
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ generated CF3CHN2 with 3-ylideneoxindoles to access CF3-containing pyrazolo[1,5-c]quinazolines derivatives†
Ming-Cheng Hua,
Hai-Tao Zhoua,
Yu-Chen Fanga,
Li-Ren Zhanga,
Bao-Dong Cuiab,
Yong-Zheng Chen *ab and
Mei Bai*ab
*ab and
Mei Bai*ab
aKey Laboratory of Biocatalysis & Chiral Drug Synthesis of Guizhou Province, School of Pharmacy, Zunyi Medical University, Zunyi 563000, China
bKey Laboratory of Basic Pharmacology of Ministry of Education, Joint International Research Laboratory of Ethnomedicine of Ministry of Education, Zunyi Medical University, Zunyi 563000, China
First published on 14th November 2024
Abstract
Toward a selective and facile method for the synthesis of CF3-containing pyrazolo[1,5-c]quinazolines, we developed a [3 + 2] cycloaddition/1,3-H shift/rearrangement/dehydrogenation cascade involving in situ generated CF3CHN2 and 3-ylideneoxindoles with DBU as a base. The reaction is distinguished by its mild conditions, metal-free process, operational simplicity, and broad functional group tolerance, thus presenting a convenient protocol for the construction of pyrazolo[1,5-c]quinazolines that are of interest in medicinal chemistry.
Introduction
The trifluoromethyl group, which can be regarded as a classical group in drugs and drug candidates, was introduced to modulate physicochemical properties and increase the binding affinity of molecules. For instance, the drugs Prozac, Celebrex and Januvia are all active agents.1 Therefore, the introduction of the trifluoromethyl group is of great interest in various areas of organic and medicinal chemistry.2 In particular, direct trifluoromethylation plays a pivotal part in organic chemistry. Numerous methods have been demonstrated to be able to promote C(sp2)–CF3 bond formation, including nucleophilic trifluoromethylation,3 electrophilic trifluoromethylation,4 and radical trifluoromethylation.5 In recent years, radical trifluoromethylation has been explored as a powerful tool for the construction of the C(sp2)–CF3 bond.6 Trifluoromethyl free radicals serve as active species in radical trifluoromethylation.7Alternatively, other routes based on the chemical conversions of CF3-containing building blocks,8 especially trifluorodiazoethane (CF3CHN2),9 have emerged as a valuable reagent for introducing trifluoromethyl groups into heterocyclic compounds. CF3CHN2 readily participates in [2 + 1]-, [3 + 2]-, and [3 + 3]-cycloaddition reactions as a trifluoromethyl-containing dipole, thereby creating new approaches to synthesize CF3-containing heterocycles.10 This impressive work stemmed from a study by the group of Xiao and Lu,11 who reported a [3 + 2] cycloaddition/ring contraction sequence of 3-ylideneoxindoles with in situ generated CF3CHN2 without a transition-metal catalyst. Initial [3 + 2] cycloaddition occurred smoothly to give corresponding cycloadducts, and the subsequent ring contraction reaction worked very well in refluxing PhMe to afford the final CF3-substituted 3,3′-cyclopropyl spirooxindoles.
Owing to the significant biological activities of both quinazoline and pyrazole derivatives, pyrazolo[1,5-c]quinazolines formed by N-fusing quinazolines with pyrazoles have occupied an important position in drug design. Examples of the biological activities of pyrazolo[1,5-c]quinazolines include Gly/NMDA antagonists,12 phosphodiesterase 10A inhibitors,13 benzodiazepine/adenosine receptors,14 and AMPA receptors (Fig. 1).15 Accordingly, considerable efforts have been undertaken to develop efficient synthetic strategies for these fascinating bioactive molecules.16 However, some involved multistep synthesis, harsh reaction conditions, lower yields, or poor chemoselectivity. Therefore, the development of readily available and easily accessible pyrazolo[1,5-c]quinazolines is exciting and practically useful.
 | ||
| Fig. 1 Some biologically active compounds with a pyrazolo[1,5-c]quinazoline core. | ||
In 2021, we reported a visible-light-induced [3 + 2] cycloaddition of α-ketoacids with a hypervalent iodine(III) reagent derived from in situ generated CF3CHN2 for the synthesis of 5-CF3-1,3,4-oxadiazoles (Scheme 1A).17 Recently, a novel sulfonium salt derived from in situ generated CF3CHN2 was prepared by our group, and it was applied in Rh-catalyzed [2 + 1 + 2] cycloadditions to afford imidazo[1,5-a] N-heterocycles (Scheme 1B).18 As part of our continuing interests in organic fluorine chemistry,19 as well as our recent progress in cycloadditions for the synthesis of N-heterocycles from in situ generated CF3CHN2 and its derivatives,17–19 we further applied a [3 + 2] cycloaddition/1,3-H shift/rearrangement/dehydrogenation cascade process starting from easily available 3-ylideneoxindoles and CF3CHN2 to efficiently form pyrazole[1,5-c]quinazoline containing a CF3 group. Herein, we wish to report our preliminary studies on this subject (Scheme 1C).
