
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIncorporating azaheterocycle functionality in intramolecular aerobic, copper-catalyzed aminooxygenation of alkenes†
Ethan M. DeCicco ,
Neively Tlapale-Lara and
Shauna M. Paradine*
,
Neively Tlapale-Lara and
Shauna M. Paradine*
Department of Chemistry, University of Rochester, 120 Trustee Road, Rochester, NY 14627, USA. E-mail: sparadin@ur.rochester.edu
First published on 10th September 2024
Abstract
Despite the maturity of alkene 1,2-difunctionalization reactions involving C–N bond formation, a key limitation across aminofunctionalization methods is incompatibility with substrates bearing medicinally relevant N-heterocycles. Using a cooperative ligand-substrate catalyst activation strategy, we have developed an aerobic, copper-catalyzed alkene aminooxygenation method that exhibits broad tolerance for β,γ-unsaturated carbamates bearing aromatic azaheterocycle substitution. The synthetic potential of this methodology was demonstrated by engaging a densely-functionalized vonoprazan analogue and elaborating an amino oxygenated product to synthesize a heteroarylated analogue precursor of the FDA-approved antibiotic chloramphenicol.
Introduction
Alkene 1,2-aminofunctionalization is a powerful tool for synthetic chemists, rapidly constructing useful difunctionalized building blocks and targeted scaffolds.1 This includes such transformations as diamination,2 aminooxygenation,3 aminohalogenation,4 carboamination,5 and less commonly, aminoborylation,6 aminocyanation,7 aminophosphorylation8 and thioamination9 (Fig. 1A). While diverse inter- and intramolecular aminofunctionalization methods are available to construct a host of highly functionalized products, there is a notable lack of functional group tolerance in most methods with respect to substrates bearing N-heterocyclic scaffolds. Although several aminofunctionalization methods have shown isolated examples of N-heterocycle-bearing substrates,6,10 a method that demonstrates broad tolerance across a range of azaheterocycles has not yet been reported in the literature. These coordinating moieties can lead to catalyst poisoning, and reactions with substrates bearing these groups are prone to undesired side reactions under the oxidizing conditions typically necessary for alkene aminofunctionalization. This means that many otherwise useful methods cannot be confidently used in medicinal chemistry endeavors due to the lack of this medicinally relevant functionality in substrate scopes. This is consequential as over 85% of biologically active molecules contain at least one heterocycle,11 and more than 75% of FDA approved therapeutics contain a nitrogen heterocycle.12 Heterocycle incorporation into drug targets is often associated with more beneficial therapeutic and pharmacokinetic properties.13 Thus, it is essential to develop methodologies that tolerate N-heterocyclic moieties to facilitate their application in the synthesis of biologically active compounds.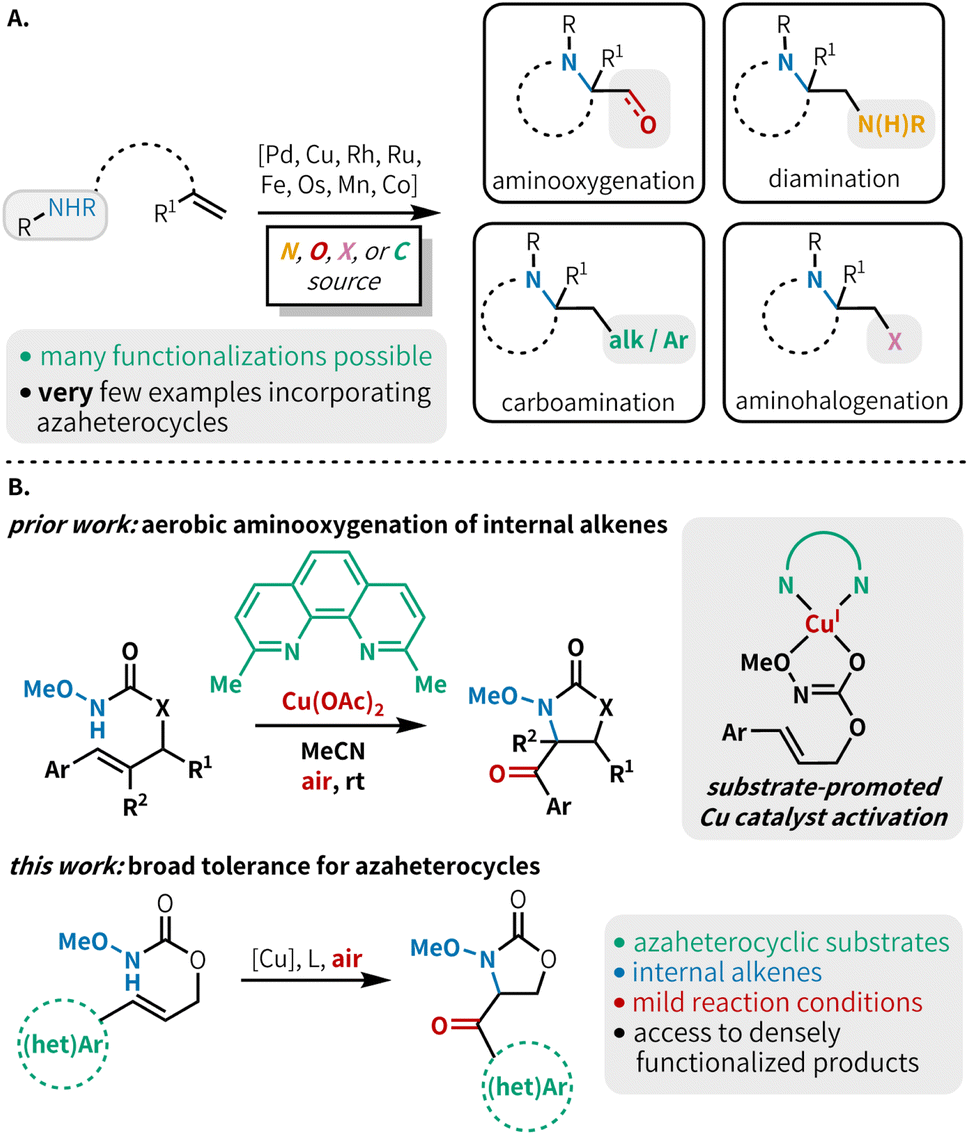 | ||
