
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyacrylamide-based hydrogel electrolyte for modulating water activity in aqueous hybrid batteries†
Damira Rakhmana,
Dauren Batyrbekulya,
Bauyrzhan Myrzakhmetovab,
Karina Zhumagaliac,
Kuralay Issabekb,
Orazaly Sultan-Akhmetov ab,
Nurzhan Umirovad,
Aishuak Konarov*ab and
Zhumabay Bakenov*abd
ab,
Nurzhan Umirovad,
Aishuak Konarov*ab and
Zhumabay Bakenov*abd
aNational Laboratory Astana, Nazarbayev University, Astana, Kazakhstan
bDepartment of Chemical and Materials Engineering, School of Engineering and Digital Sciences, Nazarbayev University, Astana, Kazakhstan
cSchool of Mining and Geosciences, Nazarbayev University, Astana, Kazakhstan
dInstitute of Batteries, LLP, Astana, Kazakhstan. E-mail: aishuak.konarov@nu.edu.kz; zbakenov@nu.edu.kz
First published on 23rd December 2024
Abstract
While zinc-ion and hybrid aqueous battery systems have emerged as potential substitutes for expensive lithium-ion batteries, issues like side reactions, limited electrochemical stability, and electrolyte leakage hinder their commercialization. Due to their low cost, high stability, minimal leakage risks, and a wide variety of modification opportunities, hydrogel electrolytes are considered the most promising solution compared to liquid or solid electrolytes. Here, we synthesized a dual-function hydrogel electrolyte based on polyacrylamide and poly(ethylene dioxythiophene):polystyrene (PPP). This electrolyte reduces water content and enhances stability by minimizing side reactions while swelling in a binary ethylene glycol and water solution (EG 10%) further stabilizes the battery system. The developed hydrogel exhibits relatively good ionic conductivity (1.6 × 10−3 S cm−1) and excellent electrochemical stability, surpassing 2.5 V on linear sweep voltammetry tests. The PPP-based system reached a value of 119.2 mA g−1, while the aqueous electrolyte reached only 80.4 mA g−1 specific capacity. The rechargeable PPP hydrogel electrolyte-based hybrid aqueous battery with zinc anode achieved more than 600 cycles. Coulombic efficiency (CE) remained at 99%, indicating good electrochemical reaction stability and reversibility. This study highlights the potential of polyacrylamide-based hydrogel electrolytes with dual functionality as the electrolyte and separator, inspiring further development in hydrogel electrolytes for aqueous battery systems. This study highlights the potential of polyacrylamide-based hydrogel electrolytes with dual functionality as the electrolyte and separator, inspiring further development in hydrogel electrolytes for aqueous battery systems.
1. Introduction
With the increasing demand for electric cars and the massive shift to renewable energy sources such as solar and wind, the demand for energy storage systems, especially lithium-ion batteries (LIBs), has increased.1,2 The increased consumption of lithium for battery production, a non-renewable and rare element in the Earth's crust, has led to a sharp rise in the price of lithium and consequently LIBs. Now, LIBs are firmly established at the top of the battery market due to their high capacity, light weight, and long life. This system, however, faces several notable challenges. A key limitation is the high manufacturing cost, largely attributed to the requirement for low-moisture assembly conditions to accommodate lithium's sensitivity to atmospheric humidity. Furthermore, the use of flammable organic electrolytes introduces a significant safety hazard, increasing the risk of fire in lithium-ion batteries. To solve the problems mentioned above, aqueous electrolytes, in combination with several alternative metal ions (Na+, Ca2+, Al3+, Zn2+ and etc.) were suggested as a promising solution. Among them, hybrid aqueous zinc-ion batteries3,4 hold the greatest potential for energy storage. Using an aqueous electrolyte solves the safety and production difficulty while zinc as an anode is a cheap, stable, and abundant material with relatively high theoretical capacity (820 mA h g−1 and 5855 mA h cm−3)5 and low redox potential (−0.763 V vs. standard hydrogen electrode (SHE)). Yet their widespread use is restricted by several issues such as electrolyte leakage, zinc dendrites, corrosion, small electrochemical stability window (ESW) (1.23 V vs. SHE) and side reactions. The majority of the rechargeable hybrid aqueous LIBs with zinc anode are associated with the electrolyte and could be improved by electrolyte engineering.6–10One of the solutions – the ‘water in salt’ principle was based on the idea of increasing the amount of salt to the limit, which strengthens the hydrogen bonds that keep the water molecules occupied.11,12 This method achieved an impressive increase in the ESW up to 3 V; however, it proved impractical due to the significant quantities of salt required, which adversely impact the overall cost of the system. Moreover, a massive decrease in ionic conductivity caused by salt precipitation negatively affected the electrochemical performance of such systems.13–15 Therefore, researchers introduced hydrogel electrolytes that could become a promising solution by effectively reducing water content and providing controlled ion deposition.16–18 Hydrogels are chemically and/or physically cross-linked polymers that retain water molecules to form a 3D network structure. Hydrogels have several advantages as an electrolyte for battery systems: (1) cheap and easily accessible, (2) prevent leakage and ensure safety, (3) chemically and mechanically stable, (4) perform both separator and electrolyte function, (5) reduce water content and widen an electrochemical potential window, and (6) have a wide pool of modification opportunities (self-healing,19,20 biodegradable,21,22 flexible,23,24 mechanically strong,25 zwitterion26–28 etc.).
Recently, among hydrogel electrolytes, the double network hydrogels29,30 have gained massive attention due to their combinations of properties. For example, Qiuhong Chen and colleagues presented PMAZn – a double-network self-healing zwitterion hydrogel electrolyte for high operating voltages.31 Weak non-covalent intertwined network between PMS and SA in synergy with chemical cross-link provided good mechanical stability and inhibited side reactions. Another example is the covalent organic frameworks (COF) based TCOF-Zn hydrogel electrolyte presented by Dr Tianyu Qiu and colleagues showed remarkable results of stable cycling for around 1400 cycles.32
Herein, in this study, the acrylamide monomer undergoes radical polymerization, creating a chemically cross-linked net-like matrix that entraps poly(ethylene dioxythiophene):polystyrene that forms another physical cross-link. Polyacrylamide (PAM) is a highly water-absorbent polymer that forms a soft gel when hydrated. It can be cross-linked using N,N-methylene bisacrylamide, resulting in a highly porous structure. The elasticity of this polymer can be fine-tuned by adjusting the ratio between the monomer and cross-linking agent.33 Poly(ethylene dioxythiophene):polystyrene is a well-studied transparent, colourless material with a reputation for high flexibility, making it a popular choice in electronic devices. It is a polymer consisting of two ionomers, charged macromolecules. Some of the sulfonyl groups within polystyrene are deprotonated and carry a negative charge, while the other component, poly(3,4-ethylenedioxythiophene), is a conjugated polymer carrying positive charges. The presence of amide (CONH2) groups within the PAM backbone, positioned at alternating carbon atoms, significantly enhances hydrogen bonding, thereby influencing water encapsulation. Simultaneously, additional physical cross-link contributes to excellent ionic conductivity, strengthens hydrogen bonding, and augments mechanical stability. Additionally, ethylene glycol is frequently utilized as an anti-freezing agent and has been successfully employed in both liquid and gel electrolytes for wide-temperature supercapacitors and zinc-ion batteries.26,34 The introduction of ethylene glycol as an additive enhances the thermal stability of the system, and forms an organic protective layer on the anode surface, thereby reducing the formation of dendrites and mitigating other side reactions. The poly(ethylene dioxythiophene):polystyrene and polyacrylamide (PPP) hydrogel electrolyte shows a wider electrochemical potential window on LSV tests with no oxygen evolution up to 2.5 V, while assembled symmetric Zn‖Zn cells demonstrated long cycling over 1300 h. Moreover, full Zn‖LiFePO4 (LFP) cells showed promising cycling performance over 600 cycles.