 | ||
| Scheme 1 Applications of in situ generated CF3CHN2 in the construction of CF3-containing N-heterocycles. | ||
Results and discussion
We commenced our investigation by screening reaction parameters for the coupling between in situ generated CF3CHN2 (2) and 3-ylideneoxindole 3a (Table 1). Using DBU as a base, a coupling occurred, and the desired CF3-containing pyrazole[1,5-c]quinazoline product 4a was isolated in 40% yield in the presence of Et2O (Table 1, entry 1). Switching the solvent to DCM slightly decreased the yield (Table 1, entry 4). Different solvents were also investigated; however, this led to no desired reaction (Table 1, entries 2 and 3). The reaction time is significantly prolonged when utilizing alternative solvents, such as Et2O and DCM, which require a duration of 120 h. Interestingly, employing PhMe as the reaction solvent enables the completion of the entire reaction within a span of 48 h. PhMe was found to be superior to the others, and product 4a was obtained in 78% yield within the optimal reaction time (48 h) (Table 1, entry 5). We also screened the bases required for the reaction and found that Cs2CO3, K2CO3, Et3N, NaOH, and tBuOK (Table 1, entries 9–13) could not perform the expected reaction. Only DBN provides the required product 4a in 50% yield (Table 1, entry 14). Screening with base loading showed that 0.5 equiv. was better than other methods, with a yield of 78% (Table 1, entries 15–17 vs. entry 5). The effect of temperature was also examined, and all resulted in slightly reduced yields, and room temperature was confirmed to be the best one (Table 1, entries 18–20 vs. entry 5). Therefore, the optimal reaction conditions were summarized: CF3CH2NH2·HCl (203.25 mg, 1.5 mmol, 6.0 equiv.), NaNO2 (115.5 mg, 1.65 mmol, 6.6 equiv.) and H2O (0.2 mL) were stirred in 5.0 mL of PhMe at 0 °C for 2 h in a 100 mL double-necking bottle under Ar. After drying Na2SO4, 3a (57.6 mg, 0.25 mmol, 1.0 equiv.) and DBU (0.5 equiv.) were added, and the reaction mixture was stirred at room temperature under an O2 atmosphere (balloon) for 48 h.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temperature | Base (x equiv.) | Yieldb (%) |
| a Standard conditions: performed with CF3CH2NH2·HCl (1.5 mmol, 6.0 equiv.), NaNO2 (1.65 mmol, 6.6 equiv.) 0.2 mL H2O at 0 °C for 2 h. The mixture was dried, and later 3a (0.25 mmol, 1.0 equiv.), DBU (0.5 equiv.) and 5 mL of drying solvent were added at room temperature under an O2 atmosphere (balloon) for 48 h.b Isolated yields based on 3a are given.c The reaction was performed for 120 h.d The reaction was performed in 3 mL of dry solvent.e The reaction was performed in 10 mL dry solvent.f The reaction was performed in 15 mL dry solvent. | ||||
| 1c | Et2O | r.t. | DBU (0.5) | 40 |
| 2c | 1,4-Dioxane | r.t. | DBU (0.5) | N.R. |
| 3c | THF | r.t. | DBU (0.5) | N.R. |
| 4c | DCM | r.t. | DBU (0.5) | 33 |
| 5 | PhMe | r.t. | DBU (0.5) | 78 |
| 6d | PhMe | r.t. | DBU (0.5) | 39 |
| 7e | PhMe | r.t. | DBU (0.5) | 55 |
| 8f | PhMe | r.t. | DBU (0.5) | 68 |
| 9 | PhMe | r.t. | Cs2CO3 (0.5) | N.R. |
| 10 | PhMe | r.t. | K2CO3 (0.5) | N.R. |
| 11 | PhMe | r.t. | Et3N (0.5) | N.R. |
| 12 | PhMe | r.t. | NaOH (0.5) | N.R. |
| 13 | PhMe | r.t. | tBuOK (0.5) | N.R. |
| 14 | PhMe | r.t. | DBN (0.5) | 50 |
| 15 | PhMe | r.t. | DBU (0.25) | 60 |
| 16 | PhMe | r.t. | DBU (0.75) | 60 |
| 17 | PhMe | r.t. | DBU (1.0) | 50 |
| 18 | PhMe | −10 °C | DBU (0.5) | 66 |
| 19 | PhMe | 0 °C | DBU (0.5) | 59 |
| 20 | PhMe | 40 °C | DBU (0.5) | 46 |

With the optimized reaction conditions in hand, the scope and generality concerning CF3CHN2 were next examined in the coupling of compound 4 (Scheme 2). The introduction of fluorine, chlorine, and bromine substituents at different positions of 3-ylideneoxindole was tolerated under mild conditions, and the products were isolated in 35–86% yield (4a–w). The reaction was also compatible with disubstituted ethyl (E)-2-(1,5,7-trimethyl-2-oxoindolin-3-ylidene) acetate that contains an electron-donating substituent on the phenyl ring, affording 4p in 57% yields. Steric hindrance seems to play an important factor in affecting the results, with 3-ylideneoxindole bearing different protecting groups (Ph, Bn, and allyl) on nitrogen reacting quite well with 2, giving 4r, 4s, and 4t in moderate yields. In contrast, the addition of a Boc-protecting group to 3-ylideneoxindole results in the removal of the Boc-protecting group in 37% yield (4q), and the same product can be obtained without any group on nitrogen, but the yield is relatively high (71%). The 3-ylideneoxindoles bearing variation of the ester moiety (nPr, iPr, or tBu) could also undergo this [3 + 2] cycloaddition/1,3-H shift/rearrangement/dehydrogenation cascade process, though leading to the corresponding products in moderate yields (4u–w). The structure of product 4a was also unambiguously confirmed by single-crystal X-ray analysis.
