
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydroxyapatite supported molybdenum oxide catalyst for selective dehydrogenation of cyclohexane to cyclohexene: studies of dispersibility and chemical environment
Mingxiao Zhenga,
Feng Zhoub,
Huixia Mab,
Xuefeng Songa and
Guang Wu *a
*a
aSchool of Chemistry and Materials Sciences, Research Institute of Crop Science, Heilongjiang University, Harbin 150080, China. E-mail: Wu.guang@163.com
bDalian Reserch Institute of Petroleum and Petrochemicals, SINOPEC, Dalian 116045, China
First published on 15th November 2024
Abstract
The selective oxidative dehydrogenation of cyclohexane to cyclohexene was conducted using molybdenum oxide (MoOx) as a catalyst and hydroxyapatite (HAP) and Ca5(OH)(PO4)3 as carriers. Two series of MOx/HAP catalysts with varying MoOx loading capacity and calcination temperature were prepared via the co-impregnation method. The impact of dispersibility and chemical environment on the catalytic performance of MoOx was investigated. The catalysts were characterized using XRD, XPS, H2-TPR, and UV-Vis spectra. These MoOx/HAP catalysts were employed for the oxidative dehydrogenation (ODH) of cyclohexane to cyclohexene. MoOx/HAP catalysts with lower loading capacity exhibited higher dispersion of MoOx and selectivity towards cyclohexane. The calcination temperature directly influenced the chemical environment of MoOx, thereby affecting its catalytic performance. Samples calcinated at lower temperatures (500 °C and 600 °C) demonstrated higher conversion rates for cyclohexane, while samples calcinated at higher temperatures (above 700 °C) displayed greater selectivity towards cyclohexane. At 430 °C, when the conversion rate of cyclohexane reached 13.1%, the selectivity of cyclohexene over MHAP-0.05-800 catalyst reached 58.2%.
Introduction
Cyclohexene is an ideal raw material for the synthesis of cyclohexanol, cyclohexanone, and adipic acid, which are in high demand in the chemical fiber industry. Due to its active double bond and six-membered ring structure, cyclohexene is also widely used in the production of fine chemicals such as pharmaceuticals, pesticides, dyes, detergents, feed additives, and polyester.1,2 Currently, the main methods for producing cyclohexene include catalytic dehydration of cyclohexanol, dehalogenation of halogenated cyclohexane, and selective hydrogenation of benzene.3,4 The raw material costs associated with the first two routes are relatively high and involve highly corrosive inorganic acids during the production processes. This increases equipment costs as well as environmental expenses. The selective hydrogenation of benzene to synthesize cyclohexene offers both availability and cost-effectiveness while aligning with the principles of atomic economy and environmental sustainability. However, its selectivity for producing pure cyclohexene is relatively low resulting in approximately 30% by-product formation of cyclohexane. Oxidative dehydrogenation (ODH) of cyclohexane to produce cyclohexene has gained significant attention due to its potential practical applications.5–7 In theory, cyclohexane exhibits thermodynamic stability compared to benzene; however, cyclohexane demonstrates higher reactivity making it prone to hydrogenate into cyclohexane or further dehydrogenate into benzene. The direct dehydrogenation process requires high reaction temperatures which can lead to carbon chain fragmentation or extensive coking. On top that, the process suffers from poor product selectivity along with short catalyst lifespan.8,9 Conversely, the ODH method reduces both reaction temperature requirements and carbon deposition on catalyst surfaces thereby extending catalyst lifespan. Hence, it has become a viable research approach for synthesizing cyclohexane. Therefore, it has become a feasible research approach for cyclohexene synthesis.The catalyst system serves as the fundamental component of the ODH process. Various catalytic systems have been reported for cyclohexene production through ODH, with particular attention given to vanadium-containing systems, molybdenum oxide systems, and other metal oxide catalysts.10–12 However, undesired COx and benzene byproducts often result from deep oxidation and dehydrogenation.13–15 While V5+ exhibits excellent C–H bond activation in hydrocarbon molecules, the presence of V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in its structure promotes the formation of C–O bonds and its high affinity for C–C bond dissociation, leading to a significant amount of COx in the final product.16–19 MoO3 possesses an acidic non-stoichiometric octahedral oxide structure with numerous oxygen vacancies. The REDOX reactions between Mo5+ and Mo6+ maintain a dynamic balance between structural defects and gas-phase oxygen. The octahedral structure of Mo6+ facilitates deep oxidation reactions, while the saturation coordination structure of Mo5+ enhances olefin selectivity in ODH reactions.20 Therefore, molybdenum-based catalysts not only offer potential for highly selective olefin production during ODH reactions but also their composition significantly influences cycloalkane ODH reactions.
O in its structure promotes the formation of C–O bonds and its high affinity for C–C bond dissociation, leading to a significant amount of COx in the final product.16–19 MoO3 possesses an acidic non-stoichiometric octahedral oxide structure with numerous oxygen vacancies. The REDOX reactions between Mo5+ and Mo6+ maintain a dynamic balance between structural defects and gas-phase oxygen. The octahedral structure of Mo6+ facilitates deep oxidation reactions, while the saturation coordination structure of Mo5+ enhances olefin selectivity in ODH reactions.20 Therefore, molybdenum-based catalysts not only offer potential for highly selective olefin production during ODH reactions but also their composition significantly influences cycloalkane ODH reactions.
It has been reported that a supported MoOx catalyst is an important catalytic system for alkane ODH.21–23 Effective reactant activation directly depends on the local structure of the catalyst, distribution of metal oxides on the carrier surface, and parameters related to catalyst preparation processes.24–26 The supported MoOx catalyst undergoes a transition from isolated monomers to larger polymolybdates due to submonolayer coverage on supports. Depending on different surface densities of Mo and sample preparation conditions, large crystals of MoOx form on carriers when single-layer coverage is exceeded.27,28 Hydroxyapatite (HAP), as a carrier with both acid–base properties, high stability and easy substitution of heterogeneous elements, has been deeply studied in acid–base dependent catalytic reactions.29,30 In addition, it was found that the phosphate in HAP can change the electronic environment of heterogeneous metals, promote their high dispersion, and promote the stability of the catalyst through strong metal–support interactions.31,32 Based on the above characteristics, HAPs as catalyst support has shown excellent performance in catalytic reactions.33,34
The dispersity of MoOx on various surfaces is dependent on the specific carrier, the surface density of Mo, and the calcination temperature of the sample. This dispersity has a significant impact on catalytic reaction performance. Although there have been reports on MoOx in REDOX reactions, there are limited studies on the synthesis of HAP-supported MoOx catalysts and their catalytic ODH reactions. Furthermore, the influence of MoOx content and chemical environment on ODH catalytic performance remains largely unexplored. In this study, a molybdenum oxide catalyst supported by HAP was synthesized using coprecipitation method under different MoOx content and calcination temperatures. The effects of synthesis method on catalyst structure and its impact on ODH catalytic performance were investigated. Additionally, the role of different MoOx species in cyclohexane ODH reaction was revealed.
