
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel enantiopure δ-thiolactones: synthesis, structural characterization, and reactivity studies†
Magdalena Rodríguez Saravia ab,
Verónica Martíneza,
Franco Vairolettiab,
Mario Macíasc,
Danilo Davyta,
Gonzalo Hernández Dossi*d and
Graciela Mahler*a
ab,
Verónica Martíneza,
Franco Vairolettiab,
Mario Macíasc,
Danilo Davyta,
Gonzalo Hernández Dossi*d and
Graciela Mahler*a
aDepartamento de Química Orgánica, Laboratorio de Química Farmacéutica, Facultad de Quimica, Universidad de la República, Gral Flores 2124, Montevideo 11800, Uruguay. E-mail: gmahler@fq.edu.uy
bPrograma de Posgrado en Quimica, Universidad de la República Uruguay, Gral Flores 2124, Montevideo 11800, Uruguay
cCristalografía y Química de Materiales, CrisQuimMat, Departamento de Química, Universidad de los Andes, Carrera 1 No. 18A-10, Bogotá 111711, Colombia
dDepartamento de Química Orgánica, Laboratorio de Resonancia Magnética Nuclear, Facultad de Química, Universidad de la República, Gral Flores 2124, Montevideo 11800, Uruguay. E-mail: gonzalohd@fq.edu.uy
First published on 24th December 2024
Abstract
A new series of chiral δ-thiolactone derivatives have been prepared. These compounds exemplify the acetalic N–C–S reversibility of fused thiazolidines toward the thermodynamic product. The stereochemistry of the synthesized compounds was elucidated using X-ray crystallography, NOESY spectroscopy, and DFT calculations. The aminolysis reaction of the δ-thiolactone was studied with various alkyl amines, which can open the thioester to yield amido thiols in a single step. This reaction has the potential to be applied in the synthesis of bioactive compounds, polymer chemistry, and dynamic combinatorial chemistry, among others fields.
Introduction
Chemically, thioesters exhibit distinctive characteristics attributed to their thermodynamic stability. Positioned at the center of the nucleophilic addition reactivity scale, thioesters surpass unreactive amides and poorly reactive esters but fall below anhydrides and acyl chlorides. They also display higher reactivity compared to oxoesters. One contributing factor for this behaviour is the C–S bond's reduced tendency to participate in electron delocalization through the carbonyl group, when compared to the C–O bond. The reduced orbital overlap between the sulfur's 3p orbital and the carbon's 2p orbital, contributes to the weaker C–S bond, resulting in lower stability and greater susceptibility to nucleophilic attack.1Due to their ability to react under low energetic barriers, thioesters play a central role in various biological processes as acylation intermediates in biochemical reactions, making them essential intermediates in biomolecular synthesis and potentially vital to the origin of life. They remain a part of the synthesis of essential cellular components such as peptides, fatty acids, sterols, and others.2
The thioester-amide exchange is one of the predominant processes in which this motif is involved, as in the acetyl-CoA mediated S-acylation of amino acid residues,3 and peptide synthesis via native chemical ligation.4
Thiolactones, cyclic thioesters, named analogously to lactams, display relevant biological functions. For example, the γ-thiolactone of homocysteine (Hcy-thiolactone), is involved in the post-translational modification of proteins, and acts as an allosteric dopamine D2 receptor antagonist (Fig. 1).5 Additionally, γ-thiolactones have also been suggested as precursors of life, owing to their substantial involvement in prebiotic chemistry.5
 | ||
| Fig. 1 Biologically relevant γ, δ and ε-thiolactones. | ||
Thiolactones are also present in several natural products such as the antibiotic thiolactomycin, a γ-thiolactone with anti-mycobacterial activity. Thiolactomycin exerts its activity by selectively inhibiting fatty acid and mycolic acid biosynthesis.6 This motif could also be found in isothiochroman-3-ones, described as metallo-amino-peptidase inhibitors,7 and agrochemical fungicides and herbicides.8,9 Lastly, thiosters are used in prodrug strategies, masking the thiol moiety and preventing disulfide formation. Examples of this strategy are the δ-thiolactone described as a glutamate carboxypeptidase II (GCPII) inhibitor,10 and the ε-thiolactone described as Angiotensin-Converting Enzyme (ACE) inhibitor11 (Fig. 1).
Indeed, the ability of thiolactones to undergo ring-opening via reaction with nucleophiles, such as amines, is the key to their use as reagents in synthetic chemistry and polymer science.12,13 For example, the one-pot aminolysis of thiolactones, followed by thiol–ene conjugation, leads to double functionalized diverse polyamines and polyurethanes with high atom efficiency. With the development of post-polymerization modification and iterative protocols based on solid supports, multimers and oligomers of defined sequence could be obtained. The versatility of thiolactones as functionalization tools, led to potential applications in biomedical14,15 and materials fields.16,17 Thiolactone chemistry has also been employed to overcome specific synthetic challenges, as enabling the coupling of sterically hindered peptide sites in native chemical ligation,18 or synthesizing disulfide-linked side-chain-to-tail cyclic peptides in a straightforward, high-yield strategy.19
In this work, we report the preparation, structural characterization, and reactivity of different enantiopure tricyclic δ-thiolactones 1, Fig. 1. Interestingly, thiolactones could serve as a prodrug of bisthiazolidine 2, which was reported as a potent cross-class metallo-β-lactamase inhibitor, since the highly reactive thiol is protected.20,21
Results and discussion
Thioester can be prepared using different methodologies like base-initiated thioesterification of amides with various thiols.22 During the preparation of analogs of the bisthiazolidine 2, the formation of three tricyclic δ-thiolactones 1a–c was noticed. This occurred when employing peptide coupling reagent DCC, which was able to activate the carboxylic acid, and lead to the formation of δ-thiolactones 1, Fig. 2. We observed the formation of three δ-thiolactones, out of the four possible compounds, namely (2R,5S,8S)-1a (38%), (2R,5R,8S)-1b (2%) and (2R,5R,8S)-1c (5%). All these compounds retain the same chiral configuration at C8 as bisthiazolidine (2R,5S,8R)-2, but because of the thioester formation the relative priority between substituents at C8 makes the configuration change from R to S. Given the unusual and attractive biological and chemical properties of thiolactones previously mentioned, we decided to focus on their structural and chemical characterization. | ||
| Fig. 2 Synthesis of δ-thiolactones 1a–c starting from bisthiazolidine 2, method A. | ||
Determination of the absolute configuration of δ-thiolactones 1a–c
First, we aimed to characterize these three isolated δ-thiolactones 1a–c from the range of possible isomers. The absolute configuration of the isolated δ-thiolactones 1a–c was assessed based on a combination of methodologies using crystal structures, two-dimensional Nuclear Overhauser Effect (NOESY) NMR experiments and theoretical calculations.X-ray diffraction data of thiolactones 1a and 1b
