
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in the synthesis of highly substituted imidazolidines
Abolfazl Olyaei
 *a and
Mahdieh Sadeghpour
b
*a and
Mahdieh Sadeghpour
b
aDepartment of Chemistry, Faculty of Science, Imam Khomeini International University, Qazvin, Iran. E-mail: olyaei_a@sci.ikiu.ac.ir
bDepartment of Chemistry, Qazvin Branch, Islamic Azad University, Qazvin, Iran
First published on 26th September 2024
Abstract
Imidazolidine is a saturated heterocycle with a cyclic aminal core that can be found in natural products and biologically active molecules. Additionally, these heterocyclic compounds have been utilized as chiral ligands, N-heterocyclic carbene precursors, and catalysts in organic synthesis. This review is an attempt to compile the literature of various synthetic procedures of highly substituted imidazolidines, chiral imidazolidines with high diastereoselectivities and enantioselectivities, bis-imidazolidines, and spiro-imidazolidines, as well as their pharmacological properties during the period from 1949 to 2023.
 Abolfazl Olyaei | Associate professor Dr Abolfazl Olyaei was born in Tabriz, Iran in 1975. He received the BSc degree in pure chemistry from Tabriz University, Tabriz, Iran in 1999 and the MSc degree in organic chemistry from Tehran University, Tehran, Iran under the supervision of professor Mohammad Raouf Darvich in 2001. He obtained PhD degree in organic chemistry from Tehran University, Tehran, Iran under the supervision of professor Mehdi Ghandi, in 2007. He is as an associate professor in Imam Khomeini International University, Qazvin, Iran. His research interests include organic synthesis, synthesis of heterocyclic compounds, multi-component reactions, green chemistry, catalysis and organocatalysis and applications of materials and organomaterials in different sciences. |
 Mahdieh Sadeghpour | Associate professor Dr Mahdieh Sadeghpour was born in Qazvin, Iran in 1978. She received the BSc degree in pure chemistry from Alzahra University, Tehran, Iran in 2001 and the MSc degree in organic chemistry from Tehran University, Tehran, Iran under the supervision of assistance professor Nikoo Sedighi in 2004. She obtained PhD degree in organic chemistry from Kharazmi University, Tehran, Iran under the supervision of professor Abbas Shokravi and associate professor Abolfazl Olyaei, in 2009. She is as an associate professor in Islamic Azad University of Qazvin, Iran. Her research field is on the synthesis of organic compounds, multi-component reactions, synthetic methodology, green chemistry and applications of materials and nanomaterials in different sciences. |