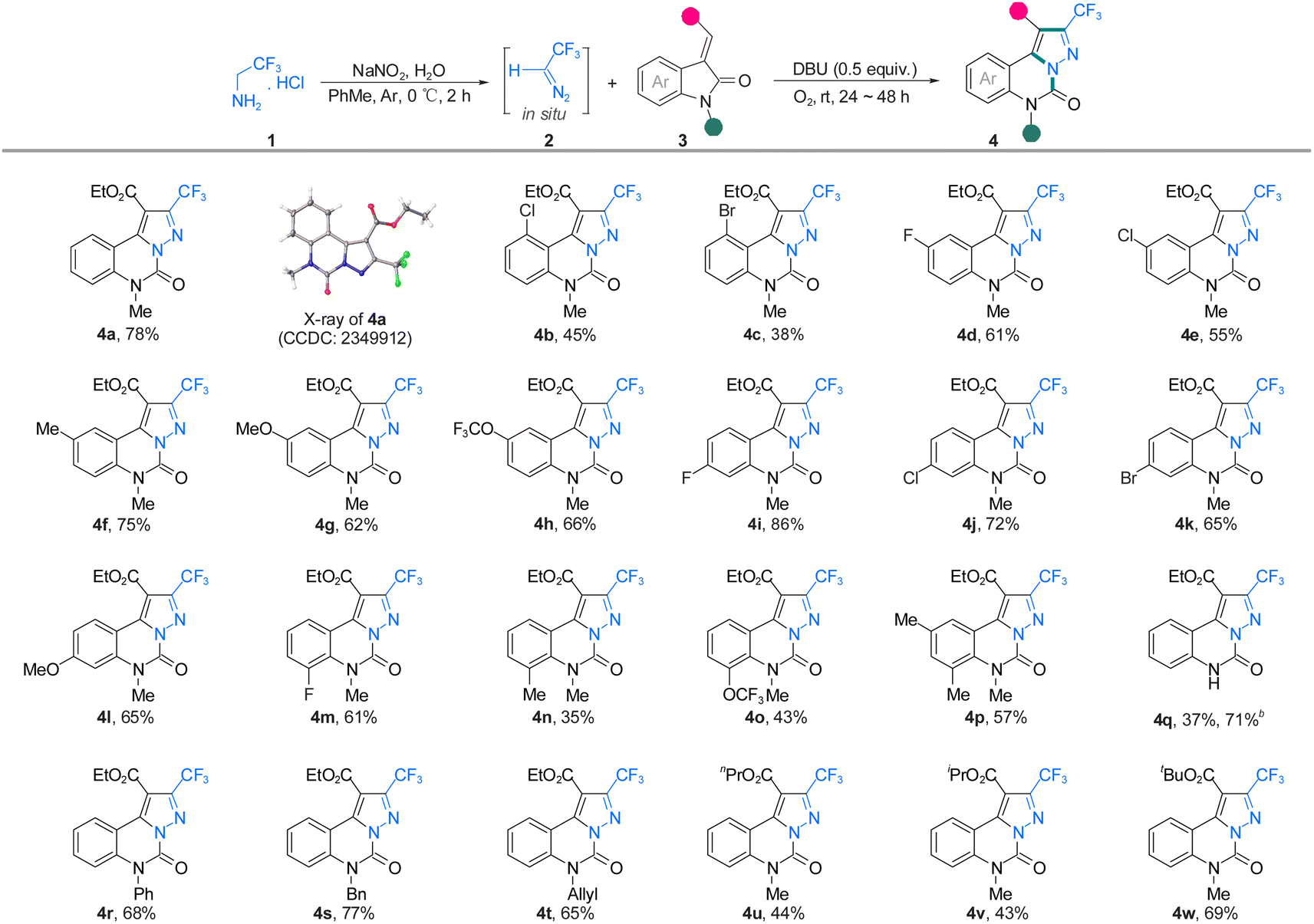 | ||
| Scheme 2 Substrate scopea. aCH3CH2NH2·HCl (1.5 mmol), NaNO2 (1.65 mmol) 3-acyldioxoindole (0.25 mmol), DBU (0.125 mmol) were stored in PhMe (5 mL) at room temperature under an O2 atmosphere (balloon) for 48 h. bThe substrate for this reaction is ethyl (E)-2-(2-oxoindolin-3-ylidene) acetate instead of tert-butyl (E)-3-(2-ethoxy-2-oxoethylidene)-2-oxoindoline-1-carboxylate. | ||
To demonstrate the high efficiency of the reaction, 4a and 4k were scaled up under optimized conditions. As shown in Scheme 3, the final yield of 4a is 1.02 g with a 70% yield. The yield of 4k was 65% to 1.64 g. At the same time, the practicability of the reaction is further analyzed. The resulting product 4k could be further transformed to 5 in 76% yield, 6 in 88% yield, 7 in 81% yield, 8 in 85% yield, 9 in 80% yield, and 10 in 92% yield through transition-metal-catalyzed Sonogashira coupling, Suzuki–Miyaura coupling, Heck coupling, Miyaura borylation, Ullmann–Ma amination and Stille coupling, respectively (Scheme 4).20
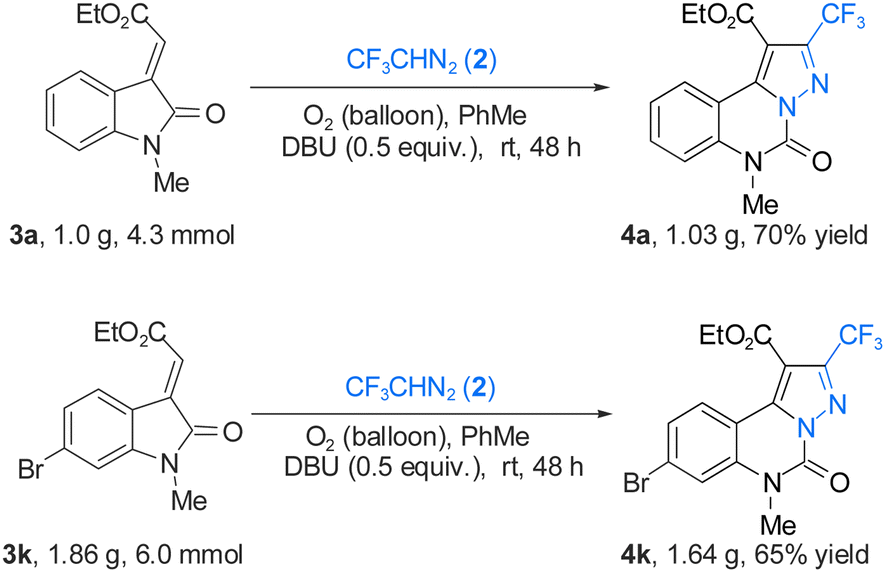 | ||
| Scheme 3 Gram-scale reactions. | ||
 | ||
| Scheme 4 Transformation of 4k. (a) Pd(PPh3)2Cl2, CuI, Et3N, phenylacetylene, DMF, Ar, 60 °C, and 24 h. (b) PhB(OH)2, Cs2CO3, Pd(OAc)2, nBuPAd2, DCE, Ar, 80 °C, and 12 h. (c) Styrene, Pd(PPh3)4, PPh3, Et3N, DMF, Ar, 