2. Experimental section
2.1 Materials
Acrylamide (AM), poly(ethylene dioxythiophene):polystyrene, N,N′-methylene bisacrylamide (MBA), ammonium persulfate (APS), zinc sulfate heptahydrate (ZnSO4·7H2O), lithium sulfate monohydrate (Li2SO4·H2O), ethylene glycol (EG), lithium iron phosphate (LiFePO4 or LFP), manganese sulfate monohydrate (MnSO4 H2O), lithium manganese oxide (LiMn2O4 or LMO) and N-methyl pyrrolidone (NMP) were purchased from Sigma-Aldrich Co. Zinc foil was provided by Goodfellow, acetylene black (AB) from Gelon lib., polyvinylidene fluoride (PVDF) from Kynar, HSV900, carbon paper from Alfa Aesar Co., and glass nanofiber filter paper from Whatman. The chemicals were used as received.2.2 Preparation of PPP hydrogel electrolyte
The 1.5 g of acrylamide monomer was thoroughly dissolved in 4.5 mL of deionized water. Then, 250 mg of 1.3 wt% poly(ethylene dioxythiophene):polystyrene solution was poured into the monomer solution. After stirring for at least 30 minutes the 15 mg of crosslinker N,N′-methylene bisacrylamide (MBA), and the same amount of a thermal initiator – ammonium persulfate (APS) were added and stirred until dissolved. The solution was quickly poured between two glass plates divided by a spacer, sealed, and heated in the oven at 60 °C for at least two hours. The polymerized hydrogel was peeled off from the glass mold and soaked in the electrolyte solution for at least 12 hours.To compare, the polyacrylamide hydrogel was prepared as a reference under the same conditions without poly(ethylene dioxythiophene):polystyrene addition.
2.3 Preparation of LiFePO4 electrode
For LiFePO4 electrode preparation semi-automatic pilot line was used, consisting of a mixer, a casting machine with a built-in drying chamber, a calendaring machine, a sealing machine, and a vacuum sealer (Fig. 1a). | ||
| Fig. 1 (a) Schematic illustration of the one-pot PPP hydrogel polymerization and chemical and physical cross-link formation. (b) Schematic of the hydrogel electrolyte with an ethylene glycol additive working principle preventing side reactions and modulating water activity. (c) Photos of prepared PPP hydrogel. | ||
The positive cathode paste was prepared by mixing 80 wt% LiFePO4, 10 wt% polyvinylidene fluoride (PVDF) dissolved in 1-methyl-2-pyrrolidone (NMP) as binder and 10 wt% acetylene black as a conductive agent. The casting procedure parameters have been optimized. Several meters of double-coated LiFePO4-based cathode were obtained, as shown in Fig. 4a.
2.4 Preparation of LiMn2O4 electrode
LiMn2O4 positive electrodes were prepared by mixing 80 wt% of active material, 10 wt% polyvinylidene fluoride (PVDF, Kynar, HSV900) as a binder, 10 wt% acetylene black (Gelon lib.) as conductive agent dissolved in 1-methyl-2- pyrrolidinone (NMP, Sigma-Aldrich). The prepared slurry was cast on carbon paper (Alfa Aesar Co.), and vacuum dried at 70 °C for 6 h. The mass loading of active material was 5 mg cm−2.2.5 Battery assembly
For full Zn‖LFP or Zn‖LMO cell assembly, the electrolyte was sandwiched between zinc foil anode and LiFePO4 or LiMn2O4 cathode without a separator. For Zn‖LFP cell, zinc foil with a thickness of 0.05 mm was used as the negative electrode, while the LFP cast on SS foil (mass loading 4.5 mg cm−2) was used as a positive site. Before use, the surface of the zinc foil was cleaned with acetone and ethanol to remove contamination and factory oil grease. The comparison aqueous batteries were assembled with a commercial filter paper separator soaked in the 2 M of ZnSO4·7H2O, 1 M Li2SO4·H2O, and 10% ethylene glycol (EG) additive solution. For Zn‖LMO cell zinc foil (Goodfellow, USA) was used as a negative electrode. For comparison, a similar pouch cell was assembled by using a cellulose nitrate filter (Whatman) as a separator. The electrolyte was prepared by dissolving 2 M of ZnSO4, 2 M of Li2SO4, and 0.1 M of MnSO4 in distilled water, and pH was adjusted to 4 by titration with 0.5 M of H2SO4. The prepared electrolyte was bubbled with nitrogen gas (N2) to remove dissolved oxygen. Both the anode and cathode on the SS foil were cut to 1.5 × 6 strips (1.5 × 3 cm active material), while the soaked hydrogel was cut to a size around 2 × 3.5 cm. A similar procedure was followed for symmetric Zn‖Zn cells. The only difference was the polymer electrolyte soaking in 2 M of zinc sulfate heptahydrate instead of the previously mentioned solution and placing the electrolyte (or separator) between two zinc anode plates. All batteries were assembled in a pouch cell format with an aluminium case or clear plastic case.2.6 Material characterization
Fourier transform infrared spectroscopy (FTIR Analysis) was performed on a Nicolet iS10 FT-IR Spectrometer in a 4000–500 cm−1 range. Scanning electron microscopy (SEM) images and X-ray energy dispersive analysis (EDS) used for elemental mapping of the polymer and zinc anodes were taken on a JEOL JSM-IT200 (LA). The samples were freeze-dried and coated with gold (20 nm) to increase their conductivity and obtain high-resolution images.Density Functional Theory with dispersion corrections (DFT+D3) was performed to optimize the electronic ground state geometries and calculate various molecular properties such as molecular electrostatic potential maps, non-covalent interactions, and binding energy (ΔEbinding). A widely used B3LYP (Becke's three-parameter Lee–Yang–Parr) functional35–37 and a more comprehensive and accurate 6-311++G(d,p) basis set with polarization and diffusion functions were used to better describe the electron distribution. To approximate the influence of the solvent, the Conductor-like Polarizable Continuum Model (C-PCM)38 was chosen, which better represents the electrostatic interactions of a molecule in a solvent. ΔEbinding was calculated according to the following equation:
| ΔEbinding = EAB − (EA + EB) + BSSE | (1) |
All DFT calculations and post-analyses were performed using the graphical user interface Gauss View (v6.0) and the powerful software package Gaussian16 (Gaussian Inc., Wallingford CT),39 and Quantum Theory of Atoms in Molecules (QTAIM) analysis using Multiwfn.40
2.7 Electrochemical characterization
The ionic conductivity (σ) of the PPP polymer electrolyte was obtained by performing electrochemical impedance spectroscopy (EIS) following the equation:| σ = l/RA | (2) |
3. Results and discussion
3.1 Material characterization of PPP hydrogel electrolyte
The hydrogel electrolyte was successfully prepared by an easy, one-pot radical polymerization method (Fig. 1a). The acrylamide monomer chemically cross-linked 3D structure in the presence of the thermal initiator and cross-linker at 60 °C. At the same time, the newly structured polyacrylamide network entraps the second polymer in its structure forming the entanglement, hydrogen bond and electrostatic interaction between their functional groups. Those forces create additional physical cross-links and double-network increasing the water retention strength and enhancing the hydrogel structure. The amide (CO–NH2) group within the polyacrylamide backbone, positioned at alternating carbon atoms, significantly enhances hydrogen bonding, which helps trap water molecules within the structure, while the poly(ethylene dioxythiophene):polystyrene component contributes to excellent ionic conductivity, strengthens hydrogen bonding, and improves mechanical stability.Additionally, the presence of additive ethylene glycol in the electrolyte solution protects the anode surface from side reactions and potentially enhances the thermal stability of the system as shown in Fig. 1b. This is because ethylene glycol, having a different surface tension compared to regular water electrolytes, forms an organic protective layer on the metallic anode surface.34 The final hydrogel is a transparent film with a faint deep blue shade, prepared by using a glass mold and an adjustable spacer (Fig. 1c). Later, it is soaked in the electrolyte solution of 2 M ZnSO4 and 1 M Li2SO4.