| Fig. 1 (A) Overview of alkene aminofunctionalization strategies. (B) Our prior work: aminooxygenation of cinnamyl N-alkoxy carbamates featuring an atypical amidyl radical pathway. This work: alkene aminooxygenation of γ-heteroaryl-β,γ-unsaturated carbamates. | ||
Among the many aminofunctionalization variants, alkene aminooxygenation is an excellent strategy to access 1,2-aminoalcohols and 1,2-aminocarbonyls, which are prevalent functional motifs within building blocks for the construction of bioactive compounds (Fig. 1A).14 Seminal reports detail the use of transition metal catalysts, often paired with a stoichiometric oxidant for catalyst turnover, and/or as a source of oxygen functionality.15 Recent reports have demonstrated an attractive alternative approach, using molecular oxygen to serve the dual role of oxidant and source of oxygen functionality; this approach also avoids the generation of stoichiometric byproducts and expensive reagents.3h,10d,16 Advantageously, aerobic aminooxygenation methods are typically mediated by earth-abundant metals such as Mn,17 Co,17 and Cu.18 Despite these notable advances in aerobic transition metal-catalyzed alkene aminooxygenation, typically monosubstituted or 1,1-disubstituted alkenes are used. Further, the incorporation of N-heteroaromatic substitution in substrates for these reactions is scarce.
Our lab recently developed an aerobic, copper-catalyzed aminooxygenation method for internal alkenes in the synthesis of 4-benzoyl-oxazolidin-2-ones from cinnamyl N-alkoxy carbamates (Fig. 1B).10d This reaction occurs under mild conditions, uses low catalyst loadings, and operates under an ambient atmosphere at room temperature. Our mechanistic findings were consistent with C–N bond formation proceeding via an amidyl radical cyclization pathway, as opposed to the aminometallation mechanism that is commonly observed in other transition metal-catalyzed alkene aminofunctionalizations.3c,16a,19 This mechanistic divergence was made possible through chelation of the substrate to the copper metal center, triggering a rapid reduction of Cu(II) to Cu(I) and subsequent generation of a potent one-electron copper oxidant. We hypothesized that the preference for O-binding of the substrate and the mild reaction conditions would allow for efficient reactivity in the presence of N-heterocyclic substituents. Herein, we report the successful implementation of this strategy through the synthesis of a variety of carbonyl-linked, heteroaryl-oxazolidin-2-ones (Fig. 1B). This method is compatible with a broad range of 5- and 6-membered N-heterocycles, including those containing multiple heteroatoms.