140 °C, and 8 h. (d) Pd(OAc)2, PCy3, (BPin)2, KOAc, PhMe, Ar, 70 °C, and 2 h. (e) Pd2(dba)3, XPhos, CsCO3, NH2Boc, PhMe, Ar, 110 °C, and 12 h. (f) Pd(PPh3)4, allyltributylstannane, DMF, Ar, 90 °C, and 1 h. | ||
To acquire more information on the mechanism of this cascade reaction, the presence of an intermediate in the reaction liquid under argon was detected by HRMS (Fig. 2), and the template reaction was monitored by 1H NMR. By monitoring the reaction mixture for 12 h, the reaction raw material 3a, and the reaction intermediate 11a, it was found that the corresponding characteristic peak appeared in 1H NMR of the mixture (Fig. 3). By monitoring 1H NMR of the mixed solution 12 h after the reaction, we found that substrate 3a was completely consumed, and the characteristic peaks of intermediate product 11a and product 4a appeared. To investigate the reaction mechanism based on the key intermediates 13a, 11a, and 12a, three control experiments were explored. However, it is troublesome to obtain the intermediate 13a owing to its scarcity, but the presence of 11a can be detected by 1H NMR. Notably, 11a can transform to intermediate 12a under the Ar atmosphere, and 4a has been realized from 11a in 80% yield under the O2 atmosphere (Scheme 5). These results indicate that 11a, 12a, and 13a are possible intermediates for the formation of 4a.