1 Introduction
Heterocyclic compounds are important structural motifs commonly found in natural products and biologically active molecules, playing a significant role in medicinal chemistry due to their diverse applications and potent effects. Among these heterocyclic compounds, imidazolidine, tetrahydroimidazole, frameworks are prevalent in numerous natural alkaloids (e.g., (−)-chaetominine)1 and an intrinsic part of various naturally occurring bioactive substances such as chaetominine, fumiquinazolines A, tryptoquivalin G, and kifunensine.2–7 They serve as chiral ligands, auxiliaries, and N-heterocyclic carbene (NHC) precursors and catalysts in organic catalysis (e.g., MacMillan's catalyst).8–11 They also form essential structural motifs in synthetic biologically active compounds12 and hold significant potential in drug discovery, including applications as antipyretic agents and cannabinoid CB2 receptor agonists.13,14 Moreover, they have shown fungicidal, antiparasitic, antibacterial, antiamebic, and antiviral activities.15,16 Due to their unique structural characteristics and impressive applications, a number of synthetic strategies have been devoted to the preparation of diversely substituted imidazolidines. In 1954, the first review article on the chemistry of 2-imidazolines and imidazolidines was published.17 Since then, no separate review article on the synthesis of imidazolidine derivatives has been published. In this review, we aim to describe the synthesis strategies of imidazolidine derivatives, including poly substituted imidazolidines, chiral imidazolidines with high diastereoselectivities and enantioselectivities, bis-imidazolidines, and spiro-imidazolidines, as well as their pharmacological properties from 1949 to 2023.2 Synthesis of imidazolidines
2.1. Synthesis of highly substituted imidazolidines
In 1949, Donia and co-workers reported the first synthesis of 1,3-dialkylimidazolidines 1 in 37–90% yields by the condensation reaction of aldehydes (formaldehyde, butyraldehyde, and benzaldehyde) with N,N′-disubstituted ethylenediamines 2 in which the substituents were ethyl, allyl, isopropyl, n-butyl, l-methylbutyl, cyclohexyl, phenyl and 2-ethylhexyl. This condensation proceeded most readily with formaldehyde, with or without a solvent at temperature below 50 °C, afforded the corresponding 1,3-imidazolidines. With butyraldehyde and benzaldehyde, the reactions producing 2-substituted imidazolidines proceeded more slowly, and were aided by warming in the presence of toluene (50–60 °C or reflux conditions) and separation of the water formed (Scheme 1).18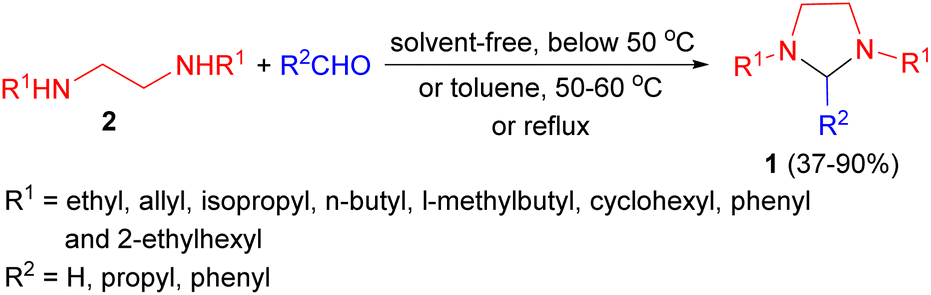 | ||