Materials and methods
Materials
Ammonium molybdate ((NH4)6Mo7O24·4H2O, ≥99%), HAP (Ca10(PO4)6(OH)2, ≥99%), were obtained from Alpha Chemical Reagent Co., Ltd (Tianjin, China).Preparation of catalysts
The support material was impregnated using an incipient wetness method. Corresponding amounts of (NH4)6Mo7O24·4H2O to the aimed nominal MoO3 loadings were dissolved in 5 mL demineralized water. The impregnation solution was thoroughly mixed with commercial HAP (Ca/P = 1.67) using glass rods for 5 minutes and dried overnight at 80 °C. The impregnated sample was then calcined in a Muffle furnace under still air at 550 °C for 4 hours at a heating rate of 5 °C min−1.Two series of catalysts were prepared based on different loading amounts and calcination temperatures: MHAP-x-y (where x represents the Mo/Ca ratio, x = 0.025, 0.05, 0.075, and 0.1; y represents the calcination temperature, y = 500 °C, 600 °C, 700 °C, 800 °C).
Characterization
The chemical composition of the sample was determined using an SRS-3400 X-ray fluorescence analyzer (XRF). The nitrogen adsorption–desorption isotherm of the sample was measured using a Quantachrome AUTOSORB-1-MP physical adsorption instrument to obtain information about its surface and structure characteristics. The surface area (SBET) of the sample was calculated using the BET method. Powder X-ray diffraction (XRD) patterns were recorded for synthesized MoOx/HAP samples using CuKα radiation intensity at ARL X'TRA powder diffractometer. X-ray photoelectron spectroscopy (XPS) measurements were conducted using PHI1600 XPS system radiated by AlKα radiation to determine binding energies for elements such as Mo (3d) and O (1s). A diffuse reflectance UV-Vis spectrum was obtained on a HITECHI U-3900 UV-Vis-NIR spectrometer, with BaSO4 as a reference material. The samples were ground, then placed in a diffuse sample tank, pressed with a cover glass and placed in the sample card tank, and recorded at room temperature in the test range of 200–1000 nm. The reflectivity of the standard carrier was used as the basis for the determination of the sample. Temperature programmed reduction (H2-TPR) experiments were conducted on a chemisorption analyzer provided by Dalian University of Technology. The samples were pretreated at 300 °C in an Ar (40 mL min−1) for 2 hours, followed by cooling to 25 °C. Subsequently, the sample underwent reduction from 25 °C to 850 °C at a heating rate of 10 °C min−1 under a flow rate of 10% H2/Ar (40 mL min−1). The amount of H2 consumed by the sample was measured using a TCD detector to obtain the H2-TPR curve. Transmission electron microscopy (TEM) analysis was performed using a JEOL-JEM-2100F electron microscope operating at 200 kV. The acidity properties of the sample were studied through NH3-temperature-programmed desorption (NH3-TPD) technique. Prior to testing, the catalyst underwent pretreatment in Ar stream at 500 °C for 2 hours and then cooled down to 100 °C for NH3 adsorption before being purged with argon again. During TPD experiment, temperature increased from 100 °C to 850 °C at a heating rate of 10 °C min−1 while monitoring desorbed NH3 with TCD detector.Catalyst performance
The catalytic reaction took place in a miniature fixed bed reactor with an inner diameter of 8 mm. The catalyst weighing 0.5 g was compressed into uniform particles ranging between 20–40 mesh before being placed inside the reaction tube's constant temperature zone. Prior to running any reactions, the system underwent air purification at a rate of 40 mL min−1 for one hour at 500 °C for 1 h. After being cooled to the reaction temperature, cyclohexane was introduced via pumping, and samples were analyzed at intervals of 1 hour. The reaction operated with a space speed of 6 h−1 and a molar ratio of cyclohexane to O2 of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The experiments were conducted under atmospheric pressure within the temperature range of 400–490 °C.
:1. The experiments were conducted under atmospheric pressure within the temperature range of 400–490 °C.
For quantitative analysis of product composition, an on-line gas chromatography (GC) system Agilent 6890 equipped with a flame detector (FID) and an HP-5 column (Agilent, dimensions: 60 m × 0.32 mm i.d × 0.5 μm) was employed. Cyclohexane conversion, product selectivity, and TOF values are calculated using eqn (1)–(3) respectively.
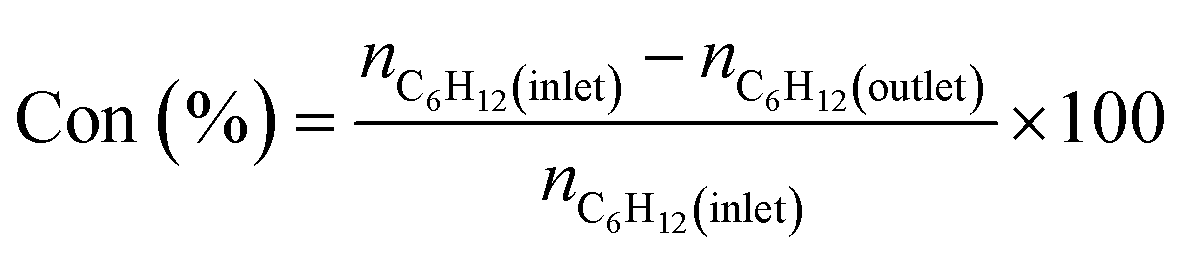 | (1) |
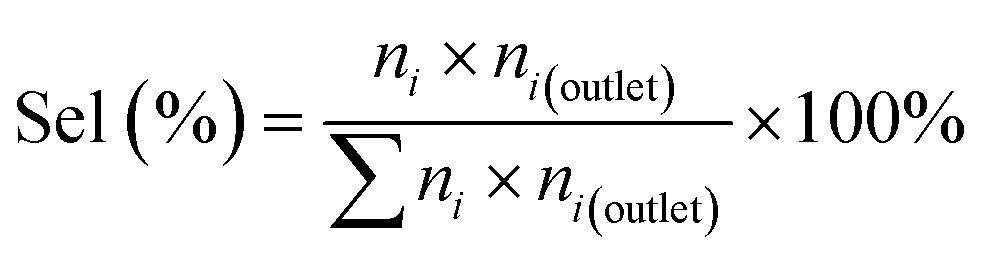 | (2) |
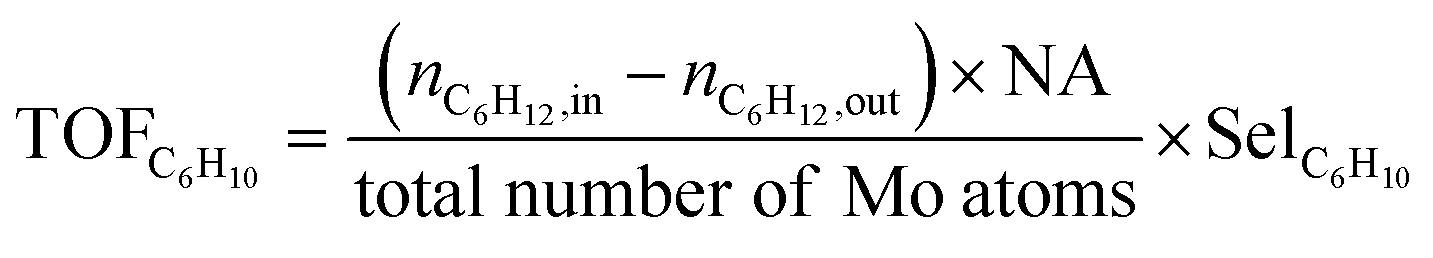 | (3) |
Results and discussion
Physical and textural properties
XRF spectroscopy was employed to analyze the elemental composition of all samples, and the results are presented in Table 1. All samples exhibited similar Ca/P ratios, ranging from 1.7 to 1.8. The Mo/Ca ratio of MOx/HAP samples increased with increasing Mo addition, while the Mo/Ca ratio remained relatively consistent across different roasting temperatures.| Samples | Ca/P ratios | Mo/Ca ratios | Surface area (m2 g−1) | Eg (eV) |
|---|---|---|---|---|
| MHAP-0.025-550 | 4.3 | 0.03 | 49.5 | 3.23 |
| MHAP-0.05-550 | 4.3 | 0.06 | 41.6 | 3.25 |
| MHAP-0.075-550 | 4.3 | 0.09 | 37.3 | 3.11 |
| MHAP-0.1-550 | 4.3 | 0.11 | 30.6 | 3.03 |
| MHAP-0.05-500 | 4.3 | 0.06 | 48.6 | 3.20 |
| MHAP-0.05-600 | 4.3 | 0.06 | 26.5 | 3.41 |
| MHAP-0.05-700 | 4.3 | 0.06 | 10.7 | 3.70 |
| MHAP-0.05-800 | 4.3 | 0.06 | 8.4 | 3.43 |