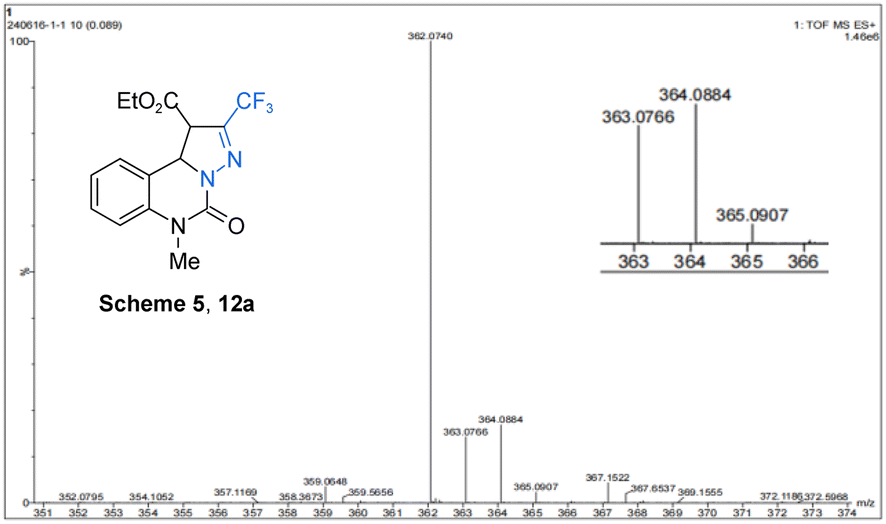 | ||
| Fig. 2 HRMS of intermediate 12a. | ||
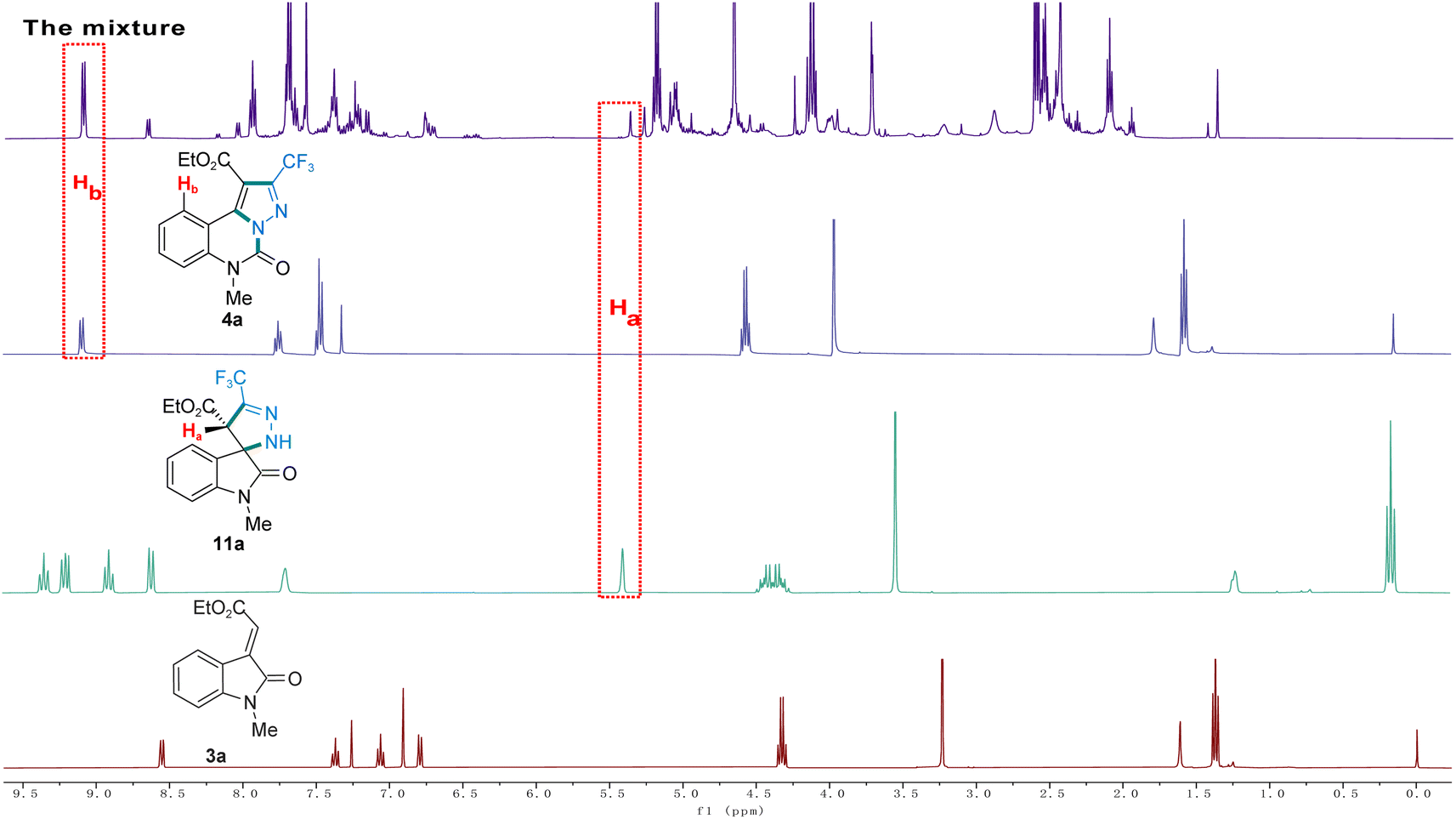 | ||
| Fig. 3 1H NMR determination of the possible intermediates. | ||
 | ||
| Scheme 5 Control experiments. | ||
Based on the above results and our previous reports,19c we postulate a plausible mechanism, which is depicted in Scheme 6. In situ generated CF3CHN2 with 3-ylideneoxindoles via [3 + 2] cycloaddition forms intermediate 13, followed by a 1,3-H shift process to yield the intermediate 11. Intermediate 11 is deprotonated and closed by DBU to obtain intermediate 14, followed by ring-opening and [1,5]-σ rearrangement to obtain intermediate 15. Intermediate 12 is obtained by protonation of intermediate 15. Finally, intermediate 12 is dehydrogenated in the presence of oxygen to obtain compound 4. In particular, due to the conjugated system of compound 4, the dehydrogenation step might be a driving force for the transformation of intermediate 11 into fused heterocycles 4.
 | ||
| Scheme 6 Proposed reaction mechanism. | ||
Xiao and Lu's group have developed the example of a sequential [3 + 2] cycloaddition/ring contraction reaction of 3-ylideneoxindoles with in situ-generated CF3CHN2, carried out in PhMe under Ar atmosphere, to deliver CF3-containing 3,3′-cyclopropyl spirooxindole derivatives.11 In contrast, our synthetic strategy opens the route to pyrazolo[1,5-c]quinazolines by a 1,3-dipolar cycloaddition in the presence of DBU in PhMe under the O2 atmosphere. The desired spirooxindole 4 was afforded in moderate to 86% yields. The reaction proceeded under mild conditions with broad moderate to well yields. The mild reaction conditions, broad substrate scope, and simple and convenient handling make this method of accessing functionalized CF3-containing N-heterocycle practical and fascinating.
Conclusions
An atom-economical protocol of valuable and versatile pyrazolo[1,5-c]quinazolines has been achieved by a sequential [3 + 2] cycloaddition/1,3-H shift/rearrangement/dehydrogenation process, proceeding in moderate to good yields at room temperature. The reaction proceeded under mild conditions with good functional group tolerance, which also constructed pyrazolo[1,5-c]quinazolines from easily accessible 3-ylideneoxindoles and CF3CHN2 without the need for multistep parallel synthesis. Efforts toward expanding this methodology to a broader 3-ylideneoxindole scope are currently underway. Further research on this topic with in situ generated CF3CHN2 is underway in our laboratory.21Experimental section
General experimental information
Unless otherwise specified, all commercially available reagents are utilized without further purification. Silica gel column chromatography (300–400 mesh) is employed. 1H, 19F, and 13C NMR spectra were measured on a 400, 376, and 100 MHz spectrometer, respectively. CDCl3 was used as a solvent. 19F chemical shifts were given as δ in ppm downfield from CFCl3. High-resolution mass spectra HRMS measurements were obtained on a TOF analyzer.The preparation of 3-ylideneoxindoles was conducted following the previously reported method.
Representative procedure for the synthesis of compound 4a
A 50 mL flask with a stir bar was charged with CF3CH2NH2·HCl (1.5 mmol, 6.0 equiv.), NaNO2 (1.65 mmol, 6.6 equiv.), 0.2 mL H2O at 0 °C for 2 h. The mixture was then dried, then 3a (0.25 mmol, 1.0 equiv.), DBU (0.5 equiv.) and 5 mL of the drying solvent were added at room temperature under an O2 atmosphere (balloon) for 48 h (as monitored by TLC). Finally, the mixture was concentrated under reduced pressure and then purified by silica gel flash column chromatography (using a solvent system of petroleum ether/ethyl acetate = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to obtain the target compound 4a (65.9 mg, yield 78%).
:1) to obtain the target compound 4a (65.9 mg, yield 78%).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful for financial support from the Science and Technology Department of Zunyi (ZSKHHZ-[2020]70, ZSKRPT-2020-5), Science and Technology Department of Guizhou Province (QKHPTRC-GCC[2022]032-1), and Science and Technology Department of Guizhou Province (QKHPTRC-CXTD[2022]012).Notes and references
- (a) K. A. Lyseng-Williamson, Drugs, 2007, 67, 587–597 CrossRef CAS; (b) I. Egashira, F. Takahashi-Yanaga, R. Nishida, M. Arioka, K. Igawa, K. Tomooka, Y. Nakatsu, T. Tsuzuki, Y. Nakabeppu, T. Kitazono and T. Sasaguri, Cancer Sci., 2017, 108, 108–115 CrossRef CAS PubMed; (c) J. B. Buse, M. A. Bethel, J. B. Green, S. R. Stevens, Y. Lokhnygina, P. Aschner, C. R. Grado, T. Tankova, J. Wainstein, R. Josse, J. M. Lachin, S. S. Engel, K. Patel, E. D. Peterson and R. R. Holman, Diabetes Care, 2017, 40, 164–170 CrossRef CAS PubMed; (d) M. Imamura, Y. Okamoto, T. Nishikawa, T. Yoneyama, Y. Yamasaki, J. Kawamura and Y. Kawano, Pediatrics, 2019, 144, e20190319 CrossRef PubMed; (e) M. Kazi, A. Alqahtani, A. Ahmad, O. M. Noman, M. S. Aldughaim, A. S. Alqahtani and F. K. Alanazi, Drug Delivery, 2021, 28, 100–114 CrossRef CAS.