| Scheme 1 Solvent-free synthesis of 1,3-dialkylimidazolidines 1. | ||
In 1957, Billman described synthesis a series of 2-substituted-1,3-bis (p-chlorobenzy1)imidazolidines 3 in 21–85% yields from 1,2-bis(p-chlorobenzylamino)ethane 4 and numerous aldehydes in absolute alcohol under shaken occasionally at room temperature for 10 to 15 minutes or 65 °C for 10 minutes. Treatment of the imidazolidines with acid regenerates the diamine and aldehyde in nearly quantitative yields. However, compound 4 did not react with ketones under the experimental conditions used. From this evidence, it appears that compound 4 can be used as a specific reagent for aldehydes in the presence of ketones (Scheme 2).19
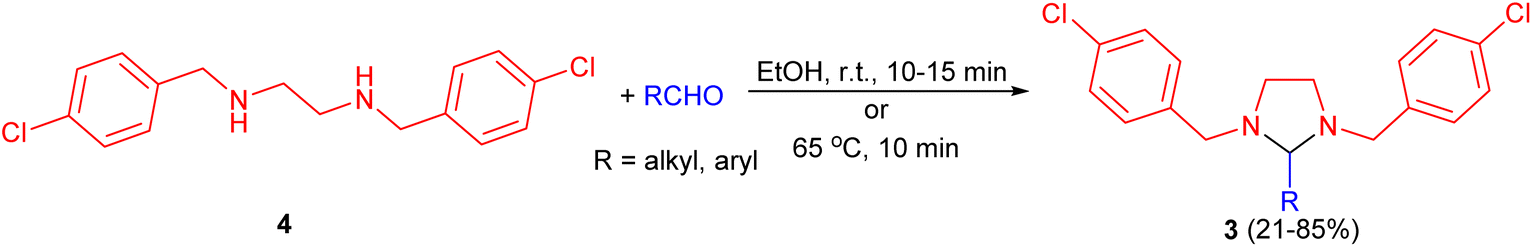 | ||
| Scheme 2 Synthesis of 2-substituted-1,3-bis(p-chlorobenzy1)imidazolidines 3. | ||
In 1959, Jaenlcke and Erode developed synthesis of 1,3-diarylimidazolidine derivatives 5 in 75–91% yields by the reaction of substituted N,N′-diphenylethylenediamines 6 with formaldehyde (30%) in CH3OH or ethyl acetate in the presence of acetic acid (Scheme 3).20
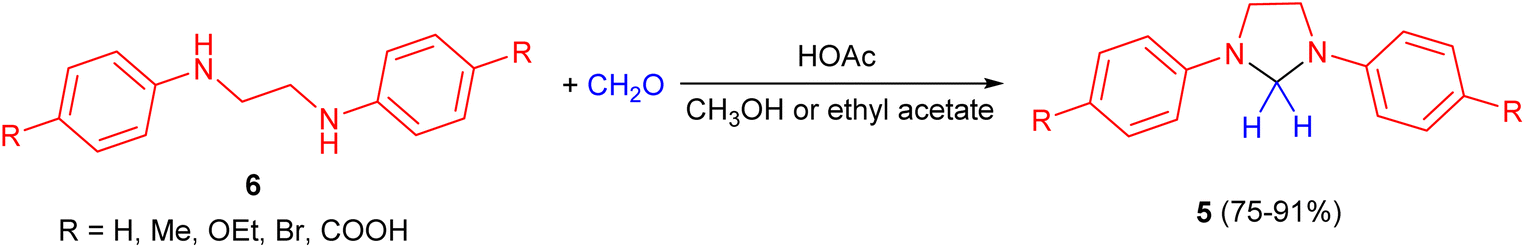 | ||
| Scheme 3 HOAc catalyzed synthesis of 1,3-diarylimidazolidine derivatives 5. | ||
In 1967, the Joullie group described the reaction of 1,2-ethylenediamine (7) with ethyl trifluoroacetoacetate (8) in xylene refluxing in a flask equipped with a Dean–Stark trap. The solution was refluxed for 1 hour, after the separation of water was completed and left standing overnight. During this procedure, the products, namely 1,2,3,4-tetrahydro-7-trifluoromethyl-1,4-diazepin-5-one (9) and ethyl 2-(trifluoromethyl)-2-imidazolidineacetate (10) were obtained with yields of 16% and 25%, respectively. Moreover, l,4-diazaspiro[4,5]decan (11) synthesized by the same group in 37% yield via the reaction of cyclohexanone with 1 in benzene under reflux condition in a flask with a Dean–Stark trap (Scheme 4).21
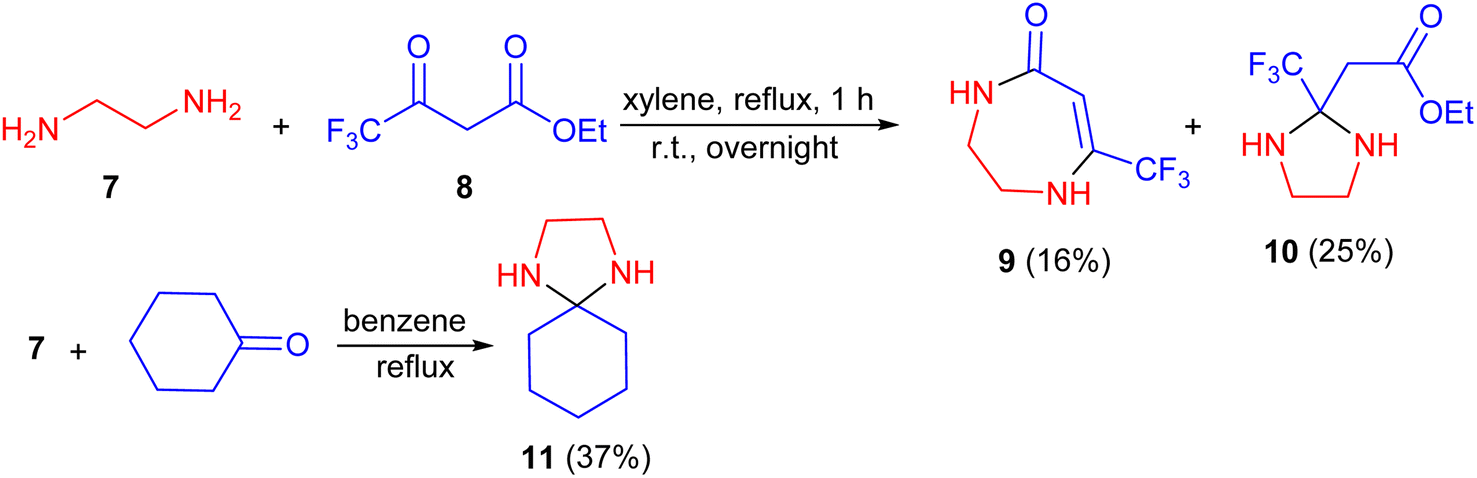 | ||
| Scheme 4 Synthesis of imidazolidine derivatives 10 and 11. | ||
In 1969, Lown and co-workers explored the synthesis of 1-alkyl-2,4-diaryl-5-aroyl-3-(N-arylsulfonyl)-imidazolidines 12 in 14.5–94% yields through the reaction of 3-aroylaziridines 13 with aryl-N-sulfonylimines 14 in refluxing benzene. The orientation of the [2 + 3] cycloaddition of the intermediate azomethine ylids to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond was proven by synthesis of specifically 5-deuterated imidazolidines (Scheme 5).22