The polymerization of the hydrogel and its structure were confirmed using FTIR analysis (Fig. 2a). The peak at 2924 cm−1 indicates the formation of –C–H bonds' stretching vibrations on the PAM (yellow) and PPP (blue), typically associated with the polymer long chain. The spectrum also exhibits absorption bands typical for polyacrylamide. Peaks at 3179 cm−1 (N–H stretching), 1652 cm−1 (CO stretching, primary amide), and 1605 cm−1 (N–H bending) peaks are all associated with the PAM's hydrophilic functional groups.41 Their presence is essential for ion transport within the polymer network structure. It is commonly agreed that the cations in the electrolyte systems travel through the polymer electrolytes by ‘hopping’ between electron donors,42 which are, in our case, located in functional groups or on the polymer backbones.
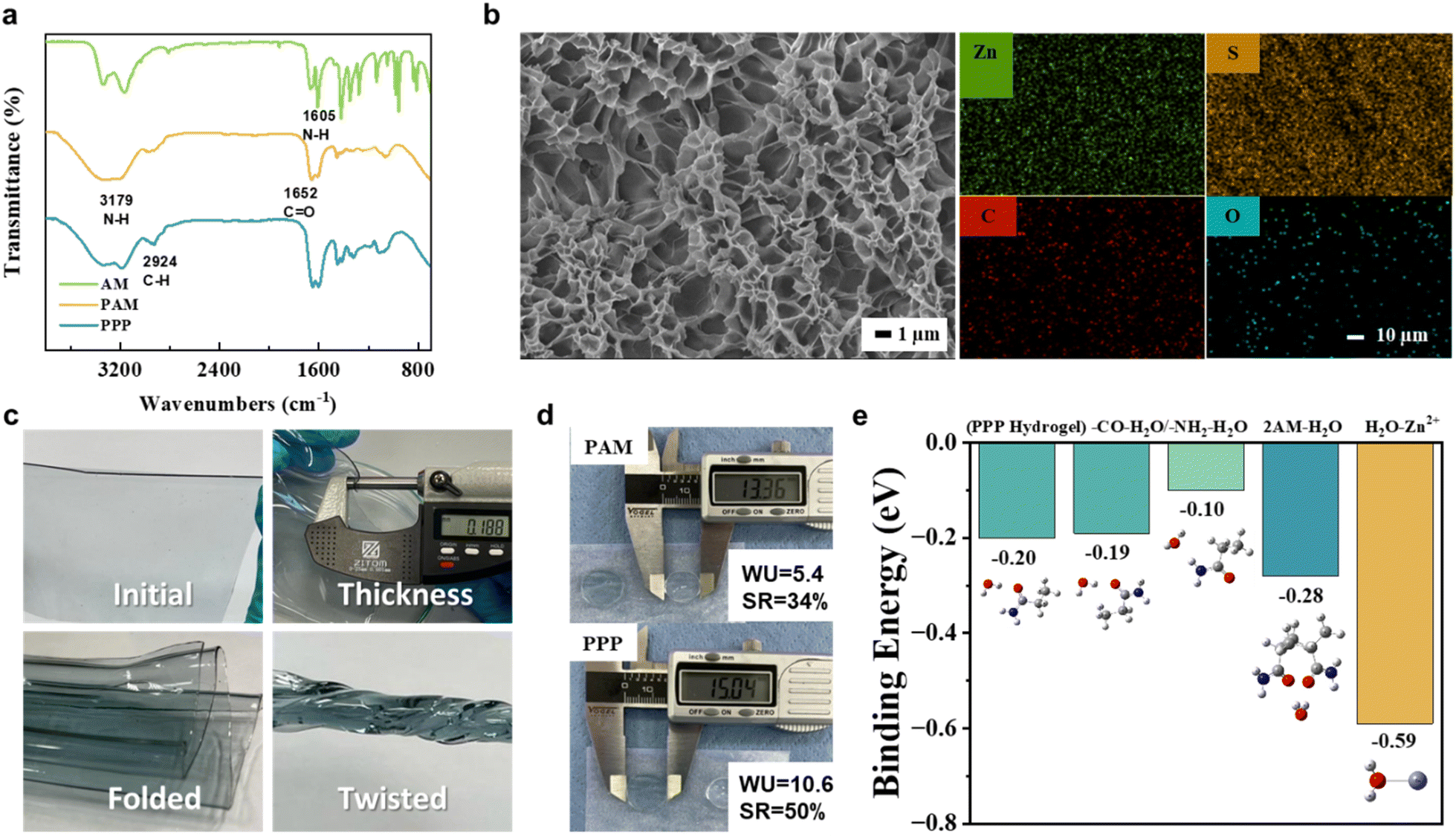 | ||
| Fig. 2 (a) FTIR spectroscopy of acrylamide monomer, comparative PAM and PPP hydrogels. (b) SEM images (average pore size is 2.15 μm. Scale: 10 μm) of freeze-dried PPP hydrogel. EDS mapping (scale: 10 μm) of PPP hydrogel after soaking the polymer in ZnSO4 and Li2SO4 solution with the EG additive. (c) Demonstration of thickness and mechanical flexibility of the PPP hydrogel (thickness = 0.188 mm). (d) Water Uptake (WU) and Swelling Ratios (SR) of PPP and comparative PAM hydrogel electrolytes. (e) Binding energies of different PPP hydrogel sites and zinc with water molecules. | ||
While the electrolyte facilitates ion movement, the separator acts as a physical barrier between electrodes. In the case of hydrogel electrolytes, one of its advantages is that the hydrogel can serve as both an electrolyte and a separator due to its excellent mechanical stability. This dual function of the electrolyte has the potential to reduce separator expenses, assembly time, and save space. Good separators need to meet certain requirements, such as good mechanical stability and high porosity, to prevent contact between electrodes and ensure ion permeability.43,44 The PPP hydrogel electrolyte was designed to perform the dual function of an electrolyte and separator. Therefore, scanning electron microscopy (SEM) was conducted to observe the surface morphology, and examine the hydrogel's porosity and permeability. The SEM images in Fig. 2b revealed a uniform, highly porous structure in the PPP hydrogel. This hierarchical porous network morphology enhances water encapsulation within the polymer network and promotes ion movement by facilitating the free movement of Zn2+ and Li+ cations.