Table 1 provides BET surface area values for all samples. With an increase in Mo content, the surface area of MOx/HAP samples gradually decreased with varying amounts of MoOx, which is consistent with the behavior observed in most supported catalyst materials regarding specific surface area variation. The BET surface area of MOx/HAP samples with different calcination temperatures decreased from 48.6 m2 g−1 (MHAP-0.05-500) to 8.4 m2 g−1 (MHAP-0.05-800) as the calcination temperature increased, indicating particle agglomeration due to calcination.
Fig. 1(a) and (b) depict XRD patterns of MOx/HAP catalysts with different MoOx amounts and calcination temperatures respectively, alongside references showing XRD patterns of CaMoO4 and HAPs samples in Fig. 1. No presence of a MoO3 phase was detected in any sample, suggesting that MoOx was effectively dispersed on the carrier's surface. However, a diffraction peak corresponding to CaMoO4 appeared in the sample due to partial substitution by Mo atoms occupying P positions in HAP structure. The strength of this peak increased with higher levels of added MoOx content signifying greater incorporation into HAP's crystal structure. As shown in Fig. 1(b), CaMoO4 began appearing at a low calcination temperature of 500 °C and its crystal diffraction peak intensified as calcination temperature rose. This indicates that elevated calcination temperatures facilitated more significant infiltration of Mo into the HAP lattice, resulting in P–O–Mo or Ca–O–Mo structures. Fig. 1(b) demonstrates that when calcination temperature reached 700 °C, Ca3(PO4)2 crystals emerged, suggesting that high-temperature calcination removes part of the hydroxyl group in HAP.
 | ||
| Fig. 1 XRD patterns of HAPs, CaMoO4, Ca3(PO4)2 and MOx/HAP with different MoOx loading amounts (a) and calcination temperature (b). | ||
The valence state of Mo in the catalyst was further determined by XPS analysis. The XPS spectra of Mo 3d (Fig. 2) reveal that the double peaks at 233.1 eV and 236.3 eV can be attributed to Mo 3d5/2 and Mo 3d2/3, respectively, indicating the presence of Mo6+ species (Fig. 2(a) and (b)).35–37 According to previous studies, there is an additional peak at 233.7 eV, which corresponds to the Mo5+ species.38 By classifying and integrating these peaks, changes in the distribution of different oxidation states of Mo were discussed. The results are summarized in Table 2. Although the diffraction peak corresponding to crystalline MoO3 is not observed in the XRD pattern due to its small particle size, it can be concluded that under low calcination temperature, most of the Mo on HAP surface are still associated with dispersed MoOx compounds. Table 2 demonstrates that as the loading amount of MoOx increases, there is a successive decrease in the proportion of Mo5+ species accompanied by an increase in the content of Mo6+. This suggests a direct relationship between dispersity and Mo valence distribution; highly dispersed samples contain more amounts of Mo5+. Furthermore, increasing calcination temperature significantly reduces the proportion of Mo5+, with a sharp decline observed at 700 °C. The XRD results indicate that as calcination temperature rises, the incorporation of Mo into HAP lattice increases, resulting in a decrease in the amount of Mo5+. The most significant reduction occurs at 700 °C.
 | ||
| Fig. 2 Mo 3d XPS spectra of MOx/HAP with different MoOx loading amounts (a) and calcination temperature (b). | ||
| Band | BE (eV) | Proportion of area (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Mo-0.025-550 | Mo-0.05-550 | Mo-0.075-550 | Mo-0.1-550 | Mo-0.05-500 | Mo-0.05-600 | Mo-0.05-700 | Mo-0.05-800 | ||
| Mo5+ | 233.7 | 25.2 | 24.1 | 17.4 | 17.0 | 18.7 | 18.3 | 11.0 | 8.4 |
| Mo6+ | 235.2 and 231.1 | 74.8 | 75.9 | 82.6 | 83.0 | 81.3 | 81.7 | 89.0 | 91.6 |
| Mo–O | 530.4 | 24.1 | 39.4 | 38.5 | 44.4 | 27.2 | 30.2 | 38.6 | 40.1 |
| Lattice oxygen | 531.1 | 41.8 | 36.4 | 36.8 | 35.7 | 41.9 | 36.7 | 41.3 | 41.7 |
| Oxygen vacancy | 532.0 | 20.0 | 14.6 | 14.8 | 12.1 | 17.3 | 16.4 | 11.9 | 11.2 |
| O–H | 533.1 | 14.1 | 9.6 | 9.8 | 7.8 | 13.6 | 16.6 | 8.2 | 7.1 |
The deconvolution XPS spectra of O 1s shown in Fig. 3(a) and (b) exhibit four distinct peaks located at binding energies 530.5 eV, 531.2 eV, 532.1 eV, and 533.1 eV, respectively. These peaks are attributed to Mo–O,39 lattice oxygen, oxygen vacancy, and O–H.40–42 Table 2 summarizes the area ratios for each binding energy range obtained from different catalysts. The XPS spectrum of O reveals that the Mo–O bond increases with an increase in MOx loading amount. This demonstrates an increase in the number of MOx crystals on the carrier surface. Lattice oxygen primarily originates from the P–O bond within the PO43−. As MOx amount increases, both lattice oxygen and O vacancy decrease, indicating a greater substitution of Mo at the P position and penetration into O vacancy. For samples subjected to different calcination temperatures, a gradual reduction in OH concentration (533.1 eV) are observed as calcination temperature rises, suggesting dehydroxylation of HAP during this process. Dehydroxylation leads to the creation of new defect sites upon removal of hydroxyl groups. At calcination temperatures between 500–600 °C, only a slight increase in O vacancy is observed. However, when reaching 700 °C, there is a significant decrease in OH content without a corresponding increase in O vacancy due to MoOx filling these vacancies instead. In other words, formation of O vacancy promotes MoOx infiltration into these regions resulting in better dispersion and simultaneous formation of P–O–Mo or Ca–O–Mo structures.