N double bond was proven by synthesis of specifically 5-deuterated imidazolidines (Scheme 5).22
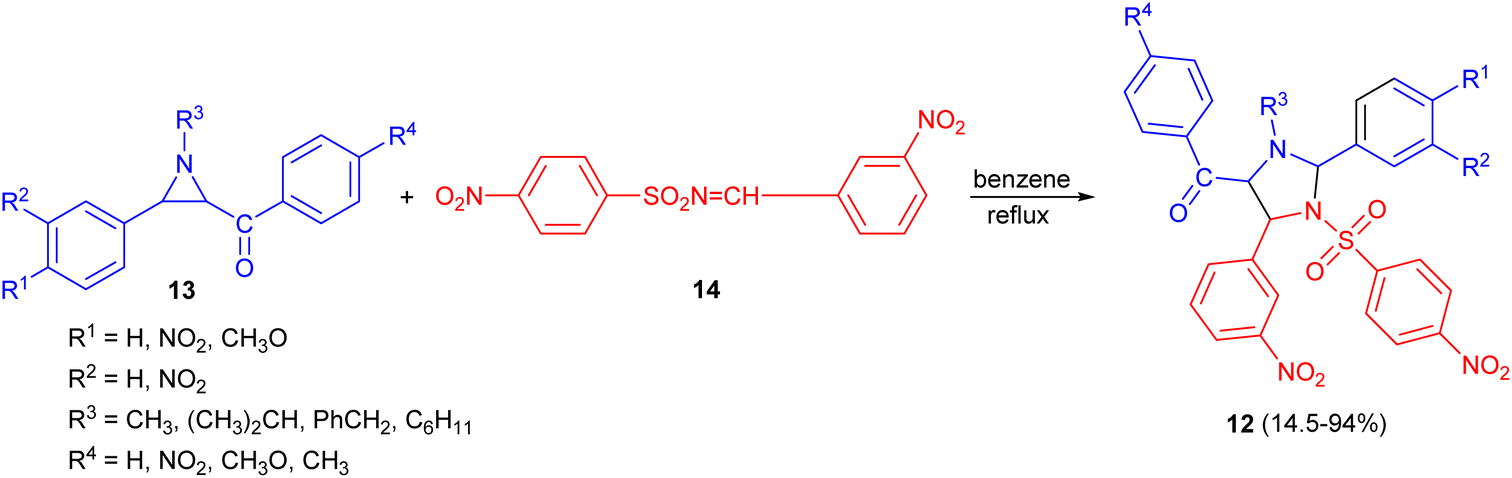 | ||
| Scheme 5 Synthesis of 1-alkyl-2,4-diaryl-5-aroyl-3-(N-arylsulfonyl)-imidazolidines 12. | ||
In 1973, Birch and Dastu demonstrated a method for the synthesis of dihydroimidazolidine. In this process, at first, the reaction of 4-isopropylbenzaldehyde with N,N′-dimethylethylenediamine (15) in benzene at 60 °C for 2 hours resulted 1,3-dimethyl-2-(4′-isopropylphenyl)imidazolidine 16 in 69% yield followed by the reduction of 16 with lithium/ammonia in dry tetrahydrofuran and t-butyl alcohol under reflux with stirring for 2 hours afforded the desired imidazolidine 17 (Scheme 6).23
 | ||
| Scheme 6 Synthesis of dihydroimidazolidines 16 and 17. | ||
Next, Hine and his group reported the reaction of carbonyl compounds with N,N′-dialkylethylenediamine 18 in pentane under reflux conditions gave imidazolidine derivatives 19 in 44–72% yields as depicted in Scheme 7.24
 | ||
| Scheme 7 Preparation of imidazolidine derivatives 19. | ||
In 1975, Itoh group synthesized imidazolidine derivatives 20 in 33–98% yields by condensation reaction of N,N′-bis(trimethylsilyl)1,2-diamines 21 with carbonyl compounds via the elimination of hexamethyldisiloxane. The reactions were conducted with cyclohexanone and benzaldehyde at 20 °C for 2 minutes, and with diethyl ketone and DMF at 120–180 °C for 1–24 hours (Scheme 8).25
 | ||
| Scheme 8 Synthesis of imidazolidine derivatives 20. | ||
The downward deviations in the pH-rate profiles between 4–6 for the reactions of a series of symmetrically meta- or para-substituted N,N′-diphenylethylenediamines 22 with formaldehyde in dioxane–water afforded the imidazolidines 23 (Scheme 9). In this process, kinetic studies of the reactions were investigated.26
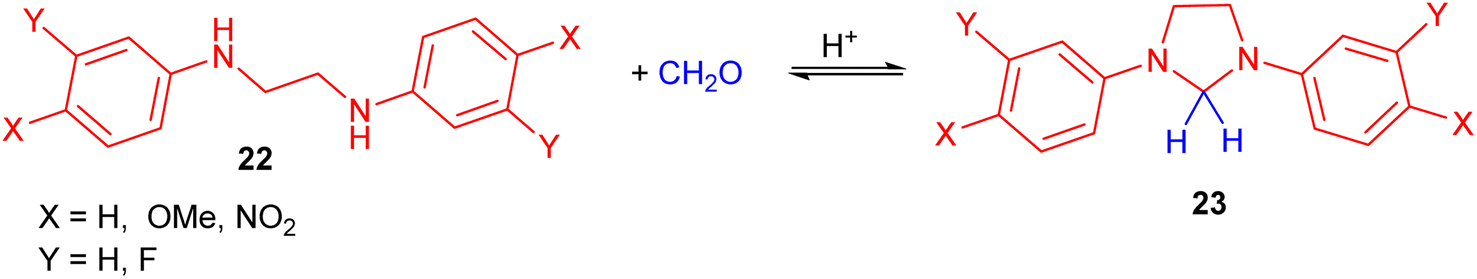 | ||
| Scheme 9 Acid catalyzed preparation of imidazolidines 23. | ||
In 1978, Keana and co-workers described a new series of mono- and dinitroxide spin labels derived by oxidation of 2,2,4,4,5,5-hexasubstituted imidazolidines. Condensation of 2,3-diamino-2,3-dimethylbutane 24 with ketones in the presence of TsOH in benzene under reflux conditions for 48 hours, led to the corresponding imidazolidines 25 in 32–76% yields. Oxidation with 1.5 equiv. of m-chloroperoxybenzoic acid (MCPA) in ether at 0 °C for overnight gave the corresponding mononitroxides 26. Catalytic hydrogenation (Pd/C, THF) of 26a at 25 °C gave 27 which, without isolation, was acetylated (AcCl, Et3N, THF) to give 28 in 73% yield. Oxidation of 28 with MCPA in ether gave nitroxide 29 in 80% yield. Hydrolysis in the presence of KOH in MeOH yielded 30 in 75% yield. Oxidation of 30 in the presence of KO-t-Bu in t-BuOH gave dinitroxide 31 in 84% yield. Using a procedure analogous to the preparation of 27 and 28, compound 31 gave diacetate 32 (Scheme 10).27
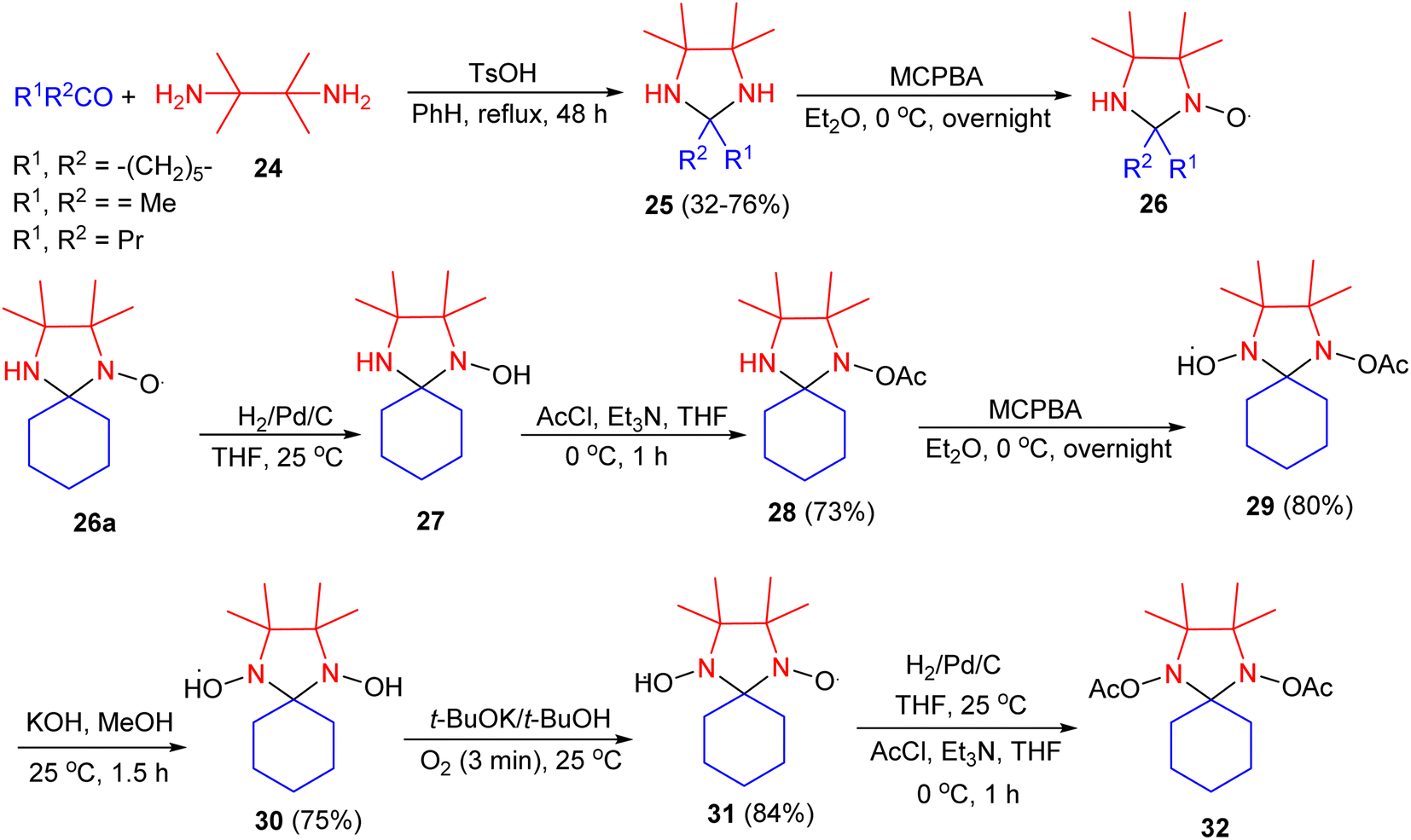 | ||
| Scheme 10 Synthesis of imidazolidine derivatives 25–32. | ||
In 1980, Amornraksa and Grigg demonstrated the reaction of arylaldehydes with diethyl aminomalonate hydrochloride 33 in the presence of 1 equiv. of sodium ethoxide in ethanol at 40 °C gave the imidazolidines 34 (Scheme 11).28
 | ||
| Scheme 11 Synthesis of imidazolidines 34 in the presence of sodium ethoxide in ethanol. | ||
In 1985, Chadwick and his group developed synthesis of imidazolidine derivatives 35 in 70–81% yields by the reaction of thiophene-2-carboxaldehyde/furan-2-carboxaldehyde/1-methyl-pyrrole-2-carboxaldehyde with N,N′-dimethylethylenediamine in benzene under reflux for 5 hours with azeotropic removal of water. Also, treatment of 36 and 37 with N,N′-dimethylethylenediamine in the same reaction conditions afforded imidazolidines 38 and 39 in 78 and 96% yields, respectively (Scheme 12).29
 | ||
| Scheme 12 Synthesis of imidazolidine derivatives 35, 38 and 39. | ||
In 1993, a one-step synthesis of 1,3-bis[2′-hydroxy-5′-substituted-benzyl]imidazolidines 40 in 21.4–28.1% yields using a Mannich type reaction in basic media was described by Rivera and his group. The reaction was carried out by condensation of tetraazatricyclo[4.4.1.13,8]dodecane (TATD) (41) with a number of p-substituted phenols by heating the reactants in aqueous dioxane solution at 40–42 °C. A plausible mechanism is illustrated in Scheme 13. First, when phenol is added to TATD the initially formed hydrogen bond could undergo mono protonation of any of the four nitrogen atoms. Introduction of a proton between nitrogens leads to polarization of the adjacent methylene (aminalic) groups. In agreement with known electrophilic substitution to aromatic rings, the reaction involves the successive attack of two molecules of the phenol. The possible intermediate 42, not yet isolated, undergoes intramolecular condensation to gain stability.30
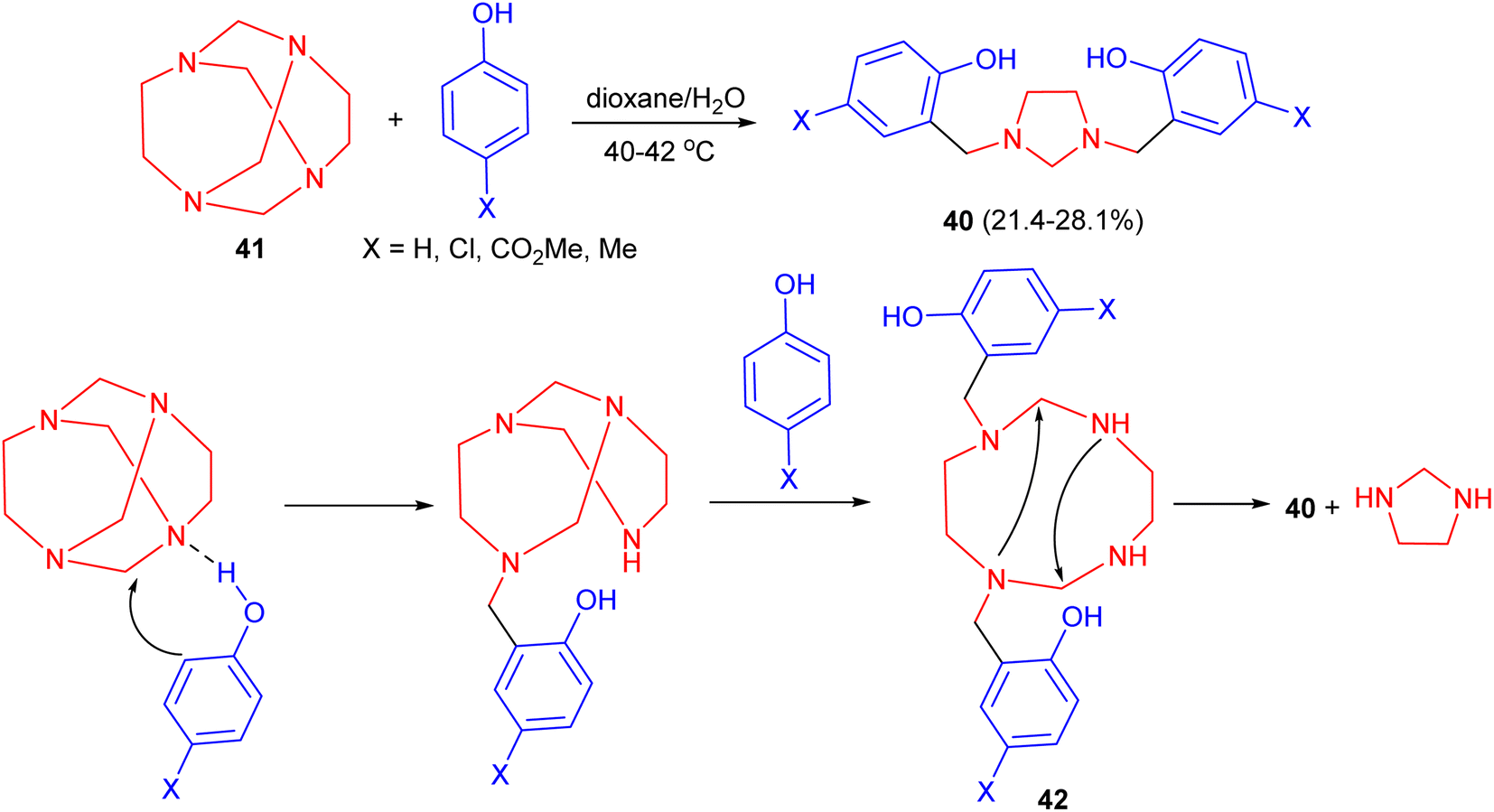 | ||
| Scheme 13 Synthesis of 1,3-bis[2′-hydroxy-5′-substituted-benzyl]imidazolidines 40. | ||
In 1996, Perisamy and co-workers revealed the reaction of low valent titanium reagent, prepared in situ in THF by the reduction of TiCl4 by Mg powder, with certain imines in the presence of 1,2-dibromoethane at 25 °C led to the formation of imidazolidine derivatives 43 in 54–74% yields after 10 hours. The 1,2-dibromoethane was utilized in order to activate Mg. A tentative mechanism is shown in Scheme 14. A possibility is that the TiCl4/Mg system may generate certain reactive species of titanium and/or Mg–X species which might metallate THF leading to cleavage of THF.31
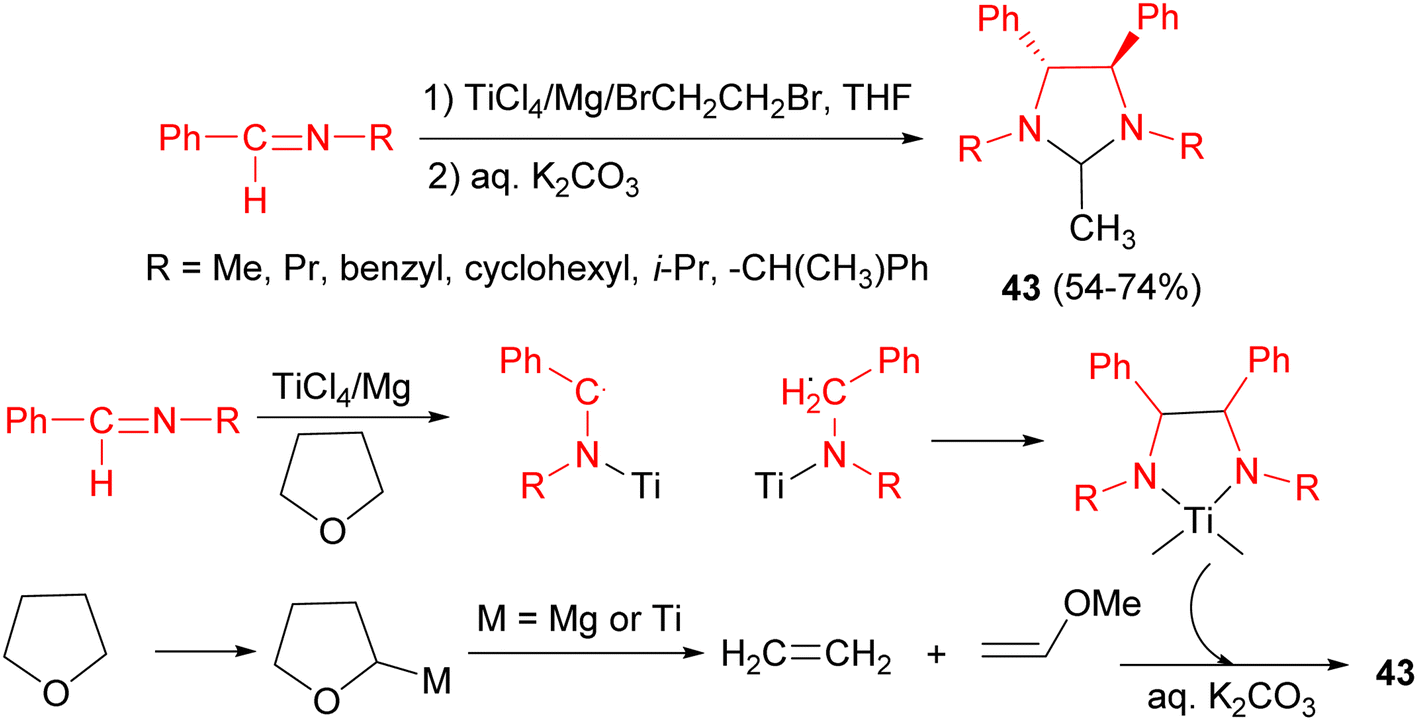 | ||
| Scheme 14 TiCl4/Mg promoted synthesis of imidazolidine derivatives 43. | ||
The Coldham group developed a one-pot, two stage process for the preparation of imidazolidines 44 and 45 in 36–73% yields bearing acyl groups on the nitrogen atoms. 1,2-Diamines were condensed with a variety of aldehydes in CHCl3 in the presence of K2CO3/MgSO4 at room temperature for 1–17 hours and the subsequent N,N′-bisunsubstituted imidazolidines 46 were acylated with a selection of acid chlorides at room temperature in the presence of pyridine or Et3N for 1–2 hours or acid anhydrides in the absence of base for 17–20 hours (Scheme 15).32
 | ||
| Scheme 15 Preparation of imidazolidines 44 and 45 in the presence of K2CO3/MgSO4. | ||
In 1999, Chen and co-workers revealed under the action of a low-valent titanium/Zn reagent, imidazolidine derivatives 47 were synthesized in 45–68% yields from imines and triethyl orthoformate in THF for 50 hours. During this procedure, two isomers (meso and dl) were obtained in different ratios (Scheme 16). The pure isomer can be obtained by recrystallization.33
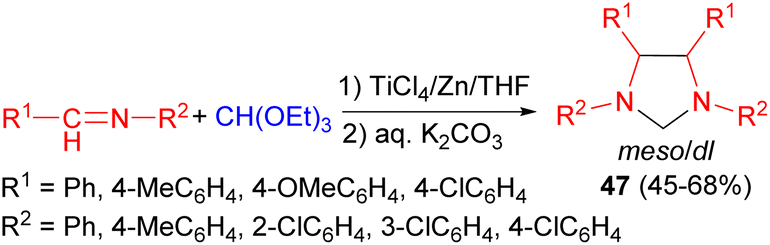 | ||
| Scheme 16 Preparation of imidazolidine derivatives 47 in the presence of TiCl4/Zn. | ||
After that, μ-bis (tetradentate)hydroxybenzamidoimidazolidine ligand 48 synthesized in 65% yield by the reaction of ligand H4L 49 with salicylaldehyde in methanol in ice-bath for 1 hour, then another period of 0.5 hours at ambient temperature (Scheme 17).34
 | ||
| Scheme 17 Synthesis of μ-bis (tetradentate)hydroxybenzamidoimidazolidine ligand 48. | ||
Further, starting from glyoxal, 1,3-diarylimidazolinium chlorides 50 were obtained in a three-step sequence via the diimines 51 and ethylene diamine dihydrochlorides 52. Reduction of 1,3-diarylimidazolinium chlorides 50 with lithium aluminium hydride in ether at 23 °C for 2 hours furnished the 1,3-diarylimidazolidines 53 in 52–74% yields (Scheme 18).35
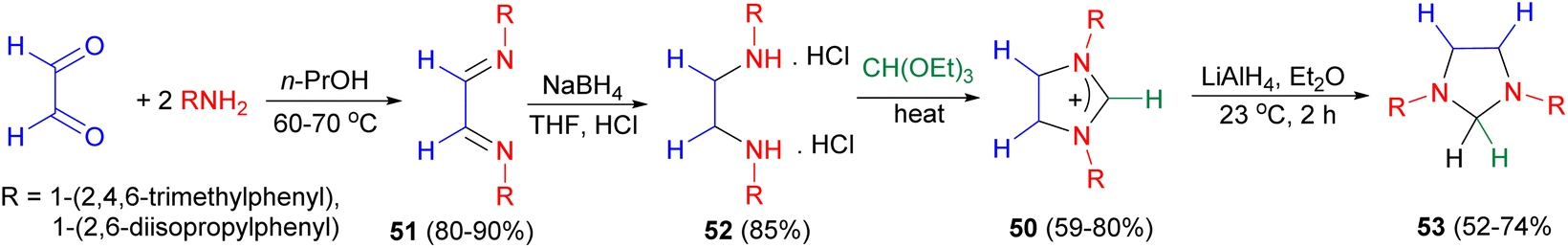 | ||
| Scheme 18 Synthesis of 1,3-diarylimidazolidines 53 starting from glyoxal. | ||
Next, the Sosnovskikh group explored the reactions of aromatic and heteroaromatic β-amino-β-polyfluoroalkylvinyl ketones 54 with ethylenediamine at 20 °C for 1–3 hours resulted in the formation of N,N′-unsubstituted imidazolidines 55 in 65–95% yields; on refluxing in ethanol for 3 hours. These products were converted into thermodynamically more stable dihydrodiazepines 56 with liberation of water (Scheme 19).36
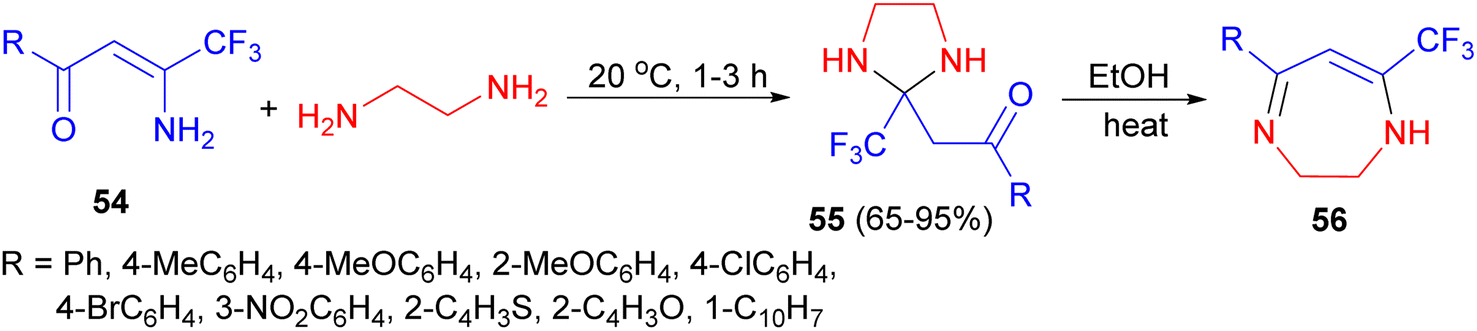 | ||
| Scheme 19 Synthesis of N,N′-unsubstituted imidazolidines 55. | ||
In 2000, Tanaka et al. developed a simple and green procedure for the synthesis of various kinds of tetrahydroimidazoles 57 in 86–95% yields by condensation reactions of aldehydes with N,N′-disubstituted ethylenediamines in a water suspension medium at room temperature for 0.5–4 hours (Scheme 20).37
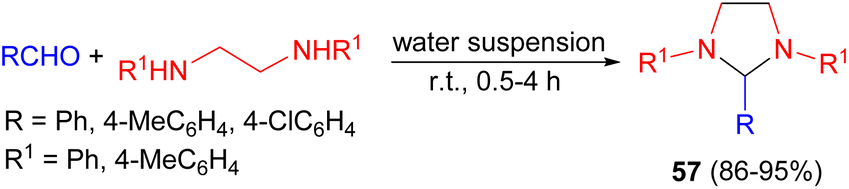 | ||
| Scheme 20 Water suspension medium synthesis of tetrahydroimidazoles 57. | ||
The Lyapova group demonstrated the quantitative reduction of compounds 58, achieved by utilizing LiAlH4 in diethyl ether and benzene at room temperature for 5 hours. This process led to the formation of trans-imidazolidines 59 (Scheme 21).38
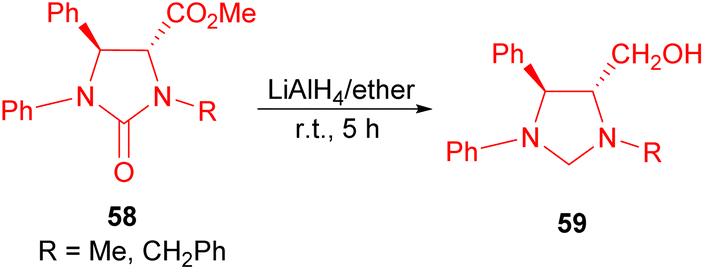 | ||
| Scheme 21 Synthesis of trans-imidazolidines 59. | ||
A one-step synthesis of imidazolidines 60 from 1,3-bis(2′-hydroxy-5′-substituted benzy1)imidazolidines 61 and aromatic aldehydes has been reported by Rivera and coworkers. The reaction was carried out in 1,4-dioxane at 60–101 °C for 50–72 hours, led to the formation of the desired products in 18–40% yields (Scheme 22). The relative stereochemistry of the five-membered ring was evident from 1H-NMR measurements combined with MMX calculations.39
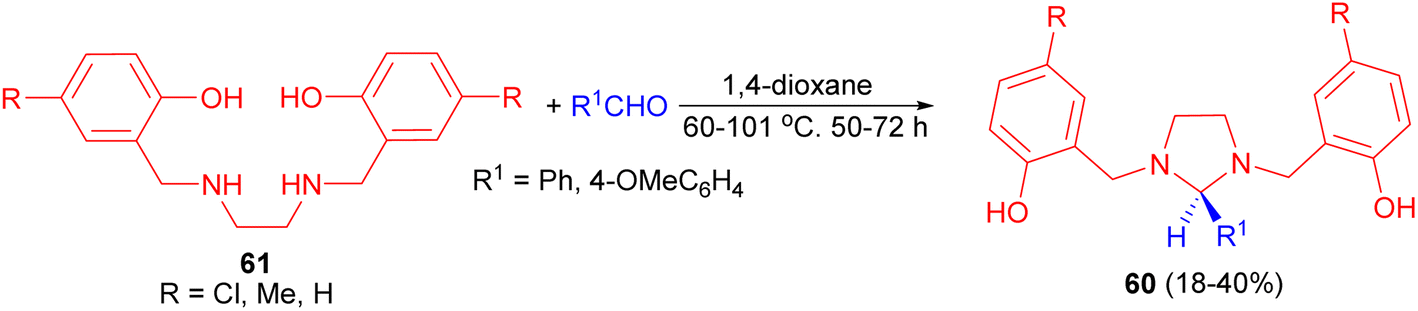 | ||
| Scheme 22 Preparation of imidazolidines 60. | ||
Next, a method for the synthesis of 2-substituted N,N′-diacylimidazolidines 62 in 10–85% yields was developed. The method based on the reactions of acylating reagents (carboxylic acid chlorides and anhydrides, sulfonic acid chlorides, a carbamic acid chloride and ethyl chlorocarbonate) with Schiff bases 63 in the presence of Et3N in CH3CN at room temperature for 1–6 hours (Scheme 23).40
 | ||
| Scheme 23 Synthesis of 2-substituted N,N′-diacylimidazolidines 62. | ||
In 2001, the Sharma and Khan reported synthesis of tetrahydroimidazoles 64 from ethylenediamine and aromatic aldehydes. In this reaction, initially, ethylenediamine was reacted with suitable aromatic aldehydes in order to prepare their respective bis-Schiff bases 65. Then, these compounds were reduced to give the corresponding tetrahydro bis-Schiff bases 66. Finally, these derivatives were condensed with different aromatic aldehydes to give the desired tetrahydroimidazoles 64 in 62–79% yields (Scheme 24). These tetrahydroimidazoles showed promising anti-inflammatory, anti-bacterial property against Staphylococcus aureus and Escherichia coli and analgesic activities.41