Additionally, the average pore size of less than 2 μm indicates that the hydrogel is well-suited to be used as a separator. Furthermore, after soaking the hydrogel in the electrolyte solution, elemental mapping through energy-dispersive X-ray spectroscopy (EDS) was used to assess the ion distribution in the hydrogel structure. Well-dispersed Zn and S elements shown on the right side of the SEM images in Fig. 2b are associated with zinc and sulfate ions from the electrolyte solution in the hydrogel. At the same time, C and O represent the polymer and its functional groups. The fact that the elements were not accumulated in one specific location supports good permeability and homogenous ion distribution, making it highly suitable for use as an electrolyte and separator.
Another supporting factor is demonstrated in the images in Fig. 2c. The hydrogel electrolyte is highly flexible and can be bent, folded, and twisted freely. Such adaptability showcases the great stability of the system driven by the double-network structure. The ability to withstand mechanical deformations promotes its promising applications in flexible electronics. Moreover, the water uptake and swelling ratio coefficient is another critical factor greatly affected by the employment of physical and chemical cross-links (Fig. 2d). Compared to pure PAM hydrogel, the PPP exhibited twofold water uptake (10.6 for PPP and 5.4 for PAM) and a slightly increased swelling ratio (50% for PPP and 34% for PAM). Remarkably, despite the doubled water uptake, the swelling ratio difference is less than expected, underscoring the enhanced stability of the polymer structure facilitated by strengthened hydrogen bonding from additional physical cross-linking. The hydrogel electrolyte has a great advantage over conventional aqueous electrolytes by demonstrated characteristics, such as mechanical stability and electrolyte solution leakage prevention. The 3D network of the structure additionally affects the cathode material dissolution pressing it toward the current collector.
Finally, Fig. 2e presents the binding energy calculated through simulations. The DFT analysis confirms earlier assumptions, revealing that the hydrogel electrolyte forms stronger bond with water molecules. The arrangement of water molecules around the acrylamide (AM) group influences how these bonds form. The first four blue bars represent the interaction of the water molecules with various hydrogel functional groups, while the last bar corresponds to the zinc–water binding energy. The hydrophilic groups on the polymer backbone play a crucial role in the hydrogel electrolyte's water retention and ion transport abilities. This difference in binding energies suggests that the water molecules are effectively trapped within the polymer structure and do not actively participate in parasitic side reactions.
A closer look at the specific interactions reveals why water forms a stronger bond with certain functional groups in the hydrogel. Notably, water has a stronger affinity for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group than for the NH2 group in AM. This is because oxygen in the carbonyl group is more electronegative and has a higher capability of accepting hydrogen bonds compared to nitrogen in the NH2 group. As a result, water molecules establish more stable hydrogen bonds with the CO group, as confirmed by QTAIM results (Fig. 3). This difference in bonding strength highlights the role of hydrogel functional groups in contributing to its overall water retention and functionality.
O group than for the NH2 group in AM. This is because oxygen in the carbonyl group is more electronegative and has a higher capability of accepting hydrogen bonds compared to nitrogen in the NH2 group. As a result, water molecules establish more stable hydrogen bonds with the CO group, as confirmed by QTAIM results (Fig. 3). This difference in bonding strength highlights the role of hydrogel functional groups in contributing to its overall water retention and functionality.
 | ||
| Fig. 3 (a) Non-covalent interactions and (b) reduced density gradient of PPP hydrogel sites. | ||
Fig. 3 illustrates non-covalent interactions (NCI) and reduced density gradient (RDG) between the AM group and water molecules. The NCI surfaces feature blue, green and red regions indicating strong attractive forces (likely hydrogen bonding), moderate attractive forces (which are consistent with van der Waals interactions), and steric repulsion, respectively. As seen in Fig. 3a, subtle changes in interaction strength and spatial distribution of NCIs are observed. For instance, the interaction between AM with two functional groups (CO and NH2) and water molecules exhibits stronger bonding interactions, evidenced by a sharper peak in the RDG plot. Larger peaks at negative values indicate stronger hydrogen bonding interactions. This suggests that the orientation of the AM group plays a key role in determining the strength and nature of its interaction with water molecules.
To provide a comprehensive understanding of the interaction between CO, NH2 and H2O molecules, bond critical points (BCP) at these bonds were analyzed using electron density (ρ(r)), and the Laplacian of electron density (∇2ρ(r)). A high ρ(r) value suggests strong covalent bonding, while the Laplacian reveals whether charge is accumulating ∇2ρ(r) < 0 or depleting ∇2ρ(r) > 0. The electron localization function (ELF) for the BCP at CO⋯H2O and NH2⋯H2O bonds was also quantitatively determined using Atoms in Molecules (AIM) approach, with the results presented in Table 1. ELF values close to 1 indicate highly localized electrons and values near 0 suggest electron delocalization.45
| Bond | Electron density, ρ(r) | Laplacian electron density, ∇2ρ(r) | ELF |
|---|---|---|---|
| 1 (CO⋯H) | 0.035 | 0.115 | 0.120 |
| 2 (CO⋯H) | 0.032 | 0.117 | 0.098 |
| 3 (NH⋯O) | 0.025 | 0.087 | 0.085 |
| 4 (CO⋯H) | 0.028 | 0.097 | 0.095 |
| 5 (CO⋯H) | 0.025 | 0.089 | 0.084 |
Table 1 shows that the ELF values for all bonds are relatively low, ranging from 0.084 to 0.120, which indicates that the interactions are primarily hydrogen bonding and van der Waals in nature. Bonds 4 and 5, associated with AM groups containing two CO functionalities, exhibit the highest overall ρ(r), ∇2ρ(r), and ELF values. As a results, these bonds correspond to higher binding energies in the PPP hydrogels.