X-ray diffraction data was used to determine the crystal structures of compounds 1a and 1b, Fig. 3. The results confirmed the absolute configurations of the isolated δ-thiolactones (2R,5S,8S)-1a and (2R,5R,8S)-1b, Fig. 2. Both compounds crystallized from enantiomerically pure solutions, and their crystals were resolved in the monoclinic and orthorhombic Sohncke space groups P21 and P212121, for 1a and 1b, respectively. These spaces groups lack of mirror or inversion symmetry which is characteristic of enantiomerically pure compounds.23,24 The crystal structure of 1a presents two independent molecules due to conformational effects. The five membered rings S3–C2–N1–C5–C4 and S6–C5–N1–C8–C7 present envelope conformations on S3 (Q = 0.518(4) − φ = 174.3(5)°) and C7 (Q = 0.465(4) − φ = 323.6(5)°) for one of the independent molecules while these same rings present twisted (on C4–S3; Q = 0.481(5) − φ = 169.2(6)°) and envelope (on C7; Q = 0.436(5) − φ = 318.7(6)°) conformations for the other independent molecule. The six membered rings S10–C9–C8–N1–C2–C11 in both independent molecules have different conformations, with puckering amplitude (Q), theta (θ) and phi (φ) values of Q = 0.597(4), θ = 37.4(4)°, φ = 194.8(8)°, and Q = 0.619(5), θ = 48.5(4)°, φ = 198.8(6)°, respectively (Fig. 3a).25,26 In the case of compound 1b, only one independent molecule is detected. For this molecule, the five membered rings S3–C2–N1–C5–C4 and S6–C5–N1–C8–C7 present twisted (on C5–C4; Q = 0.401(4) − φ = 304.5(6)°) and envelope (on C8; Q = 0.372(4) − φ = 102.8(6)°) conformations (Fig. 3b), showing different conformations compared to the same five membered rings in compound 1a. In fact, the strongest conformational difference is observed in the six membered ring S10–C9–C8–N1–C2–C11, which in compound 1b has puckering parameters of Q = 0.754(4), θ = 95.3(3)°, φ = 115.5(3)°. The conformational differences in the S10–C9–C8–N1–C2–C11 ring are also detected in the C2–C11–S10–C9 torsion angle with values of 24.3° and 16.8° for the two independent molecules in compound 1a, and 50.8° for compound 1b. Due to the diverse conformations, the supramolecular structures are assembled differently. In the case of compound 1a, the two independent molecules form separate molecular chains connected by C8–H8⋯O1 (H⋯O = 2.72 Å) and C11–H11⋯O1 (H⋯O = 2.85 Å) hydrogen interactions along [010] direction (Fig. 3a). In 1b, these chains are assembled along [100] directions through C8–H8⋯O1 (H⋯O = 2.64 Å), C7–H7⋯O1 (H⋯O = 2.55 Å), and C4–H4⋯N1 (H⋯O = 2.74 Å) hydrogen interactions (Fig. 3b). In both compounds, C–H⋯S hydrogen interactions contribute to the formation of the crystals with H⋯S distances between 2.86–3.20 Å (Fig. 3b), which are values that seem long but in the case of C–H⋯S contacts, such values have already been reported in literature.27 | ||
| Fig. 3 Molecular and ORTEP diagram of δ-thiolactones: (a) 1a and (b) 1b showing the conformations of the respective enantiomers and the C–H⋯O, C–H⋯N and C–H⋯S hydrogen interactions. Atom color code: C (gray), H (white), O (red), N (purple), S (yellow). | ||
DFT calculation and NOESY-NMR to determine δ-thiolactone 1c structure
Once the absolute configuration of δ-thiolactones 1a and 1b was determined based on the crystal structures, we aimed to determine if the absolute configuration of the remaining diastereomer, which could not be crystallized, was either (2S,5R,8S)-1c or (2S,5S,8S)-1d.To assist in the assignment, DFT calculations of 1H-RMN shifts were performed with GIAO (Gauge Including Atomic Orbitals) method and compared with the experimental data. The method was validated using the X-ray-confirmed assignment of the other two isomers 1a–b. Based on the correlation coefficients (R2) between the calculated and the experimental data, and the mean absolute error (MAE), the best-fitting configuration (lowest MAE, highest R2) was stablished.
The comparison of the predicted 1H-NMR spectra of the two δ-thiolactones 1c and 1d is shown in Table 1. According to the results, the configuration (2S,5R,8S)-1c fits better the experimental data, with the lowest MAE between the experimental and calculated values (0.14) and best R2 (0.95).
|
|||||
|---|---|---|---|---|---|
| H | Experimental δ (ppm) | Calculated δ (ppm) | |||
| 1c (2S,5R,8S) | |Δδ| | 1d (2S,5S,8S) | |Δδ| | ||
| a MAE = mean absolute error.b R2 = correlation coefficient; Δδ = δexperimental − δcalculated. | |||||
| H2 | 4.41 | 4.50 | 0.09 | 5.08 | 0.67 |
| H4a | 3.31 | 3.83 | 0.12 | 3.02 | 0.68 |
| H4b | 3.73 | 3.28 | 0.05 | 3.01 | 0.32 |
| H5 | 5.27 | 5.34 | 0.07 | 4.79 | 0.48 |
| H7a | 3.45 | 3.37 | 0.02 | 3.36 | 0.09 |
| H7b | 3.35 | 3.33 | 0.12 | 3.49 | 0.14 |
| H8 | 4.09 | 4.05 | 0.04 | 4.11 | 0.02 |
| H11a | 3.22 | 3.58 | 0.36 | 3.59 | 0.37 |
| H11b | 3.58 | 3.20 | 0.38 | 3.30 | 0.28 |
| MAEa | — | — | 0.14 | — | 0.34 |
| R2b | — | — | 0.91 | — | 0.68 |
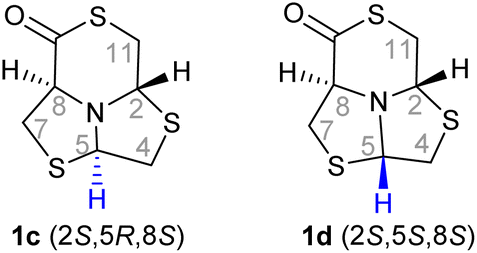
To confirm this assignment, the NOESY correlation between H2, H5 and H8 protons for each compound was studied. The assignment of the structures of 1a and 1b were confirmed based on the crystal structure and were used to set up the conditions for the NOESY experiment, Fig. S14–S16.†
The NOESY-NMR experiment of the non-assigned compound revealed the existence of a NOE interaction between H5 and H8 protons. No interactions were observed between H5–H2 and H2–H8, (Fig. S1c†). Considering all these results collectively, it becomes apparent that the data aligns more closely with (2S,5R,8S) as the absolute configuration of the third diastereomer. Overall considering the X-ray diffraction techniques, DFT theoretical calculations and NOESY-NMR, it was possible to assign the absolute configuration of the three δ-thiolactones obtained from L-2: (2R,5S,8S)-1a, (2R,5R,8S)-1c and (2S,5R,8S)-1b.
Optimization of the preparation of δ-thiolactones
After establishing the configuration of the thiolactones, we proceeded to optimize their preparation. To identify the best conditions for the synthesis of δ-thiolactone 1, we conducted an initial analytical-scale screening based on recommendations from a previous report on thioester preparation.28 For the intramolecular cyclization, we tested five coupling agents with their respective additives: two second-generation carbodiimides, DIC and the water-soluble EDCI, as alternatives to DCC, which forms a hard-to-remove by-product precipitate; CDI, useful for ester and thioester formation; HATU, a more reactive iminium salt than carbodiimides;29 and COMU, a third-generation derivative with enhanced solubility and safety.30 We chose diethyl carbonate (DEC) and acetonitrile (ACN) for their sustainability and performance, using dichloromethane (DCM) as a reference solvent due to its effectiveness with the selected coupling agents.31 Out of the 15 tested conditions, the results indicated that the highest yields were achieved using combinations of EDCI/4-DMAP and COMU/DIPEA, as shown in ESI Table S1.† The best results were obtained using DCM as solvent and EDCI (80%) or COMU (40%) as coupling reagents.Based on these results the reaction was scaled up using DCM as solvent and EDCI or COMU as coupling reagents. The additions of base or not were also explored. The results were compared with the previously obtained using HATU and DCC, as shown in Table 2, entries 1–7.
|
|||||
|---|---|---|---|---|---|
| Thiolactone preparation (TP) method | [SM]: (mM) | Isomer, yieldb (%) | |||
| 2S,5S,8S 1a | 2R,5R,8S 1b | 2S,5R,8S 1c | |||
| a Methods conditions, A: DCC, HOBt, AcOEt, 24 h; B: HATU, DIPEA, 4-DMAP, DCM, 24 h; C: EDCI, HOBt, DCM, 24 h; D: EDCI, DIPEA, DCM, 24 h; E: COMU, DIPEA, DCM, 24 h; F: EDCI, 4-DMAP, DCM, 24 h.b Isolated yields. | |||||
| 1 | TP-A: DCC, HOBT | [2]: 0.20 | 1a (38) | 1b (2) | 1c (5) |
| 2 | TP-B: HATU, DIPEA | [2]: 0.08 | — | — | 1c (14) |
| 3 | TP-C: EDCI, HOBT | [2]: 0.14 | 1a (40) | 1b (8) | 1c (10) |
| 4 | TP-D: EDCI, DIPEA | [2]: 0.14 | — | — | 1c (32) |
| 5 | TP-E: COMU, DIPEA | [2]: 0.20 | — | — | 1c (37) |
| 6 | TP-F: EDCI, 4-DMAP | [2]: 0.28 | — | — | 1c (38) |
| 7 | TP-F | [2]: 0.20 | — | — | 1c (32) |
| 8 | TP-E: COMU, DIPEA | [2]: 0.20 | — | 1b (8) | 1c (30) |
| 9 | TP-B | [3]: 0.08 | 4a (22) | 4c (2) | — |
| 10 | TP-D | [3]: 0.14 | 4a (75) | — | — |

Interestingly, different distribution of diastereomers 1 were observed depending on the conditions employed for its preparation. When a base like DIPEA or 4-DMAP was used as additive 1c was the main isolated product, with yields from 14 to 38% (Table 2, entries 2, 4–8). In contrast, when HOBt was used as carbodiimide additive,29 the diastereomer 1a was isolated as the main isomer, together with minor proportions of 1b and 1c (Table 2, entries 1 and 3). All together those results underscore the crucial role of the basic medium in the isomerization process of thiolactones 1.