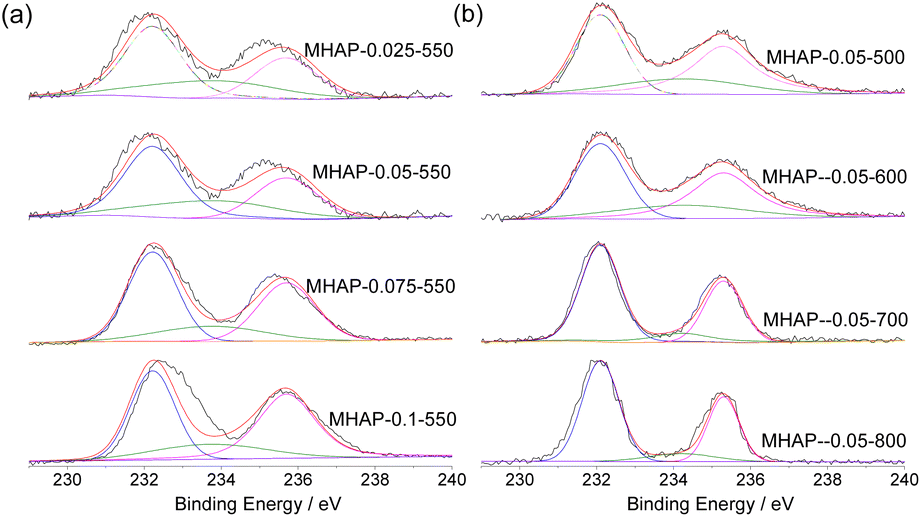 | ||
| Fig. 3 O 1s XPS spectra of MOx/HAP with different Mo loading amounts (a) and calcination temperature (b). | ||
The effect of weight loading and calcination temperature on the formation of catalytic sites was further investigated by UV-Vis diffuse reflectance spectroscopy to determine bandgaps for MOx/HAP samples (Fig. 4). As shown by Chakrabarti and Wachs, lower bandgap values of Mo supported on Al2O3 indicate larger molybdenum clusters (>1 nm) on the surface, and higher bandgaps translate to smaller dimeric and monomeric (<1 nm) MoOx species on the support.43 A broad feature at 320 nm is assigned to polymeric MoOx while 245 nm is attributed to oligomeric MoOx species. At lower loadings (MHAP-0.025-550), the isolated, oligomeric species dominated while at higher loadings (MHAP-0.05-550, MHAP-0.075-550 and MHAP-0.1-550) there was more polymeric species in addition to oligomeric MoOx. As weight loading was increased, the bandgap fell, due to larger oxide clusters forming, as also shown in Fig. 4(a). The band of MoOx with the high-temperature calcinated sample, as shown in Fig. 5(b), exhibits enhanced sharpness, thereby contributing to the increased crystallinity of MoOx on the HAP surface. However, the band intensity of high temperature calcinated samples decreased significantly, which was due to the increase of Mo entering HAP lattice under high temperature calcination.
 | ||
| Fig. 4 Diffuse reflectance UV-Vis spectra for HAPs and MOx/HAP samples with different Mo loading amounts (a) and calcination temperature (b). | ||
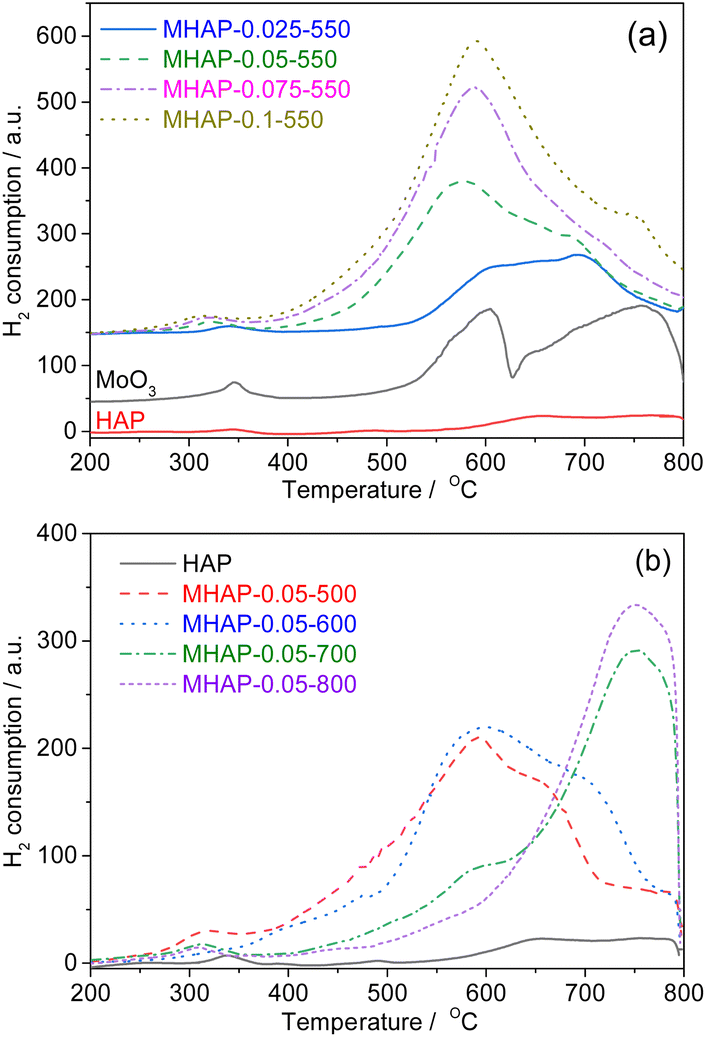 | ||
| Fig. 5 H2-TPR patterns of HAP, MoO3 and MOx/HAP samples with different Mo loading amounts (a) and calcination temperature (b). | ||
Fig. 5(a) illustrates the H2-TPR profiles of MoO3, HAP, and supported molybdenum oxide samples. Notably, there is minimal reduction peak observed in HAP. Generally, the reduction pathway of molybdenum species can be described as follows:44
| MoO3 + H2 → MoO2 + H2O |
| MoO2 + H2 → Mo0 + 2H2O |
The first reaction involves the reduction of MoO3 to MoO2 within a temperature range of 500–630 °C. On the other hand, the second reaction occurs at temperatures between 630–800 °C and leads to the reduction of MoO2 to Mo0.45 In contrast, HAP-loaded MoOx exhibits a distinct low temperature reduction peak and lacks an obvious high temperature reduction peak, indicating a strong interaction between MoOx and the carrier. As the load increases, it is reasonable for the peak area to gradually increase. The disparity between samples prepared at different calcination temperatures becomes more pronounced due to the significant influence of oxide structure on the reducibility of molybdenum oxides.46 A distinct low-temperature reduction peak is observed in samples calcined below 600 °C, while this peak disappears when the calcination temperature exceeds 700 °C, indicating an almost complete disappearance of independent MoO3. Reduction peaks around 750 °C are attributed to strongly interacting MoOx species with the carrier and partially incorporated Mo species within the HAP lattice. This suggests that the oxidation state of Mo in the catalyst is significantly influenced by high-temperature treatment.