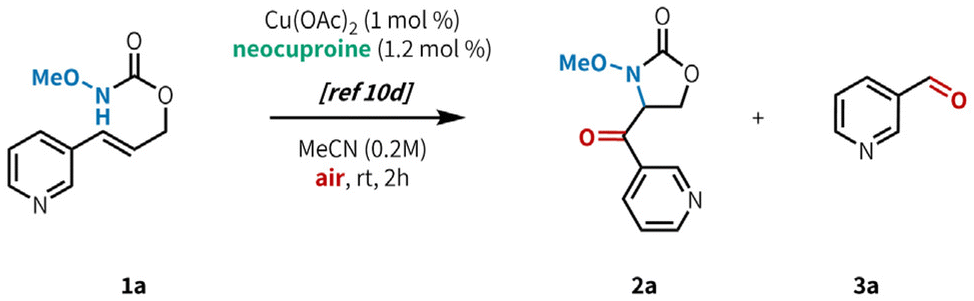
In our initial investigations (Table 1), we applied our previously published conditions to the reaction with 3-pyridyl substituted allyl carbamate 1a, and found that aminooxygenated product 2a was formed in 54% yield (entry 1). Similar Cu sources bearing acetate counterions performed well (entries 3–4), but a slight improvement in yield was found when using CuTC (entry 4, 59%). It is worth noting that while the original conditions from our previous report worked well in this case, Cu(OAc)2 is highly sensitive to trace impurities in the starting material. Indeed, in that report we tested several azaheterocycle-containing substrates and found them all to be unreactive under those conditions.10d In contrast, CuTC is much more robust to minor impurities, giving us more consistent reactivity. Model substrate 1a reacted well even at 1 mol% loading of CuTC (entry 5), but 5 mol% loading gave better results across a wider range of substrates (Fig. 2, see below).20 Base and acid additives both hindered the reaction (entries 6–7). Further increasing catalyst loading to 20 mol% did not provide any benefit (entry 8), furnishing 2a in a slightly diminished yield (42%). Increasing the reaction temperature (entry 9) led to decomposition of the reaction components, presumably from overoxidation. Similar results were observed when increasing both temperature and catalyst loading (entry 10). Investigation into ligand structure–reactivity relationships revealed 2,9-dimethyl-1,10-phenanthroline (neocuproine) was optimal, consistent with our previous studies.10d,20 Other 1,10-phenanthroline ligands bearing alkyl substitution adjacent to the nitrogen centers (i.e. ortho) showed similar results, in addition to 2,2′-bipyridyl ligands bearing 6,6′-dialkyl substitution.20
|
||||
|---|---|---|---|---|
| Entry | Changes to conditions | Yield 2a | Yield 3a | Recovered 1a |
| a 6 mol% of neocuproine was used.b 24 mol% of neocuproine was used. | ||||
| 1 | None | 54% | 12% | 0% |
| 2a | 5 mol% Cu(OAc)2 | 50% | 11% | 0% |
| 3a | 5 mol% Cu(EH)2 | 51% | 10% | 0% |
| 4a | 5 mol% CuTC | 59% | 13% | 0% |
| 5 | 1 mol% CuTC | 59% | 12% | 0% |
| 6a | As in entry 4, 1 eq. K2CO3 added | 0% | 3% | 61% |
| 7a | As in entry 4, 1 eq. PivOH added | 0% | 0% | >95% |
| 8b | 20 mol% CuTC | 2% | 10% | 0% |
| 9a | 5 mol% CuTC, 70 °C | 3% | 8% | 42% |
| 10b | 20 mol% CuTC, 70 °C | 4% | 8% | 41% |
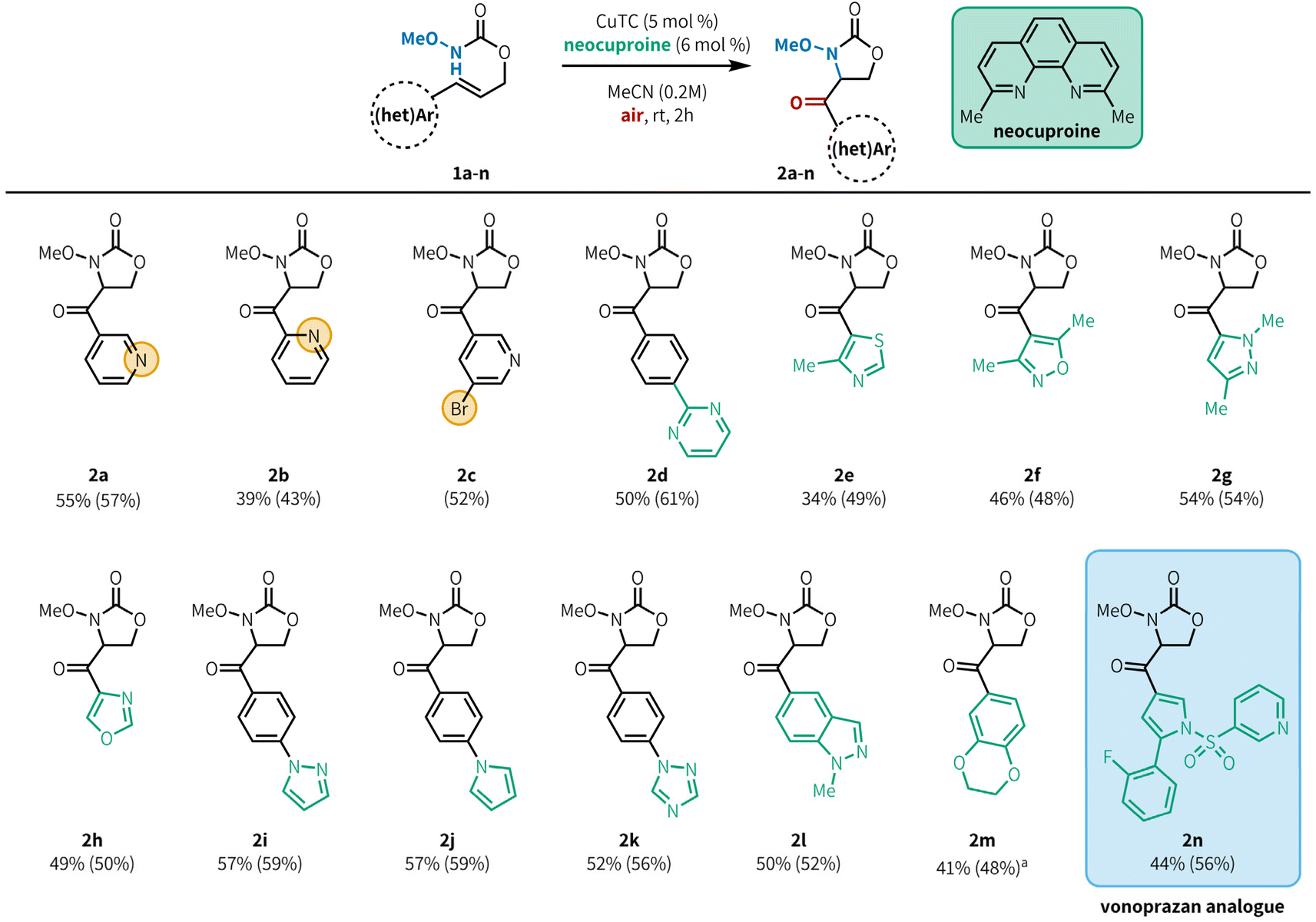 | ||
| Fig. 2 Substrate scope. Yields correspond to isolated products and are averages of three runs at 0.500 mmol scale. Quantitative NMR yields in parentheses, determined using 1,3,5-trimethoxybenzene as internal standard. Reaction conditions: 1 (0.500 mmol), CuTC (25.0 μmol), neocuproine (30.0 μmol), MeCN (0.2 M), rt, 2 h.a70 °C. CuTC = copper(I) thiophene 2-carboxylate. | ||