3.2 Electrochemical characterization of the PPP hydrogel-based batteries
Several alternate cathode materials were well studied for their performance such as manganese-based oxide,46–48 vanadium-based oxide,49 and Prussian blue analogues (PABs).50 However, the LFP cathode was mainly used due to its excellent performance and high cycling stability.51Fig. 4a and b shows the images of the cathode preparation. The cathode material was prepared on a prototyping pilot line for large-scale applications. After that, it was cut into desired dimensions and used in battery assembly. During battery assembly, the PPP hydrogel was sandwiched between the zinc anode and LFP or LMO cathode which was cast on the SS current collector. For the electrochemical characterization tests, symmetric and full pouch cell battery types were assembled, as illustrated in Fig. 4c. A pouch cell is a type of battery characterized by its flexible, flat pouch-shaped design, which is commonly used in portable devices. Compared to cylindrical and prismatic cells, pouch cells are generally less expensive to produce, lighter, and more adaptable in design. However, they are also more susceptible to being pierced and damaged. Pouch cells are typically assembled in a multilayered structure, which includes anode, cathode, and separator. However, in our case with the hydrogel electrolyte, no separator is used because the hydrogel itself provides sufficient mechanical protection against the electrodes coming into contact with each other (Fig. 4c).
 | ||
| Fig. 4 (a) The pilot line for pouch cell preparation. (b) Preparation of the cathode. (c) Schematic illustration of the symmetric Zn‖Zn (left) and full Zn‖LiFePO4 (right) pouch cell assembly. | ||
The ionic conductivity of the PPP hydrogel electrolyte was determined by performing EIS measurements, shown in Fig. 5a. Using the EIS analysis, the ionic conductivity was calculated using formula (2) and equals 1.6 × 10−3 S cm−1. While this value may seem modest compared to some high-performance hydrogel electrolytes, it aligns with several reported systems that exhibit excellent electrochemical performance. Studies have shown that hydrogel electrolytes with relatively low ionic conductivities can still deliver remarkable electrochemical behavior. For instance, a hydrogel based on carboxymethyl cellulose and zinc acetate52 achieved an ionic conductivity of 2.10 × 10−3 S cm−1 or a hydrogel electrolyte synthesized with iota carrageenan and acrylamide exhibited an ionic conductivity of 2.15 × 10−3 S cm−1 but sustained a stable capacity of 127.5 mA h g−1 over 500 cycles at 5C in a Zn–V2O5 battery, underscoring superior cycling stability and Zn2+ transference.30
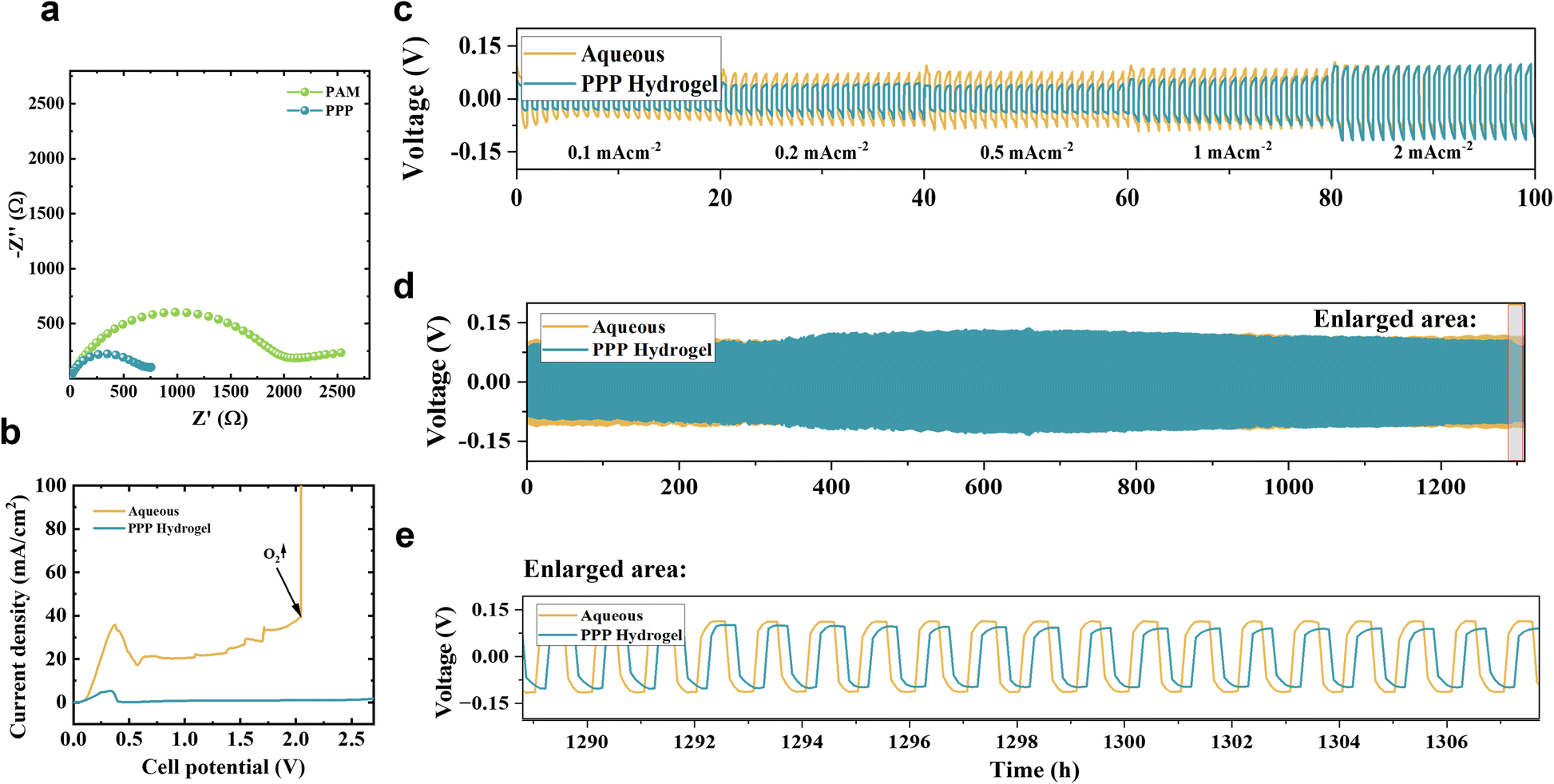 | ||
| Fig. 5 (a) EIS spectra of PPP and PAM hydrogel in symmetric Zn‖Zn cell. (b) LSV curves of the PPP hydrogel and aqueous symmetric Zn‖Zn cells at 1 mV s−1. (c) Zn plating and stripping behavior of PPP hydrogel and aqueous Zn‖Zn symmetric cells at different current densities (0.1–2 mA cm−2). (d) Zn plating and stripping long cycling of PPP hydrogel and aqueous electrolyte-based Zn‖Zn symmetric cells at 0.1 mV h−1. (e) An enlarged demonstration of the zone is indicated on the above Zn plating/stripping long cycling behavior test. | ||
The PPP hydrogel electrolyte's ionic conductivity outperformed the comparative pure PAM hydrogel electrolyte, which had a performance of 0.5 × 10−3 S cm−1. This increased ionic conductivity is credited to the higher water content and improved water encapsulation due to the additional physical cross-link, high porosity, permeability, and sulfate groups from the polystyrene sulfonate. Sulfonate groups were previously mentioned to facilitate ionic conductivity by controlling zinc ion placement.32,53,54 The properties of the PPP hydrogel electrolyte position it as a competitive candidate among advanced hydrogel systems, particularly when compared to emerging solid-state electrolytes, which typically exhibit ionic conductivities below 10−4 to 10−5 S cm−1 at room temperature. However, its relatively low ionic conductivity compared to liquid electrolytes remains a limitation that could be addressed in future research.
To further boost its ionic conductivity, several strategies could be implemented. Incorporating ionic liquids like 1-ethyl-3-methylimidazolium trifluoromethane sulfonate ([Emim]OTF) can enhance both ionic conductivity and mechanical properties by acting as a plasticizer and reducing resistance.55 Additionally, the use of dual-ion zwitterionic polymers or advanced cross-linked networks has been shown to significantly improve water retention and ionic mobility, with recent studies achieving conductivities as high as 24.32 mS cm−1 through such innovations.28 These advancements could elevate the performance of the PPP hydrogel electrolyte even further, enabling its use in cutting-edge applications.
The LSV test was employed to assess the effectiveness of the PPP hydrogel in modulating water activity. As shown in Fig. 5b a wider stability window is observed on the hydrogel's curve with no changes even after 2.5 V, with a small peak at the beginning of the LSV curve associated with zinc dissolution. In contrast, the aqueous electrolyte-based system demonstrated an oxygen evolution at 2 V. This expanding stability resulted from several factors our system was designed to achieve. By using hydrogel and adding ethylene glycol, we reduce the amount of water in the system. A double network with chemical and physical cross-links strengthens hydrogen bonds, improving water entrapment, all to restrict reactive water molecules from participating in the oxygen evolution reaction.
The voltage profile of Zn plating and stripping is shown in Fig. 5c, which was measured at a current density range of 0.1–2 mA cm−2. The voltage profiles exhibit small polarization (voltage deviation from 0 V), indicating efficient and reversible zinc plating/stripping. At lower current densities, the potential approaches 0 V, reflecting minimal overpotential. At higher current densities, the potential deviates further due to increased polarization.
Compared to aqueous electrolyte the PPP hydrogel electrolyte-based system exhibited lower polarization at each current density, with a 50% difference observed at 0.5 mA cm−2. At the same time, in a long cycling experiment at 0.1 mV h−1, shown in Fig. 5d, the Zn|PPP|Zn system displayed a slight expansion in the middle, followed by a return to lower polarization compared to the aqueous system with glass nanofiber. The increased voltage in the middle is suspected to be due to unstable contact between the electrode surface and the electrolyte. Both batteries lasted for more than 1500 cycles. The obtained results suggest that the PPP hydrogel electrolyte system improved the reversibility of the zinc plating and stripping process.
The zinc stripping and plating process in a slightly acidic environment can be represented by the following reversible reaction:
| Zn(s) ⇌ Zn(aq.) + 2e− | (3) |
Zinc plating and stripping involve activation energy, small overpotential is required:
Cathodic (negative) overpotential during plating (deposition of Zn on the electrode).
Anodic (positive) overpotential during stripping (dissolution of Zn from the electrode).
The overpotential typically ranges between tens of millivolts (mV) depending on the current density, electrode surface condition, and electrolyte composition.
Plating (Zn2+ + 2e− → Zn): occurs slightly below 0 V (e.g., ∼−10 to −50 mV vs. Zn).
Stripping (Zn → Zn2+ + 2e−): occurs slightly above 0 V (e.g., ∼+10 to +50 mV vs. Zn).
Uniform zinc deposition is critical for battery performance. The formation of zinc dendrites and undesired side reactions on the anode side is a challenge that impedes the wider adoption of rechargeable aqueous batteries with a zinc anode. Undesired side reactions include hydrogen evolution reactions and irreversible formation of byproducts. In the case of sulfate-based electrolytes, there is a common formation of the black-coloured solid – Zn4SO4(OH)6·5H2O. The byproduct formation process is as follows:
| 4Zn2+ + 6OH− + SO42− + 5H2O = Zn4 (SO4)(OH)6·5H2O | (4) |
The formation of such byproducts on the anode surface can disrupt ion intercalation and result in permanent capacity loss due to the anode's deterioration caused by the irreversibility of the process.46–48
For further investigation of the applicability of the PPP hydrogel electrolyte, rechargeable aqueous Zn|PPP|LFP batteries were assembled in a pouch cell format. During the charging process, due to the applied current, zinc ions dissolved and dispersed in the PPP hydrogel network travel toward the zinc anode and are plated on its surface. At the same time, on the cathode electrode Li+ ions leave from the LiFePO4 structure and are released into the electrolyte. Reversely, during discharge, lithium ions are inserted back into the FePO4 structure from the electrolyte while the zinc ions are stripped from the anode surface to the electrolyte media. The electrochemical reactions happening in the system are as follows:
| Anode: Zn(aq.)2+ + 2e− ⇌ Zn(s) | (5) |
| Cathode: Li1−xFePO4 ⇌ xLi+ + xe− + Li1−xFePO4 | (6) |
| Overall: Zn(aq.)2+ + 2LiFePO4 ⇌ Zn(s) + 2Li+ + 2FePO4 | (7) |
To characterize the electrochemical reactions, cyclic voltammetry (CV) was performed at a scan rate of 0.1 mV s−1 in the range of 0–2.4 V (Fig. 6a). The Li+ extraction/insertion from the LFP structure were present on the curve which signifies the reversibility of the process. It also suggests that the developed PPP hydrogel electrolyte is a good medium that ensures sufficient intercalation for the electrochemical reactions. It is noticeable that past 2 V, the curve representing the aqueous electrolyte begins to rise. This indicates the onset of the oxygen evolution reaction. In contrast, the hydrogel electrolyte remains stable beyond 2.2 V. In a concentrated aqueous electrolyte system (pH = 4) the practical maximum voltage range is likely around 1.9–2.0 V (instead of 1.23 V) due to reduced water activity and suppressed decomposition reactions.3
 | ||
| Fig. 6 (a) CV of the PPP hydrogel and aqueous electrolyte-based full Zn‖LFP pouch cells at 0.1 mV s−1. (b) Discharge capacity and coulombic efficiency, and (c) charge–discharge curve comparison PPP hydrogel and aqueous electrolyte-based hybrid Zn‖LFP pouch cell battery system tested. (d) SEM images of the Zn anode surface after 100 charge–discharge cycles. | ||
Galvanostatic charge–discharge cycling was performed on the aqueous hybrid Zn‖LFP pouch cells to evaluate their performance. The beginning few cycles in both PPP and comparison systems showed a gradual rise of the specific capacity. Associated with the activation step – before stabilization of the electrode–electrolyte interface, it was removed to not affect calculations. Fig. 6b compares the discharge capacities and coulombic efficiencies of aqueous and PPP systems at 0.1C for 100 cycles. From the beginning, the PPP hydrogel showed superior specific capacity to the aqueous system.
Regarding coulombic efficiency, the hydrogel-based system exhibited higher consistency, while the aqueous system showed greater deviation. This difference in performance is attributed to the better distribution of ions through the polymer network and the system's overall higher stability. The charge–discharge curve is shown in Fig. 6c where the PPP-based system reached a value of 119.2 mA g−1, while the aqueous electrolyte reached only 80.4 mA g−1 specific capacity. Notably, the significant thickness of the hydrogel increases ionic resistance and reduces ion transport, resulting in slightly higher voltage polarization during charge and discharge.
Additionally, uniform Zn deposition was studied using SEM images taken after extended cycling and Zn stripping/plating behavior. To compare changes on the anode surface and observe dendrite formation, we disassembled rechargeable aqueous hybrid batteries after 100 cycles of galvanostatic charge–discharge cycling. The zinc anodes of both the PPP hydrogel and aqueous electrolyte-based systems were washed with DI water and dried before SEM imaging. As expected, the surface morphology of the zinc anode changed significantly after 100 cycles, as shown in Fig. 6d. Compared to the initial smooth surface, both foils became rougher with visible zinc accumulations in various areas and some holes stripped of zinc. However, the degree of deformation is much greater on the anode surface images taken from the aqueous solution, with its large-sized corrosion sites and mountains of migrated zinc depositions. Changes in the anode surface lead to further acceleration of zinc growth on top of dendrites and promote the hydrogen evolution reaction. Differences in the surface levels lead to unstable contact between the battery elements and affect performance. When compared, the surface of the anode in a PPP hydrogel-based battery also exhibits grain-like dendrites and occasional larger buildups. However, the degree of deformation is much less, compared to when in aqueous conditions. This demonstrates that the PPP hydrogel inhibits significant deformations and dendrite formations on the anode surface even after extended cycling periods. Notably, the grain-like formations on the anode surface from the PPP hydrogel system suggest that the zinc ions were deposited in a more ordered and well-aligned manner as if they moved back and forth through the hydrogel network pores. The uniform nucleation of zinc cations is aided by the hydrogen bonds that form between the PPP hydrogel and water. The hydrophilic functional groups tightly bind water molecules, preventing the formation of unwanted byproducts while allowing safe transit of cations through the polymer backbone, which is filled with electronegative oxygen atoms. Moreover, evenly located sulfate groups trapped in the polyacrylamide matrix support the movement of zinc ions and enhance stability.
Long cycling stability (Fig. 7a) and galvanostatic charge/discharge curves (Fig. 7b) were also assessed for the PPP hydrogel-based battery system. The rechargeable PPP hydrogel electrolyte-based hybrid aqueous battery with zinc anode achieved more than 600 cycles. CE remained at 99%, indicating good electrochemical reaction stability and reversibility. Similarly, good capacity stability was supported in galvanostatic charge/discharge curves even after the 125th cycle with no visible shoulders that could signify side reactions.
 | ||
| Fig. 7 (a) Long cycling performance and (b) galvanostatic charge/discharge curves of PPP hydrogel-based Zn‖LFP hybrid pouch cell battery at 0.1C. (c) Discharge capacity and (d) voltage/capacity curves of the PPP hydrogel and aqueous electrolyte-based Zn‖LiMn2O4 hybrid pouch cell batteries. | ||
Additionally, the LMO cathode material was tested as an alternative. In this case, the cathode was cast on the SS foil manually, while the electrolyte solution contained the additional manganese sulfate to prevent cathode dissolution. The LMO‖Zn cells were cycled galvanostatically between 2.3 V and 1.5 V using a multichannel battery tester (BT2000, Arbin) at C/10. As seen in Fig. 7c and d the PPP-based system showed higher capacity than the aqueous electrolyte with the highest 150.6 mA g−1 and 88.6 mA g−1 respectively. However, capacity fading couldn't be avoided after the 35th cycle associated with the manganese dissolution from the cathode material. M. Minakshi et al. have extensively studied methods to control manganese dissolution in aqueous electrolytes, a key challenge in improving the performance and longevity of MnO2-based electrodes. Their work emphasizes the use of additives like Bi2O3, TiS2, and CeO2, which mitigate dissolution through distinct mechanisms.49–51
To evaluate the real-life performance of the prepared batteries, a Zn|PPP|LFP clear pouch cell was created to demonstrate its multilayered structure. An LED light was used for testing at room temperature (Fig. 8). Furthermore, to showcase the flexibility of the battery system, the pouch cell was bent at a 90-degree angle and folded. The provided image shows that the battery is operational despite various deformations.
 | ||
| Fig. 8 Images of LED light applications of the PPP-based hybrid pouch cells in clear polyethylene packaging under various deformations (bending and folding). | ||
Significant progress has been made in developing hydrogels, but further optimization is needed for practical use in energy storage systems. Our study introduced a new hydrogel formulation, previously unexplored, especially in the context of hybrid batteries. This work aims to boost interest in using hydrogel-based electrolytes by highlighting their versatility. Hydrogels can be customized using various component combinations to improve their properties and electrochemical performance, leading to enhanced functionality in future applications.
4. Conclusion
In summary, the PPP hydrogel electrolyte showed good prospects in expanding the working electrochemical potential window of the rechargeable hybrid aqueous battery systems. The PPP hydrogel is easily synthesized by one-pot radical polymerization suggesting scalability opportunities. Easy atmospheric assembly conditions and accessibility of the materials could reduce costs significantly. The abundance of hydrophilic groups and highly porous structure successfully entraps water molecules in the polymer network, as shown through experiments and DFT calculations. Moreover, the porous structure of the hydrogel is in the range to be used as a separator suggesting the dual function of the electrolyte. Due to the increased hydrogen bonds the water molecules were prevented from participating in side reactions, therefore, the LSV test revealed no oxygen evolution up to 2.7 V. Benefitting from these advantages, zinc plating/stripping behavior showed less polarization and the SEM images showed lesser anode degradation compared to the aqueous electrolyte. While assembled rechargeable hybrid aqueous batteries demonstrated good cycling over 600 cycles maintaining good coulombic efficiency of around 100%. The highest specific capacity demonstrated in hydrogel-based hybrid LFP‖Zn pouch cells reached 119.2 mA g−1 while in LMO‖Zn systems it showed 150.6 mA g−1. This research opens a pathway for the future development of hydrogel electrolytes for modulating water activity in large-scale battery applications.Data availability
All data supporting findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Research Targeted Program “Development of new technologies of materials and energy storage systems for a green economy” BR21882402 from the Ministry of Science and Higher Education of the Republic of Kazakhstan.References
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef PubMed.
- A. Konarov, N. Voronina, J. H. Jo, Z. Bakenov, Y.-K. Sun and S.-T. Myung, ACS Energy Lett., 2018, 3, 2620–2640 CrossRef.
- J. Yan, J. Wang, H. Liu, Z. Bakenov, D. Gosselink and P. Chen, J. Power Sources, 2012, 216, 222–226 CrossRef.
- P. Ayyanusamy, S. D. Tharani, R. Alphonse, M. Minakshi and R. Sivasubramanian, Chem.–Eur. J., 2024, 30, e202402325 CrossRef.
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 CrossRef.
- A. Li, J. Li, Y. He and M. Wu, J. Energy Chem., 2023, 83, 209–228 CrossRef.
- H. Li, S. Guo and H. Zhou, Energy Storage Mater., 2023, 56, 227–257 CrossRef.
- L. Fan, X. Hu, Y. Jiao, L. Cao, S. Xiong, F. Gu and S. Wang, Chem. Eng. J., 2024, 479, 147763 CrossRef.
- X. Gao, J. Yang, Z. Xu, Y. Nuli and J. Wang, Energy Storage Mater., 2023, 54, 382–402 CrossRef.
- M. Minakshi, Electrochim. Acta, 2010, 55, 9174–9178 CrossRef.
- D. Chao and S.-Z. Qiao, Joule, 2020, 4, 1846–1851 CrossRef.
- T. Zhang, Y. Tang, S. Guo, X. Cao, A. Pan, G. Fang, J. Zhou and S. Liang, Energy Environ. Sci., 2020, 13, 4625–4665 RSC.
- R. Qi, W. Tang, Y. Shi, K. Teng, Y. Deng, L. Zhang, J. Zhang and R. Liu, Adv. Funct. Mater., 2023, 33, 2306052 CrossRef.
- X. Li, D. Wang and F. Ran, Energy Storage Mater., 2023, 56, 351–393 CrossRef.
- V. Vandeginste and J. Wang, Energies, 2024, 17, 310 CrossRef.
- Y. Guo, J. Bae, Z. Fang, P. Li, F. Zhao and G. Yu, Chem. Rev., 2020, 120, 7642–7707 CrossRef.
- P. Yang, J.-L. Yang, K. Liu and H. J. Fan, ACS Nano, 2022, 16, 15528–15536 CrossRef PubMed.
- Y. Guo, J. Bae, F. Zhao and G. Yu, Trends Chem., 2019, 1, 335–348 CrossRef.
- H. Liao, W. Zhong, T. Li, J. Han, X. Sun, X. Tong and Y. Zhang, Electrochim. Acta, 2022, 404, 139730 CrossRef.
- Y. Liu, A. Gao, J. Hao, X. Li, J. Ling, F. Yi, Q. Li and D. Shu, Chem. Eng. J., 2023, 452, 139605 CrossRef.
- Z. Liu, R. Wang, Q. Ma, H. Kang, L. Zhang, T. Zhou and C. Zhang, Carbon Neutralization, 2022, 1, 126–139 CrossRef.
- Z. Zheng, S. Yan, Y. Zhang, X. Zhang, J. Zhou, J. Ye and Y. Zhu, Chem. Eng. J., 2023, 475, 146314 CrossRef.
- P. Zhang, K. Wang, P. Pei, Y. Zuo, M. Wei, X. Liu, Y. Xiao and J. Xiong, Mater. Today Chem., 2021, 21, 100538 CrossRef.
- Z. Wang, H. Li, Z. Tang, Z. Liu, Z. Ruan, L. Ma, Q. Yang, D. Wang and C. Zhi, Adv. Funct. Mater., 2018, 28, 1804560 CrossRef.
- C. Ji, D. Wu, Z. Liu, H. Mi, Y. Liao, M. Wu, H. Cui, X. Li, T. Wu and Z. Bai, ACS Appl. Mater. Interfaces, 2022, 14, 23452–23464 CrossRef CAS PubMed.
- C. Yuan, X. Zhong, P. Tian, Z. Wang, G. Gao, L. Duan, C. Wang and F. Shi, ACS Appl. Energy Mater., 2022, 5, 7530–7537 CrossRef.
- Q. Fu, S. Hao, X. Zhang, H. Zhao, F. Xu and J. Yang, Energy Environ. Sci., 2023, 16, 1291–1311 RSC.
- O. Zhanadilov, H. J. Kim, H.-J. Lai, J.-C. Jiang, A. Konarov, A. Mentbayeva, Z. Bakenov, K.-S. Sohn, P. Kaghazchi and S.-T. Myung, Small, 2023, 19, 2302973 CrossRef.
- S. Liu, Y. Zhong, X. Zhang, M. Pi, X. Wang, R. Zhu, W. Cui and R. Ran, ACS Appl. Mater. Interfaces, 2022, 14, 15641–15652 CrossRef PubMed.
- C. Y. Chan, Z. Wang, Y. Li, H. Yu, B. Fei and J. H. Xin, ACS Appl. Mater. Interfaces, 2021, 13, 30594–30602 CrossRef PubMed.
- Q. Chen, J. Zhao, Z. Chen, Y. Jin and J. Chen, Int. J. Hydrogen Energy, 2022, 47, 23909–23918 CrossRef.
- T. Qiu, T. Wang, W. Tang, Y. Li, Y. Li, X. Lang, Q. Jiang and H. Tan, Angew. Chem., Int. Ed., 2023, 62, e202312020 CrossRef.
- Y. Wang, G. Nian, J. Kim and Z. Suo, J. Mech. Phys. Solids, 2023, 170, 105099 CrossRef.
- X. Li, H. Wang, X. Sun, J. Li and Y.-N. Liu, ACS Appl. Energy Mater., 2021, 4, 12718–12727 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef.
- P. M. W. Gill, B. G. Johnson, J. A. Pople and M. J. Frisch, Chem. Phys. Lett., 1992, 197, 499–505 CrossRef CAS.
- E. V. R. de Castro and F. E. Jorge, J. Chem. Phys., 1998, 108, 5225–5229 CrossRef CAS.
- B. Mennucci, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2012, 2, 386–404 CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, Gaussian, Inc., Wallingford, CT.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- X. Hu, L. Feng, A. Xie, W. Wei, S. Wang, J. Zhang and W. Dong, J. Mater. Chem. B, 2014, 2, 3646–3658 RSC.
- M. Zhang, S. Yu, Y. Mai, S. Zhang and Y. Zhou, Chem. Commun., 2019, 55, 6715–6718 RSC.
- S. Mallick and C. R. Raj, ChemSusChem, 2021, 14, 1987–2022 CrossRef CAS.
- L. Li and Y. Duan, Polymers, 2023, 15, 3690 CrossRef CAS PubMed.
- N. A. F. Abdullah, S. Hasan and S. A. Lee, Sci. Lett., 2021, 15, 90–100 CrossRef.
- Y. Zhang, A. Chen and J. Sun, J. Energy Chem., 2021, 54, 655–667 CrossRef.
- Z. Li, W. Lu, N. Zhang, Q. Pan, Y. Chen, G. Xu, D. Zeng, Y. Zhang, W. Cai, M. Yang, Z. Yang, Y. Sun, H. Ke and H. Cheng, J. Mater. Chem. A, 2018, 6, 14330–14338 RSC.
- K. Wu, J. Yi, X. Liu, Y. Sun, J. Cui, Y. Xie, Y. Liu, Y. Xia and J. Zhang, Nano-Micro Lett., 2021, 13, 79 CrossRef PubMed.
- M. Minakshi, P. Singh, D. R. G. Mitchell, T. B. Issa and K. Prince, Electrochim. Acta, 2007, 52, 7007–7013 CrossRef.
- M. Minakshi, J. Solid State Electrochem., 2009, 13, 1209–1214 CrossRef.
- M. Minakshi, K. Nallathamby and D. R. G. Mitchell, J. Alloys Compd., 2009, 479, 87–90 CrossRef.
- I. Dueramae, M. Okhawilai, P. Kasemsiri and H. Uyama, Sci. Rep., 2021, 11, 13268 CrossRef PubMed.
- S. Park, I. Kristanto, G. Y. Jung, D. B. Ahn, K. Jeong, S. K. Kwak and S.-Y. Lee, Chem. Sci., 2020, 11, 11692–11698 RSC.
- Z. Wang, J. Hu, L. Han, Z. Wang, H. Wang, Q. Zhao, J. Liu and F. Pan, Nano Energy, 2019, 56, 92–99 CrossRef.
- D. Wang, X. Guo, Z. Chen, Y. Zhao, Q. Li and C. Zhi, ACS Appl. Mater. Interfaces, 2022, 14, 27287–27293 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07551j |
| This journal is © The Royal Society of Chemistry 2024 |