The intramolecular cyclization was also studied for analogous bisthiazolidine 3, derived from penicillamine,21 giving place to the δ-thiolactones 4. Using HATU and DIPEA compound 4a (22%) was obtained as the main product together with a minor proportion of compound 4c (2%) (Table 2 entry 9). The stereochemistry assignment was done based on 2D NOESY NMR, Fig. S17 and S18.† When using EDCI and DIPEA the only product was the kinetic product 4a, in good yield (75%, Table 2, entry 10). The different behavior observed for the thiolactone bearing a gem-dimethyl group at C7, could be due to a high isomerization barrier between 4a and 4c, resulting in a difference in energetic profile of the possible isomers.
Isomerization studies of δ-thiolactones 1
Bisthiazolidines bear two fused thiazolidine rings with acetal bonds at N–C2–S and N–C5–S. Thiazolidines are recognized for the reversible nature of their acetalic bonds, demonstrating a capacity to accommodate substituents under specific conditions and promoting the formation of the most stable product.32 Despite the potential of tricyclic δ-thiolactones 1 to generate four diastereomers (1a–d), only three isomers 1a–c were detected in 1H-NMR analysis of the crude reactions. In order to explain this results, we performed ab initio calculations for the free energy of the four possible δ-thiolactones 1a–d, Fig. 4. | ||
| Fig. 4 Hartree free energy of compounds 1a–d and their interconversion calculated using DFT calculations (B3LYP 311+G(d,p) level of theory). | ||
The highest energy enantiomer, 1a, bearing the same configuration in carbons 2, 5 and 8 as the starting material 2, is the kinetic product, the first being formed. The isomerization process towards the thermodynamically more stable product, 1c, with lower energy, may encompass the diastereomer 1b, as outlined in the proposed isomerization mechanisms (Fig. 4). Interestingly, the free energy difference (ΔE2) between 1b and 1c is −2.28 kcal mol−1, enabling the isolation of both isomers. On the other hand, the energy gap between 1d and 1c (ΔE4) is −0.64 kcal mol−1, however, despite this, the presence of 1d was not detected. It is plausible to hypothesize that the transient state between 1c and 1d is energetically unfavorable and therefore not attainable at room temperature.
The thiolactones interconversion was experimentally investigated, as outlined in Fig. 5. The kinetic thiolactone 1a could be converted to 1b (54% yield) using Lewis acid In(OTf)3 at 40 °C using microwave heating for 10 min. Alternatively, when DIPEA was used, 1a was isomerized to the thermodynamic product 1c (60% yield) by heating at 65 °C in microwave for 40 min (Fig. S19†).
 | ||
| Fig. 5 (i) In(OTf)3 (10% molar), AcOEt, MW, 40 °C, 10 min, 54%; (ii) DIPEA (1.1 eq.) AcOEt, MW, 65 °C, 35 min, 60%. | ||
We proposed a mechanism for the interconversion of 1a to 1c via thiolactone 1b, with the consecutively ring opening and closing towards the most stable compound, 1c (Fig. S20†). Anew, when a base is used, the interconversion between 1b and 1c is accomplished quickly and the last is the only product observed.
Aminolysis of δ-thiolactones
Bearing in mind the characteristic reactivity of this motif, we studied the ring opening reaction of 1c by using simple nucleophiles like primary amines or amino acid esters to get amide analogs of bisthiazolidine 2 bearing a different stereochemistry from the previously prepared analogs.33First, the ring opening was studied using benzyl amines in presence of DABCO as a base and AgOTf to prevent thiol dimerization, as described recently for the ring opening of γ-thiolactones.34 Compound 1c was opened smoothly with p-F-Bn-NH2 or p-OMe-Bn-NH2 but the isolated products 6a and 6b (Fig. 6), obtained in 52% and 45% respectively, were the result of isomerization in C2 and C5 and homodimer formation. Based on these results, to prevent thiol dimerization we incorporate n-propanethiol in large excess to act as a scavenger.35 For p-OMe-benzyl and n-octyl amines, the obtained product resulted in a disulfide formation (–S–S–) between propanethiol and the mercapthomethyl-bisthiazolidine, compounds 6c–d (47% and 35% yields respectively). Otherwise, when p-Cl-benzylamine was used the free thiol was obtained as the main product 5a in 56% yield. We also focus on the use of aminoacidic derivatives like L-H2N-Gly-OEt, L-H2N-Phe-OMe and L-NH2-Thr-OMe as nucleophiles to open the δ-thiolactone 1c. The free thiol products 5b (35%), 5c (56%) and 5d (8%) were obtained in moderate to poor yields. Due to the intense odor of propanethiol, we explored its substitution by β-mercaptoethanol (βME), a less volatile thiol. The ring opening of 1c using p-OMe-benzylamine and this scavenger, gave the heterodisulfide of βME, compound 6e (52% yield). These results show neither βME nor propanethiol prevent thiol oxidation. The disulfide formation seems to be influenced in greater extent by the used amine. Additionally, when dithiothreitol, a potent thiol scavenger broadly used in biology,36 was used with p-OMe-Bn-NH2 and L-NH2-Thr-OMe as nucleophiles, surprisingly 1c undergo ring opening and free thiol products were obtained. The 1H-NMR analysis of the reaction crude showed a varied mixture of diastereomers (Fig. S64–S66†). In case of L-NH2-Thr-OMe three diastereomers were identified and product maintaining the configuration of the starting material 5d was the minor product, Fig. 6 and S66.†