With optimized conditions in hand, we evaluated the scope of heteroaryl alkenes in the reaction (Fig. 2). Model product 2a was isolated in 55% yield at 0.500 mmol scale. The position of the pyridine nitrogen relative to the tether did not substantially impact the reaction, with 2-pyridyl substitution providing 2b in 39% yield, and synthetically versatile bromide substituents were well tolerated (2c, 52%). Pyrimidines were effective as substituents on phenyl rings (2d, 50%), although product yields were poor when the pyrimidine was directly attached to the alkene.20 This method is also compatible with a variety of 5-membered azaheterocycles, including thiazole (2e, 34%), isoxazole (2f, 46%), pyrazole (2g, 54%), and oxazole (2h, 49%). Substrates bearing arenes with heteroaromatic substitution are also effective in this reaction (2i–2k, 52–57%). We had previously demonstrated that protected indoles were compatible with aerobic aminooxygenation;10d under our revised conditions, the related but more challenging indazole 2l is also a good substrate (50%). Oxygen-containing heterocycles can also be employed,10d exemplified by benzodioxine 2m (41%), although elevated temperatures were required for efficient reaction of this substrate within 2 h. Notably, a precursor compound in the synthesis of the ulcer medication vonoprazan could be elaborated to generate analogue 1n, which underwent the desired aminooxygenation. Product 2n was isolated in reasonable yield (44%) showcasing this method's tolerance for densely functionalized, drug-like substrates. Some heterocycles bearing reactive C–X bonds, such as α-halopyridines, were not compatible in the reaction. The primary limitation of this method was our ability to access the substrate; substrates containing azaheterocycles such as imidazole, pyrroles, pyrazoles, halopyrimidines, and pyrrolopyridine were incompatible with the substrate synthesis and were unable to be tested in the aminooxygenation reaction.20
To showcase the synthetic utility of our aerobic aminooxygenation method, we synthesized a heteroaryl analogue precursor of the amphenicol antibiotics, which includes FDA-approved therapeutics chloramphenicol and thiamphenicol. Aminooxygenation product 2i was selected by analogy to the chloramphenicol structure (Fig. 3). Preparation of 2i via aerobic aminooxygenation scaled readily to 5.0 mmol, proceeding in identical yield to the scope investigation scale (0.500 mmol), and the reaction was performed in an open beaker (57% yield, 826 mg). Reduction with NaBH4 fashioned alcohol 4 in 68% yield. Subsequent N–O cleavage with SmI2 generated free carbamate 5 (57%), which underwent base mediated ring opening in 52% yield (21% isolated) to generate heteroarylated amphenicol analogue 6. Subsequent N-acetylation of free amines in the presence of alcohols has been reported in previous chloramphenicol syntheses.21