The HRTEM images of the MHAP catalyst after calcination at 600 °C and 800 °C are presented in Fig. 6. The MHAP-0.05-600 sample (Fig. 6(a)) exhibits a uniform rice-shaped morphology with a particle size ranging from 50 to 200 nm. The HRTEM image (Fig. 6(b)) clearly reveals the lattice structure of HAP. Moreover, numerous surface pits can be observed on the MHAP-0.05-600 sample, which is consistent with N2 physical adsorption measurements and previous studies.47 It should be noted that HAP samples do not possess an interconnected pore structure; hence, the white regions in these TEM images correspond to surface pits on the particles. In Fig. 6(b), distinct lattice fringes are visible for MHAP-0.05-600, with lattice spacings of 0.345 nm matching the crystallographic planes of MoO3 indexed as {210} and 0.242 nm matching the crystallographic planes of CaMoO4 indexed as {202}, respectively.48,49 However, no diffraction peak corresponding to MoO3 appears in the XRD pattern due to its small grain size. When the calcination temperature is increased to 800 °C, the particle size of the catalyst undergoes significant enlargement and the pores on the catalyst surface become indiscernible, indicating that high temperature calcination leads to particle agglomeration. Fig. 6(g) clearly exhibits both the lattice fringe of HAP and CaMoO4, while no lattice fringe of MoO3 was observed in samples calcinated at 800 °C, further suggesting that high temperature calcination facilitates Mo incorporation into the HAP lattice and nearly eliminates MoO3. The findings are in accordance with this observation of XRD, XPS, and H2-TPR. Fig. 6(c–f) and (i–l) present EDS spectra for MHAP-0.05-600 and MHAP-0.05-800 respectively. The results demonstrate even distribution of Mo, Ca, and P in the both samples. Notably, after calcination at high temperatures, molybdenum dispersion in MHAP-0.05-800 is enhanced compared to that in MHAP-0.05-600. These findings indicate an increase in molybdenum dispersion with rising calcination temperature.
 | ||
| Fig. 6 TEM images and EDS mappings of the MHAP-0.05-600 (a)–(f) and MHAP-0.05-800 (g)–(l) samples. | ||
Acidities of the MHAP samples
In order to investigate the role of acidity in the ODH reaction, NH3-TPD spectra of MoOx/HAP samples were acquired, as depicted in Fig. 7. Two distinct HAP peaks were observed within the temperature range of 130–350 °C and 350–580 °C (Fig. 7(a)), which is consistent with the TPD results of HAP reported in the literature.50,51 Phosphate groups in HAP are responsible for the acidity of the catalyst, whereas the Ca2+ ions are responsible for the basicity. The strong acid site can be ascribed to protons originating from the HPO42− group, whereas the weak acid site results from adsorbed water interacting with ammonia through dipole interactions.52 The low temperature peak of MHAP samples with different MoOx loading amounts, as shown in Fig. 8(a), exhibits a similar trend to that of HAP, albeit with a slight increase in peak area. In the temperature range of 350–580 °C, the high temperature peak of MHAP samples decreases significantly with an increasing Mo/Ca ratio, and eventually disappears for MHAP-0.1-550 sample due to the coverage of acidic sites on the HAP surface by MoOx. In addition, a new strong peak was observed in the temperature range of 600–800 °C for samples with high loading amounts (MHAP-0.075-550 and MHAP-0.1-550). This high temperature peak can be attributed to the chemical reaction of MoOx with NH3 at high temperatures. | ||
| Fig. 7 NH3-TPD profiles of MHAP samples with different MoOx loading amounts (a) and calcination temperature (b). | ||
 | ||
| Fig. 8 Catalytic activity of MHAP catalysts with different MoOx loading amounts (a) and calcination temperature (b). | ||
For the samples obtained under different calcination conditions, the intensity of the low temperature peak (130–350 °C) gradually decreases while the high temperature peak (350–580 °C) nearly disappears. This phenomenon can be attributed to the increasing dehydroxylation of HAP with rising calcination temperatures, resulting in a reduction in surface acidity of the sample. Additionally, the enhanced interaction between MoOx and HAP leads to the formation of P–O–Mo or Ca–O–Mo, resulting in a reduction of the HPO42− group. The NH3 desorption peaks of MHAP-0.05-500 and MHAP-0.05-600 samples are observed within the temperature range of 600–800 °C, while these peaks disappear for MHAP-0.05-700 and MHAP-0.05-800 samples, further confirming the disappearance of MoOx species under high temperature roasting conditions.
Catalytic performance
The ODH of cyclohexane was investigated over the MoOx/HAP catalyst in a fixed bed reactor under atmospheric pressure at various temperatures. Fig. 8 illustrates the comparison of activity of MOx/HAP catalyst. HAP exhibited negligible activity for the ODH of cyclohexane (no shown). The activity of MHAP catalyst initially increased and then slight decreased with increasing Mo loading amounts, indicating that the primary source of ODH activity is derived from MOx. MHAP-0.75-550 demonstrated the highest activity with a conversion rate of 19.5% at 490 °C. XPS and UV-Vis analysis revealed that isolated, oligomeric MOx species were predominant at low MoOx loading amount (MHAP-0.025-550), while there was more polymeric species in addition to oligomeric MoOx at higher loadings (MHAP-0.05-550, MHAP-0.075-550 and MHAP-0.1-550). This suggests that the activity of isolated, oligomeric MoOx species is superior to that of MoOx particles in polymeric state.The catalyst conversion of MoOx/HAP decreased significantly with increasing calcination temperature, indicating a strong correlation between the two factors. The conversion of cyclohexane over MHAP-0.05-500 catalyst exhibited an upward trend with the elevation of reaction temperature. Specifically, as the reaction temperature rose from 400 °C to 490 °C, the conversion escalated from 4.8% to 11.8%. When the calcination temperature was increased to 700°, the catalyst activity decreased significantly to only 5.8% at 490 °C. However, when the calcination temperature was further raised to 800 °C, there was minimal change observed in the catalytic activity. These observed catalytic properties can be attributed to the specific type and structure of the active components present in the catalyst. The form of Mo in the sample changed from Mo5+ to Mo6+ as the calcination temperature increased from 500 °C to 600 °C, indicating that Mo5+ exhibited higher ODH activity compared to Mo6+. Furthermore, with an increase in calcination temperature up to 700 °C, there was a gradual strengthening of interaction between MoOx and the carrier. The interaction between MoOx and the carrier gradually intensified as the calcination temperature increased to 700 °C, leading to a higher substitution of P in HAP by Mo and resulting in the formation of well-dispersed P–O–Mo or Ca–O–Mo species. However, it should be noted that both MHAP-0.05-700 and MHAP-0.05-800 samples displayed low conversion rates due to lower ODH activity associated with P–O–Mo or Ca–O–Mo. In addition, the NH3-TPD results revealed a significant reduction in the surface acidity of MHAP-0.05-800. Since the cleavage of C–C bonds during ODH of cyclohexane is closely associated with acidity, the observed decrease in conversion rate can be attributed to the decline in acidity.