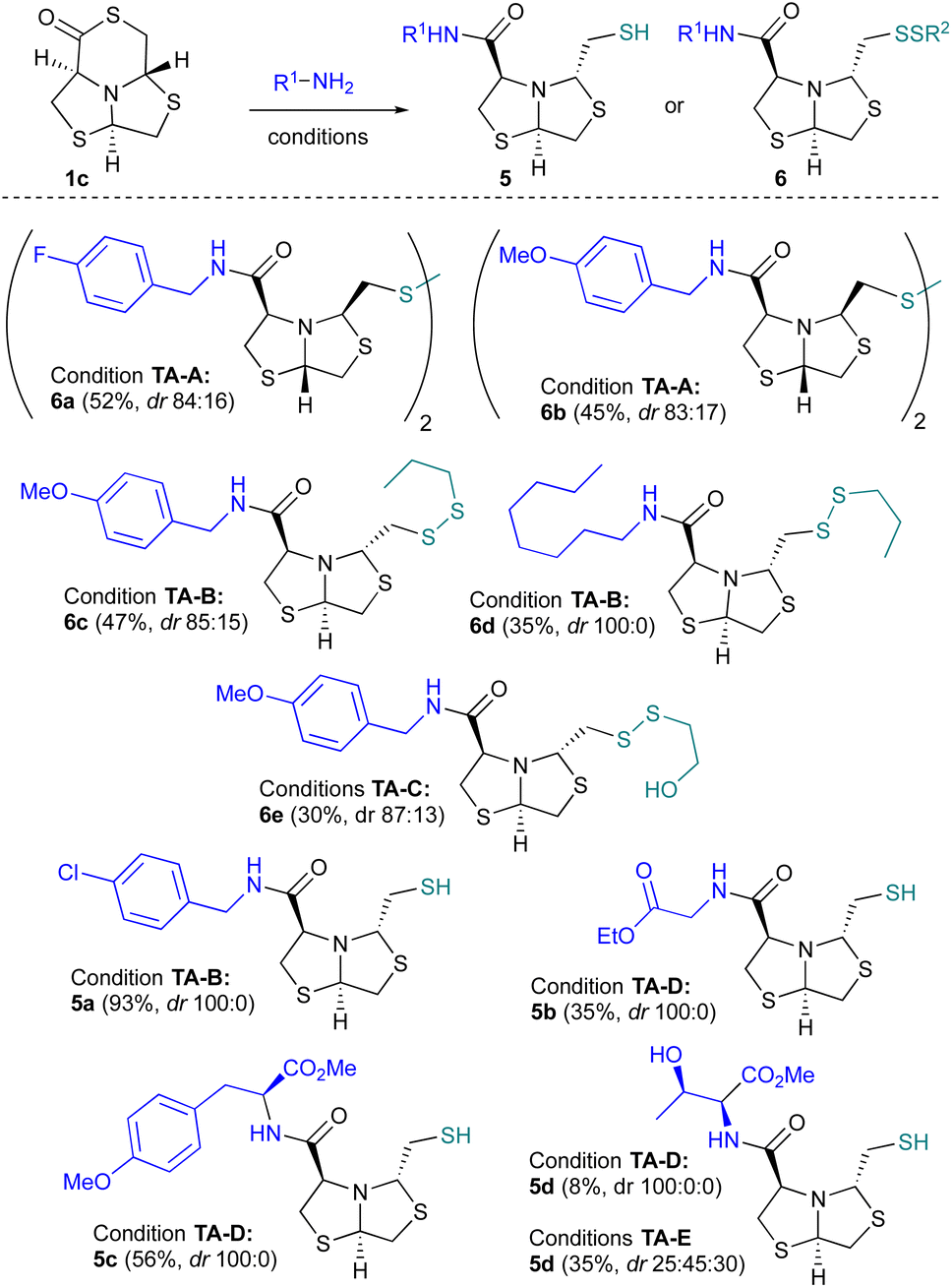 | ||
| Fig. 6 Thiolactone amine ring opening conditions: (TA-A) R1NH2 (2eq.), MeCN AgOTf (1 eq.) DABCO (1.2 eq.); (TA-B) R1NH2 (1.1 eq.), THF, n-PropSH (30 eq.); (TA-C) R1NH2 (1.2 eq.), MeCN, β-ME (2 eq.); (TA-D) R1NH2 (1.2 eq.), 4-DMAP (5%) n-PropSH (30 eq.) MeCN; (TA-E) R1NH2 (1.2 eq.), DTT (2 eq.), MeCN. | ||
In most of the cases, under the assayed conditions, the main product retain the stereochemistry of the starting material. However, for compound 5b in CDCl3 we observed the interconversion towards the most stable compound (2R,5R,8S)-5b, as previously reported for similar systems,37 achieving a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio after 24 h.
:1 ratio after 24 h.
In summary the opening of δ-thiolactone 1c was successfully achieved using eight amines under different conditions. When a base, AgOTf and benzylic amines were employed, the primary product was the dimeric amides 6a and 6b. When the base was omitted and an excess of thiol scavenger was used, the predominant isolated products were 6c–e, resulting from the ring opening by the amine and subsequent disulfide formation with the scavenger. The isolation of the free thiol was observed in products 5a–d, suggesting a possible influence of the substituent on the reduction potential of the formed products.
When δ-thiolactone 4a was assayed for ring opening using conditions TA-B: p-Cl-benzylamine as nucleophile the disulfide 8a was obtained in 34% yield, see Fig. 7. When glycine ethyl ester was used as nucleophile conditions TA-D, no opening product was observed, and the starting material was recovered, see Fig. 7 conditions TA-D.
 | ||
| Fig. 7 Thiolactone amine ring opening conditions: (TA-B) R1NH2: p-Cl-Bn-NH2 (1.1 eq.), THF, n-PropSH (30 eq.), 8a (34%); (TA-D) R1NH2: L-H2N-Gly-OEt (1.2 eq.), 4-DMAP (5%) n-PropSH (30 eq.) MeCN, no reaction. | ||
Thiolysis of δ-thiolactones
Thiol-thioester exchange is a well know reaction used in the preparation of dynamic covalent libraries.38 We further explore the ring opening of 1c and 4a using β-mercaptoethanol in large excess (35 equivalents) and Gly (1.2 equivalents) across different solvent mixtures. Under these conditions, it was observed the ring opening by the thiol of the β-mercaptoethanol, instead of the amino acid, and products 9a–b were obtained in good yields. These results can be explained by the fact that amino acids are in a zwitterionic form and therefore are weaker nucleophiles than the previously used amines, Fig. 8.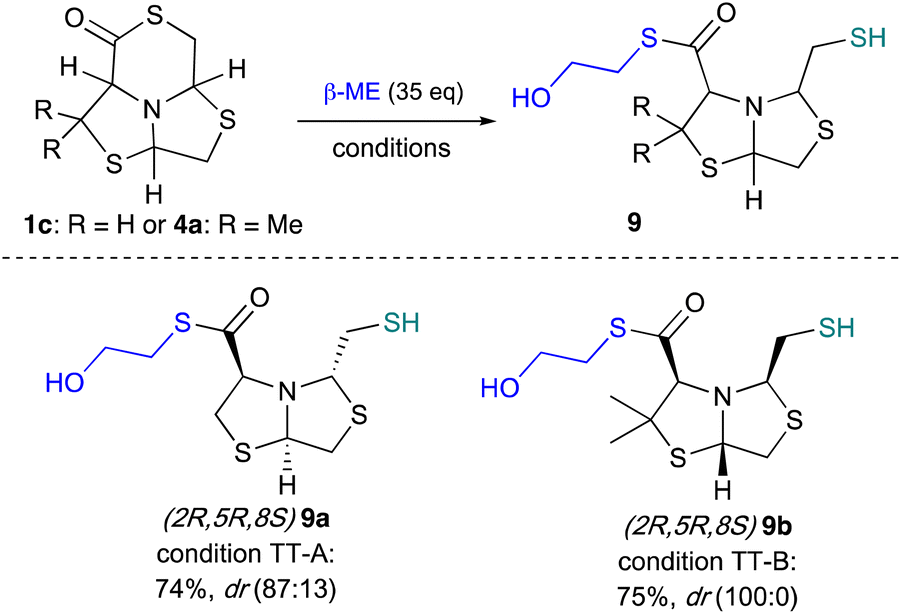 | ||
| Fig. 8 Thiolysis of δ-thiolactones 1c and 4a. Condition TT-A: Gly (1.2 eq.), MeCN:DMF (3:1), gave: 9a 74%, dr (87:13); condition TT-B: Gly (1.2 eq.), H2O:AcOEt (1:1), gave: 9b 75%, dr (100:0). | ||
Products obtained by the ring opening reaction are prime examples of another widely exploited chemoselective reaction, namely the S-to-N acyl transfer reaction,39 which forms the basis of the native chemical ligation methodology, which revolutionized peptide and protein synthesis.
Multicomponent reaction of δ-thiolactones
Taking advantage of the high reactivity of the thiol, we explored the potential of three component reaction using δ-thiolactone 1c, amines as nucleophiles and N-methylmaleimide as a Michael acceptor,40 Fig. 9. | ||
| Fig. 9 Three component reaction of δ-thiolactones 1c, benzylamines and, methylmaleimide. | ||
The addition product was obtained in both cases in moderate yields 10a (50%, dr 55:45) or 10b (48%, dr 59:41) as diastereomeric mixtures. These results evidence the versatility of the δ-thiolactone 1c towards amine ring opening and thiol–ene conjugation.
Conclusions
We have described the preparation of five new δ-thiolactones. Its structural elucidation was achieved using a combination of X-ray crystallography, NMR spectroscopy and ab initio calculations.The ring opening of the new tricyclic δ-thiolactones was investigated, using amines and thiols as nucleophiles. Compound 1c exhibited a high reactivity against both types of nucleophiles, and the ring opening products strongly depended on the presence or not of a thiol scavenger.
The δ-thiolactones are useful scaffolds for the preparation of new structurally diverse bisthiazolidines bearing different substituents and stereochemistry in the bicycle core from previously reported ones. Together, these results demonstrate the versatility of thiolactone's chemistry and its usefulness in addressing synthetic challenges.