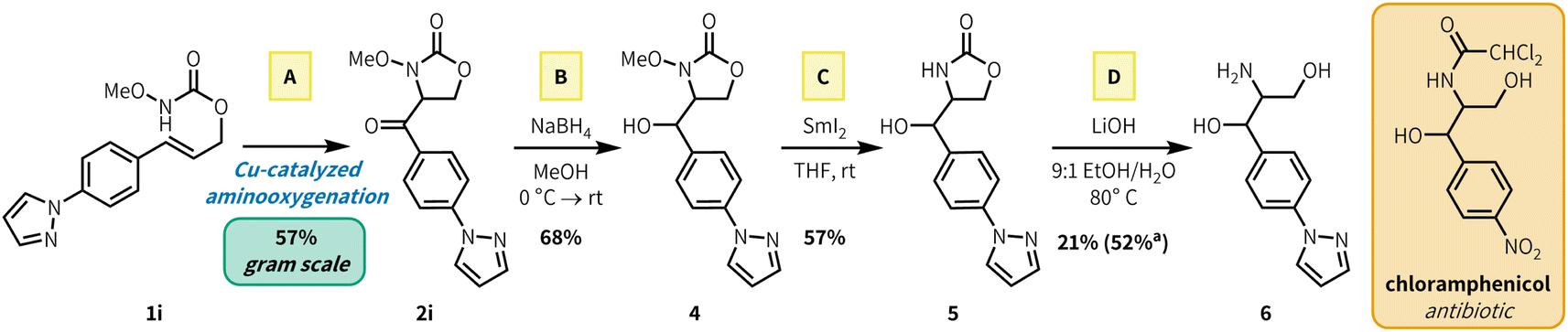 | ||
| Fig. 3 Synthesis of amphenicol antibiotic analogues. Reaction conditions: (A): 1i (1.37 g, 5.00 mmol, 1.0 equiv.) CuTC (47.7 mg, 0.250 mmol, 0.05 equiv.), neocuproine (62.5 mg, 0.300 mmol, 0.06 equiv.), MeCN (25 mL, 0.2 M), rt, open beaker, 2 h. (B): 2i (318 mg, 1.11 mmol, 1.0 equiv.), NaBH4 (46.0 mg, 1.1 equiv.), MeOH (0.2 M), 0 °C → rt, 1 h. (C): 4 (180 mg, 0.622 mmol, 1.0 equiv.), SmI2 (0.1 M in THF) (50 mL, 5.0 mmol, 8.0 equiv.), THF (16 mL, 0.4 M), rt, 2 h. (D): 5 (40.0 mg, 0.154 mmol, 1.0 equiv.), LiOH·H2O (19.4 mg, 0.462 mmol, 3.0 equiv.), 16 h.aQNMR yield using 1,3,5-trimethoxybenzene as internal standard. | ||
Conclusions
By employing the catalytic strategy of substrate-promoted catalyst activation, we have developed an operationally simple, mild, and efficient approach to use N-heteroaryl alkene substrates in copper-catalyzed alkene aminooxygenation. This method displays broad compatibility across a diverse range of heteroaromatic groups that has not previously been demonstrated in alkene aminofunctionalization reactions. Further, the practicality and applicability of our strategy to the synthesis of drug-like compounds shows promise for its adoption in the rapid preparation of valuable α-amino-oxygenated motifs for diverse synthetic applications.Data availability
The data supporting this article, including experimental procedures, additional reaction screening data, and full compound characterization, have been included as part of the ESI.†Author contributions
E. M. D. and S. M. P. conceived of and designed the project. E. M. D. and N. T.-L. conducted all the experimental work, and all authors analyzed the data and discussed the results. E. M. D. and S. M. P. wrote the manuscript with support from all authors. S. M. P. directed the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Institutes of Health (R35GM150584) for financial support. The authors acknowledge the use of JEOL NMR spectrometers acquired with support from the National Science Foundation (MRI-2215973). We thank Shannon O'Neil for checking the experimental procedure for preparation of compound 2i in Fig. 2.References
- (a) Z. Wu, M. Hu, J. Li, W. Wu and H. Jiang, Org. Biomol. Chem., 2021, 19, 3036–3054 RSC; (b) X. Chen, F. Xiao and W.-M. He, Org. Chem. Front., 2021, 8, 5206–5228 RSC.