Fig. 9(a) and (b) depict the product selectivity of the cyclohexane ODH reaction at a temperature of 460 °C. There are three potential reactions in the ODH of cyclohexane:4 (1) partial ODH of cyclohexane yields cyclohexene, which can subsequently undergo further dehydrogenated to benzene; (2) direct dehydrogenation of cyclohexane to benzene, followed by peroxidation to COx; (3) direct peroxidation of cyclohexane to COx. Cyclohexene can be considered as an intermediate product in the ODH process of cyclohexane, thus the selectivity towards cyclohexene is influenced by both the catalyst's dehydrogenation ability and the desorption capability of cyclohexene. The results depicted in Fig. 9(a) demonstrate that MHAP with a low MoO3 loading amount exhibits high selectivity towards cyclohexene and COx. This suggests that the reaction rates of route (1) and route (3) are elevated in the MHAP-catalyzed ODH reaction system, indicating that the MHAP catalyst possesses an appropriate ODH capacity. However, the high COx selectivity of MHAP with a low MoO3 loading amount also indicates its strong capability in breaking C–H bonds, which is closely associated with the strong acidity of MHAP. The MoOx/HAP samples with low loading amount exhibited a pronounced preference for cyclohexene production, suggesting that the high dispersion of MoOx and Mo5+ species played a crucial role in enhancing selectivity. The MoOx/HAP samples exhibited high selectivity towards benzene, with an increase in benzene selectivity observed as the amount of MoOx impregnation increased. This suggests that MoOx/HAP possesses a strong oxidative dehydrogenation capacity.
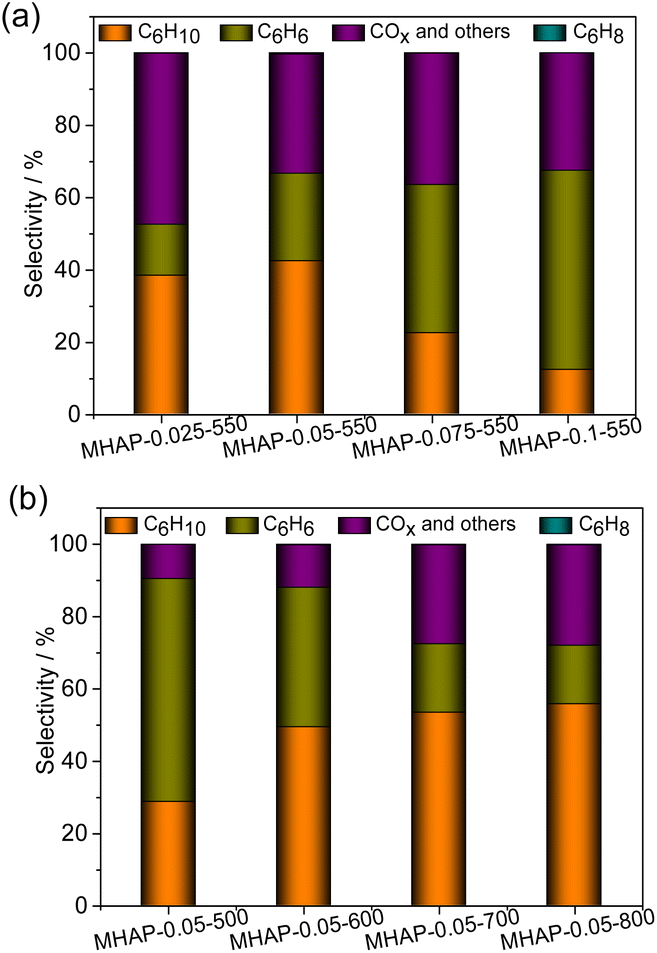 | ||
| Fig. 9 The product distribution patterns of the MHAP catalysts with different MoOx loading amounts (a) and calcination temperature (b). | ||
The selectivity of cyclohexene exhibited significant variations among MoOx/HAP samples calcined at different temperatures, with the low-temperature calcination samples (MHAP-0.05-500 and MHAP-0.05-600) differing notably from the high-temperature calcination samples (MHAP-0.05-700 and MHAP-0.05-800). This discrepancy can be attributed to the distinct chemical environments surrounding the molybdenum species. The selectivity of cyclohexene increased proportionally with the rise in calcination temperature, ultimately reaching 58.2% in the MHAP-0.05-800 sample. The selectivity of cyclohexene is significantly higher compared to the previously reported cyclohexane ODH reaction.10,53–55 The impact of Mo5+ and Mo6+ on the activity and selectivity of oxidative dehydrogenation of cyclohexane has been demonstrated in various studies. The internal structure of MoO3 contains a significant number of oxygen vacancies, and it maintains a dynamic equilibrium between the structural defects and gas phase oxygen through REDOX reactions involving Mo5+ and Mo6+.56,57 The octahedral structure of Mo6+ promotes deep oxidation reactions, and catalysts with higher Mo5+ content exhibit excellent catalytic activity in oxidative dehydrogenation reactions. Moreover, the unsaturated coordination structure of Mo5+ contributes to enhanced olefin selectivity. Although the proportion of Mo6+ in highly calcined samples increased, the elevated calcination temperature facilitated the incorporation of Mo into HAP lattice, resulting in the formation of P–O–Mo or Ca–O–Mo species. This observation implies that highly dispersed P–O–Mo or Ca–O–Mo species are more favorable for olefin production. The successive increase in calcination temperature leads to a decrease in acidic sites, thereby facilitating the desorption of cyclohexene and inhibiting its further oxidation to benzene and COx. The MHAP-0.05-700 and MHAP-0.05-800 catalysts exhibited high selectivity towards cyclohexene and COx, indicating that pathways (1) and (3) had higher reaction rates in the MHAP-catalyzed ODH reaction system. This suggests that the MHAP catalyst possesses suitable ODH capacity. However, the elevated COx selectivity also implies that MHAP exhibits a strong ability to break C–H bonds, which is attributed to its pronounced acidity.
