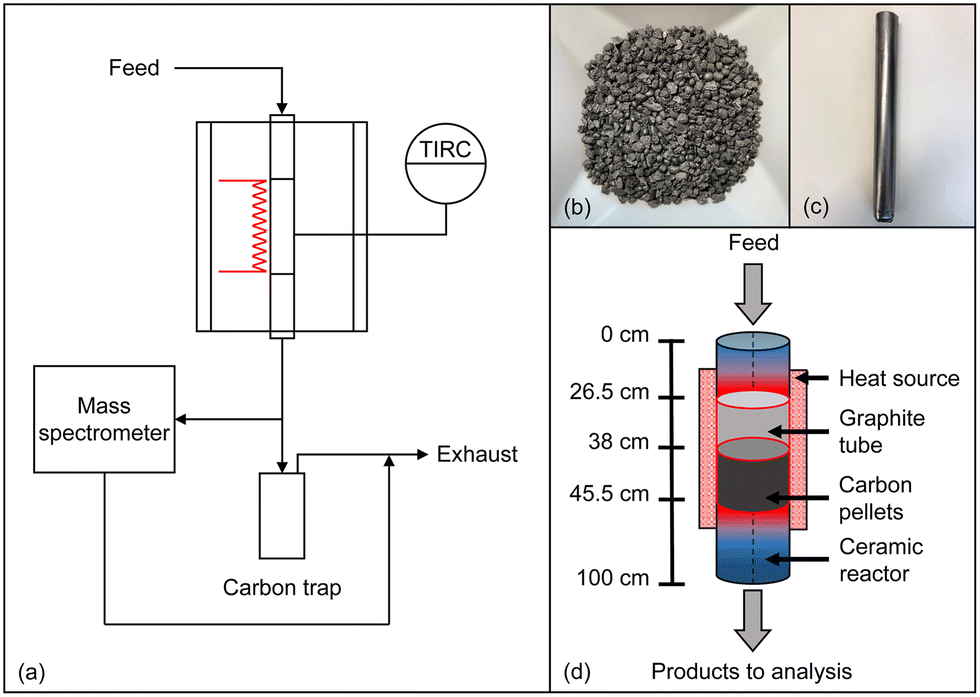 | ||
| Fig. 1 Schematic flow diagram of the experimental setup (a), acetylene coke used as a base material of the fixed bed (b), a graphite container with flow holes at the bottom (c), and a scheme of the positioning of the carbon fixed bed container in the reactor (d). | ||
In addition to experiments with an empty reactor tube, experiments with a carbonaceous fixed bed were conducted, for which 20 g of acetylene coke pellets (Carbolux, provided by BASF SE, Fig. 1b) with an average pellet diameter of 2 mm to 3 mm were filled into a 190 mm high container made of graphite foil (Fig. 1c). To ensure that the feed gas is heated to the respective reaction temperature before reaching the fixed bed, the container was positioned in the reactor so that the distance between the top of the fixed bed (length of 75 mm) and the reactor inlet was 380 mm (Fig. 1d). Note that a slight conical shape of the graphite container provided a seal at the top edge of the container and prevented bypass: the top seal forced the reaction gases to flow through the fixed bed, and the gases exited the fixed bed through holes at the bottom of the container (Fig. 1c).
The reactor was purged with argon (Ar) prior to each measurement and then continuously flushed with H2 during the heating phase. During the experiments, the reactants were diluted with H2. Although high H2 levels were reported to inhibit CH4 conversion,53 the H2 content is also a valuable parameter that can be used to control the formation of undesired byproducts, soot, and carbon deposits.14 Hence, a carefully chosen H2 dilution allows a fast pressure increase or even clogging of the reactor due to carbon deposition to be avoided. If not indicated otherwise, a molar H2:reaction gas ratio of 2:1 was chosen throughout this study, as this controls side-reactions to a certain extent while allowing a reasonable CH4 conversion.
Once the desired reaction temperature was reached, the reactants diluted with H2 were fed into the reactor for 20 minutes and the concentrations of the respective product gases were recorded with the mass spectrometer. Subsequently, the reactor was purged with Ar until all H2 was removed and then carbonaceous deposits were burned off by flushing the reactor with synthetic air after each empty tube experiment. The burn-off was considered complete once no CO and CO2 species in the exhaust gas were detected anymore. After another Ar purging phase to remove all oxygen from the reactor, the next experiment (20 min) with a new reaction gas mixture was conducted. In contrast, only one experiment was conducted if the reactor was loaded with a fixed bed and the reactor was cooled down to room temperature while purging with Ar, before the fixed bed could be removed.
Results and discussion
Following the aforementioned procedure, the influence of temperature (1000 °C, 1200 °C, 1400 °C, and 1600 °C), residence time (1 s, 3 s, 5 s, and 7 s), and biogas composition (molar CH4:CO2 ratio 1:1, 2:1, and 4:1) was systematically investigated. In the focus were CH4 and CO2 conversion, H2 selectivity, product composition, and solid carbon yield (definition of each is given in the ESI†). Note that the biogas compositions tested herein mimic typical CH4:CO2 ratios as found for real-world biogas40 and the set of reaction parameters applied throughout our experimental measurement campaign is based on previous studies that identified promising conditions for (industrially viable) H2 production.14,53 Furthermore, the effect of a carbonaceous fixed bed in the reactor was evaluated.
Influence of temperature
To investigate the effect of temperature on the reaction process in an empty reactor configuration, the temperature is varied when pyrolyzing either pure CH4 or biogas with a molar CH4:CO2 ratio of 2:1 in a gas mixture with a molar H2 dilution ratio of 2:1 and a residence time of 5 s. Fig. 2 shows the CH4 conversion (Fig. 2a) and H2 selectivity (Fig. 2b) for both feeds during reactor operation at temperatures between 1000 °C and 1600 °C.
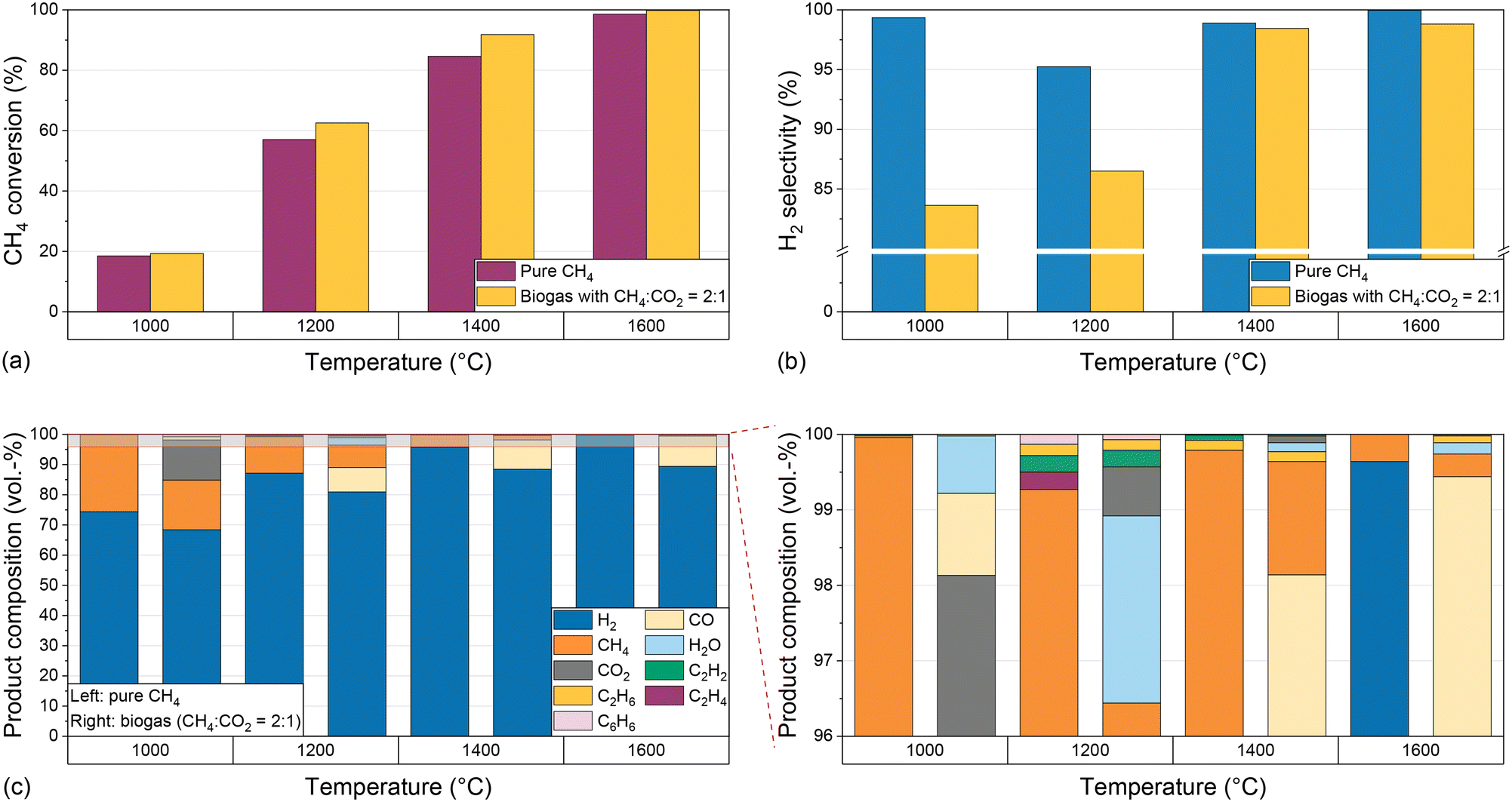 | ||
| Fig. 2 Molar CH4 conversion (a), molar H2 selectivity (b), and product composition (c) for pure CH4 and biogas (CH4:CO2 ratio 2:1) as a feed at temperatures from 1000 °C to 1600 °C, a residence time of 5 s, a molar H2:CH4 ratio of 2:1, and a molar H2:biogas ratio of 2:1. | ||
Irrespective of the feed gas composition, rising temperatures result in a significant increase in CH4 conversion from approximately 20% at 1000 °C to almost 90% for pure CH4 and more than 90% for biogas at 1400 °C; at 1600 °C, almost full conversion is achieved (Fig. 2a). Notably, the CH4 conversion is higher when biogas is dosed, which can be attributed to a multitude of additional reaction pathways coming into play due to the presence of CO2 (eqn (2)–(4)). This will be discussed in more detail below.
Although the temperature strongly influences the conversion, the H2 selectivity (Fig. 2b) always exceeds 95% when pure CH4 is used as a feed due to kinetic inhibition of most side reactions above 1000 °C.53 Hence, the formation of byproducts hardly plays a role. For biogas as a feed, a rising reaction temperature promotes the H2 selectivity, which is as high as 83% at 1000 °C and rises to 98% at 1400 °C. A further temperature increase to 1600 °C is only beneficial in terms of CH4 conversion (Fig. 2a), but has only a marginal effect on the H2 selectivity (Fig. 2b). The higher CH4 conversion when using biogas as a feed may be due to a kinetic promotion of dry reforming (eqn (2)) at high temperatures. The lower H2 selectivity for the biogas feed, however, may be due to a kinetic promotion of the RWGS reaction (eqn (3)).
Fig. 2c shows the product concentrations for a H2/CH4 gas feed. At 1000 °C, H2 exhibits the highest share of approximately 74%, and unconverted methane with a volumetric share of approx. 25% is the predominant C-containing gas species. With increasing temperature, an increasing H2 proportion and a decreasing methane content can be observed, corresponding to the increasing methane conversion and the comparably constant H2 selectivity. Ethane (C2H6), ethylene (C2H4), acetylene (C2H2), and benzene (C6H6) can be identified as further byproducts. At 1000 °C and 1600 °C their total concentration is below 0.1%. At 1200 °C all four components mentioned above are formed in a concentration range between 0.1% and 0.2% each, and at 1400 °C only acetylene and ethane can be observed in a significant amount. The formation of these byproducts, which also play a role as essential intermediates during soot formation,19 was also observed in previous studies at temperatures above 1000 °C.53
If biogas (CH4/CO2 diluted with H2) is used as a feed, unreacted CO2 can be observed in the product stream in addition to H2 and unreacted CH4. Its volume fraction drops from over 10% at 1000 °C to less than 1% at temperatures of 1200 °C and above (Fig. 2c). The decreasing CO2 concentration correlates with the formation of CO, which is detected in significant amounts of up to 10% in the product gas stream at a temperature of 1200 °C and above, and whose origin we attribute to the dry reforming (eqn (2)) or the Boudouard reaction (eqn (4)). Furthermore, similar to experiments with a CH4/H2 feed gas, acetylene and ethane are formed in a significant amount, with volume fractions of 0.1% to 0.2% at 1200 °C and 1400 °C, which is significantly more than that at 1000 °C and 1600 °C. Last but not least, up to 2% of water (H2O) is formed if the feed contains CO2, which is due to the RWGS reaction (eqn (3)).
Influence of residence time and CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :CO2 ratio
:CO2 ratio
As previously mentioned, CO2 can be consumed via dry reforming (eqn (2)) or via RWGS (eqn (3)), which both results in the formation of H2 and CO. Since H2/CO mixtures with various stoichiometries are widely used as syngas in industry, the H2:CO ratio in the effluent product gas stream is of particular importance. Fig. 3 shows CH4 conversion (a), H2 selectivity (b), and CO2 conversion (c) for both H2-diluted feeds, pure CH4 and biogas, as a function of the residence time and CH4:CO2 ratio of the biogas at 1200 °C and 1400 °C.
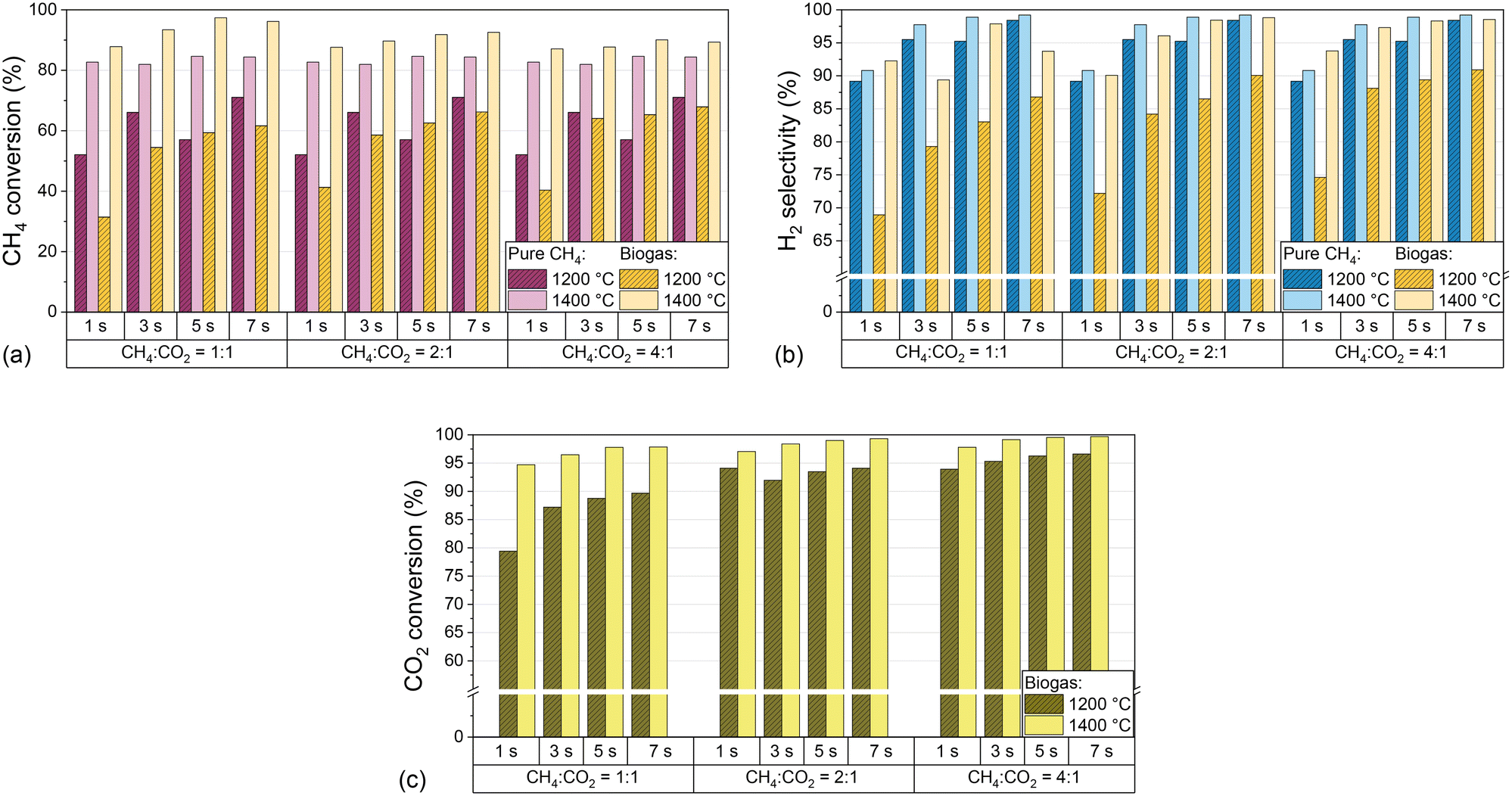 | ||
| Fig. 3 Molar CH4 conversion (a), molar H2 selectivity (b), and molar CO2 conversion (c) as a function of residence time and CH4:CO2 ratio of biogas at 1200 °C to 1400 °C, and a molar H2:biogas ratio of 2:1. As reference, data obtained with a feed gas that contains only CH4 (in H2 as a dilutant with a molar H2:CH4 ratio of 2:1) are also plotted in (a) and (b). | ||
The data point to a beneficial effect of an increasing residence time on CH4 conversion, H2 selectivity, and CO2 conversion, although above 1400 °C residence time variations have a lower impact compared to temperatures as low as 1000 °C or 1200 °C. In analogy to previous findings on methane pyrolysis,54 dry reforming,55 and the RWGS reaction,56 a longer exposure of the reactants to high temperatures enhances the thermocatalytic conversion of CH4 and CO2 and, in the case of CH4, benefits the decomposition of intermediate species via dehydrogenation to form H2 and solid carbon.14 Moreover, the variations of the CO2 content in the biogas mixtures uncovered that CH4 conversion and H2 selectivity increase with higher CH4 content in the feed, but with a lower impact of the CH4:CO2 ratio at 1400 °C than at 1200 °C (Fig. 3a and b). Notably, the CO2 conversion at 1400 °C exceeds 94% even under the most unfavorable conditions, namely a residence time of 1 s and a CH4:CO2 ratio of 1:1, and is higher for any other operational point (Fig. 3c). Even at 1200 °C, CO2 conversion values of more than 95% can be achieved if the CH4:CO2 ratio is set to 4:1 and a residence time of 3 s or higher is chosen. These findings emphasize the huge potential of gas-phase biogas pyrolysis for efficient CO2 transformation and utilization.
The substantial increase in CO2 conversion with increasing temperature can be explained by the endothermicity of the equilibria. Furthermore, the conversion of CO2 during dry reforming additionally promotes the conversion of CH4, which explains the higher CH4 conversion in biogas feeds compared to measurements with pure CH4. In contrast, the lower H2 selectivity with biogas compared to measurements with pure CH4 indicates a consumption of H2, most likely via the RWGS reaction according to eqn (3), which also accounts for the formation of H2O, i.e. as depicted in Fig. 4. In order to understand to what extent competing reactions influence the overall process, the product composition, in particular the CO concentration, must be examined in more detail. Hence, Fig. 4 shows the volume fractions of the product gas components when using either CH4 only (a) or biogas (b) as the feed (both with H2 dilution) at different residence times, H2:CH4 ratios, and biogas compositions at a temperature of 1400 °C.
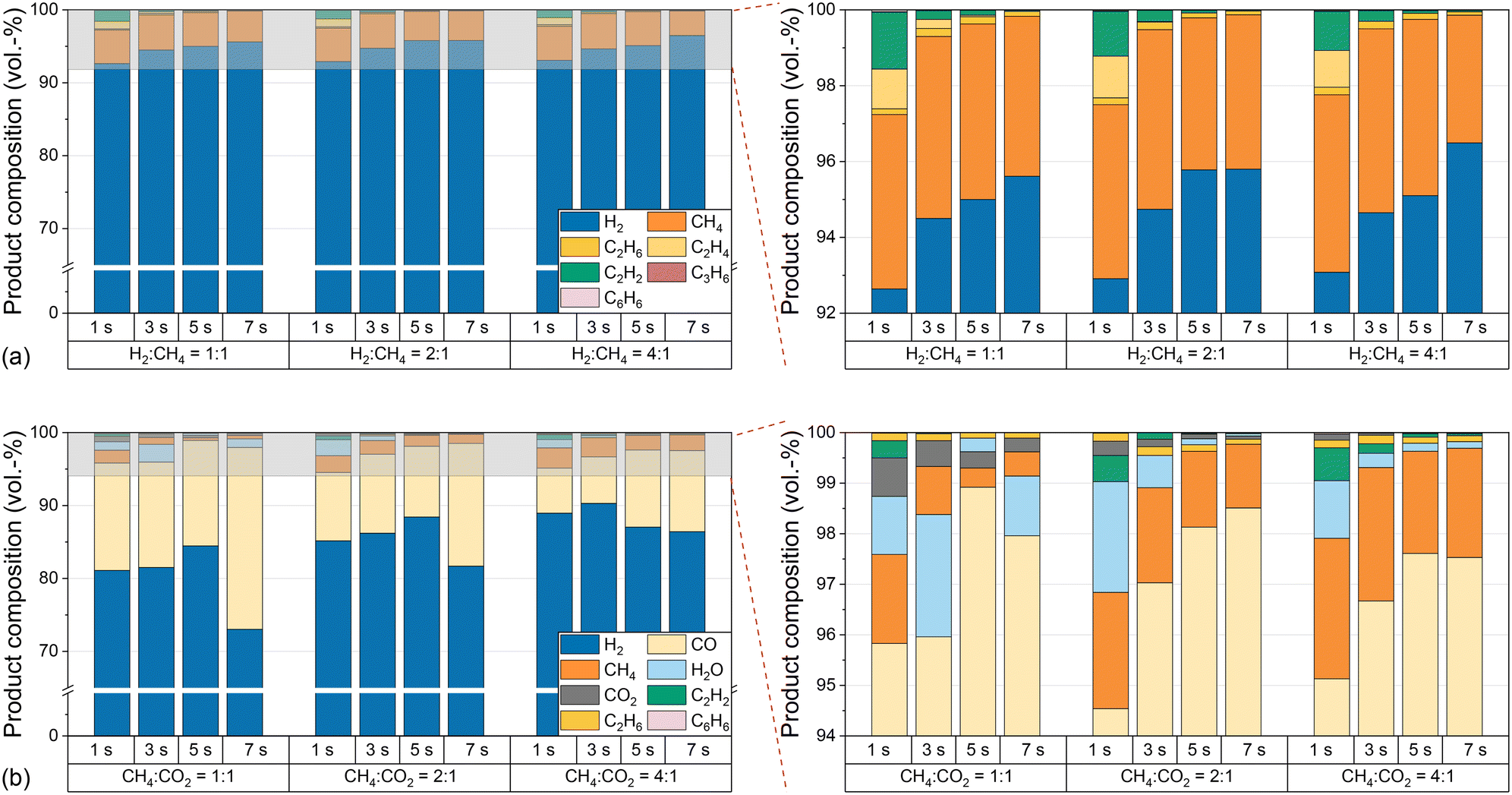 | ||
| Fig. 4 Product composition for pure CH4 (a) and biogas (b) as a function of residence time, molar H2:CH4 dilution, and molar CH4:CO2 ratio of biogas (in H2 as a dilutant with a molar H2:biogas ratio of 2:1) at 1400 °C. | ||
With amounts of at least 92% in the product stream, H2 is the main product when pure CH4 is used in the feed, irrespective of the H2:CH4 ratio or the residence time; byproducts such as ethane, ethylene, acetylene, propylene or benzene form only to a small extent. These findings are consistent with previously postulated hydrocarbon decomposition mechanisms, where the aforementioned species act as intermediates for the formation of solid carbon.14,19,37,38,57 Complementary to the CO2 conversion data shown in Fig. 3c, only minor amounts of unconverted CO2 are found in the effluent gas stream when using biogas as a feed (Fig. 4b). Instead, CO contents of up to 25% are found. Although some purification and process adaption may be necessary, for instance to remove humidity (H2O contents of up to 2.5% are found, cf.Fig. 4b) or to tune the H2:CO ratio, the high CO content may allow a direct use of the effluent product gas stream as syngas. For instance, a H2:CO ratio of 1:1 is required for oxo synthesis or from 1:1 to 2:1 for the synthesis of alcohols.58,59 Since H2 serves as a diluent that is added to the feed gas stream, the reaction conditions subject to this work yield relatively H2-rich syngas. As mentioned in the experimental part, dilution generally inhibits the formation of solids and unwanted byproducts. However, a dilution with H2 in particular offers the advantage that no purification of the product gas is required afterwards, since it is part of the product itself. Note that the diluent H2 from the feed is included in all figures showing product compositions. However, for the calculation of H2 selectivity only the H2 formed during the reaction was considered, as specified in the supporting information. In particular, the lowest H2:CO ratio of approximately 3:1 is observed at a temperature of 1400 °C when choosing a CH4:CO2 ratio of 1:1 and a residence time of 7 s. Since lower temperatures may result in syngas formation with lower H2:CO ratios, but at the expense of a drop in CH4 and CO2 conversion, downstream conditioning of the syngas would be more appropriate if lower H2:CO ratios are desired.60
In addition to CH4 conversion, H2 selectivity, and product gas composition, the amount of produced solid carbon in relation to the carbon entry in the form of CH4 and CO2 is of particular interest for an evaluation of the process with respect to its potential as a carbon sink. Thus, Fig. 5 shows the solid carbon yield as a function of residence time and CH4:CO2 ratio of the biogas at 1200 °C and 1400 °C; data for a feed gas stream containing only CH4 diluted with H2 are given as a reference. Note that the carbon amount was calculated from a carbon balance that includes all C-containing gas-phase species. PAHs that may deposit in minor quantity on the carbon accrued during methane pyrolysis40,61 are not analyzed quantitatively. Hence, along with the uncertainty in gas-phase species quantification with the mass spectrometer, the minor yet unknown amount of PAHs contributes to the error bar. Generously estimated, we assume an error bar for the solid carbon yield data depicted in Fig. 5 of approx. 3% for experiments with pure CH4. Since the experiments with biogas yield more different C-containing gas species with individual uncertainties in quantification, we assume a higher error of approx. 5%.
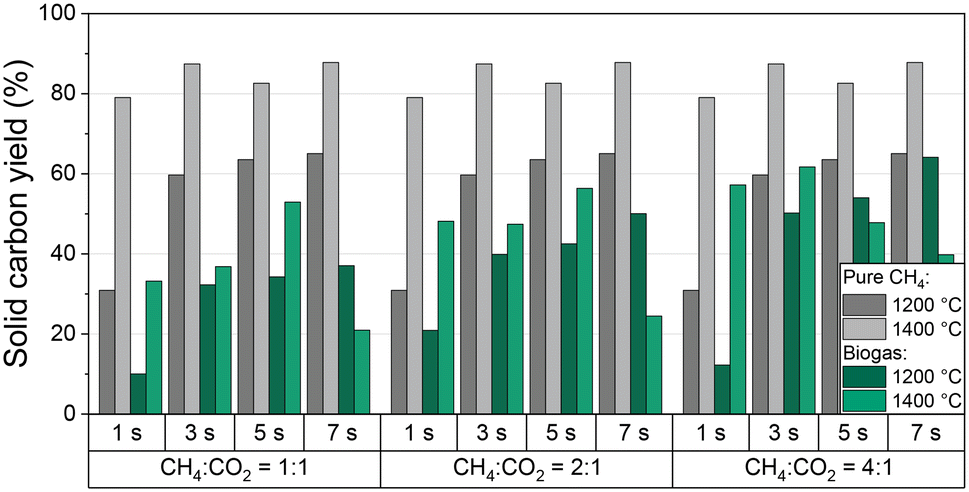 | ||
| Fig. 5 Mass-based solid carbon yield as a function of residence time and molar CH4:CO2 ratio of biogas at 1200 °C to 1400 °C, and a molar H2:biogas ratio of 2:1. As reference, data obtained with a feed gas that contains only CH4 (in H2 as a dilutant with a molar H2:CH4 ratio of 2:1) is also plotted. | ||
For pure methane, the solid carbon yield is generally promoted either by increased temperature or residence time. A maximum carbon yield of almost 90% is found at a temperature of 1400 °C and a residence time of 7 s. These findings correlate well with the trends observed for methane conversion (Fig. 3a) that were already discussed above.
When feeding biogas, on the other hand, the carbon yield is always lower than for the feed gas with pure CH4. Moreover, the solid carbon yield increases with an increasing proportion of methane in the feed gas, both at 1200 °C and 1400 °C. The maximum carbon yield of 65% was achieved at a CH4:CO2 ratio of 4:1, a residence time of 7 s, and a temperature of 1200 °C. A residence time-induced promotion of the solid carbon yield predominantly occurs at 1200 °C, whereas the solid carbon yield correlates directly with the volume fraction of CO in the product gas stream (Fig. 4b) at 1400 °C.
These observations indicate that in the case of a biogas feed the CH4 molecules mainly participate in the pyrolysis reaction, while CO2 primarily reacts in the reactions shown in eqn (2)–(4). Notably, in addition to CO2, CH4 is also consumed during dry reforming (eqn (2)), which increases the proportion of carbonaceous species that do not participate in the pyrolysis reaction, hereby decreasing the overall carbon yield.
In summary, the use of biogas offers the possibility of synthesis gas production and simultaneous fixation of a considerable proportion of carbon that enters the reactor via gas-phase species. Since the carbon produced during pyrolysis can also support the pyrolysis reaction,14,62 the influence of a carbon-containing fixed bed is of particular interest and is therefore investigated in more detail in the following section.
Influence of a carbonaceous fixed bed
In addition to the process parameters that were already extensively discussed above, the introduction of carbon into the reactor, e.g. in the form of graphitic or amorphous carbon, can change the product composition.32,39,62 As described in the experimental section, the reactor was loaded with a carbon particle fixed bed and its impact regarding CH4 conversion (Fig. 6a), H2 selectivity (Fig. 6b), and CO2 conversion (Fig. 6c) was evaluated for H2-diluted CH4 and biogas feed streams. For this, the temperature and the CH4:CO2 ratio of the biogas were varied while keeping the residence time of 5 s and the H2 dilution ratio of 2:1 constant.
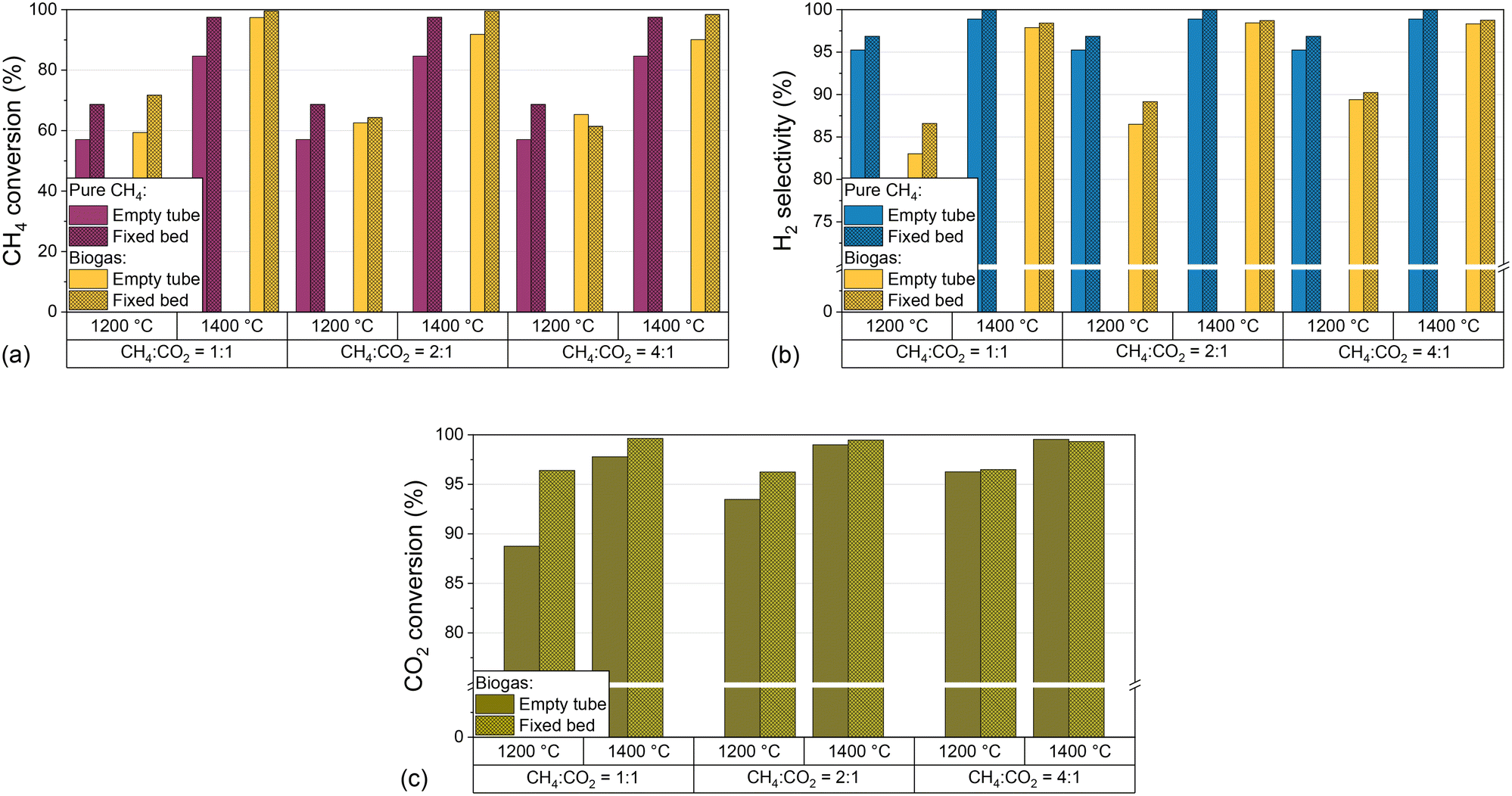 | ||
| Fig. 6 Empty tube and fixed bed results for molar CH4 conversion (a), molar H2 selectivity (b) for pure CH4 and biogas and molar CO2 conversion (c) for biogas as a function of temperature and molar CH4:CO2 ratio of biogas at a constant residence time of 5 s and a molar H2 dilution ratio of 2:1. | ||
The data depicted in Fig. 6 underscore that the introduction of a carbonaceous fixed bed significantly promotes CH4 conversion (Fig. 6a) as well as H2 selectivity (Fig. 6b), irrespective of the feed gas composition. At 1400 °C, both methane conversion and H2 selectivity exceed 95%, with the highest CH4 conversions observed when the feed gas contains biogas instead of only methane. On the other hand, the absence of CO2 benefits the product selectivity towards H2. With regard to the CO2 conversion, the promoting effect of the carbonaceous fixed bed depends on the CH4:CO2 ratio and the temperature. In particular, the fixed bed increases the CO2 conversion from 87% to 97% at a CH4:CO2 ratio of 1:1 and a temperature of 1200 °C. However, the promoting effect decreases with increasing temperature and CH4:CO2 ratio and is almost negligible at a CH4:CO2 ratio of 4:1.
Our results with a feed gas that contains only (H2-diluted) CH4 underscore the beneficial effect of carbon on methane pyrolysis, which is in accordance with previous findings.14,34,36,62 In this context, the catalytic effect of carbon is particularly dependent on structural and surface properties. It is assumed that surface defects, or more precisely high-energy sites of the carbon surface, are capable of activating the methane molecule.39 These defects are found primarily in disordered, amorphous materials, such as the acetylene coke used in this work.
More importantly, our experiments with a biogas feed stream suggest that a carbonaceous fixed bed is not only beneficial for CH4 conversion and H2 selectivity, but also enhances CO2 conversion as uncovered by the results presented in Fig. 6c. At all temperatures and CH4:CO2 ratios, the fixed bed promotes the conversion of CO2. While the conversion increase is most pronounced for the experiments with high CO2 content in the feed gas and at 1200 °C, the difference between the results for an empty reactor and a fixed bed configuration becomes smaller with rising CH4 content and at 1400 °C. We attribute this converging behavior to a promotion of the forward reactions of eqn (2)–(4) in gas compositions with high CO2 contents, both due to the endothermic nature of these reactions and Le Chatelier's principle.63–65
Furthermore, since the product stream composition is a key parameter for understanding and optimizing the overall process, especially when using biogas as a feed, Fig. 7 provides further details on the product gas composition as a function of temperature and CH4:CO2 ratio. Compared to empty tube tests, the introduction of a carbonaceous fixed bed does not only decrease the CH4 and CO2 concentrations in the effluent gas stream, especially at a temperature of 1400 °C, but also suppresses the formation of the byproducts ethane, acetylene, benzene, and water.
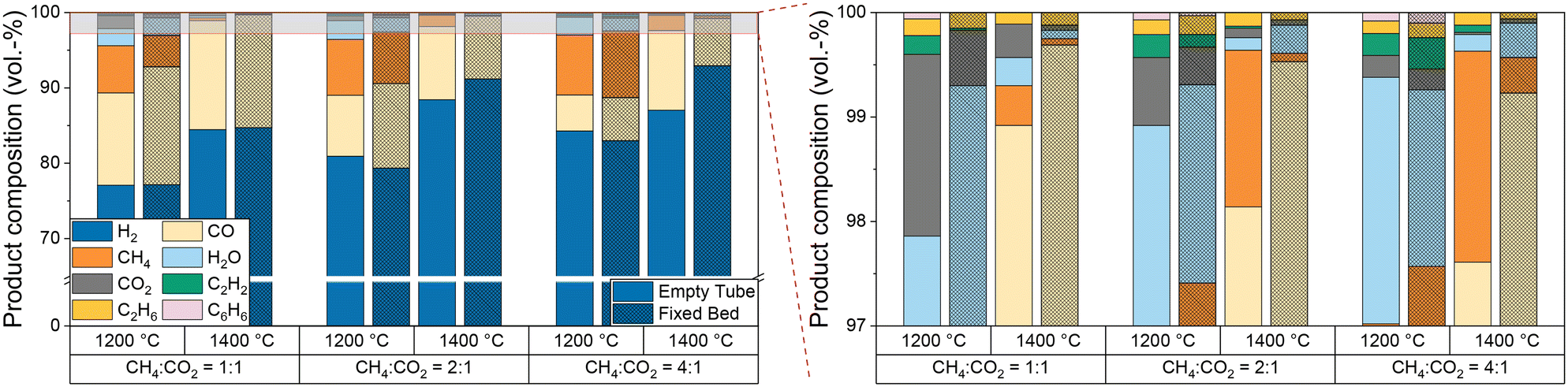 | ||
| Fig. 7 Empty tube and fixed bed results for product composition for biogas as a function of temperature and molar CH4:CO2 ratio of biogas at a constant residence time of 5 s and a molar H2 dilution ratio of 2:1. | ||
At a CH4:CO2 ratio of 1:1, our data suggest that the use of a fixed bed mainly promotes CO formation. Although the H2 content in the product gas stream is almost the same, a lower methane content is found in the product gas stream compared to the empty reactor experiments. This composition of the product gas stream indicates that dry reforming (eqn (2)) consumes methane and carbon dioxide over a carbonaceous fixed bed, resulting in the formation of CO and H2. As suggested by the H2O content in the product gas stream, the reverse water-gas shift reaction (eqn (3)) converts considerable amounts of H2 and CO2 into CO and H2O at 1200 °C. This observation matches with the equilibrium constant of the RWGS reaction at temperatures above 1100 °C.56 Despite its endothermic nature, RWGS seems to become significantly less relevant at 1400 °C, as less steam is observed in the effluent gas stream. This apparent mismatch may be explained by a reaction between H2O and CH4 to form CO (or CO2) and H2, which is essentially a reverse methanation reaction. As methanation itself is strongly exothermic, temperatures above 700 °C promote the reverse reaction.66 However, since a lower methane content is always accompanied by a higher H2 content due to the pyrolysis reaction itself although a possible in situ consumption of H2O formed via the RWGS reaction would result in a comparably lower H2 evolution, more detailed experiments are necessary to uncover the mechanistic details in the future. An increasing CH4:CO2 ratio (namely 2:1 and 4:1) diminishes the effect of the fixed bed on the methane content, but still a beneficial effect on the CO2 conversion remains. The higher CO content found during experiments with the fixed bed reactor configuration is desirable when the product stream is supposed to be used as syngas.
In order to assess the suitability of methane and biogas pyrolysis as a process acting as a carbon sink, Fig. 8 summarizes experiments conducted with an empty tube and a fixed bed reactor configuration by showing the H2 selectivity (Fig. 8a) and the solid carbon yield (Fig. 8b) as a function of CH4 conversion and with varying H2 dilution.
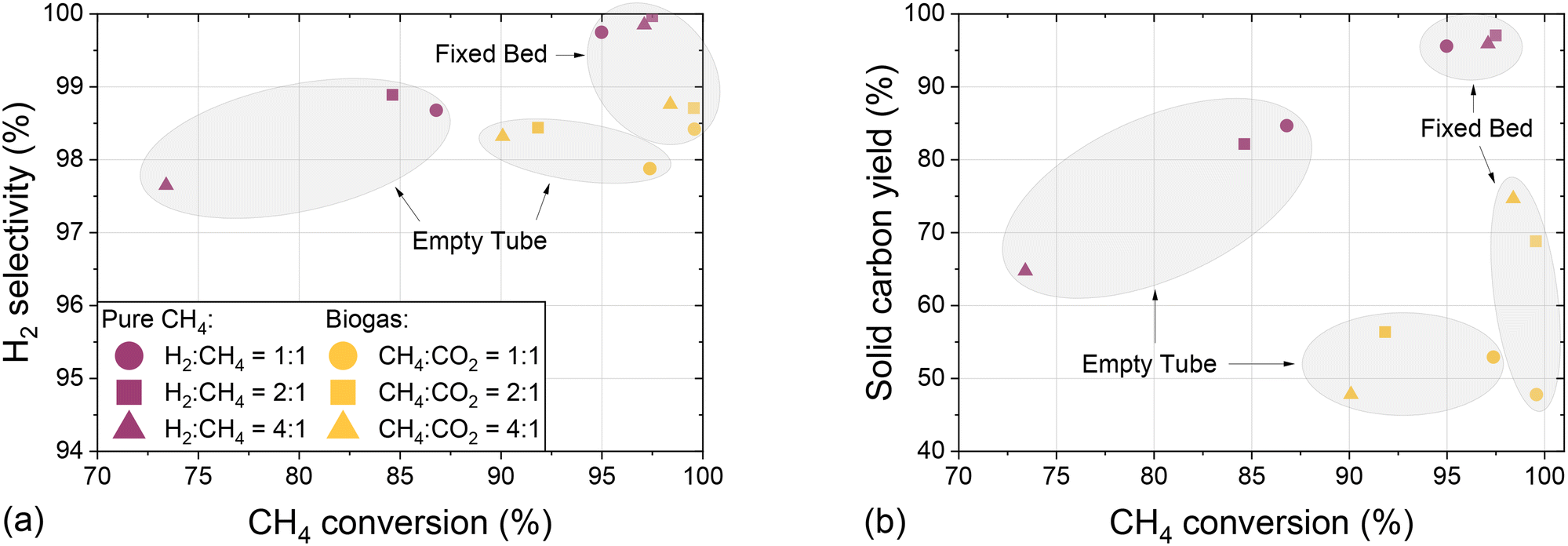 | ||
| Fig. 8 Molar H2 selectivity (a) and mass-based solid carbon yield (b) as a function of molar CH4 conversion for pure CH4 and biogas feeds in an empty tube and fixed bed configuration at a temperature of 1400 °C, a residence time of 5 s, and a constant molar H2:biogas ratio of 2:1. | ||
The data presented in Fig. 8a clearly emphasize that the use of a fixed bed increases both methane conversion and H2 selectivity, irrespective of the H2:CH4 dilution ratio and biogas composition. Compared to experiments with pure methane, the use of CO2-containing biogas barely reduces the selectivity towards hydrogen and allows for even higher methane conversion. These observations underscore the flexibility of the studied pyrolysis process in terms of feed gas composition. Similarly, the data presented in Fig. 8b reveal that the use of a carbonaceous fixed bed benefits the formation of solid elemental carbon not only in a feed gas containing solely CH4, but also in a biogas-based feed stream. The feed with a CH4:CO2 ratio of 1:1 is the only exception, which we assume is due to the high CO2 content that benefits the Boudouard reaction (eqn (4)). For both reactor configurations, the carbon yields for experiments with pure methane as a feed always exceed those for experiments with a biogas feedstock. This observation substantiates the above-mentioned hypothesis that it is primarily the carbon from the methane molecules in the feed gas that can be fixed in solid form, whereas CO2 is rather reacting to CO. Since under the conditions subject to the present study the carbon yield in a fixed bed reactor configuration varies between 47% and 75% when using biogas, corresponding to CH4:CO2 ratios of 1:1 and 4:1, respectively, pyrolysis is an auspicious process for carbon fixation in elemental solid carbon. Non-solid carbon is predominantly bound in CO, which along with H2 in the product gas stream can serve as synthesis gas.
Conclusions
Our work that was conducted in a lab-scale high-temperature pyrolysis reactor evaluates the thermocatalytic decomposition of biogas at high temperatures and compares the results with results obtained for conventional CH4 pyrolysis. Hereby, we analyze the suitability of biogas pyrolysis for H2 and syngas production and simultaneous carbon capture. Our tests identified the main reaction parameters that govern CH4 conversion, H2 selectivity, CO2 conversion, and product composition, namely temperature, residence time, H2 content in the feed gas, and the molar CH4:CO2 ratio of the biogas used as a feedstock.
For H2-diluted feed gas streams containing either pure CH4 or biogas, CH4 conversions, H2 selectivities, and CO2 conversions of more than 90% are achieved at temperatures of 1400 °C and above. Herein, an increase of the residence time from 1 s to 7 s does not only promote the conversion of CH4 and CO2, but also enhances the selectivity to H2. Moreover, a high CH4 content and low amounts of CO2 in the feed promote CH4 conversion and H2 selectivity especially at temperatures as low as 1200 °C, whereas the impact of the CH4:CO2 ratio diminishes at temperatures of 1400 °C and above. Although without doubt higher temperatures further increase operating costs, they also allow the process to be operated with higher H2 dilutions while still maintaining a sufficiently high CH4 conversion. In terms of process design, a high H2 dilution is very attractive as it ensures a safe operation with reduced byproduct formation and improved control of the solid formation, hereby resulting in less reactor clogging.
In addition to the primary product H2, the usage of biogas as a feed results in considerable CO formation; reactor operation at 1400 °C, a residence time of 7 s, and a CH4:CO2 ratio of 1:1 yields the highest CO content and results in a H2:CO ratio of approximately 3:1. Although the product gas stream can be used directly as syngas, a further tuning of the H2:CO ratio may be mandatory in order to account for downstream follow-up processes. For instance, if lower H2:CO ratios are needed, lower temperatures could be used, however, at the expense of CH4 and CO2 conversion. Although the design of a real-world process would require a profound techno-economic analysis, downstream conditioning of the syngas could be more appropriate, considering the trade-off between feed stream conversion and product stream composition.
Moreover, the formation of solid carbon is promoted by high temperatures and high residence time. At a temperature of 1400 °C and a residence time of 7 s, a solid carbon yield of almost 90% is achieved when using pure methane as a feed gas. When using biogas instead, high methane contents in the feed promote the formation of elemental carbon with a maximum carbon yield of 65% at a CH4:CO2 ratio of 4:1, a residence time of 7 s, and a temperature of 1200 °C. Herein, the solid carbon yield directly correlates with the volume fraction of CO in the product gas stream, indicating that mainly the methane molecules participate in the pyrolysis reaction, whereas CO2 is predominantly converted to CO via dry reforming and reverse water-gas shift reactions.
Finally yet importantly, the presence of a carbonaceous fixed bed enhances heterogeneous reactions during carbon deposition, hereby promoting the conversion of both CH4 and CO2, increasing the selectivity to H2, and suppressing the formation of undesired byproducts such as ethane, ethylene, or benzene. In addition, the fixed bed promotes the yield of solid carbon, allowing a carbon fixation of up to 95% for a feed gas stream containing pure CH4 to be achieved. When using biogas as a feedstock, the carbon yield directly correlates with the methane content in the feed, which can be attributed to the pyrolytic reaction pathways for CH4 and the dry reforming and reverse water-gas shift reactions consuming CO2. Remarkably, at a temperature of 1400 °C and a residence time of 5 s, the solid carbon yield in a biogas-based feed with a CH4:CO2 ratio of 4:1 is as high as 75%, which is an encouraging value regarding carbon capture and simultaneous syngas production without any direct CO2 emissions.
In addition, the use of comparably cheap carbon as the material for the fixed bed suggests a high economic appeal, particularly considering that metal impurities in the deposited carbon are irrelevant, which otherwise cannot be avoided if conventional metal-based catalysts are used for CH4 and CO2 activation. Beyond a simple sequestration of accrued carbon, the commercialization of the solid carbon product is considered essential for establishing an economically competitive process.7 Hence, further characterization, especially in terms of structural parameters or particle size distribution as a function of the operational points, is imperative. In addition to a detailed deconvolution of competing reaction pathways in the gas-phase, i.e. pyrolysis, dry reforming, reverse water-gas shift, and the Boudouard reaction, future studies also need to clarify whether the presence of oxygen-containing species such as CO2 has an impact on the accrued carbon and therefore on its further usage.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank M. Bender J. Bode, K. Ehrhardt, D. Flick, F. Scheiff, and D. Schlereth (BASF) for fruitful discussions, M. Berg and C. Kroll (Berg-idl GmbH) for their support in engineering the high-temperature vessel of the experimental setup, and S. Lichtenberg (KIT) for technical support.Notes and references
- C. A. Horowitz, International Legal Materials, 2016, 55, 740–755 CrossRef.
- M. van der Spek, C. Banet, C. Bauer, P. Gabrielli, W. Goldthorpe, M. Mazzotti, S. T. Munkejord, N. A. Røkke, N. Shah, N. Sunny, D. Sutter, J. M. Trusler and M. Gazzani, Energy Environ. Sci., 2022, 15, 1034–1077 RSC.
- O. Machhammer, A. Bode and W. Hormuth, Chem. Eng. Technol., 2016, 39, 1185–1193 CrossRef CAS.
- V. M. Maestre, A. Ortiz and I. Ortiz, Renewable Sustainable Energy Rev., 2021, 152, 111628 CrossRef CAS.
- S. Griffiths, B. K. Sovacool, J. Kim, M. Bazilian and J. M. Uratani, Energy Res. Soc. Sci., 2021, 80, 102208 CrossRef.
- P. Lott and O. Deutschmann, Proc. Combust. Inst., 2023, 39, 3183–3215 CrossRef CAS.
- B. Parkinson, P. Balcombe, J. F. Speirs, A. D. Hawkes and K. Hellgardt, Energy Environ. Sci., 2019, 12, 19–40 RSC.
- K. Christopher and R. Dimitrios, Energy Environ. Sci., 2012, 5, 6640–6651 RSC.
- N. Sánchez-Bastardo, R. Schlögl and H. Ruland, Ind. Eng. Chem. Res., 2021, 60, 11855–11881 CrossRef.
- S. Schneider, S. Bajohr, F. Graf and T. Kolb, ChemBioEng Rev., 2020, 7, 150–158 CrossRef CAS.
- N. Muradov and T. Veziroǧlu, Int. J. Hydrogen Energy, 2005, 30, 225–237 CrossRef CAS.
- H. F. Abbas and W. Wan Daud, Int. J. Hydrogen Energy, 2010, 35, 1160–1190 CrossRef CAS.
- U. Ashik, W. Wan Daud and H. F. Abbas, Renewable Sustainable Energy Rev., 2015, 44, 221–256 CrossRef CAS.
- P. Lott, M. B. Mokashi, H. Müller, D. J. Heitlinger, S. Lichtenberg, A. B. Shirsath, C. Janzer, S. Tischer, L. Maier and O. Deutschmann, ChemSusChem, 2023, 16, e202201720 CrossRef CAS PubMed.
- G. Fau, N. Gascoin, P. Gillard and J. Steelant, J. Anal. Appl. Pyrolysis, 2013, 104, 1–9 CrossRef CAS.
- C.-J. Chen, M. H. Back and R. A. Back, in Industrial and Laboratory Pyrolyses, ed. L. F. Albright and B. L. Crynes, American Chemical Society, 1976, pp. 1–16 Search PubMed.
- A. Holmen, O. Olsvik and O. A. Rokstad, Fuel Process. Technol., 1995, 42, 249–267 CrossRef CAS.
- W. Benzinger, A. Becker and K. J. Hüttinger, Carbon, 1996, 34, 957–966 CrossRef CAS.
- J. Appel, H. Bockhorn and M. Frenklach, Combust. Flame, 2000, 121, 122–136 CrossRef CAS.
- A. B. Shirsath, M. Mokashi, P. Lott, H. Müller, R. Pashminehazar, T. Sheppard, S. Tischer, L. Maier, J.-D. Grunwaldt and O. Deutschmann, J. Phys. Chem. A, 2023, 127, 2136–2147 CrossRef CAS PubMed.
- M. R. Kholghy, G. A. Kelesidis and S. E. Pratsinis, Phys. Chem. Chem. Phys., 2018, 20, 10926–10938 RSC.
- M. Frenklach, Symp. (Int.) Combust., [Proc.], 1996, 26, 2285–2293 CrossRef.
- A. Böttcher, F. Hennrich, H. Rösner, S. Malik, M. M. Kappes, S. Lichtenberg, G. Schoch and O. Deutschmann, Carbon, 2007, 45, 1085–1096 CrossRef.
- T. I. Korányi, M. Németh, A. Beck and A. Horváth, Energies, 2022, 15, 6342 CrossRef.
- S. Timmerberg, M. Kaltschmitt and M. Finkbeiner, Energy Convers. Manage.: X, 2020, 7, 100043 CAS.
- N. Muradov, F. Smith, C. Huang and A. T. Raissi, Catal. Today, 2006, 116, 281–288 CrossRef CAS.
- L. Zhou, L. R. Enakonda, M. Harb, Y. Saih, A. Aguilar-Tapia, S. Ould-Chikh, J.-L. Hazemann, J. Li, N. Wei, D. Gary, P. Del-Gallo and J. Basset, Appl. Catal., B, 2017, 208, 44–59 CrossRef CAS.
- S. H. Sharif Zein, A. R. Mohamed and P. S. Talpa Sai, Ind. Eng. Chem. Res., 2004, 43, 4864–4870 CrossRef.
- N. Muradov, Energy Fuels, 1998, 12, 41–48 CrossRef CAS.
- T. Keipi, K. E. Tolvanen, H. Tolvanen and J. Konttinen, Energy Convers. Manage., 2016, 126, 923–934 CrossRef CAS.
- A. Abánades, E. Ruiz, E. M. Ferruelo, F. Hernández, A. Cabanillas, J. M. Martínez-Val, J. A. Rubio, C. López, R. Gavela, G. Barrera, C. Rubbia, D. Salmieri, E. Rodilla and D. Gutiérrez, Int. J. Hydrogen Energy, 2011, 36, 12877–12886 CrossRef.
- N. Muradov, Catal. Commun., 2001, 2, 89–94 CrossRef CAS.
- D. P. Serrano, J. A. Botas and R. Guil-Lopez, Int. J. Hydrogen Energy, 2009, 34, 4488–4494 CrossRef CAS.
- D. P. Serrano, J. Botas, P. Pizarro and G. Gómez-Pozuelo, Int. J. Hydrogen Energy, 2013, 38, 5671–5683 CrossRef CAS.
- D. P. Serrano, J. A. Botas, J. Fierro, R. Guil-López, P. Pizarro and G. Gómez, Fuel, 2010, 89, 1241–1248 CrossRef CAS.
- J. Zhang, X. Li, H. Chen, M. Qi, G. Zhang, H. Hu and X. Ma, Int. J. Hydrogen Energy, 2017, 42, 19755–19775 CrossRef CAS.
- K. Norinaga and O. Deutschmann, Ind. Eng. Chem. Res., 2007, 46, 3547–3557 CrossRef CAS.
- K. Norinaga, O. Deutschmann, N. Saegusa and J.-I. Hayashi, J. Anal. Appl. Pyrolysis, 2009, 86, 148–160 CrossRef CAS.
- N. Muradov, F. Smith and A. T. Raissi, Catal. Today, 2005, 102, 225–233 CrossRef.
- A. Calbry-Muzyka, H. Madi, F. Rüsch-Pfund, M. Gandiglio and S. Biollaz, Renewable Energy, 2022, 181, 1000–1007 CrossRef CAS.
- S. Kim, Y. T. Kim, L. S. Oh, H. J. Kim and J. Lee, J. Mater. Chem. A, 2022, 10, 20024–20034 RSC.
- W. Yang, K.-H. Kim and J. Lee, J. Cleaner Prod., 2022, 376, 134292 CrossRef.
- C. Park, H. Lee, N. Lee, B. Ahn and J. Lee, J. Hazard. Mater., 2022, 440, 129825 CrossRef CAS.
- J. Kim, C. Park, H. Park, J. Han, J. Lee and S.-K. Kim, Energy, 2022, 258, 124877 CrossRef CAS.
- H. Park, J. Joo, J. Kim, J. Lee and S.-K. Kim, Energy, 2023, 278, 127797 CrossRef CAS.
- C. H. Geissler and C. T. Maravelias, Energy Environ. Sci., 2022, 15, 2679–2689 RSC.
- H. Liu, N. Agrawal, A. Ganguly, Y. Chen, J. Lee, J. Yu, W. Huang, M. Mba Wright, M. J. Janik and W. Li, Energy Environ. Sci., 2022, 15, 4175–4189 RSC.
- R. M. Navarro, M. C. Sánchez-Sánchez, M. C. Alvarez-Galvan, F. Del Valle and J. L. G. Fierro, Energy Environ. Sci., 2009, 2, 35–54 RSC.
- X. Zhao, B. Joseph, J. Kuhn and S. Ozcan, iScience, 2020, 23, 101082 CrossRef CAS.
- R. Sivaranjani, S. Veerathai, K. Jeoly Jenifer, K. Sowmiya, K. J. Rupesh, S. Sudalai and A. Arumugam, Int. J. Hydrogen Energy, 2023, 48, 23785–23820 CrossRef CAS.
- M. Ni, D. Y. Leung, M. K. Leung and K. Sumathy, Fuel Process. Technol., 2006, 87, 461–472 CrossRef CAS.
- S. D. Angeli, S. Gossler, S. Lichtenberg, G. Kass, A. K. Agrawal, M. Valerius, K. P. Kinzel and O. Deutschmann, Angew. Chem., 2021, 60, 11852–11857 CrossRef CAS.
- C. Guéret, M. Daroux and F. Billaud, Chem. Eng. Sci., 1997, 52, 815–827 CrossRef.
- O. Olsvik, O. A. Rokstad and A. Holmen, Chem. Eng. Technol., 1995, 18, 349–358 CrossRef CAS.
- U. Olsbye, T. Wurzel and L. Mleczko, Ind. Eng. Chem. Res., 1997, 36, 5180–5188 CrossRef.
- F. Bustamante, R. M. Enick, A. V. Cugini, R. P. Killmeyer, B. H. Howard, K. S. Rothenberger, M. V. Ciocco, B. D. Morreale, S. Chattopadhyay and S. Shi, AIChE J., 2004, 50, 1028–1041 CrossRef CAS.
- M. Frenklach, Phys. Chem. Chem. Phys., 2002, 4, 2028–2037 RSC.
- H. Weber and J. Falbe, Ind. Eng. Chem., 1970, 62, 33–37 CrossRef CAS.
- H. T. Luk, C. Mondelli, D. C. Ferré, J. A. Stewart and J. Pérez-Ramírez, Chem. Soc. Rev., 2017, 46, 1358–1426 RSC.
- M. M. Yung, W. S. Jablonski and K. A. Magrini-Bair, Energy Fuels, 2009, 23, 1874–1887 CrossRef CAS.
- G. F. Glasier, R. Filfil and P. D. Pacey, Carbon, 2001, 39, 497–506 CrossRef CAS.
- V. Shilapuram, N. Ozalp, M. Oschatz, L. Borchardt and S. Kaskel, Carbon, 2014, 67, 377–389 CrossRef CAS.
- M. Jafarbegloo, A. Tarlani, A. W. Mesbah and S. Sahebdelfar, Int. J. Hydrogen Energy, 2015, 40, 2445–2451 CrossRef CAS.
- M. Wenzel, L. Rihko-Struckmann and K. Sundmacher, AIChE J., 2017, 63, 15–22 CrossRef CAS.
- P. Lahijani, Z. A. Zainal, M. Mohammadi and A. R. Mohamed, Renewable Sustainable Energy Rev., 2015, 41, 615–632 CrossRef CAS.
- J. Gao, Y. Wang, Y. Ping, D. Hu, G. Xu, F. Gu and F. Su, RSC Adv., 2012, 2, 2358–2368 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00360d |
| This journal is © The Royal Society of Chemistry 2024 |