- (a) R. Kumar, Y. Khanna, P. Kaushik, R. Kamal and S. Khokhar, Chem.–Asian J., 2023, 18, e202300017 CrossRef CAS PubMed; (b) Z.-L. Tao and S. E. Denmark, Synthesis, 2021, 53, 3951–3962 CrossRef CAS.
- (a) K. Hashimoto, T. Watari and T. Uchida, Tetrahedron Lett., 2023, 123, 154542 CrossRef CAS; (b) G. Kirby, G. Prestat and F. Berhal, J. Org. Chem., 2023, 88, 4720–4729 CrossRef CAS PubMed; (c) C. Liu, J. Wu, X. Tan, J. Zhang, W. Wu and H. Jiang, ACS Catal., 2023, 13, 11339–11344 CrossRef CAS; (d) X. Li, H. Song, S. Yu, R. Mi and X.-X. Li, Angew. Chem., Int. Ed., 2023, 62, e202305669 CrossRef CAS PubMed; (e) X. Nie, C. W. Ritter, M. Hemming, S. I. Ivlev, X. Xie, S. Chen and E. Meggers, Angew. Chem., Int. Ed., 2023, 62, e202314398 CrossRef CAS PubMed; (f) M. He, C. Shi, M. Luo, C. Yang, L. Guo, Y. Zhao and W. Xia, J. Org. Chem., 2024, 89, 1967–1979 CrossRef CAS PubMed; (g) R. Madiu, B. Dellosso, E. L. Doran, J. M. Doran, A. A. Pinarci, T. M. TenHoeve, A. M. Howard, J. L. Stroud, D. A. Rivera and D. A. Moskovitz, et al., Org. Biomol. Chem., 2024, 22, 2300–2306 RSC; (h) R. L. L. Carmo, S. L. Galster, T. Wdowik, C. Song and S. R. Chemler, J. Am. Chem. Soc., 2023, 145, 13715–13729 CrossRef CAS PubMed.
- (a) X. Zhan, G. Gao, Y. Liang, F. Li, K. Liu, W. Fan, S. Zhang and M.-B. Li, Org. Chem. Front., 2023, 10, 3353–3360 RSC; (b) Z. Wang, C. Hou and P. Chen, Org. Lett., 2023, 25, 2685–2690 CrossRef CAS PubMed; (c) E. Mejri, K. Higashida, Y. Kondo, A. Nawachi, H. Morimoto, T. Ohshima, M. Sawamura and Y. Shimizu, Org. Lett., 2023, 25, 4581–4585 CrossRef CAS PubMed; (d) C. T. Constantinou, P. L. Gkizis, O. T. G. Lagopanagiotopoulou, E. Skolia, N. F. Nikitas, I. Triandafillidi and C. G. Kokotos, Chem.–Eur. J., 2023, 29, e202301268 CrossRef CAS PubMed; (e) A. U. Rahman, N. Zarshad, I. Khan, F. Faiz, G. Li and A. Ali, Front. Chem., 2021, 9, 742399 CrossRef PubMed; (f) W.-H. Ng, Y.-P. Lam, R.-B. Hu, W.-L. Ng and Y.-Y. Yeung, Asian J. Org. Chem., 2021, 10, 1131–1140 CrossRef CAS; (g) Y. Luo, S. Zhou, A. A. Nkingwa and Q. Zeng, Eur. J. Org Chem., 2024, 27, e202301178 CrossRef CAS; (h) M. Schäfer, T. Stünkel, C. G. Daniliuc and R. Gilmour, Angew. Chem., Int. Ed., 2022, 61, e202205508 CrossRef PubMed; (i) B. Wang, J. Li, W. Wu and H. Jiang, Chin. J. Chem., 2024, 42, 464–470 CrossRef CAS.