The relationship between selectivity and conversion of C6H10, C6H6, and COx at different reaction temperatures on MoOx/HAP is illustrated in Fig. 10. The selectivity of various catalysts for C6H6 and COx decreases as the conversion rate increases, while the selectivity of different catalysts for cyclohexene exhibits a distinct correlation with the conversion rate. The selectivity of C6H10 increases with the increase in conversion rate for MHAP-0.05-500 catalysts, indicating that benzene is primarily formed through cyclohexene intermediates. The selectivity of C6H6 increases with the rise in conversion, attributed to the elevated reaction temperature. The enhanced lattice oxygen mobility at elevated temperatures suggests that the nucleophilic lattice oxygen in the catalyst becomes more advantageous for the dehydrogenation process, thereby promoting the conversion of intermediate cyclohexene into thermodynamically stable benzene. The selectivity of C6H10 on the MHAP-0.05-600 catalyst exhibits irregular variations with increasing conversion. The catalytic properties of MHAP-0.05-700 and MHAP-0.05-800 samples demonstrate the influence of P–O–Mo and Ca–O–Mo on product selectivity. In the case of MHAP-0.05-800 samples, the selectivity towards C6H6 and C6H10 increases with increasing conversion, while the selectivity towards COx decreases, suggesting that C6H6 is formed through direct dehydrogenation of cyclohexane, whereas COx may be generated through oxidation of cyclohexane and benzene.
 | ||
| Fig. 10 The selectivity–conversion relationships for cyclohexane ODH over different catalysts. (a) C6H10, (b) C6H6 and (c) COx. | ||
Conclusions
Two series of MoOx/HAP catalysts with different MoOx loading amounts and calcination temperature were prepared by impregnation method. The MoOx/HAP catalyst was characterized using XRD, XPS, UV-Vis, and H2-TPR. MoOx/HAP catalysts with lower loading exhibited higher dispersion and a greater proportion of Mo5+ species, whereas those with higher loading showed an increased presence of Mo6+ species. The MoOx/HAP obtained at a low calcination temperature primarily consists of surface MoOx and contains a higher proportion of Mo5+ species. However, upon high calcination, the Mo5+ species undergo transformation to Mo6+, and the interaction between MoOx and HAP carrier leads to the substitution of P positions in the HAP lattice by Mo, resulting in the formation of P–O–Mo or Ca–O–Mo structures. The MoOx/HAP catalysts were employed for the catalysis of cyclohexane ODH to cyclohexene. The MoOx/HAP catalysts with lower loading amount exhibited reduced cyclohexane conversion and enhanced selectivity towards cyclohexene, suggesting that the presence of Mo5+ species promotes olefin production. The conversion of catalysts calcined at low temperature was found to be higher than that of catalysts calcined at high temperature, whereas the selectivity towards cyclohexene was observed to be higher for MHAP-0.05-700 and MHAP-0.05-800 catalysts. At a reaction temperature of 430 °C, when the conversion rate of cyclohexane reached 13.1%, the selectivity of MHAP-0.05-800 towards cyclohexene achieved 58.2%. The high selectivity is directly influenced by the chemical environment of MoOx species, where dispersed P–O–Mo or Ca–O–Mo species are more favorable for olefin production. Moreover, the high-temperature calcination-induced decrease in acidity results in a reduction of the adsorption capacity of MoOx/HAP for cyclohexene, thereby promoting its desorption on the catalyst surface, inhibiting deep oxidation and enhancing selectivity.Data availability
All data and related metadata underlying the findings reported in a submitted manuscript should be deposited in an appropriate public repository, unless already provided as part of the submitted article.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 201905081).References
- H. Yu, F. Peng, J. Tan, X. Hu, H. Wang, J. Yang and W. Zheng, Angew. Chem., Int. Ed., 2011, 50, 3978–3982 CrossRef CAS PubMed.
- A. P. Unnarkat, T. Sridhar, H. Wang, S. Mahajani and A. K. Suresh, AlChE J., 2016, 62, 4384–4402 CrossRef CAS.
- C. Resini, M. Panizza, F. Raccoli, M. Fadda, M. M. Carnasciali, G. Busca, E. F. Lopez and V. S. Escribano, Appl. Catal., A, 2003, 251, 29–38 CrossRef CAS.
- H. Feng, J. W. Elam, J. A. Libera, M. J. Pellin and P. C. Stair, J. Catal., 2010, 269, 421–431 CrossRef CAS.
- Y. Gambo, S. Adamu, A. A. Abdulrasheed, R. A. Lucky, M. S. Ba-Shammakh and M. M. Hossain, Appl. Catal., A, 2021, 609, 117914 CrossRef CAS.
- Z. Fu, D. Z. Li, L. D. Zhou, Y.-M. Li, J. W. Guo, Y. Q. Li, H.-M. Liu and Q. J. Zhang, Pet. Sci., 2023, 20, 2488–2498 CrossRef CAS.
- X. Song, F. Zhou, H. Ma, Y. Liu and G. Wu, Mol. Catal., 2023, 542, 113105 CrossRef CAS.
- S. Kumar, A. Qadir, F. Maury and N. Bahlawane, ACS Appl. Mater. Interfaces, 2017, 9, 21447–21456 CrossRef CAS.
- D. Contreras, V. Melin, K. Márquez, G. Pérez-González, H. D. Mansilla, G. Pecchi and A. Henríquez, Appl. Catal., B, 2019, 251, 17–24 CrossRef CAS.
- I. A. Samek, N. S. Bobbitt, R. Q. Snurr and P. C. Stair, J. Catal., 2020, 384, 147–158 CrossRef CAS.
- V. Vaiano, D. Sannino, A. R. Almeida, G. Mul and P. Ciambelli, Catalysts, 2013, 3, 978–997 CrossRef.
- S. A. Kadam, S. Sandoval, Z. Bastl, K. Simkovičová, L. Kvítek, J. Jašík, J. E. Olszówka, S. Valtera, M. Vaidulych, J. Morávková, P. Sazama, D. Kubička, A. Travert, J. A. van Bokhoven, A. Fortunelli, A. Kleibert, M. Kalbáč and Š. Vajda, ACS Catal., 2023, 13, 13484–13505 CrossRef CAS PubMed.
- S. Lee, A. Halder, G. A. Ferguson, S. Seifert, R. E. Winans, D. Teschner, R. Schlögl, V. Papaefthimiou, J. Greeley, L. A. Curtiss and S. Vajda, Nat. Commun., 2019, 10, 954 CrossRef PubMed.
- B. Sarkar, C. Pendem, L. N. S. Konathala, T. Sasaki and R. Bal, Catal. Commun., 2014, 56, 5–10 CrossRef CAS.
- X. Chen, X. Su, H. Duan, B. Liang, Y. Huang and T. Zhang, Catal. Today, 2017, 281, 312–318 CrossRef CAS.
- G. J. Hutchings, A. Desmartin-Chomel, R. Olier and J.-C. Volta, Nature, 1994, 368, 41–45 CrossRef CAS.
- A. Ebadi, N. Safari and M. H. Peyrovi, Appl. Catal., A, 2007, 321, 135–139 CrossRef CAS.
- S. Goergen, C. Yin, M. Yang, B. Lee, S. Lee, C. Wang, P. Wu, M. B. Boucher, G. Kwon, S. Seifert, R. E. Winans, S. Vajda and M. Flytzani-Stephanopoulos, ACS Catal., 2013, 3, 529–539 CrossRef CAS.