Experimental
Synthesis
Unless otherwise stated, all reagents and solvents were purchased at the highest quality from commercial suppliers and used as received without further purification. Experiments involving moisture and/or air-sensitive compounds were performed in oven or flame dried glassware with rubber septa under a positive pressure of nitrogen. Nonaqueous reagents were transferred by hypodermic syringe. Heating was accomplished by silicon oil bath using a temperature controller. Organic solutions were concentrated under reduced pressure at 30 °C (water bath temperature) using a Büchi rotary evaporator, unless otherwise noted. Reactions were magnetically stirred and monitored by thin-layer chromatography (TLC). Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous (>95%) materials, unless otherwise stated. TLC was performed on E. Merck 60-F254 precoated plates (0.25 mm), and spots were visualized by UV fluorescence (254 nm) quenching, iodine vapor, ninhydrin. Flash chromatography was carried out with Merck silica gel 60 (200–400 mesh) according to the procedure of Still et al.41Analytical data of compounds
Melting points were measured using a Fisher-Johns apparatus and are uncorrected. Optical rotation was measured using a Jasco p-2000 polarimeter with a 2.0 mL cell (3.5 × 100 mm), optical path length of 100 mm, and sodium lamp (λ = 589 nm) at 20 °C. The concentration c is given as g/100 mL, αD values are given in 10−1 deg cm2 g−1. 1H and 13C NMR spectra were recorded on a Bruker Avance Neo 400 (400 MHz and 101 MHz, respectively) spectrometer. Chemical shifts of the 1H NMR (CDCl3: 7.26 ppm, MeOD: 4.78 ppm, DMSO-d6: 2.50 ppm) and 13C NMR (CDCl3: 77.00 ppm, MeOD: 49.15 ppm, DMSO-d6: 39.50 ppm) spectra were referenced to residual solvent peaks or Me4Si (0.00 ppm) as an internal standard. 1H NMR spectra are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quarter, quin = quintet, sext = sextet, sept = septet, m = multiplet, br = broad), coupling constant (J) in Hz and integration. High-resolution mass spectra were acquired at Institut Pasteur Montevideo, in a Q Exactive Plus mass spectrometer (Thermo Scientific, USA) by direct injection (10 μL min−1) using 100% methanol HPLC grade as solvent and an Ion Max API source with a HESI-II probe. The mass spectrometer was operated in a positive mode, ion spray voltage was set at 3.5 kV and capillary temperature at 250 °C.Synthetic procedures
Compounds L-2 and L-3 were prepared as previously reported.21Conditions for δ-thiolactone preparation (TP)
:AcOEt 90:10), to give δ-thiolactones 1a (92 mg, 0.42 mmol, 20% yield) and 1c (82 mg, 0.38 mmol, 18%).:AcOEt 90:10) to give 1c (12 mg, 0.056 mmol, 14% yield).Using condition TP-B starting from L-3 (105 mg, 0.4 mmol), and purified by column chromatography (nHex:EtOAc 80:20) the thiolactones 4a (22 mg, 0.08 mmol, 22% yield) and 4c (2 mg, 0.008 mmol, 2% yield) were obtained.
:AcOEt 90:10 to 70:30) to give 1a (184 mg, 0.84 mmol, 40% yield), 1b (37 mg, 0.17 mmol, 8% yield) and 1c (46 mg, 0.21 mmol, 10% yield).Using condition TP-C starting from L-3 (105 mg, 0.4 mmol) and purified by column chromatography (nHex:EtOAc 80:20) 4a (148 mg, 0.60 mmol, 75% yield) was obtained.
:AcOEt 90:10) to give 1c (147 mg, 0.67 mmol, 32%).:AcOEt 90:10) to give 1c (89 mg, 0.41 mmol, 37% yield).:AcOEt 90:10) to give 1c (67 mg, 0.30 mmol, 38% yield).Thiolactone aminolysis (TA)
:AcOEt 80:20–30:70) to give 6a (64 mg, 0.09 mmol, 52%, dr: 84:16) as a yellow to brown solid; mp: 168–170 °C; 1H-NMR (CDCl3) δ: 7.26–7.18 (m, 2H), 7.08–6.90 (m, 2H), 4.87 (dt, J = 6.0, 3.8 Hz, 1H), 4.62–4.44 (m, 2H), 4.27 (dd, J = 14.9, 5.3 Hz, 1H), 4.07 (dd, J = 7.2, 3.1 Hz, 1H), 3.50 (ddd, J = 12.0, 7.4, 4.4 Hz, 2H), 3.34–3.26 (m, 1H), 3.10 (dd, J = 11.8, 4.4 Hz, 1H), 2.96–2.85 (m, 2H); 13C NMR (CDCl3) δ 170.17, 160.97, 129.94, 129.35, 115.73, 71.42, 71.23, 70.93, 46.97, 43.42, 39.76, 33.61; HRMS calculated for C28H32F2N4NaO2S6+ [M + Na]+: 709.0710, found 709.0704.:EtOAc 80:20) to give dimer 6b (80 mg, 0.11 mmol, 45%, dr (83:17)) as a brown oil: 1H-NMR (CDCl3) δ: 7.19 (t, J = 8.6 Hz, 2H), 6.89–6.74 (m, 2H), 4.91–4.79 (m, 1H), 4.58–4.36 (m, 2H), 4.25 (dd, J = 14.6, 5.1 Hz, 1H), 4.06 (dd, J = 7.1, 3.3 Hz, 1H), 3.79 (s, 3H), 3.57–3.39 (m, 2H), 3.29 (dd, J = 11.1, 7.3 Hz, 1H), 3.09 (dd, J = 11.9, 4.2 Hz, 1H), 2.96–2.77 (m, 1H); 13C-NMR (CDCl3) δ 169.93, 159.07, 130.05, 129.04, 114.15, 73.06, 72.50, 70.94, 55.35, 47.10, 42.99, 39.73, 33.63; HRMS calculated for C30H38N4NaO4S6+ [M + Na]+: 733.1110, found: 733.1089.:AcOEt 80:20) to give 5a (60 mg, 0.17 mmol, 93%, dr (100:0)) as a white solid: mp: 148–150 °C; [α]20D = −46 (c 0.59 CHCl3); 1H-NMR (CDCl3) δ: 7.33 (d, J = 8.4 Hz, 1H), 7.27–7.22 (m, 4H), 6.41 (s, 1H), 5.19 (dd, J = 4.4, 1.8 Hz, 1H), 4.50 (dd, J = 6.7, 4.8 Hz, 1H), 4.44 (t, J = 5.7 Hz, 1H), 3.93 (dd, J = 9.5, 5.5 Hz, 1H), 3.57 (ddd, J = 11.8, 4.4, 1.1 Hz, 1H), 3.37 (dd, J = 10.9, 9.5 Hz, 1H), 3.20–3.10 (m, 1H), 3.01 (dd, J = 11.9, 1.9 Hz, 1H), 2.54 (ddd, J = 8.4, 5.8, 3.3 Hz, 1H), 1.62 (dd, J = 8.8, 8.0 Hz, 1H); 13C-NMR (CDCl3) δ: 165.50, 133.35, 131.44, 127.10, 126.69, 74.11, 68.65, 66.05, 40.84, 34.95, 32.17, 31.41. HRMS calculated for C14H17ClN2NaOS3+ [M + Na]+: 383.0084, found: 383.0078.:EtOAc 80:20) to give 6c (34 mg, 0.08 mmol, 47%, dr (85:15)) as a yellow solid: mp 120–122 °C; 1H-NMR (CDCl3) δ: 7.25 (d, J = 8.8 Hz, 2H), 6.88 (d, J = 8.7 Hz, 2H), 6.36 (s, 1H), 5.17 (dd, J = 4.3, 2.1 Hz, 1H), 4.64 (dd, J = 6.7, 4.8 Hz, 1H), 4.51 (dd, J = 14.3, 6.0 Hz, 1H), 4.35 (dd, J = 14.3, 5.0 Hz, 1H), 3.93 (dd, J = 9.4, 5.6 Hz, 1H), 3.81 (s, 3H), 3.57 (ddd, J = 11.9, 4.3, 1.0 Hz, 1H), 3.39 (dd, J = 10.9, 9.4 Hz, 1H), 3.16 (dd, J = 10.8, 5.6 Hz, 1H), 3.04 (dd, J = 11.8, 2.1 Hz, 1H), 2.92 (dd, J = 11.3, 5.8 Hz, 2H), 2.67 (t, J = 7.2 Hz, 2H), 1.77–1.63 (m, 2H), 0.99 (t, J = 7.3 Hz, 3H); 13C-NMR (CDCl3) δ: 166.79, 158.46, 128.71, 113.43, 75.55, 70.28, 64.74, 54.50, 49.16, 42.69, 40.45, 37.11, 33.76, 21.59, 12.30. HRMS calculated for C18H27N2O2S4+ [M + H]+: 431.0950, found: 431.0942.:EtOAc 80:20) to give 6d (23 mg, 0.05 mmol, 30%, dr (100:0)) as a brown solid: mp 120–122 °C; [α]20D = −7 (c 0.29 CHCl3); 1H-NMR (CDCl3) δ: 6.12 (s, 1H), 5.19 (dd, J = 4.3, 2.1 Hz, 1H), 4.63 (dd, J = 6.7, 4.9 Hz, 1H), 3.91 (dd, J = 9.4, 5.6 Hz, 1H), 3.65–3.56 (m, 1H), 3.42–3.33 (m, 2H), 3.26 (dt, J = 13.3, 6.6 Hz, 1H), 3.15 (dd, J = 10.9, 5.6 Hz, 1H), 3.06 (dd, J = 11.8, 2.1 Hz, 1H), 2.99–2.93 (m, 2H), 2.69 (t, J = 7.2 Hz, 2H), 1.71 (q, J = 7.3 Hz, 2H), 1.39–1.17 (m, 14H), 0.99 (t, J = 7.3 Hz, 3H), 0.93–0.85 (m, 3H); 13C-NMR (CDCl3) δ: 167.81, 76.42, 71.14, 65.55, 50.04, 41.29, 39.92, 37.83, 34.66, 31.79, 29.36, 29.20, 27.05, 22.65, 22.44, 14.10, 13.13. HRMS calculated for C18H34N2NaO2S4+ [M + Na]+: 445.1446, found: 445.1427.:AcOEt 60:40–30:70) to give 6e (23 mg, 0.05 mmol, 30%, dr (87:13)) as a brown solid: mp 100–103 °C; 1H-NMR (CDCl3) δ: 7.25 (d, J = 8.7 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 6.48 (s, 1H), 5.19 (dd, J = 4.2, 1.7 Hz, 1H), 4.70 (dd, J = 7.7, 4.1 Hz, 1H), 4.48 (dd, J = 14.3, 5.8 Hz, 1H), 4.39 (dd, J = 14.3, 5.4 Hz, 1H), 3.97–3.89 (m, 1H), 3.85 (dt, J = 8.1, 5.7 Hz, 2H), 3.81 (s, 3H), 3.57 (ddd, J = 12.0, 4.3, 1.1 Hz, 1H), 3.40 (t, J = 10.3 Hz, 1H), 3.13 (dd, J = 10.8, 5.5 Hz, 1H), 3.07–2.98 (m, 2H), 2.90–2.85 (m, 2H), 2.81 (dd, J = 13.6, 7.6 Hz, 1H); 13C-NMR (CDCl3) δ: 165.37, 157.01, 127.25, 126.76, 111.96, 74.50, 68.66, 63.35, 57.70, 53.02, 52.98, 47.30, 41.30, 40.26, 34.84, 32.06. HRMS calculated for C17H24N2NaO2S4+ [M + Na]+: 455.0562, found: 455.0553.:AcOEt 60:40) to give 5b (20 mg, 0.06 mmol, 35%, dr (100:0)) as an oil: 1H-NMR (CDCl3) δ: 6.59 (s, 1H), 5.22 (dd, J = 4.3, 1.9 Hz, 1H), 4.52 (dd, J = 7.3, 4.4 Hz, 1H), 4.25 (q, J = 7.1 Hz, 2H), 4.08 (dd, J = 11.9, 5.3 Hz, 2H), 4.01–3.96 (m, 1H), 3.34 (dd, J = 10.8, 9.5 Hz, 1H), 3.17 (dd, J = 10.9, 5.6 Hz, 1H), 3.06 (dd, J = 11.9, 2.0 Hz, 1H), 2.77 (td, J = 9.0, 4.5 Hz, 1H), 2.68–2.61 (m, 1H), 1.71 (dd, J = 9.1, 8.0 Hz, 1H), 1.30 (t, J = 7.2 Hz, 3H); 13C-NMR (CDCl3) δ: 172.04, 169.68, 75.36, 74.25, 62.54, 61.89, 61.65, 41.14, 39.44, 35.29, 26.29, 14.17. HRMS calculated for C11H18N2NaO3S3+ [M + Na]+: 345.0372, found: 345.0363. After 12 h in CDCl3 the diasteromeric ratio changed to 50:50.:EtOAc 70:30) to give 5c (40 mg, 0.10 mmol), 56%, dr (100:0) as a white solid: mp 150–153 °C; [α]20D = −10 (c 0.51 CHCl3); 1H-NMR (CDCl3) δ: 7.30 (ddd, J = 11.6, 7.7, 5.9 Hz, 2H), 7.13–7.08 (m, 2H), 6.47–6.40 (m, 1H), 5.16 (dd, J = 4.2, 1.8 Hz, 1H), 4.89 (dt, J = 7.6, 6.0 Hz, 1H), 4.36 (dd, J = 7.6, 4.2 Hz, 1H), 3.85 (dd, J = 9.7, 5.4 Hz, 1H), 3.78 (s, 3H), 3.55 (ddd, J = 12.0, 4.2, 1.1 Hz, 1H), 3.29–3.19 (m, 2H), 3.16–3.06 (m, 2H), 2.99 (dd, J = 11.9, 1.9 Hz, 1H), 2.66 (ddd, J = 13.5, 9.2, 4.2 Hz, 1H), 2.55 (dt, J = 13.7, 7.8 Hz, 1H), 1.65 (dd, J = 9.2, 8.0 Hz, 1H); 13C-NMR (CDCl3) δ: 170.22, 166.41, 134.15, 127.90, 127.76, 127.59, 126.20, 75.18, 69.91, 67.79, 51.97, 51.42, 36.42, 36.30, 33.36, 32.64. HRMS calculated for C17H21N2NaO3S3+ [M + Na]+: 421.0685, found: 421.0671.:EtOAc 40:60) to give 5d (5 mg, 0.014 mmol), 8%, dr (100:0) as a brown oil: 1H-NMR (CDCl3) δ: 6.79 (d, J = 8.4 Hz, 1H), 5.24 (d, J = 4.1 Hz, 1H), 4.59 (dd, J = 8.6, 2.3 Hz, 1H), 4.46 (dd, J = 8.0, 4.0 Hz, 2H), 4.04 (dd, J = 9.7, 5.3 Hz, 1H), 3.80 (s, 3H), 3.62 (dd, J = 12.1, 4.2 Hz, 1H), 3.34 (t, J = 10.3 Hz, 1H), 3.16 (dd, J = 11.0, 5.4 Hz, 1H), 2.85 (ddd, J = 13.3, 9.3, 4.1 Hz, 1H), 2.67 (dd, J = 13.8, 7.8 Hz, 1H), 1.73 (dd, J = 9.2, 8.0 Hz, 1H), 1.31–1.23 (m, 3H); 13C-NMR (CDCl3) δ: 170.92, 168.55, 77.23, 71.51, 69.48, 67.67, 57.35, 52.92, 37.78, 34.54, 34.03, 20.23. HRMS calculated for C12H20N2NaO4S3+ [M + Na]+: 375.0477, found: 375.0470.:EtOAc 80:20) to give 8a (26 mg, 0.068 mmol, 34%, dr (100:0)) as a yellow oil; [α]20D = −140 (c 0.32 CHCl3); 1H-NMR (CDCl3) δ: 7.28 (t, J = 2.6 Hz, 4H), 4.86 (dd, J = 6.8, 5.6 Hz, 1H), 4.71 (dd, J = 14.5, 7.7 Hz, 1H), 4.57 (dd, J = 10.0, 4.5 Hz, 1H), 4.19 (dd, J = 14.4, 4.5 Hz, 1H), 3.64 (s, 1H), 3.41 (dd, J = 11.5, 6.9 Hz, 1H), 3.06 (dd, J = 11.6, 5.6 Hz, 1H), 2.99–2.84 (m, 2H), 2.67 (td, J = 6.9, 3.2 Hz, 2H), 1.68 (qd, J = 7.1, 2.1 Hz, 2H), 1.31 (s, 3H), 1.26 (s, 3H), 0.99 (t, J = 7.4 Hz, 3H); 13C-NMR (CDCl3) δ: 168.43, 136.71, 133.30, 129.82, 128.82, 128.77, 78.48, 71.91, 68.50, 55.30, 46.57, 42.63, 41.23, 40.71, 29.71, 29.67, 28.32, 22.43, 13.07. HRMS calculated for C19H28ClN2OS4+ [M + H]+: 463.0768 found: 436.0751.Thiolactone thiolysis by β-mercaptoethanol (TT)