- (a) M. B. Bertrand, J. D. Neukom and J. P. Wolfe, J. Org. Chem., 2008, 73, 8851–8860 CrossRef CAS PubMed; (b) D. R. White, J. T. Hutt and J. P. Wolfe, J. Am. Chem. Soc., 2015, 137, 11246–11249 CrossRef CAS PubMed; (c) P. Shi, J. Wang, Z. Gan, J. Zhang, R. Zeng and Y. Zhao, Chem. Commun., 2019, 55, 10523–10526 RSC; (d) T. Wdowik, S. L. Galster, R. L. L. Carmo and S. R. Chemler, ACS Catal., 2020, 10, 8535–8541 CrossRef CAS PubMed; (e) E. Falk, S. Makai, T. Delcaillau, L. Gürtler and B. Morandi, Angew. Chem., Int. Ed., 2020, 59, 21064–21071 CrossRef CAS PubMed.
- Z. Liu, H.-Q. Ni, T. Zeng and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 3223–3227 CrossRef CAS PubMed.
- (a) H. Jiang, H. Gao, B. Liu and W. Wu, Chem. Commun., 2014, 50, 15348–15351 RSC; (b) Y. Kwon and Q. Wang, Org. Lett., 2020, 22, 4141–4145 CrossRef CAS PubMed.
- J.-A. Li, P.-Z. Zhang, K. Liu, A. Shoberu, J.-P. Zou and W. Zhang, Org. Lett., 2017, 19, 4704–4706 CrossRef CAS PubMed.
- J. Luo, Z. Zhu, Y. Liu and X. Zhao, Org. Lett., 2015, 17, 3620–3623 CAS.
- (a) Y. Li, J. Bao, Y. Zhang, X. Peng, W. Yu, T. Wang, D. Yang, Q. Liu, Q. Zhang and J. Fu, Chem, 2022, 8, 1147–1163 CrossRef CAS; (b) Z. Liu, Y. Wang, Z. Wang, T. Zeng, P. Liu and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 11261–11270 CrossRef CAS PubMed; (c) Z. Tao, B. B. Gilbert and S. E. Denmark, J. Am. Chem. Soc., 2019, 141, 19161–19170 CrossRef CAS PubMed; (d) C. P. McNichol, E. M. DeCicco, A. M. Canfield, D. P. Carstairs and S. M. Paradine, ACS Catal., 2023, 13, 6568–6573 CrossRef CAS; (e) L. Wang and C. Wang, J. Org. Chem., 2019, 84, 6547–6556 CrossRef CAS PubMed; (f) Y. Li, Y. Liang, J. Dong, Y. Deng, C. Zhao, Z. Su, W. Guan, X. Bi, Q. Liu and J. Fu, J. Am. Chem. Soc., 2019, 141, 18475–18485 CrossRef CAS PubMed; (g) J. Zhao, H.-G. Huang, W. Li and W.-B. Liu, Org. Lett., 2021, 23, 5102–5106 CrossRef CAS PubMed; (h) J. P. Ariyarathna, N.-E. Alom, L. P. Roberts, N. Kaur, F. Wu and W. Li, J. Org. Chem., 2022, 87, 2947–2958 CrossRef CAS PubMed; (i) Z. Pan, S. Wang, J. T. Brethorst and C. J. Douglas, J. Am. Chem. Soc., 2018, 140, 3331–3338 CrossRef CAS PubMed; (j) S.-Q. Lai, B.-Y. Wei, J.-W. Wang, W. Yu and B. Han, Angew. Chem., Int. Ed., 2021, 60, 21997–22003 CrossRef CAS PubMed; (k) Y. Li, Y. Dong, X. Wang, G. Li, H. Xue, W. Xin, Q. Zhang, W. Guan and J. Fu, ACS Catal., 2023, 13, 2410–2421 CrossRef CAS; (l) Y. Zheng, Z.-J. Wang, Z.-P. Ye, K. Tang, Z.-Z. Xie, J.-A. Xiao, H.-Y. Xiang, K. Chen, X.-Q. Chen and H. Yang, Angew. Chem., Int. Ed., 2022, 61, e202212292 CrossRef CAS PubMed; (m) D. E. Holst, C. Dorval, C. K. Winter, I. A. Guzei and Z. K. Wickens, J. Am. Chem. Soc., 2023, 145, 8299–8307 CrossRef CAS PubMed; (n) W. Xue, Z. Zhu, S. Chen, B. You and C. Tang, J. Am. Chem. Soc., 2023, 145, 4142–4149 CrossRef CAS PubMed; (o) D. E. Holst, D. J. Wang, M. J. Kim, I. A. Guzei and Z. K. Wickens, Nature, 2021, 596, 74–79 CrossRef CAS PubMed; (p) H. Zeng, H. Li, C. Li, H. Jiang and C. Zhu, Org. Chem. Front., 2022, 9, 1383–1388 RSC; (q) X. Fang, P. Yu and B. Morandi, Science, 2016, 351, 832–836 CrossRef CAS PubMed; (r) G. Feng, C. K. Ku, J. Zhao and Q. Wang, J. Am. Chem. Soc., 2022, 144, 20463–20471 CrossRef CAS PubMed; (s) L. Legnani, G. Prina-Cerai, T. Delcaillau, S. Willems and B. Morandi, Science, 2018, 362, 434–439 CrossRef CAS PubMed; (t) G. Tan, M. Das, R. Kleinmans, F. Katzenburg, C. Daniliuc and F. Glorius, Nat. Catal., 2022, 5, 1120–1130 CrossRef CAS; (u) J.-J. Dai, X. Yin, L. Li, M. E. Rivera, Y.-C. Wang and M. Dai, Nat. Commun., 2023, 14, 1774 CrossRef CAS PubMed.
- M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247–44311 RSC.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- T. J. Ritchie, S. J. Macdonald, R. J. Young and S. D. Pickett, Drug Discov. Today, 2011, 16, 164–171 CrossRef CAS PubMed.
- (a) B. N. Hemric, Org. Biomol. Chem., 2021, 19, 46–81 RSC; (b) L. A. T. Allen, R.-C. Raclea, P. Natho and P. J. Parsons, Org. Biomol. Chem., 2021, 19, 498–513 RSC; (c) O. K. Karjalainen and A. M. P. Koskinen, Org. Biomol. Chem., 2012, 10, 4311–4326 RSC; (d) S. C. Bergmeier, Tetrahedron, 2000, 56, 2561–2576 CrossRef CAS.
- (a) W.-H. Rao, L.-L. Jiang, F.-Y. Chen, M. Zhang, Y.-Y. Guo, J.-J. Chen, Y. Cui, G.-D. Zou and L. Tang, Tetrahedron Lett., 2020, 61, 151540 CrossRef CAS; (b) P. H. Fuller, J.-W. Kim and S. R. Chemler, J. Am. Chem. Soc., 2008, 130, 17638–17639 CrossRef CAS PubMed; (c) M. C. Paderes and S. R. Chemler, Org. Lett., 2009, 11, 1915–1918 CrossRef CAS PubMed; (d) S. D. Karyakarte, T. P. Smith and S. R. Chemler, J. Org. Chem., 2012, 77, 7755–7760 CrossRef CAS PubMed.
- (a) K. Wen, Z. Wu, B. Huang, Z. Ling, I. D. Gridnev and W. Zhang, Org. Lett., 2018, 20, 1608–1612 CrossRef CAS PubMed; (b) X. Kou, Y. Li, L. Wu, X. Zhang, G. Yang and W. Zhang, Org. Lett., 2015, 17, 5566–5569 CrossRef CAS PubMed; (c) H.-C. Shen, Y.-F. Wu, Y. Zhang, L.-F. Fan, Z.-Y. Han and L.-Z. Gong, Angew. Chem., Int. Ed., 2018, 57, 2372–2376 CrossRef CAS PubMed.
- M. Balkenhohl, S. Kölbl, T. Georgiev and E. M. Carreira, JACS Au, 2021, 1, 919–924 CrossRef CAS PubMed.
- (a) Z. Liu, P. Wang, H. Ou, Z. Yan, S. Chen, X. Tan, D. Yu, X. Zhao and T. Mu, RSC Adv., 2020, 10, 7698–7707 RSC; (b) J. Li, J. Wei, B. Zhu, T. Wang and N. Jiao, Chem. Sci., 2019, 10, 9099–9103 RSC; (c) T. Wdowik and S. R. Chemler, J. Am. Chem. Soc., 2017, 139, 9515–9518 CrossRef CAS PubMed.
- (a) F. Wu, S. Stewart, J. P. Ariyarathna and W. Li, ACS Catal., 2018, 8, 1921–1925 CrossRef CAS; (b) M. C. Paderes, L. Belding, B. Fanovic, T. Dudding, J. B. Keister and S. R. Chemler, Chem.–Eur. J., 2012, 18, 1711–1726 CrossRef CAS PubMed; (c) M. C. Paderes, J. B. Keister and S. R. Chemler, J. Org. Chem., 2013, 78, 506–515 CrossRef CAS PubMed.
- For additional details, see (ESI).†.
- J. Liu, Y. Li, M. Ke, M. Liu, P. Zhan, Y.-C. Xiao and F. Chen, J. Org. Chem., 2020, 85, 15360–15367 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06178k |
| This journal is © The Royal Society of Chemistry 2024 |