- G. W. Coulston, S. R. Bare, H. Kung, K. Birkeland, G. K. Bethke, R. Harlow, N. Herron and P. L. Lee, Science, 1997, 275, 191–193 CrossRef CAS.
- L. E. Cadus, M. F. Gomez and M. C. Abello, Catal. Lett., 1997, 43, 229–233 CrossRef CAS.
- E. Heracleous, A. F. Lee, I. A. Vasalos and A. A. Lemonidou, Catal. Lett., 2003, 88, 47–53 CrossRef CAS.
- M. C. Abello, M. F. Gomez and O. Ferretti, Appl. Catal., A, 2001, 207, 421–431 CrossRef CAS.
- K. Chen, A. T. Bell and E. Iglesia, J. Phys. Chem. B, 2000, 104, 1292–1299 CrossRef CAS.
- E. Heracleous, J. Vakros, A. A. Lemonidou and C. Kordulis, Catal. Today, 2004, 91–92, 289–292 CrossRef CAS.
- F. R. Brown, L. E. Makovsky and K. H. Rhee, J. Catal., 1977, 50, 162–171 CrossRef CAS.
- L. Wang and W. K. Hall, J. Catal., 1983, 83, 242–244 CrossRef CAS.
- Y. C. Xie and Y. Q. Tang, Spontaneous Monolayer Dispersion of Oxides and Salts onto Surfaces of Supports: Applications to Heterogeneous Catalysis, Adv. Catal., 1990, 37, 1–43 CAS.
- J. A. Bergwerff, T. Visser, G. Leliveld, B. D. Rossenaar, K. P. de Jong and B. M. Weckhuysen, J. Am. Chem. Soc., 2004, 126, 14548–14556 CrossRef CAS.
- O. P. Oni, Y. Hu, S. Tang, H. Yan, H. Zeng, H. Wang, L. Ma, C. Yang and J. Ran, Mater. Chem. Front., 2023, 7, 9–43 RSC.
- C. Boucetta, M. Kacimi, A. Ensuque, J. Y. Piquemal, F. Bozon-Verduraz and M. Ziyad, Appl. Catal., A, 2009, 356, 201–210 CrossRef CAS.
- C. Li, G. Xu, X. Liu, Y. Zhang and Y. Fu, Ind. Eng. Chem. Res., 2017, 56, 8843–8849 CrossRef CAS.
- G. Xu, Y. Zhang, Y. Fu and Q. Guo, ACS Catal., 2017, 7, 1158–1169 CrossRef CAS.
- S. Sugiyama, T. Osaka, Y. Hirata and K. I. Sotowa, Appl. Catal., A, 2006, 312, 52–58 CrossRef CAS.
- K. E. Kabouss, M. Kacimi, M. Ziyad, S. Ammar, A. Ensuque, J. Y. Piquemal and F. Bozon-Verduraz, J. Mater. Chem., 2006, 16, 2453–2463 RSC.
- S. Huang, T. Ouyang, B.-F. Zheng, M. Dan and Z.-Q. Liu, Angew. Chem., Int. Ed., 2021, 60, 9546–9552 CrossRef CAS PubMed.
- J. Dou and H. C. Zeng, J. Am. Chem. Soc., 2012, 134, 16235–16246 CrossRef CAS.
- I. A. de Castro, R. S. Datta, J. Z. Ou, A. Castellanos-Gomez, S. Sriram, T. Daeneke and K. Kalantar-zadeh, Adv. Mater., 2017, 29, 1701619 CrossRef PubMed.
- W. Du, Y. Shi, W. Zhou, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 7051–7055 CrossRef CAS PubMed.
- K. Cui, L. Yang, Z. Ma, F. Yan, K. Wu, Y. Sang, H. Chen and Y. Li, Appl. Catal., B, 2017, 219, 592–602 CrossRef CAS.
- S. Chen, C. Pei, X. Chang, Z.-J. Zhao, R. Mu, Y. Xu and J. Gong, Angew. Chem., Int. Ed., 2020, 59, 22072–22079 CrossRef CAS.
- D. Wang, L. Jin, Y. Li and H. Hu, Fuel, 2017, 210, 803–810 CrossRef CAS.
- J. Cao, T. Nakamura, T. Kitazawa, N. Yamashiki, T. Yamamoto and T. Taneike, Prostaglandins Other Lipid Mediators, 2008, 86(1–4), 26–34 CrossRef CAS.
- A. Chakrabarti and I. E. Wachs, ACS Catal., 2018, 8, 949–959 CrossRef CAS.
- L. O. Alemán-Vázquez, F. Hernández-Pérez, J. L. Cano-Domínguez, A. Rodríguez-Hernández and J. L. García-Gutiérrez, Fuel, 2014, 117, 463–469 CrossRef.
- M. Saghafi, S. Heshmati-Manesh, A. Ataie and A. A. Khodadadi, Int. J. Refract. Met. Hard Mater., 2012, 30, 128–132 CrossRef CAS.
- E. Cheng and J. Notestein, J. Catal., 2021, 397, 212–222 CrossRef CAS.
- Y. Sun, Z. Qu, D. Chen, H. Wang, F. Zhang and Q. Fu, Chin. J. Catal., 2014, 35, 1927–1936 CrossRef CAS.
- S. Zhang, G. Wang, J. Jin, L. Zhang, Z. Wen and J. Yang, Nano Energy, 2017, 36, 186–196 CrossRef CAS.
- Y. Wu, W. Liu, Z. Zhang, Y. Zheng, X. Fu, J. Lu, S. Cheng, J. Su and Y. Gao, Energy Storage Mater., 2023, 61, 102849 CrossRef.
- B. Yan, L. Z. Tao, Y. Liang and B. Q. Xu, ACS Catal., 2014, 4, 1931–1943 CrossRef CAS.
- V. C. Ghantani, S. T. Lomate, M. K. Dongare and S. B. Umbarkar, Green Chem., 2013, 15, 1211–1217 RSC.
- N. S. Resende, M. Nele and V. M. M. Salim, Thermochim. Acta, 2006, 451, 16–21 CrossRef CAS.
- P. Du, X. X. Zhang, S. Zhang, Y. Zhao, L. Zhang, B. Zhang and B. Yang, ChemCatChem, 2021, 13, 610–616 CrossRef CAS.
- J. Wang, C. Wan, D. G. Cheng, F. Chen and X. Zhan, Appl. Surf. Sci., 2021, 565, 150609 CrossRef CAS.
- N. L. Wang, J. E. Qiu, Z. W. Wu, J. Wu, K. Y. You and H. A. Luo, Appl. Catal., A, 2015, 503, 62–68 CrossRef CAS.
- K. Murugappan, E. M. Anderson, D. Teschner, T. E. Jones, K. Skorupska and Y. Román-Leshkov, Nat. Catal., 2018, 1, 960–967 CrossRef CAS.
- T. Hu, B. Xue, F. Meng, L. Ma, Y. Du, S. Yu, R. Ye, H. Li, Q. Zhang, L. Gu, Z. Zhou, R. Liang and C. Tan, Adv. Healthcare Mater., 2023, 12, 2202911 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |