
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegrating 3D-printed hydrogels and polymeric nanofibers in a double-layered transdermal patch for the treatment of rheumatoid arthritis†
Hend A. Elshabrawy,
Ahmed S. Abo Dena ,
Mohamed H. Sobhy and
Ibrahim M. El-Sherbiny*
,
Mohamed H. Sobhy and
Ibrahim M. El-Sherbiny*
Nanomedicine Laboratories, Center for Materials Science, Zewail City of Science and Technology, 6th of October City, Giza, Egypt. E-mail: ielsherbiny@zewailcity.edu.eg
First published on 23rd December 2024
Abstract
Patients with rheumatoid arthritis (RA), an inflammatory illness that affects the synovial joints, have a much worse quality of life. Mostly, oral or injectable formulations are used to treat RA, underscoring the critical need for an innovative medication delivery method to enhance therapeutic outcomes and patient compliance. The present study integrated 3D bioprinting and electrospinning technologies to create a unique double-layered transdermal patch (TDDP) for the treatment of RA. The first layer was prepared by 3D-printing a newly developed hydrogel (HG) containing hyaluronic acid (HA) that maintains the joint components and dexamethasone (DEX), an anti-inflammatory agent. A second layer of electrospun polycaprolactone (PCL) nanofibers (NFs) loaded with naringin (NAR), an antimicrobial and anti-inflammatory natural plant extract, was fabricated and integrated with the former layer. The morphology of the NFs was investigated using SEM, and their diameter ranged from 156.28 to 220.66 nm. The physicochemical properties of the suggested TDDP were investigated using FTIR and DSC. The DEX-loaded 3D-printed HG was able to release about 98% of the loaded DEX over a period of 10 days. In vivo experiments definitively confirmed the efficacy of the newly developed TDDPs, showcasing a substantial reduction in the levels of proinflammatory cytokines (IL-6 and TNF-α) through sandwich ELISA measurements in Rattus norvegicus plasma samples. Besides, the stained photomicrographs of the ankle joints of the animals confirmed the alleviation of the RA symptoms via reducing cell infiltration with a preserved joint tissue structure.
Introduction
Synovium, the soft tissue that lines tendon sheaths, diarthrodial joints, and the bursae,1 is made up of both the underlying tissue and the continuous cell layer on the outside (intima and subintima).1,2 The intima is assembled from macrophages, along with fibroblasts; on the other hand, the subintima includes lymphatic vessels and blood, coupled with a cellular composition containing both infiltrating and resident cells embedded in a collagenous extracellular matrix.1 Collectively, these features of the structure offer a non-adhesive interface between the tissue components. Rheumatoid arthritis (RA) and other inflammatory joint diseases, such as osteoarthritis, have their pathology concentrated in the synovium.3 In patients with inflammatory arthritis, like RA, the intimal lining layer is typically noticeably thickened because of overexpression of rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPAs), which activate B cells, T cells, and macrophages and produce inflammatory molecules, in particular interleukin-6 (IL-6), and interleukin-V (IL-V), besides tumour necrosis factor (TNF).3 Rheumatoid arthritis is a chronic autoimmune disease that results in growth and inflammation of the synovial fibroblasts, a rise in inflammatory markers, and invasion of neighbouring cartilage, which leads to bone erosion and cartilage tissue loss.4 The disease has a direct link to severe cartilage deterioration that damages the joints, the most prevalent musculoskeletal ailment in the elderly, and the most widespread cause of disability all over the globe.5,6 The smaller joints in the hands and feet, such as the wrists, fingers, and toes, are all affected by RA.3 It is assessed that nearly 1% of the world's population is influenced by RA.3 The underlying mechanisms of RA are complex and involve both genetic and environmental factors, which in turn stimulate an extraordinary immune response.7 Currently, the treatment choices for RA comprise non-steroidal anti-inflammatory drugs (NSAIDs) and biologics, along with disease-modifying anti-rheumatic drugs (DMARDs).8 Nevertheless, these treatments are related to several consequences, including, but not limited to, gastrointestinal disorders, cardiovascular complications, and immunosuppression.9Despite advances in basic research and therapeutics, current RA therapy is still partially successful and has a number of limitations, including high and/or frequent doses, time of administration, drug interactions, side effects, and toxicity.4,10 In addition, anti-rheumatic drugs are associated with a significant risk of deadly and severe adverse effects. Treatment costs, the complexity of long-term management, and other variables still need to be considered.8 The ideal system should be simple to use, deliver the drug consistently, and maintain the optimal drug concentration without unpleasant side effects.10 By delivering the medication at a higher concentration to the desired site of action, microparticles, nanoparticles, nanodispersions, nanocapsules, nanoemulsions, nanosuspensions, and other new delivery materials for RA treatment increase the drug efficacy.8 The DMARDs, NSAIDs, selective cyclooxygenase-2 (COX-2) inhibitors, glucocorticoids, substances from natural origin, and biological agents are some of the therapeutic options for the treatment of RA.11 Rabiei et al. reported that all immune-modulating and anti-rheumatic drugs include a significant risk of adverse effects, such as the medications' lack of tissue specificity and quick clearance by macrophages.5 To regulate the distribution and release of medications used in RA therapy, polymeric drug delivery systems have been employed.12 Kesharwani et al. developed customized delivery systems for inflamed joints by targeting the treatment to the inflamed area and reducing the drug dose and adverse effects.8 The nanocarrier systems improve the bioavailability of anti-inflammatory drugs. Therefore, nanoparticles are utilized to enhance the drug bioavailability and to exert a sustained activity.8 As a result, the biodegradability of polymeric nanoparticles used in drug delivery systems has been enhanced, and the drug release profile, half-life time, and therapeutic efficacy of these systems are all improved. Researchers predicted that nanoparticles can improve targeting and play a significant role in cartilage repair.8 Apart from that, Norouzi developed indomethacin-loaded nanofibers (NFs) composed of cyclodextrin-grafted chitosan (CS) for the treatment of inflammatory illnesses.13,14 Recently, non-invasive systems known as transdermal drug delivery patches (TDDPs) can prevent gastrointestinal irritation caused by oral medications and avoid the liver's first-pass effect.15
The main objective of this study is to evaluate (in vitro and in vivo) a newly developed double-layered TDDP comprising naringin (NAR)-loaded PCL NFs and 3D-printed hydrogel (HG) composed of CS, PVA, and sodium alginate (ALG) containing hyaluronic acid (HA) and DEX for the topical alleviation of RA symptoms (Fig. 1). Electrospun nanofibers (NFs) and hydrogels (HGs) are promising materials for developing advanced drug delivery systems due to their unique properties. Electrospinning is a versatile technique that produces nanofibers with high surface area-to-volume ratios, facilitating efficient drug loading and controlled release. These NFs can be engineered to mimic the extracellular matrix, promoting cell adhesion and proliferation, which is beneficial for tissue engineering applications in RA. Furthermore, 3D printing allows precise control over the hydrogel's architecture, leading to better mechanical properties and drug distribution compared to conventional casting methods. In addition, the porosity of the 3D-printed hydrogel material can be controlled, while the conventionally casted ones are almost nonporous. Porosity, or mesh-like architecture, is desired in the present work to allow simultaneous contact of both layers (NFs and HG layers) with the skin. Thus, combining electrospun NFs with HGs can create systems that leverage the strength of both materials. This dual-layer system can provide an enhanced action, where the NFs offer structural support and controlled drug release, while the HG layer ensures a moist environment and further sustains the release of therapeutic agents. Such a system can be particularly advantageous in the treatment of RA, where localized and sustained delivery of anti-inflammatory drugs is crucial. The use of the natural antimicrobial compound (NAR) is very helpful since, in addition to its anti-inflammatory effect, it governs the suppression of the growth of microorganisms over the topically applied transdermal patch. In addition, during inflammation, immune cells produce reactive oxygen species (ROS) to kill the invading pathogens, thus causing a state of oxidative stress. Therefore, the antioxidant property of the loaded NAR is also very important to relieve the symptoms of inflammation.
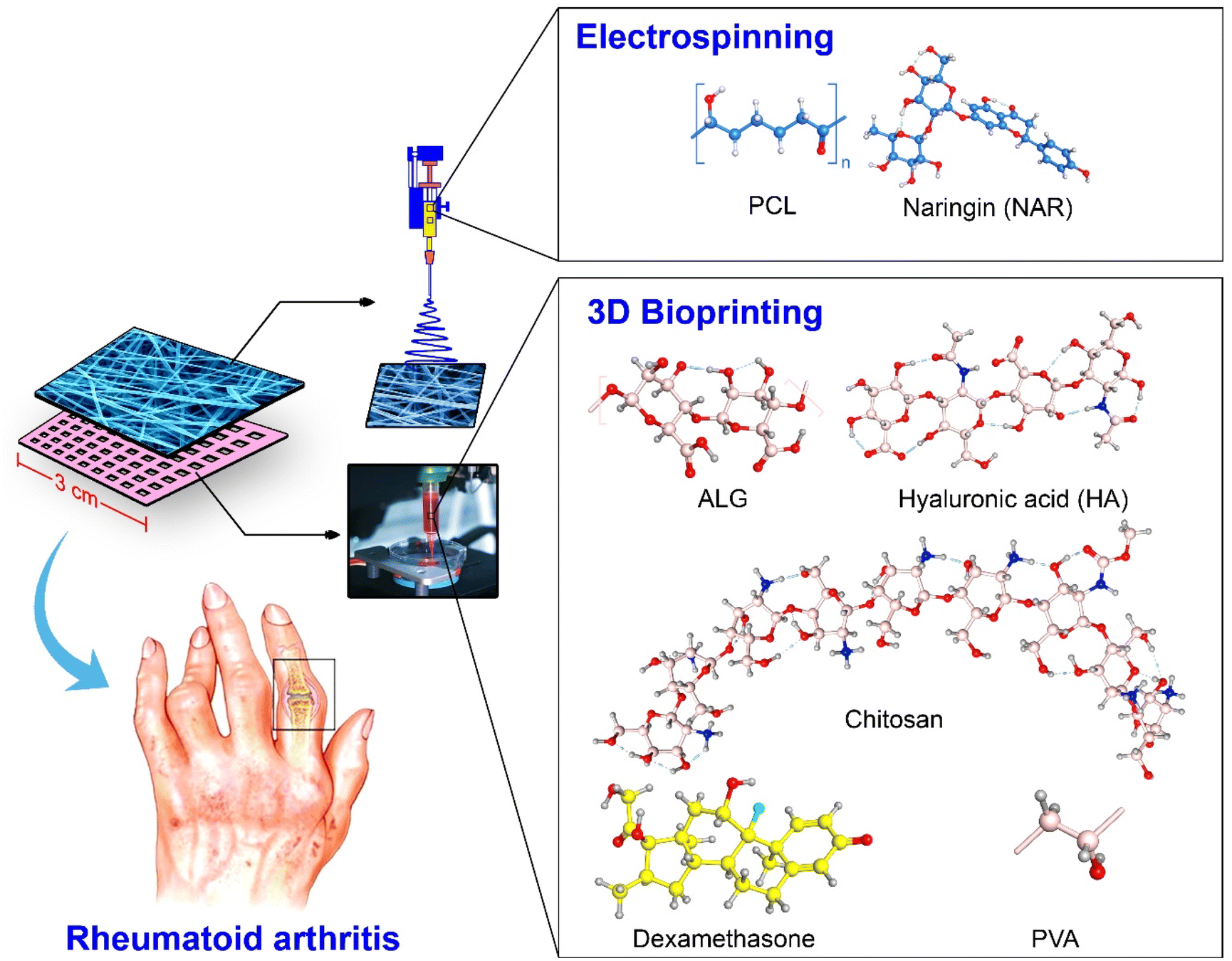 | ||
| Fig. 1 Composition and preparation procedure of the proposed double-layered transdermal patch via 3D-printing of DEX/HA-loaded HG and electrospinning of NAR-loaded PCL solution. | ||
HA and DEX were added to the 3D-printed meshes as anti-inflammatory, non-immunogenic, and modulators of the intra-articular environment. The 3D-printed HG layer was designed in such a way as to confer a sustained release of bioactive agents and extend their presence at the inflammation site. The combination of HA, analgesic, and anti-inflammatory agents has synergistic effects, inhibiting white blood cell proliferation, preserving synovial components, reducing leukocyte-induced cartilage degradation, protecting against mechanical stresses, and offering a more effective treatment for RA. NAR was loaded in the nanofibers (NF) due to its hydrophobic nature, which aligns with the polymeric fibrous matrix, while Dex was loaded in the hydrogel (HG) due to its hydrophilic properties, thus optimizing release profiles and therapeutic outcomes. In addition, the patch must be maintained as long as possible, so NAR (an antimicrobial agent) was loaded in the outermost NFs layer, which is susceptible to contamination wit microorganisms.
The newly developed topical platform represents the first integration of drug-loaded NFs and 3D-printed HG mesh in a multiple-layered transdermal patch for RA treatment. This approach addresses drawbacks of conventional treatments, emphasizing the crucial need for an effective topical remedy, as current RA treatments mainly rely on oral or injectable forms.
Experimental
Materials
Sodium phosphate monobasic (M.W. 156.01 g mol−1) and poly(vinyl alcohol) (PVA, M.W. 13–23 kDa) were acquired from ACROS, USA. Chitosan (CS, M.W. 140–220 kDa) was purchased from Alamia Company for Chemicals, Egypt. Sodium phosphate dibasic (M.W. 141.96 g mol−1), calcium chloride (CaCl2, M.W. 110.98 g mol−1), polycaprolactone (PCL, M.W. 45 kDa), hyaluronic acid sodium salt (SH) from Streptococcus equi (M.W. 8–15 kDa), and sodium alginate (ALG, M.W. ranging from 12–40 kDa) based on the viscosity measurements, and with a ratio of mannuronic to guluronic acid (M/G) in the range of 1.28–1.42, were obtained from Sigma-Aldrich (St. Louis, MO, USA). Sodium chloride (NaCl, M.W. 58.44 g mol−1, 99.5% extra pure) and potassium chloride (KCl) were purchased from Advent, India. Glacial acetic acid and dichloromethane (DCM) were purchased from Fisher Scientific, UK. The N,N-dimethylformamide (DMF) was obtained from Advent, India. Citrus limon was obtained from a local market in Cairo, Egypt. Standard dexamethasone was obtained from the Egyptian Drug Authority (EDA), Egypt. Dimethyl sulfoxide (DMSO) was obtained from Sigma-Aldrich, Germany. Müeller–Hinton broth bacterial culture medium (MHB) was purchased from Merck, Germany. Rat IL-6 (Cat. No SEA079Ra), TNF-α (Cat. No. SEA133Ra), human IL-6 (Cat. No SEA079Hu), and TNF-α (Cat. No. SEA133Hu) ELISA kits were purchased from Cloud-Clone Corp (Texas, USA).Extraction of NAR from Citrus limon fruit
The naringin (NAR) crystals were isolated from the albedos of Citrus limon according to the protocol described elsewhere.16,17 Briefly, the orange skin (flavedos) and the white, spongy inside (albedos) of the peels were cut into small pieces by hand-slicing. The fresh albedos were then separated manually from the flavedos. A weight of 26 g of the albedos and a volume of 150 mL of methanol were added into a conical flask. After 3 hours of heating the mixture at 55 °C, a rotary evaporator was used for the removal of the organic solvent. Thereafter, using 100 mL of methanol, another step of hot extraction was performed for 30 min at 55 °C. In a rotary evaporator, the organic phase was vacuum-dried at 45 °C. The collection of pure NAR crystals was performed by adding 20 mL of double-distilled water and 10 mL of DCM. Finally, the NAR crystals were filtered and collected through a filter paper and then dried in a vacuum oven. The total yield of NAR crystals was found to be ca. 78 mg.Preparation of the NAR-loaded PCL NFs
A polymeric solution of 10 g of PCL was obtained via dissolving the polymer in a DMF/DCM solvent mixture (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60, v/v) under continuous stirring. Later, about 5 mL of this PCL solution were loaded with 50 mg of NAR. Finally, a 5 mm syringe containing the above NAR-loaded PCL solution was positioned inside the specified place in the electrospinning device (NANON-01A electrospinning system, MECC Co. Ltd). The NAR-loaded NFs were fabricated by adjusting the electrospinning parameters to a voltage of 23 kV, a spinneret width of 10 cm, a feed rate of 1.5 mL h−1, and a 15 cm distance from the tip.
:60, v/v) under continuous stirring. Later, about 5 mL of this PCL solution were loaded with 50 mg of NAR. Finally, a 5 mm syringe containing the above NAR-loaded PCL solution was positioned inside the specified place in the electrospinning device (NANON-01A electrospinning system, MECC Co. Ltd). The NAR-loaded NFs were fabricated by adjusting the electrospinning parameters to a voltage of 23 kV, a spinneret width of 10 cm, a feed rate of 1.5 mL h−1, and a 15 cm distance from the tip.
Fabrication of the drug-loaded 3D-printed hydrogel mesh
The 3D-printing technique was employed to fabricate the HG mesh with the incorporation of both dexamethasone (DEX) and hyaluronic acid (HA). Concisely, a weight of 1.2 g of low-molecular-weight PVA, 233 mg of sodium hyaluronate, and 5.75 mL of DEX solution (4 mg mL−1) were dissolved using 4 mL of double distilled water. Afterwards, sodium alginate (2 g) was dissolved in a separate volume of double-distilled water (4 mL). Moreover, 1.128 g of chitosan (CS) was solubilized in 10 mL of 1% (v/v) acetic acid solution. After complete dissolution of CS, the above solutions were mixed vigorously with the aid of a mechanical mixer to form a gel with a suitable viscosity to be used in the 3D-printing step (the net volume is 23.75 mL). The gel was either left at room temperature for several hours or subjected to bath sonication to remove any air bubbles that resulted from the mechanical mixing. Finally, the 3D-printed mesh was built by additive manufacturing with the aid of a 3D bioprinter (BIO X, Cellink, Canada), and then crosslinked with a 2% CaCl2 solution for two minutes. The dimensions of the mesh were adjusted to 3 cm × 3 cm × 1 mm. The fabricated 3D-printed HG meshes were sterilized by UV light and then left to dry at room temperature for 24 hours in sterile conditions to prevent contamination by microorganisms.To bind the HG layer with the NF layer, first the HG layer was gently sprayed with deionized water. Second, the HG layer was adhered to the NFs layer, and a heavy weight was put above the resulting double layer until it was totally dry. It is worth mentioning that a large number of the double-layer patches has been prepared following the same preparation procedure, and they exhibited excellent reproducibility.
Physicochemical characterizations
 | (1) |
Regarding the HPLC method, the analysis was performed using an Agilent 1260 series Man. Inj. separation module with a photodiode array detector (Germany). Throughout the HPLC experiment, an Eclipse Plus UHPLC column (4.6 mm × 25 cm) with a high-strength silica stationary phase material with a particle size of 5 mm was utilized. A quaternary pump model 1260 (Germany) with an auto-sampler (HiPALS), a temperature-controlled column compartment (25 °C), and a multi-channel gradient valve (MCGV) made up the LC system. Agilent Chemstation software was used for data acquisition, and Agilent LabAdvisor (utility quantitative analysis software) was used for data processing. DEX was analysed by applying the US pharmacopoeia method where a mixture of methanol and acetate buffer of pH 4.0 was used as the mobile phase, and the sample injection volume was 15 μL at a sample flow rate of 1 mL min−1. The drug was detected at 254 nm in the eluted mobile phase.
Determination of the hydrophilicity of the NFs
The contact angle between the surface of a 4 μL drop of double distilled water and the surface of a sheet of the NFs (water contact angle; WCA) was measured in a digital image taken by a smartphone camera with the aid of ImageJ software.Water absorption and biodegradability of the 3D-printed HG mesh
The degree of swelling and the weight loss of the developed 3D-printed HG mesh were assessed. Briefly, in the water absorption test, firstly, the dried hydrogel samples measuring about 2 cm (length) by 2 cm (width) were recorded as the initial dry weight (Wi). The samples were then immersed in 20 mL of double-distilled water in three 50 mL Falcon tubes and incubated in a shaking incubator (VWR International, USA) at 120 rpm and 37 °C for different time intervals. After that, the samples were removed from the water and gently blotted with soft filter paper to remove excess surface water. The weight of the hydrated hydrogel (Wf) was recorded immediately after blotting. The water absorption capacity was calculated according to eqn (2).To evaluate water loss, the same sample Wi were placed in 50 mL Falcon tubes containing distilled water and incubated in a shaking incubator (VWR International, USA) at 120 rpm and 37 °C. The weight of the hydrogel samples was measured at specific time intervals (e.g., 1, 2, 4, 6, 12, and 24 hours) until a constant weight was reached. The weight recorded at each time interval was denoted as Wf. The water loss percentage was calculated according to eqn (3).
Water gain and water loss were plotted in the same figure. These distinct methods ensured accurate and separate evaluation of the hydrogel's water absorption and water loss characteristics, providing comprehensive insights into its hydration behaviour and stability.
 | (2) |
 | (3) |
Similarly, the swelling and biodegradation of the PCL NFs were investigated in double distilled water. Over a period of two weeks, NFs samples of 1 cm × 1 cm size were incubated in 10 mL of double distilled water in the shaking incubator at 120 rpm and 37 °C. The dry NF weight (Wi) was recorded prior to being immersed in the double distilled water. At different time intervals, the swelled NF samples were taken out of the solution and weighed after being gently cleaned using two soft filter papers (Wf). The percent swelling and weight loss of the NFs were calculated from eqn (2) and (3) as mentioned above.
Antimicrobial activity
The antimicrobial activity of the prepared NAR-loaded PCL NFs was tested against Staphylococcus aureus (NCTC10788), Salmonella typhimurium (NCTC12023), and Pseudomonas aeruginosa (ATCC27853) cultured in MHB culture medium at 37 °C for 24 h. The in vitro antimicrobial activity was evaluated according to the method described by Eloff with minor modifications.18 Briefly, the NFs were suspended as fragments to allow the release of NAR and determine its antimicrobial activity (a weight of the NAR-loaded NFs, containing 50 mg of NAR, was investigated). Subsequently, 100 μL of the bacterial suspension (105 CFU mL−1) were added, and the plates were incubated for 24 h at 37 °C. For bacterial count, 100 μL from each well were withdrawn, placed over Müeller–Hinton agar plates, and incubated for 24 h at 37 °C.Cytotoxicity
The MTT assay was performed to evaluate the impact of the proposed transdermal drug delivery system on the cell viability of normal human skin fibroblasts (HFB4 cell line).19 After UV sterilization, a known mass of the NFs or the 3D-printed HG meshes was incubated in a 96-well plate with HFB4 cells for 24 h at 37 °C. Once a cohesive cell layer has been formed, the monolayer was washed and the culture medium was removed. Subsequently, an aliquot of 20 μL of MTT solution was added to the cells, shaken, and then incubated. Afterwards, 200 μL of DMSO were added to dissolve the formed formazan crystals, and the absorbance was measured at 640 nm using a microplate reader (BMG LABTECH FLUOstar Omega, Ortenberg, Germany).Assay of IL-6 and TNF-α
The levels of IL-6 and TNF-α markers were measured in rat serum using ELISA kits with the aid of a microplate reader (BMG LABTECH FLUOstar Omega, Ortenberg, Germany) according to the manufacturer's instructions based on the ELISA sandwich technique at a wavelength of 450 nm. The complete procedure for sample preparation and assay is provided in the ESI materials.†In vivo arthritic model
Four groups of Rattus norvegicus weighing 130–150 g were acquired from the Animal Health Research Institute, Egypt, for in vivo experiments. Animal experiments were performed according to institutional ethical guidelines and in accordance with the ARRIVE guideline. The followed procedure of the animal model experiment was approved by the Institutional Animal Care and Use Committee (ARC-IACUC) at the Agricultural Research Centre (Protocol Number: ARC-ZCST-23-19). The animal groups were labelled as follows: (i) N (normal animals, 3 rats); (ii) C (diseased untreated animals, 4 rats); (iii) P (diseased animals treated with plain transdermal patches, 4 rats); and (iv) L (animals treated with the drug-loaded transdermal patches, 4 rats). Each rat received water and mouse edibles for nourishment. According to the Asquith et al. protocol,20 RA was induced by intradermal injection of 40 μL of Freund's adjuvant in the tail's basal region for all groups except group N. A week later, swollen and inflamed ankle joints confirmed successful RA induction. Groups P and L were treated with plain and drug-loaded transdermal patches affixed to the ankle joint using medical tape, and the patch was applied twice for the duration of the experiment. After four weeks, the animals were anesthetized, and blood samples were collected from the medial canthus, centrifuged, and the obtained plasma samples were stored at −4 °C for subsequent ELISA measurements. Animals were euthanized with a ketamine overdose, and hind limbs were collected for histopathological examination of the ankle joints.Statistical analysis
Every step was carried out three times, and the results were expressed as the mean value ± SD. Using one-way analysis of variance (ANOVA) and the Tukey HSD (the asterisk sign indicates that p < 0.001), the statistically significant difference was examined using the GraphPad Prism 7 software (GraphPad Software, San Diego, USA).Results and discussion
In the following section, the results of the characterization techniques and the biological assessments of the developed drug delivery system are presented and discussed. The materials were characterized using spectroscopic, thermal analysis, and microscopic techniques. In addition, the physical properties of the NFs and the 3D-printed HG meshes were investigated by measuring the water absorption, biodegradability, and the WCA. Moreover, the drug release profile of the 3D-printed HG mesh was measured and evaluated via HPLC analysis.FTIR
Infrared spectroscopy was employed to characterize the prepared materials. The purpose of this analysis is to identify any chemical changes in the resulting formulations because any chemical interaction between the loaded drug molecules and the carrier molecules might affect the drug release profile of the final formulation. The loading of the bioactive agents onto the 3D-printed HG mesh or the generated NFs may lead to an apparent change in the FTIR spectra.21 As shown in Fig. 2A, the FTIR spectrum of the plain PCL NFs shows the absorption bands of 2944 and 2866 cm−1 (C–H functional group in CH2) and 1721 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functional group). These bands are characteristic of pure PCL and are in coincidence with the literature.21
O functional group). These bands are characteristic of pure PCL and are in coincidence with the literature.21
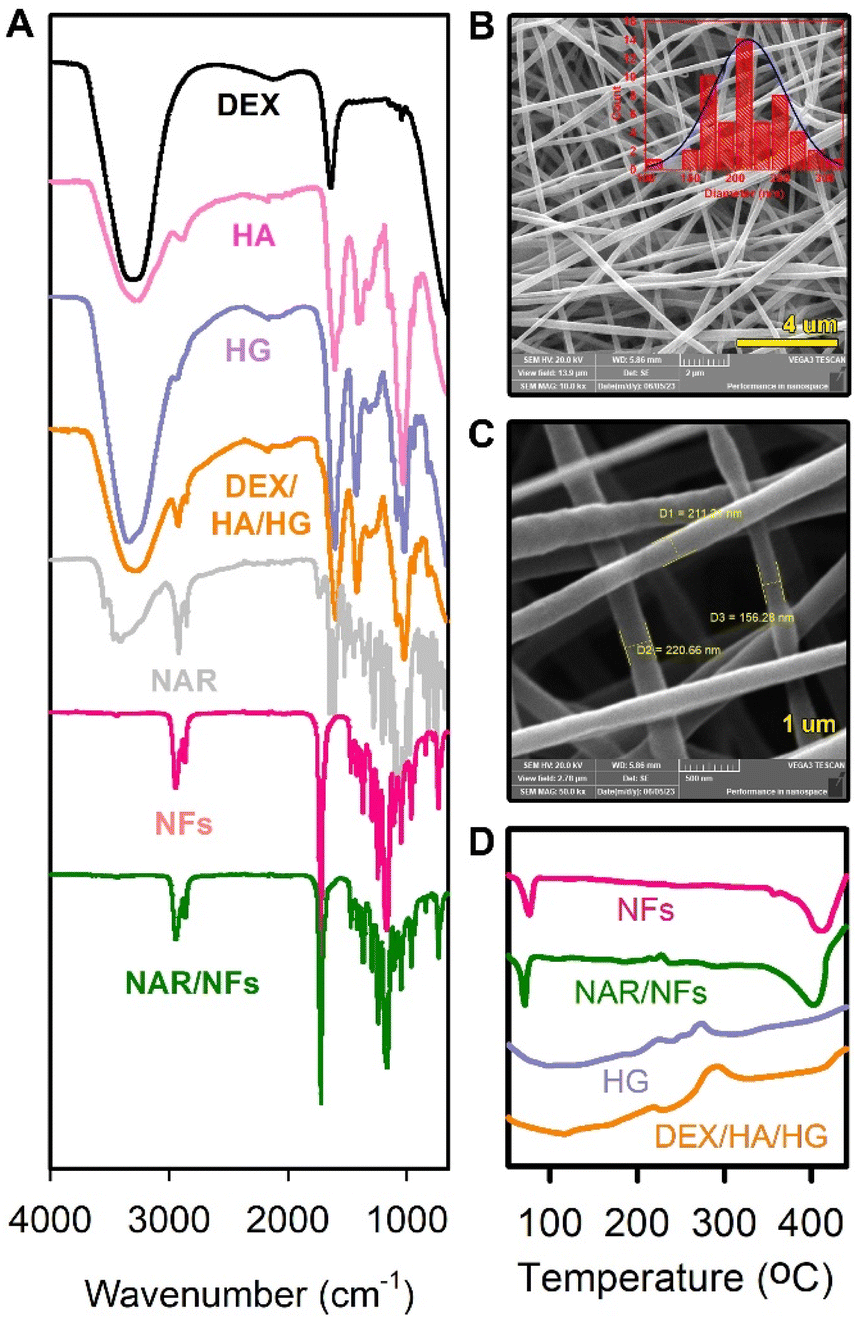 | ||
| Fig. 2 (A) FTIR spectra of the DEX, HA, plain HG, DEX/HA-loaded HG, NAR, plain NFs, and NAR-loaded NFs. (B and C) SEM images of the synthesized nanofibrous layer with different magnifications. (D) DSC thermograms of plain NFs, NAR-loaded NFs, plain 3D-printed HG, and DEX/HA-loaded 3D-printed HG. | ||
In the spectrum of free NAR, the absorption bands at 1605 and 1520 cm−1 can be assigned to aromatic bending and stretching.22 The major characteristic bands of NAR located at 1204–1444, 1646, and 3411 cm−1 are attributed to bending vibrations of the phenolic OH group, CO stretching, and the broad phenolic –OH of the NAR molecule, respectively.23,24 These bands also appeared in NAR-loaded PCL NFs with a slight shift.
The plain 3D-printed HG scaffold showed sharp bands at 711 cm−1 (out-of-plane bending NH), 1079 cm−1 (C–O–C stretching), and 3340 cm−1 (−OH stretching), which are distinctive for CS, sodium ALG, and PVA. Moreover, the vibrational mode of amide CO stretching was observed at 1601 and 1422 cm−1.
Regarding the spectrum of DEX, it exhibited a characteristic absorption band at 1635 cm−1, which is attributed to –CO bonds.25 Besides, the more characteristic absorption bands at 3319 cm−1 and 1270 cm−1 were expressed due to the stretching vibration of O–H.26 Moreover, HA exhibited the absorption bands at 1602, 1147, and 1404 cm−1 that might be related to N–H stretching, C–N stretching, and O–H bending (carboxylic acid O–H), respectively.27 These bands are considered a fingerprint for HA, and their appearance in the spectrum of the drug-loaded 3D-printed HG mesh confirms the presence of HA. No band shifting was observed in the positions of the HA characteristic bands, thus revealing no chemical interactions between HA molecules and the components of the 3D-printed HG.
All the characteristic absorption bands of the utilized polymers are present in both the plain and the drug-loaded NFs and the 3D-printed HG mesh. In addition, the absorption bands of DEX and HA are obvious in the drug-loaded HG mesh. Therefore, the results obtained from FTIR analysis suggest the successful synthesis and loading of the proposed patches and reveal that no chemical interactions have occurred between the components.
SEM imaging
Electron microscopy was used to image the developed PCL NFs as shown in Fig. 2(B and C). The SEM images of the NFs exhibit a porous nanostructure with interconnecting voids throughout the 2D NFs sheet. The randomly aligned nanofibrils demonstrate a smooth surface and a small diameter ranging from 156 to 211 nm. Further, the fabricated NFs show a homogeneous structure with no beads, indicating a good distribution of NAR in the NFs matrix. The SEM images of the prepared plain HG are shown in Fig. S1 in the ESI material.†Thermal analysis
Differential scanning calorimetry is able to reveal the thermal transitions such as crystallization (Tc), glass transition (Tg), and melting (Tm) phenomena. The results of DSC analysis of all the utilized polymeric components of the NFs as well as the 3D-printed HG are depicted in Fig. 2D. The thermal behaviour of the 3D-printed HG and the NFs before and after drug loading was investigated to assess the effect of the drug incorporation on their physical properties. The plain PCL NFs showed a sharp endothermic peak at about 65 °C attributed to its melting temperature. Additionally, another obvious endothermic trough at 410 °C and a small exothermic transition in the range of 350–380 °C are shown in the PCL thermogram, indicating the thermal decomposition of the NFs. The DSC chart of the NAR-loaded PCL NFs revealed a similar thermal behaviour to that of the plain PCL NFs with a slight change in the peaks indicating the successful incorporation of the drug. For instance, the NAR-loaded PCL NFs showed a sharp endothermic peak at about 50 °C and another slightly broader peak at about 400 °C. However, a small exothermic transition appeared at 220 °C which confirmed the presence of NAR in the PCL NFs patch.On the other hand, the DSC thermograms of 3D-printed HG with and without drug loading are shown in Fig. 2D. The plain HG has a broad endothermic peak at approximately 40–180 °C, which is characteristic for PVA and CS.28 This broad endothermic peak corresponds to the loss of moisture from the HG. Moreover, three exothermic peaks at approximately 220, 240, and 270 °C were observed and might indicate thermal decomposition of the HG polymeric components. The drug-free HG mesh also shows broad endothermic peaks at about 300 and 390 °C.
Regarding the drug-loaded 3D-printed HG, there is an exothermic peak at about 280 °C along with a reduced endothermic peak at approximately 70 °C. These peaks also indicate the dehydration of the HG. Similarly, the drug-loaded HG showed a broad endothermic peak at 320–430 °C. The appearance of the sharp exothermic peak at about 280 °C and the broad endothermic peak at 320–430 °C in the DSC thermogram of the drug-loaded HG mesh with slight shifting implies the successful loading of DEX and HA into the HG mesh.
In vitro drug release
The sustained release of the loaded DEX from the developed 3D-printed HG mesh is a desired feature for inflammation treatment because it permits the release of DEX in the inflamed tissues continually over a long period of time. Therefore, investigating the release profile of DEX from the 3D-printed HG mesh is crucial for the assessment of its bioactivity as well as cytocompatibility since the amount of the released drug may cause toxicity to the cells if released in a high concentration. The release profile of DEX was investigated with the aid of HPLC (Fig. 3A). As illustrated in Fig. 3B, the released amount of DEX was monitored over a period of more than ten days. First, the drug exhibited a burst release, where about 92% of the total loaded drug amount was released from the 3D-printed HG mesh after one day and then reached its maximum (ca. 98%) at the end of nine days. In addition, the release profile of DEX from the whole patch containing the HG and the NF layers was found to be very similar to that of the HG layer without a significant difference. From the above results, the mechanism of DEX release from the mesh can be explained as follows. First, the HG mesh absorbs a large amount of water when it comes in contact with the release solution, leading to its swelling. The swelling of the HG matrix allows for the breathing of the polymeric chains, leaving behind more voids for water penetration. Therefore, the absorbed amount of water dissolves DEX efficiently from the HG matrix, resulting in the burst release observed in the first release phase. In this phase, the main player that controls the release of DEX from the HG mesh is its ability to swell and absorb water. Second, after absorbing this large amount of water in the first phase, the HG mesh becomes unable to absorb any more water. | ||
| Fig. 3 (A) Change in area under the curve of the HPLC signal according to the change in DEX concentration. (B) Drug release profiles of free DEX, DEX-loaded 3D-printed HG, and DEX-loaded HG and NF. (C) Water absorption of plain PCL NFs. (D) Water absorption and (E) biodegradation of the 3D-printed HG mesh. (F and G) Water contact angles of NAR-loaded PCL NFs and plain PCL NFs, respectively. | ||
However, the HG mesh matrix starts to degrade and release the remaining amount of the entrapped drug. This degradation, which is the main player in the second phase, occurs at a slower rate compared to the rate of swelling taking place in the first phase. Accordingly, the rate of DEX release from the HG matrix will slow down until reaching its maximum after 9 days.
Water absorption and biodegradability
The swelling and biodegradability of the developed PCL nanofibrous layer are shown in Fig. 3C. As depicted in the figure, the changes in the weight of the fabricated plain NFs were determined over two weeks of soaking in double distilled water. The weight of the NFs samples showed a slow increase, reached a maximum after about 8 days, followed by a plateau due to the hydrophobicity of PCL. Although the PCL is a hydrophobic polymer, the developed PCL NFs are still showing a relative water absorption ability, which may be attributed to the fact that PCL NFs have a high surface area to volume ratio, which leads to a high-water penetration into its nano-mesh structure.29 Furthermore, the PCL NFs layer did not demonstrate any decline in weight during the course of the experiment (360 h), indicating its durability and low rate of degradation.The swelling and biodegradability of the developed 3D-printed HG mesh were investigated (Fig. 3D and E, respectively). As depicted in the figure, the water uptake increased and peaked after two hours and then gradually reached a plateau. The 3D-printed HG mesh gained about 12.7 folds of its initial weight after soaking for 3 h. This result implies that the produced patches have an acceptable hydrophilicity, which makes it easier to release drugs into the skin when they get into contact with the moisture coming out from the body surface. Concerning the biodegradability of the 3D-printed HG mesh, the weight began to decline after 4 days. Consequently, the mesh has a long lifetime, which allows for the continuous release of the cargo over a long period of time required for the treatment of inflammation. Furthermore, these findings support the postulated drug release mechanism, which depicts the breakdown of certain components of the mesh and the subsequent release of drugs into the surrounding tissues. Based on these results, it can be concluded that the produced drug delivery system has a relatively long lifetime suitable for the continuous release of the loaded drug molecules over the long period required for the treatment of RA.
Water contact angle of the NFs layer
The hydrophilicity of biomedical materials is an essential parameter for tissue regeneration as they promote better cell attachment, proliferation, diffusion, and nutrient exchange, thus creating a conductive environment for tissue growth.30,31 Therefore, it is crucial to analyse the WCA of the as-prepared NFs in order to evaluate their hydrophilicity and drug release ability, as the matrix permeability and solvent penetration rates may restrict the rates of medication release.32 This can be attributed to the matrix pore size distribution and the permeation pressure of the release medium, which is determined by its surface tension and WCA.33,34 In this manner, the WCA of NAR-loaded PCL NFs (Fig. 3F) and plain PCL NFs (Fig. 3G) were found to be 94.7° and 100.2°, indicating their hydrophobicity. However, the hydrophilicity of the PCL NFs was relatively enhanced upon being loaded with NAR. On the other hand, the very small change in hydrophobicity of PCL NFs is attributed to (i) the low amount of NAR that has been loaded into the NFs and (ii) the high hydrophobicity of the PCL NFs. Accordingly, these findings suggest that the proposed patches have enhanced wettability and water-absorption capacity, which would optimize and retain their drug-release ability when being attached to moist skin.Antibacterial activity
The need for new antibacterial lead molecules is driving researchers to explore the world's growing antimicrobial resistance phenomenon. Since natural products have comparable antibacterial properties and fewer side effects compared to chemically manufactured ones, they are becoming more and more popular.35,36 It has been reported that naturally occurring chemicals interfere with the enzymes involved in bacterial membrane production, compromise the integrity of bacterial membranes, resist biofilm formation, and impede autoinducer-mediated cell signalling. Considering phytochemicals as one of the alternative natural sources of antimicrobials, NAR has demonstrated antibacterial effects against different bacterial species, such as Escherichia coli, Salmonella typhimurium, and Staphylococcus aureus.In the present study, NAR did not show any inhibitory activity in the disk diffusion assay due to its slow diffusion resulting from its very low solubility in water.37 In addition, the bacterial count showed a 2-log reduction in the number of S. aureus and S. Typhimurium and showed a 1-log reduction in the number of P. aeruginosa, Fig. 4A. These results came in coincidence with those reported in the literature.38 In a study reported by Lee et al.,39 the cell number of S. aureus after treatment with NAR (25 μM) was reduced by 5.2logCFU mL−1 units. The antibacterial effect of NAR may be attributed to its ability to inhibit the microbial growth, and impairment of the bacterial membrane bilayer.40 In order to confirm that the integration of the HG and the NFs layers does not affect the antimicrobial properties of the NAR-loaded layer, the antimicrobial test was also performed on the whole patch. The results shown in Fig. 4A revealed that the antimicrobial activity of both NAR-loaded PCL NFs and the whole patch (comprising the NFs layer and the HG layer) are very close.
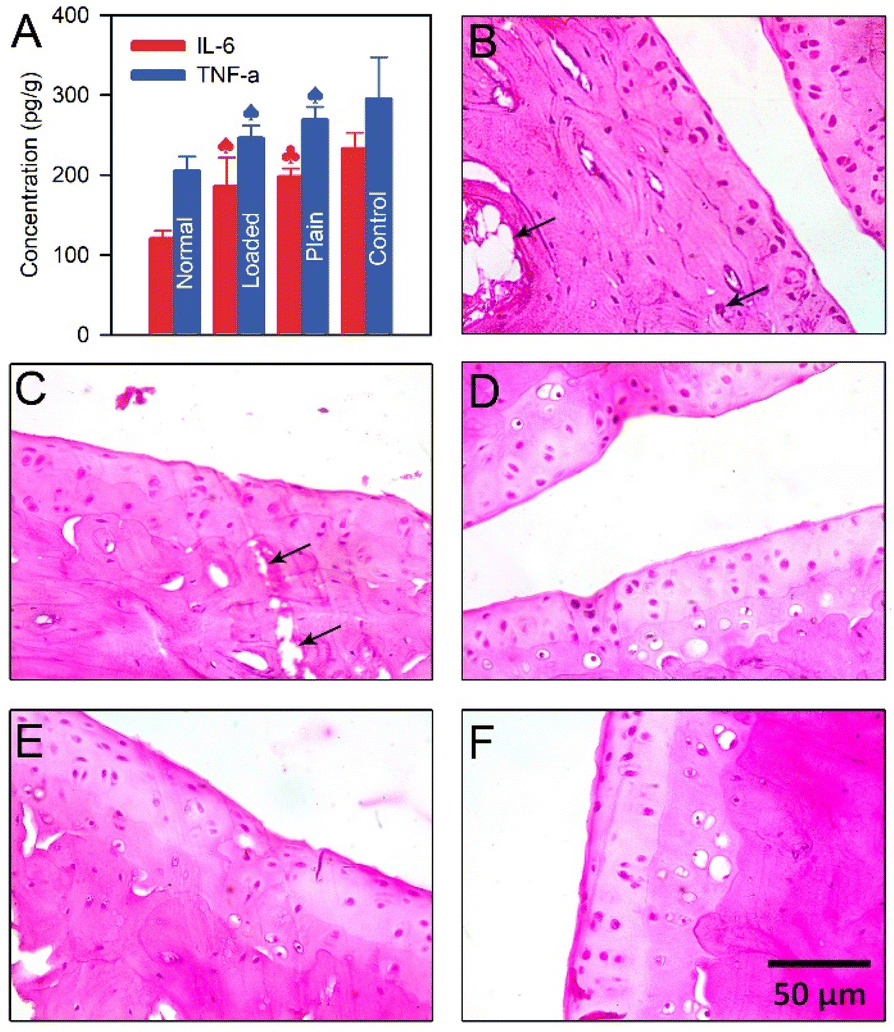 | ||
| Fig. 4 (A) In vivo levels of IL-6 and TNF-α in normal, control, plain transdermal patches-, and drug-loaded transdermal patches-treated animal groups; (B) a section in the ankle joint of control animal group; (C) a section in the ankle joint of a diseased rat treated with plain transdermal patch; (D) a section in the ankle joint of normal animal group; (E and F) a section in the ankle joint of a diseased rat treated with drug-loaded transdermal patch. Tissue damage (black arrow) and inflammation (represented by cell infiltration, blue arrow) Samples in panels B–F are light microscopic images of tissue samples stained with H&E. The asterisk symbol (*) indicates a significant difference (p < 0.001) compared to the control group using one-way ANOVA test. | ||
In vitro cytotoxicity
It is believed that the material's cytotoxicity is a crucial aspect to be considered when it comes to the development of smart drug delivery systems.41 The metabolic activity of living cells is detected to assess the cell viability as well as cell proliferation of the human skin fibroblasts (HFB4 cell line) in vitro.42 Fig. 4B shows the viability/cytotoxicity percentages of HFB4 cells on the fabricated transdermal patches, reflecting both cytotoxicity and cellular proliferation inhibition. The results showed that the loaded patch acquired a greater cell viability percentage (about 98.20%) compared to the plain counterpart. Furthermore, it was found that the loaded patch exhibits 10.8% cytotoxicity on the HFB4 cells, in contrast to the plain patches, which attained a cytotoxicity of about 51.6%. On the other hand, a noticeable corresponding decline in the count of viable cells was observed in the plain patches, indicating that the presence of DEX- and HA-loaded transdermal patches proved a more convenient transdermal application coupled with better cell viability promotion than the plain one. Accordingly, it is supposed that the low cytotoxic effect of the developed transdermal patches may be due to the biocompatibility of their components, which mimic biological environments.In vivo experiments
 | ||
| Fig. 5 (A) Antimicrobial testing results of plain PCL NFs, NAR-loaded PCL NFs; and the whole (B) cytotoxicity testing of plain and drug-loaded transdermal patches on human skin fibroblast cells. SA: Staphylococcus aureus, ST: Salmonella typhimurium, PA: Pseudomonas aeruginosa. | ||
Histological evaluation of RA treatment
The photomicrograph of H&E-stained sections of the normal ankle joint (Fig. 5D) demonstrates a normal histological structure devoid of any obvious tissue loss or inflammation, as revealed by the absence of cell infiltration.41 On the other hand, the photomicrograph of H&E-stained sections of the control ankle joint demonstrates a histological structure with an obvious tissue damage (black arrow) and inflammation (represented by cell infiltration, blue arrow) (Fig. 5B). In addition, severe destruction of the ankle joint tissue can be seen in this animal group due to the progressive inflammation of the synovium. Moreover, the diseased rats treated with plain transdermal patches showed obvious tissue damage (black arrows, Fig. 5C). Conversely, diseased animals treated with the developed TDDPs showed a relatively normal joint architecture without overt indications of tissue damage or inflammation (Fig. 5E and F) like the normal tissue shown in Fig. 5D. Additionally, the photomicrographs of the TDDPs-treated diseased animal group showed no signs of cell invasion. Accordingly, the suggested TDDPs may be useful in treating RA in humans, as evidenced by the fact that they were able to alleviate the symptoms of the condition in diseased rats. However, more research is necessary to determine the precise and sensitive analytical procedures needed to measure the amount of drug released in the synovium. Additionally, before being used in future clinical trials, the study ought to be conducted on a different species.Conclusions
The current work aimed at creating a new double-layered transdermal platform by combining two fabrication techniques, namely electrospinning and 3D bioprinting, and then investigating its potential use in treating rheumatoid arthritis in rats. In addition, the study provided a novel hydrogel comprising chitosan, alginate, and PVA for 3D bioprinting purposes. According to the obtained characterization, in vitro and in vivo results, the recently developed double-layered transdermal patch shows promise in treating RA in the examined Rattus norvegicus animal model. Extensive examinations employing electron microscopy and physicochemical characterization methods demonstrated the effective synthesis of the intended transdermal patches. Experiments conducted in vitro and in vivo on the survival of cells and the levels of inflammatory mediators (TNF-α and IL-6) showed that the fabricated patches are biocompatible and can reduce RA symptoms by lowering TNF-α and IL-6 levels. In addition, the double-layer patches discussed herein provide an advantage over our previously published triple-layer patches because they provide better compliance, shorter manufacturing time, and a minimized manufacturing complexity. Future research should be done on assaying the concentrations of the bioactive substances under study in the synovial fluid upon treating the animals with the proposed TDDPs. Moreover, before examining the use of the suggested TDDPs in clinical trials, more investigations are required to determine the effectiveness of these patches in treating RA in a different animal species.Data availability
The data supporting this article has been included as part of the ESI.†Author contributions
HAE: methodology, investigation, writing – original draft. ASAD: conceptualization, methodology, investigation, formal analysis, visualization, writing – review and editing. MHS: methodology, writing – original draft. IME: conceptualization, funding acquisition, resources, supervision, writing – review and editing, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The corresponding author extends his appreciation to the Science, Technology, and Innovation Funding Authority (STDF), Egypt, for funding and supporting this work through the funds (Capacity Building Fund, CB-22808, and STDF FLUG Call 1-Project ID 46715).Notes and references
- M. D. Smith, Open Rheumatol. J., 2012, 5, 100–106 CrossRef PubMed.
- J. A. Singh, T. Arayssi, P. Duray and H. R. Schumacher, Ann. Rheum. Dis., 2004, 63, 785–790 CrossRef PubMed.
- L. Biswas, V. Shukla, V. Kumar and A. Kamra Verma, Smart Drug-Delivery Systems in the Treatment of Rheumatoid Arthritis: Current, Future Perspectives, 2021 Search PubMed.
- Y. Zhang, Z. Gao, S. Chao, W. Lu and P. Zhang, Drug Deliv., 2022, 29, 1934–1950 CrossRef PubMed.
- M. Rabiei, S. Kashanian, S. S. Samavati, H. Derakhshankhah, S. Jamasb and S. J. P. McInnes, J. Drug Deliv. Sci. Technol., 2021, 61, 102011 CrossRef CAS.
- M. Rahimi, G. Charmi, K. Matyjaszewski, X. Banquy and J. Pietrasik, Acta Biomater., 2021, 123, 31–50 CrossRef CAS PubMed.
- D. Kesharwani, R. Paliwal, T. Satapathy and S. Das Paul, J. Pharmacopuncture, 2019, 22, 210–224 CrossRef PubMed.
- D. Kesharwani, R. Paliwal, T. Satapathy and S. Das Paul, J. Pharmacopuncture, 2019, 22, 210–224 CrossRef PubMed.
- S. Nasra, D. Bhatia and A. Kumar, Nanoscale Adv., 2022, 4, 3479–3494 RSC.
- M. Amjadi, S. Sheykhansari, B. J. Nelson and M. Sitti, Adv. Mater., 2018, 30, 1704530 CrossRef.
- M. P. Venkatesh, T. P. Kumar and D. R. Pai, Saudi Pharmaceut. J., 2020, 28, 1548–1557 CrossRef PubMed.
- M. Rahimi, G. Charmi, K. Matyjaszewski, X. Banquy and J. Pietrasik, Acta Biomater., 2021, 123, 31–50 CrossRef PubMed.
- Z. Norouzi and M. Abdouss, Int. J. Biol. Macromol., 2023, 233, 123518 CrossRef PubMed.
- Z. Norouzi and M. Abdouss, Int. J. Biol. Macromol., 2023, 233, 123518 CrossRef PubMed.
- A. Roth, J. Mollenhauer, A. Wagner, R. Fuhrmann, A. Straub, R. A. Venbrocks, P. Petrow, R. Bräuer, H. Schubert, J. Ozegowski, G. Peschel, P. J. Müller and R. W. Kinne, Arthritis Res. Ther., 2005, 7, 677–686 CrossRef PubMed.
- M. M. Victor, J. M. David, M. C. K. Sakukuma, E. L. França and A. V. J. Nunes, Green Process. Synth., 2018, 7, 524–529 Search PubMed.
- K. Sudto, S. Pornpakakul and S. Wanichwecharungruang, Int. J. Food Sci. Technol., 2009, 44, 1737–1742 CrossRef.
- X. Li, H. Wang, X. Zou, H. Su and C. Li, Eur. J. Pharm. Sci., 2022, 170, 106101 CrossRef.
- A. A. van de Loosdrecht, R. H. J. Beelen, G. J. Ossenkoppele, M. G. Broekhoven and M. M. A. C. Langenhuijsen, J. Immunol. Methods, 1994, 174, 311–320 CrossRef PubMed.
- D. L. Asquith, A. M. Miller, I. B. McInnes and F. Y. Liew, Eur. J. Immunol., 2009, 39, 2040–2044 CrossRef.
- P. Huo, X. Han, W. Zhang, J. Zhang, P. Kumar and B. Liu, Pharmaceutics, 2021, 12, 1228 CrossRef.
- S. Mohanty, V. B. Konkimalla, A. Pal, T. Sharma and S. C. Si, ACS Omega, 2021, 6, 28630–28641 CrossRef PubMed.
- P. Guo, L. Dong, W. Yan, J. Wei, C. Wang and Z. Zhang, Biomed. Chromatogr., 2015, 29, 246–253 CrossRef.
- R. L. Nagula and S. Wairkar, Int. J. Biol. Macromol., 2020, 164, 717–725 CrossRef PubMed.
- J. Long, A. V. Nand, C. Bunt and A. Seyfoddin, Pharm. Dev. Technol., 2019, 24, 839–848 CrossRef.
- S. J. Kim, Y. Choi, K. T. Min and S. Hong, Pharmaceutics, 2022, 14(5), 985 CrossRef.
- K. S. Kim, S. J. Park, J. A. Yang, J. H. Jeon, S. H. Bhang, B. S. Kim and S. K. Hahn, Acta Biomater., 2011, 7, 666–674 CrossRef PubMed.
- H. Chopra, S. Bibi, S. Kumar, M. S. Khan, P. Kumar and I. Singh, Gels, 2022, 8, 111 CrossRef.
- M. Ghorbani, P. Nezhad-Mokhtari and S. Ramazani, Int. J. Biol. Macromol., 2020, 153, 921–930 CrossRef PubMed.
- J. Ye, M. Gong, J. Song, S. Chen, Q. Meng, R. Shi, L. Zhang and J. Xue, Pharmaceutics, 2022, 14, 1273 CrossRef.
- M. Gong, C. Chi, J. Ye, M. Liao, W. Xie, C. Wu, R. Shi and L. Zhang, Colloids Surf. B, 2018, 170, 201–209 CrossRef.
- I. M. Alarifi, A. Alharbi, W. S. Khan, A. Swindle and R. Asmatulu, Materials, 2015, 8, 7017–7031 CrossRef.
- P. Singh, S. J. Desai, A. P. Simonelli and W. I. Higuchi, J. Pharm. Sci., 1968, 57, 217–226 CrossRef.
- S. Yahia, A. S. Abo Dena, R. M. El Nashar and I. M. El-Sherbiny, J. Mater. Chem. B, 2022, 10, 4984–4998 RSC.
- J. Cai, H. Wen, H. Zhou, D. Zhang, D. Lan, S. Liu, C. Li, X. Dai, T. Song, X. Wang, Y. He, Z. He, J. Tan and J. Zhang, Biomed. Pharmacother., 2023, 164, 114990 CrossRef.
- P. Dey, D. Parai, M. Banerjee, S. T. Hossain and S. K. Mukherjee, Int. J. Med. Microbiol., 2020, 310(3), 151410 CrossRef.
- S. Moreno, T. Scheyer, C. S. Romano and A. A. Vojnov, Free Radic. Res., 2006, 40, 223–231 CrossRef PubMed.
- G. Celiz, M. Daz and M. C. Audisio, J. Appl. Microbiol., 2011, 111, 731–738 CrossRef.
- K. A. Lee, S.-H. Moon, J.-Y. Lee, K.-T. Kim, Y.-S. Park and H.-D. Paik, Food Sci. Biotechnol., 2013, 22, 1725–1728 CrossRef.
- F. J. Osonga, A. Akgul, R. M. Miller, G. B. Eshun, I. Yazgan, A. Akgul and O. A. Sadik, ACS Omega, 2019, 4, 12865–12871 CrossRef.
- M. Bahadoran, A. Shamloo and Y. D. Nokoorani, Sci. Rep., 2020, 10, 7–9 CrossRef.
- S. Sa’adon, M. N. M. Ansari, S. I. A. Razak, J. S. Anand, N. H. M. Nayan, A. E. Ismail, M. U. A. Khan and A. Haider, Polymers, 2021, 13, 1–18 CrossRef.
- M. R. Bhalekar, P. G. Upadhaya and A. R. Madgulkar, Eur. J. Pharm. Sci., 2016, 84, 1–8 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07306a |
| This journal is © The Royal Society of Chemistry 2024 |