:AcOEt 60:40) to give 9a (40 mg, 0.13 mmol), 74%, dr (87:13) as a pale oil; 1H-NMR (CDCl3) δ: 5.15 (dd, J = 5.2, 3.0 Hz, 1H), 4.35 (dd, J = 6.6, 2.5 Hz, 1H), 4.31 (t, J = 6.5 Hz, 1H), 3.78 (t, J = 6.1 Hz, 2H), 3.65–3.59 (m, 1H), 3.42 (dd, J = 11.0, 2.6 Hz, 1H), 3.21 (dd, J = 11.0, 6.6 Hz, 1H), 3.14–3.05 (m, 3H), 2.98 (ddd, J = 13.8, 8.3, 6.8 Hz, 1H), 2.79–2.71 (m, 1H), 2.06 (dd, J = 9.1, 8.3 Hz, 1H); 13C-NMR (CDCl3) δ: 200.75, 78.57, 76.77, 76.46, 76.14, 75.50, 73.73, 60.95, 38.73, 33.38, 33.23, 31.50. HRMS calculated for C9H15NNaO2S4+ [M + Na]+: 319.9878, found: 319.9876.:AcOEt 90:10–70:30) to give 9b (50 mg, 0.15 mmol, 75%) as a pale oil; 1H-NMR (CDCl3) δ: 5.14 (dd, J = 6.4, 3.5 Hz, 1H), 4.30 (dd, J = 9.5, 4.4 Hz, 1H), 3.81–3.76 (m, 3H), 3.44 (dd, J = 12.1, 6.4 Hz, 1H), 3.16 (dt, J = 13.9, 6.1 Hz, 1H), 3.10–3.02 (m, 2H), 3.02–2.98 (m, 1H), 2.70 (ddd, J = 13.6, 9.5, 6.8 Hz, 1H), 1.86 (dd, J = 10.2, 6.8 Hz, 1H), 1.57 (s, 3H), 1.46 (s, 3H); 13C-NMR (CDCl3) δ: 199.37, 84.80, 78.43, 69.86, 61.04, 54.61, 38.42, 31.05, 30.95, 30.61, 27.40, 26.67; HRMS calculated for C11H20N2O2S4+ [M + H]+: 326,0371, found: 326.0361.Thiolactone multicomponent reaction (TM)
:AcOEt 70:30–30:70) to give 10a as a mayor syn diastereomer (43 mg, 0.09 mmol, 50%, dr: 55:45) as a white solid: mp = 149–152 °C; 1H-NMR (CDCl3) δ: 7.32 (d, J = 8.57 Hz, 2H), 7.28 (d, J = 8.66 Hz, 2H), 6.61 (t, J = 5.75 Hz, 1H), 5.18 (dd, J = 4.35 Hz, 1.71 Hz, 1H), 4.68 (dd, J = 7.46 Hz, 4.15 Hz, 1H), 4.52 (dd, J = 14.62 Hz, 6.16 Hz, 1H), 4.41 (dd, J = 14.61 Hz, 5.26 Hz, 1H), 3.90 (dd, J = 9.61 Hz, 5.55 Hz, 1H), 3.68 (dd, J = 9.17 Hz, 3.86 Hz, 1H), 3.60 (dd, J = 11.91 Hz, 4.30 Hz, 1H), 3.40 (dd, J = 10.83 Hz, 9.65 Hz, 1H), 3.22 (dd, J = 13.89 Hz, 4.15 Hz, 1H), 3.03 (dd, J = 11.88 Hz, 1.77 Hz, 1H), 2.99 (s, 3H), 2.85 (dd, J = 13.88 Hz, 7.48 Hz, 1H), 2.45 (dd, J = 18.76 Hz, 3.83 Hz, 1H); 13C-NMR (CDCl3) δ: 177.41, 174.58, 167.86, 136.06, 133.73, 129.71, 128.99, 76.91, 70.85, 65.45, 43.34, 41.91, 38.76, 37.33, 35.92, 34.43, 25.19. HRMS calculated for C19H23ClN3O3S3+ [M + H]+: 472.0405, found: 472.0585.:EtOAc 60:40–40:60) to give 10b as major syn diastereomer (40 mg, 0.09 mmol, 48%, dr: 59:41) as a white solid; mp = 170–172 °C; 1H-NMR (CDCl3) δ: 7.26 (d, J = 8.6 Hz, 2H), 6.87 (d, J = 8.6 Hz, 2H), 6.43 (s, 1H), 5.19 (dd, J = 4.2, 1.7 Hz, 1H), 4.69 (dd, J = 7.7, 4.0 Hz, 1H), 4.43 (t, J = 5.8 Hz, 2H), 3.87 (dd, J = 9.9, 5.5 Hz, 1H), 3.80 (s, 3H), 3.66 (dd, J = 9.2, 3.9 Hz, 1H), 3.60 (dd, J = 11.8, 4.3 Hz, 1H), 3.41 (t, J = 10.2 Hz, 1H), 3.23 (dd, J = 13.9, 4.0 Hz, 1H), 3.15–3.06 (m, 2H), 3.04 (dd, J = 11.8, 1.7 Hz, 1H), 3.00 (s, 3H), 2.84 (dd, J = 14.0, 7.6 Hz, 1H), 2.45 (dd, J = 18.7, 3.9 Hz, 1H); 13C-NMR (CDCl3) δ: 175.11, 172.41, 165.25, 157.09, 127.53, 127.31, 127.01, 111.97, 75.00, 68.76, 63.23, 53.12, 41.33, 39.76, 36.60, 35.00, 33.82, 32.11, 22.95. HRMS calculated for C20H26N3O4S3+ [M + H]+: 468.1080, found: 468.1585.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data was deposited in Cambridge Crystallographic Data Centre. CCDC 2387412–2387413.Author contributions
MR and VM contributed to experiments and product characterization. MM, FV and DD contributed to product characterization. GM conceived the project. FV run the DFT calculations. GH run the NOESY experiments and analysis. MR, FV, GH and GM, prepared the manuscript, and contributed to discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIH) to G. M. under award number R01AI100560 and Comisión Sectorial de Investigación Científica Uruguay Iniciación (CSIC I+D No. 529-2019) to VM and Programa de Desarrollo de las Ciencias Básicas AI 29242338. M. R. and F. V. are recipients of a fellowship from Comisión Académica de Posgrado, Udelar. The authors would like to thank Rosario Durán and Alejandro Leyva for kindly providing access to the HRMS equipment at UBYPA, Institute Pasteur Montevideo.Notes and references
- V. Hirschbeck, P. H. Gehrtz and I. Fleischer, Regioselective Thiocarbonylation of Vinyl Arenes, J. Am. Chem. Soc., 2016, 138, 16794–16799 CrossRef CAS PubMed.
- M. Frenkel-Pinter, M. Bouza, F. M. Fernández, L. J. Leman, L. D. Williams and N. V. Hud, et al., Thioesters provide a plausible prebiotic path to proto-peptides, Nat. Commun., 2022, 13, 2569 CrossRef CAS.
- F. Pietrocola, L. Galluzzi, J. M. Bravo-San Pedro, F. Madeo and G. Kroemer, Acetyl Coenzyme A: A Central Metabolite and Second Messenger, Cell Metab., 2015, 21, 805–821 CrossRef CAS.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J. C. M. Monbaliu and O. Melnyk, Native Chemical Ligation and Extended Methods: Mechanisms, Catalysis, Scope, and Limitations, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS.
- Y. Vallee, I. Shalayel, K. D. Ly, K. V. R. Rao, G. De Paëpe and K. Märker, et al., At the very beginning of life on Earth: the thiol-rich peptide (TRP) world hypothesis, Int. J. Dev. Biol., 2017, 61, 471–478 CrossRef CAS PubMed.
- L. Kremer, J. D. Douglas, A. R. Baulard, C. Morehouse, M. R. Guy and D. Alland, et al., Thiolactomycin and Related Analogues as Novel Anti-mycobacterial Agents Targeting KasA and KasB Condensing Enzymes in Mycobacterium tuberculosis, J. Biol. Chem., 2000, 275, 16857–16864 CrossRef CAS PubMed.
- S. Albrecht, A. Defoin, E. Salomon, C. Tarnus, A. Wetterholm and J. Z. Haeggström, Synthesis and structure activity relationships of novel non-peptidic metallo-aminopeptidase inhibitors, Bioorg. Med. Chem., 2006, 14, 7241–7257 CrossRef CAS.
- S. Arimori and T. Nakayama, Oxadiazole Compound and Use Thereof, Sumimoto Chemical Company, Limited, Japan, WO2017110863A1, 2017 Search PubMed.
- H. Tobler, H. Szczepansky and W. Fory, Herbicidal Composition, Novartis AG, Switzerland, WO1998013361A1, 1998 Search PubMed.
- D. V. Ferraris, P. Majer, C. Ni, C. E. Slusher, R. Rais and Y. Wu, et al., δ-Thiolactones as Prodrugs of Thiol-Based Glutamate Carboxypeptidase II (GCPII) Inhibitors, J. Med. Chem., 2014, 57, 243–247 CrossRef CAS PubMed.
- J. W. Skiles, J. T. Suh, B. E. Williams, P. R. Menard, J. N. Barton and B. Loev, et al., Angiotensin-converting enzyme inhibitors: new orally active 1,4-thiazepine-2,5-diones, 1,4-thiazine-2,5-diones, and 1,4-benzothiazepine-2,5-diones possessing antihypertensive activity, J. Med. Chem., 1986, 29, 784–796 CrossRef CAS.
- P. Espeel and F. E. Du Prez, One-pot multi-step reactions based on thiolactone chemistry: a powerful synthetic tool in polymer science, Eur. Polym. J., 2015, 62, 247–272 CrossRef CAS.
- N. Illy and E. Mongkhoun, Thiolactone chemistry, a versatile platform for macromolecular engineering, Polym. Chem., 2022, 13, 4592–4614 RSC.
- T. V. Popova, O. A. Krumkacheva, A. S. Burmakova, A. S. Spitsyna, O. D. Zakharova and V. A. Lisitskiy, et al., Protein modification by thiolactone homocysteine chemistry: a multifunctionalized human serum albumin theranostic, RSC Med. Chem., 2020, 11, 1314–1325 RSC.
- A. Kemmer and T. Heinze, Dextran thioparaconate – evaluation of the multifunctional thiolactone linker for easily adaptable polysaccharide modification, Carbohydr. Polym., 2023, 315, 120946 CrossRef CAS PubMed.
- C. M. Reese, B. J. Thompson, P. K. Logan, C. M. Stafford, M. Blanton and D. L. Patton, Sequential and one-pot post-polymerization modification reactions of thiolactone-containing polymer brushes, Polym. Chem., 2019, 10, 4935–4943 RSC.
- A. Mammadova, B. Gyarmati, K. Sárdi, A. Paudics, Z. Varga and A. Szilágyi, Thiolated cationic poly(aspartamides) with side group dependent gelation properties for the delivery of anionic polyelectrolytes, J. Mater. Chem. B, 2022, 10, 5946–5957 RSC.
- H. Chen, Y. Xiao, N. Yuan, J. Weng, P. Gao and L. Breindel, et al., Coupling of sterically demanding peptides by β-thiolactone-mediated native chemical ligation, Chem. Sci., 2018, 9, 1982–1988 RSC.
- D. Van Lysebetten, S. Felissati, E. Antonatou, L. L. G. Carrette, P. Espeel and E. Focquet, et al., A Thiolactone Strategy for Straightforward Synthesis of Disulfide-Linked Side-Chain-to-Tail Cyclic Peptides Featuring an N-Terminal Modification Handle, ChemBioChem, 2018, 19, 641–646 CrossRef CAS.
- V. Villamil, C. Saiz and G. Mahler, Thioester deprotection using a biomimetic NCL approach, Front. Chem., 2022, 10, 934376 CrossRef CAS.
- P. Hinchliffe, M. M. González, M. F. Mojica, J. M. González, V. Castillo and C. Saiz, et al., Cross-class metallo-β-lactamase inhibition by bisthiazolidines reveals multiple binding modes, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E3745–E3754 CrossRef CAS.
- Q. Wang, L. Liu, J. Dong, Z. Tian and T. Chen, Metal-free thioesterification of amides generating acyl thioesters, New J. Chem., 2019, 43, 9384–9388 RSC.
- A. Dey and E. Pidcock, The relevance of chirality in space group analysis: a database study of common hydrogen-bonding motifs and their symmetry preferences, CrystEngComm, 2008, 10, 1258 RSC.
- M. Nespolo, Molecular versus structural chirality, J. Appl. Crystallogr., 2023, 56, 44–47 CrossRef CAS.
- D. Cremer and J. A. Pople, General definition of ring puckering coordinates, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- D. G. Evans and J. C. A. Boeyens, Conformational analysis of ring pucker, Acta Crystallogr., Sect. B, 1989, 45, 581–590 CrossRef.
- H. A. Fargher, T. J. Sherbow, M. M. Haley, D. W. Johnson and M. D. Pluth, C–H⋯S hydrogen bonding interactions, Chem. Soc. Rev., 2022, 51, 1454–1469 RSC.
- A. Jordan and H. F. Sneddon, Development of a solvent-reagent selection guide for the formation of thioesters, Green Chem., 2019, 21, 1900–1906 RSC.
- A. El-Faham and F. Albericio, Peptide Coupling Reagents, More than a Letter Soup, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS.
- A. El-Faham and F. Albericio, COMU: a third generation of uronium-type coupling reagents, J. Pept. Sci., 2010, 16, 6–9 CrossRef CAS.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla and L. E. Shuster, et al., Updating and further expanding GSK's solvent sustainability guide, Green Chem., 2016, 18, 3879–3890 RSC.
- C. Saiz, P. Wipf, E. Manta and G. Mahler, Reversible Thiazolidine Exchange: A New Reaction Suitable for Dynamic Combinatorial Chemistry, Org. Lett., 2009, 11, 3170–3173 CrossRef CAS.
- V. Villamil, M. A. Rossi, M. F. Mojica, P. Hinchliffe, V. Martínez and V. Castillo, et al., Rational Design of Benzobisheterocycle Metallo-β-Lactamase Inhibitors: A Tricyclic Scaffold Enhances Potency against Target Enzymes, J. Med. Chem., 2024, 67, 3795–3812 CrossRef CAS.
- S. Jiang, W. Hsieh, W. Chen, J. Liao, P. Chiang and Y. A. Lin, Synthesis of Thiol-Containing Oligopeptides via Tandem Activation of γ-Thiolactones by Silver-DABCO Pair, Asian J. Org. Chem., 2020, 9, 1638–1649 CrossRef CAS.
- P. Espeel, F. Goethals, M. M. Stamenović, L. Petton and F. E. Du Prez, Double modular modification of thiolactone-containing polymers: towards polythiols and derived structures, Polym. Chem., 2012, 3, 1007 RSC.
- W. W. Cleland, Dithiothreitol, a New Protective Reagent for SH Groups, Biochemistry, 1964, 3, 480–482 CrossRef CAS.
- V. Martínez, V. Villamil, D. Duarte, C. Saiz, D. Davyt and C. Fontana, et al., Preparation and Mechanistic Studies of 2-Substituted Bisthiazolidines by Imine Exchange, Eur. J. Org. Chem., 2020, 2020, 1084–1092 CrossRef PubMed.
- B. T. Worrell, S. Mavila, C. Wang, T. M. Kontour, C. H. Lim and M. K. McBride, et al., A user's guide to the thiol-thioester exchange in organic media: scope, limitations, and applications in material science, Polym. Chem., 2018, 9, 4523–4534 RSC.
- H. M. Burke, L. McSweeney and E. M. Scanlan, Exploring chemoselective S-to-N acyl transfer reactions in synthesis and chemical biology, Nat. Commun., 2017, 8, 15655 CrossRef.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi and C. R. Fenoli, et al., The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- W. C. Still, M. Kahn and A. Mitra, Rapid chromatographic technique for preparative separations with moderate resolution, J. Org. Chem., 1978, 43, 2923–2925 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data for all new compounds 1, 4–10. CCDC 2387412–2387413. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra07780f |
| This journal is © The Royal Society of Chemistry 2024 |