- (a) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475–4521 CrossRef CAS PubMed; (b) X. Mu, S. Chen, X. Zhen and G. Liu, Chem. – Eur. J., 2011, 17, 6039–6042 CrossRef CAS PubMed; (c) X. Mu, T. Wu, H.-Y. Wang, Y.-L. Guo and G. Liu, J. Am. Chem. Soc., 2012, 134, 878–881 CrossRef CAS PubMed; (d) C. Chen, L. Chu and F.-L. Qing, J. Am. Chem. Soc., 2012, 134, 12454–12457 CrossRef CAS PubMed; (e) R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2012, 134, 12462–12465 CrossRef CAS PubMed; (f) L. Chu and F.-L. Qing, J. Am. Chem. Soc., 2012, 134, 1298–1304 CrossRef CAS; (g) Y. Zeng, L. Zhang, Y. Zhao, C. Ni, J. Zhao and J. Hu, J. Am. Chem. Soc., 2013, 135, 2955–2958 CrossRef CAS PubMed; (h) Z.-B. He, R. Zhang, M.-Y. Hu, L.-C. Li, C.-F. Ni and J.-B. Hu, Chem. Sci., 2013, 4, 3478–3483 RSC; (i) M. Huang and C. Zhang, Org. Lett., 2024, 26, 4158–4162 CrossRef CAS PubMed.
- (a) J. A. Pike and J. W. Walton, Chem. Commun., 2017, 53, 9858–9861 RSC; (b) K. Hirano, T. Saito, Y. Fujihira, D. M. Sedgwick, S. Fustero and N. Shibata, J. Org. Chem., 2020, 85, 7976–7985 CrossRef CAS; (c) Q. Wang, Q. Tao, H. Dong, C. Ni, X.-M. Xie and J.-B. Hu, Angew. Chem., Int. Ed., 2021, 60, 27318–27323 CrossRef CAS; (d) S. Mizuta, K. Kitamura, Y. Morii, J. Ishihara, T. Yamaguchi and T. Ishikawa, J. Org. Chem., 2022, 87, 5035 CrossRef CAS.
- (a) Q. Glenadel, E. Ismalaj and T. Billard, J. Org. Chem., 2016, 81, 8268–8275 CrossRef CAS PubMed; (b) J. Wang, X.-X. Lu, R.-P. Yang, B.-B. Zhang, Z.-H. Xiang, J.-C. Li, L. Liu, S. Chao and X. Shang, Org. Lett., 2023, 25, 8489–8494 CrossRef CAS; (c) Y.-Y. Hu, X.-Q. Xu, W.-C. Deng, R.-X. Liang and Y.-X. Jia, Org. Lett., 2023, 25, 6122–6127 CrossRef CAS.
- (a) H. Xiao, Z. Zhang, Y. Fang, L. Zhu and C. Li, Chem. Soc. Rev., 2021, 50, 6308–6319 RSC; (b) Y. Li, X. Liang, K. Niu, J. Gu, F. Liu, Q. Xia, Q. Wang and W. Zhang, Org. Lett., 2022, 24, 5918–5923 CrossRef CAS PubMed.
- Z.-H. Lu, H. Liu, S.-H. Liu, X.-B. Leng, Y. Lan and Q.-L. Shen, Angew. Chem., Int. Ed., 2019, 58, 8510–8514 CrossRef CAS PubMed.
- J.-A. Ma and D. Cahard, J. Fluorine Chem., 2007, 128, 975–996 CrossRef CAS.
- (a) C.-L. Zhu, L.-J. Yang, S. Li, Y. Zheng and J.-A. Ma, Org. Lett., 2015, 17, 3442–3445 CrossRef CAS PubMed; (b) Z. Chen, Y. Zheng and J.-A. Ma, Angew. Chem., Int. Ed., 2017, 56, 4569–4574 CrossRef CAS PubMed; (c) S. Qin, Y. Zheng, F.-G. Zhang and J.-A. Ma, Org. Lett., 2017, 19, 3406–3409 CrossRef CAS; (d) Z. Chen, N. Ren, X. Ma, J. Nie, F.-G. Zhang and J.-A. Ma, ACS Catal., 2019, 9, 4600–4608 CrossRef CAS; (e) X. Zhang, Z. Liu, X. Yang, Y. Dong, M. Virelli, G. Zanoni, E. A. Anderson and X. Bi, Nat. Commun., 2019, 10, 284 CrossRef; (f) X.-W. Zhao, W. Q. Zhu, Y.-R. Shi, J. Zhang, H. Li, M.-G. Yang and Y. Li, Chem. – Eur. J., 2024, 30, e202304056 CrossRef CAS PubMed.
- P. K. Mykhailiuk, Chem. Rev., 2020, 120, 12718–12755 CrossRef CAS.
- (a) Y.-J. Chen, F.-G. Zhang and J.-A. Ma, Org. Lett., 2021, 23, 6062–6066 CrossRef CAS PubMed; (b) A. Kumar, W. A. Khan, S. Ahamad and K. Mohanan, J. Heterocycl. Chem., 2022, 59, 607–632 CrossRef; (c) C.-F. Gao, Y.-J. Chen, J. Nie, F.-G. Zhang, C.-W. Cheung and J.-A. Ma, Chem. Commun., 2023, 59, 11664–11667 RSC; (d) S. Kumar, L. Fatma, N. K. Vaishanv and K. Mohanan, J. Org. Chem., 2024, 89, 761–769 CrossRef CAS.
- T.-R. Li, S.-W. Duan, Y.-Y. Liu, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, J. Org. Chem., 2014, 79, 2296–2302 CrossRef CAS PubMed.
- (a) M. Bacilieri, F. Varano, F. Deflorian, M. Marini, D. Catarzi, V. Colotta, G. Filacchioni, A. Galli, C. Costagli, C. Kaseda and S. Moro, J. Chem. Inf. Model., 2007, 47, 1913–1922 CrossRef CAS; (b) F. Varano, D. Catarzi, V. Colotta, D. Poli, G. Filacchioni, A. Galli and C. Costagli, Chem. Pharm. Bull., 2009, 57, 826–829 CrossRef CAS.
- B. Asproni, G. Murineddu, A. Pau, G. A. Pinna, M. Langgard, C. T. Christoffersen, J. Nielsen and J. Kehler, Bioorg. Med. Chem., 2011, 19, 642–649 CrossRef CAS PubMed.
- (a) V. Colotta, D. Catarzi, F. Varano, G. Filacchioni and L. Cecchi, J. Med. Chem., 1996, 39, 2915–2921 CrossRef CAS PubMed; (b) D. Catarzi, V. Colotta, F. Varano, D. Poli, L. Squarcialupi, G. Filacchioni, K. Varani, F. Vincenzi, P. A. Borea, D. D. Ben, C. Lambertucci and G. Cristalli, Bioorg. Med. Chem., 2013, 21, 283–294 CrossRef CAS PubMed.
- (a) S. S. Nikam and B. E. Kornberg, Curr. Med. Chem., 2001, 8, 155–170 CrossRef CAS; (b) F. Varano, D. Catarzi, V. Colotta, O. Lenzi, G. Filacchioni, A. Galli and C. Costagli, Bioorg. Med. Chem., 2008, 16, 2617–2626 CrossRef CAS.
- (a) M. Garg, M. Chauhan, P. K. Singh, J. M. Alex and R. Kumar, Eur. J. Med. Chem., 2015, 97, 444–461 CrossRef CAS; (b) L. Liu, L. Li, S.-K. Mao, X. Wang, M.-D. Zhou, Y.-L. Zhao and H. Wang, Chem. Commun., 2020, 56, 7665–7668 RSC.
- W.-W. Zhao, Y.-C. Shao, A.-N. Wang, J.-L. Huang, C.-Y. He, B.-D. Cui, N.-W. Wan, Y.-Z. Chen and W.-Y. Han, Org. Lett., 2021, 23, 9256–9261 CrossRef CAS PubMed.
- W.-W. Zhao, M.-Y. Tian, Y.-L. Zhou, L.-J. Liu, S.-F. Tian, C.-Y. He, X.-Z. Yang, Y.-Z. Chen and W.-Y. Han, Angew. Chem., Int. Ed., 2024, 63, e202318887 CrossRef CAS.
- (a) W.-Y. Han, J. Zhao, J.-S. Wang, G.-Y. Xiang, D.-L. Zhang, M. Bai, B.-D. Cui, N.-W. Wan and Y.-Z. Chen, Org. Biomol. Chem., 2017, 15, 5571–5578 RSC; (b) W.-Y. Han, J. Zhao, J.-S. Wang, B. -D Cui, N.-W. Wan and Y.-Z. Chen, Tetrahedron, 2017, 73, 5806–5812 CrossRef CAS; (c) W.-Y. Han, J.-S. Wang, J. Zhao, L. Long, B.-D. Cui, N.-W. Wan and Y.-Z. Chen, J. Org. Chem., 2018, 83, 6556–6565 CrossRef CAS; (d) J.-S. Wang, J. Shan, M. Bai, B.-D. Cui, N.-W. Wan, Y.-S. Wang, W.-Y. Han and Y.-Z. Chen, Tetrahedron, 2018, 74, 3904–3911 CrossRef CAS; (e) J.-S. Wang, K.-S. Huang, W.-Y. Han, B.-D. Cui, N.-W. Wan and Y.-Z. Chen, Org. Lett., 2019, 21, 8751–8755 CrossRef CAS PubMed; (f) Z.-H. Wang, J.-H. Liu, Y.-P. Zhang, J.-Q. Zhao, Y. You, M.-Q. Zhou, W.-Y. Han and W.-C. Yuan, Org. Lett., 2022, 24, 4052–4057 CrossRef CAS.
- (a) D. Arora, H. Kumar, D. Malhotra and M. Malhotra, Pharmacologyonline, 2011, 3, 659–668 Search PubMed; (b) Y.-C. Fang, J.-H. Chen, R.-F. Xiu, L.-R. Zhang, F.-H. Zheng, Y.-Z. Chen, Z.-W. Gao and W.-Y. Han, Org. Chem. Front., 2023, 10, 3752–3759 RSC.
- Z. Su, Z. Xie, S. Wang, N. Luo and C. Wang, Org. Biomol. Chem., 2019, 17, 7342–7351 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectra data of new products, and single crystal data of compound 4aa. CCDC 2349912. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra06651k |
| This journal is © The Royal Society of Chemistry 2024 |