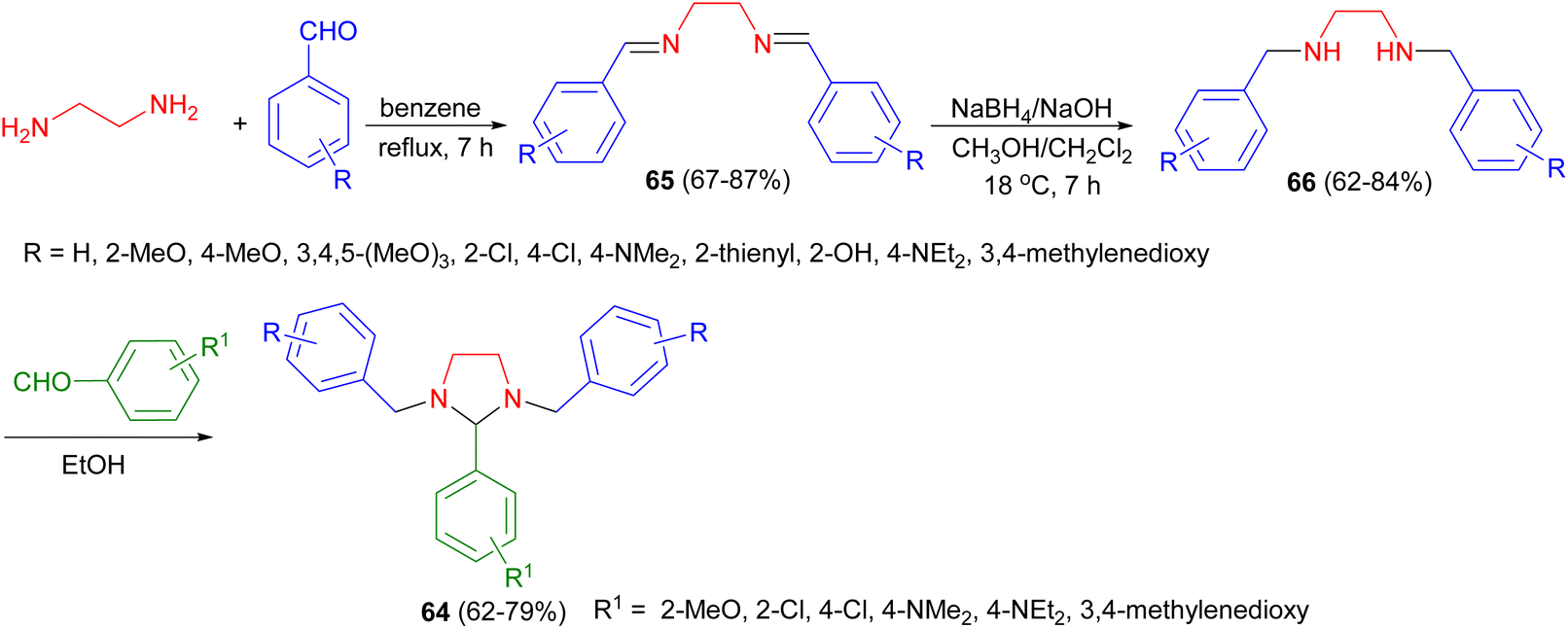 | ||
| Scheme 24 Synthesis of tetrahydroimidazoles 64 starting from ethylenediamine. | ||
Next, Coldham et al. demonstrated synthesis of imidazolidine 67 in one step from N-iso-propylethylenediamine, paraformaldehyde, MgSO4 and K2CO3, followed by addition of Boc2O. Then, imidazolidine 67 converted to imidazolidine 68 at −78 °C by using the standard conditions for asymmetric deprotonation, with sec-BuLi in Et2O and (−)-sparteine after 4–48 hours with high optical purity. Hydrolysis of imidazolidine 68 (carried out for R = SiMe3, SiMePh2, Me, allyl) using malonic acid resulted in the selective formation of the amino-carbamate 69, without loss of the N-Boc group (Scheme 25).42
 | ||
| Scheme 25 Preparation of imidazolidines 67 and 68. | ||
After that, the cross-coupling reaction of Schiff bases 70 with methylene diiodide in the presence of TiCl4/Sm in THF at room temperature for 10–14 hours to give imidazolidine derivatives 71 in 60–75% yields (Scheme 26).43
 | ||
| Scheme 26 TiCl4/Sm catalyzed synthesis of imidazolidines 71. | ||
In addition, phase-transfer catalyzed reaction of N-(benzylidene)benzylamine with arylmethyleneanilines 72 using NaOH afforded the stereoisomeric 1,2,4,5-tetraarylimidazolidines 73 and 74 via a two-step addition–cyclisation mechanism. In all cases formation of both 73 and 74 was observed, but further crystallisation of the mixtures yielded pure 73 or 74 (Scheme 27).44
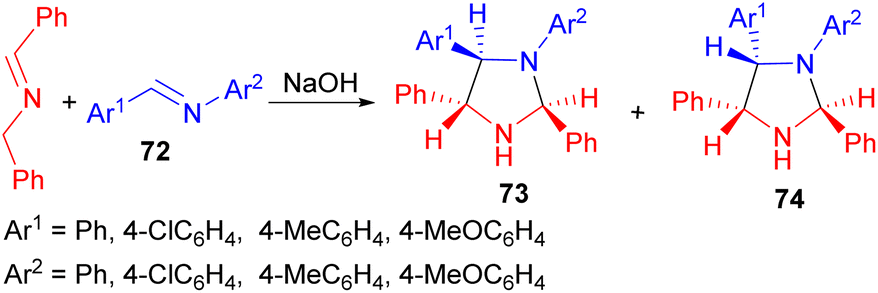 | ||
| Scheme 27 NaOH catalyzed synthesis of tetraarylimidazolidines 73 and 74. | ||
In 2002, Katritzky and co-workers developed synthesis of unsymmetrical imidazolidines 75 in 90–96% yields by Mannich reaction of 1,2-ethanediamines 76 with benzotriazole and formaldehyde in CH3OH/H2O for 4 hours at 20 °C. Then, the other imidazolidines 77 synthesized from 75 under various conditions as depicted in Scheme 28. Also, the same group reported optically active imidazolidines 78–81 in 66–99% yields. At first, reaction of diamines with benzotriazole and formaldehyde generated benzotriazol-1-yl intermediates 78 in 85–93% yields. Then, nucleophilic substitutions of 78 by Grignard reagents, triethyl phosphite, or sodium cyanide gave the desired product 79–81 (Scheme 28).45
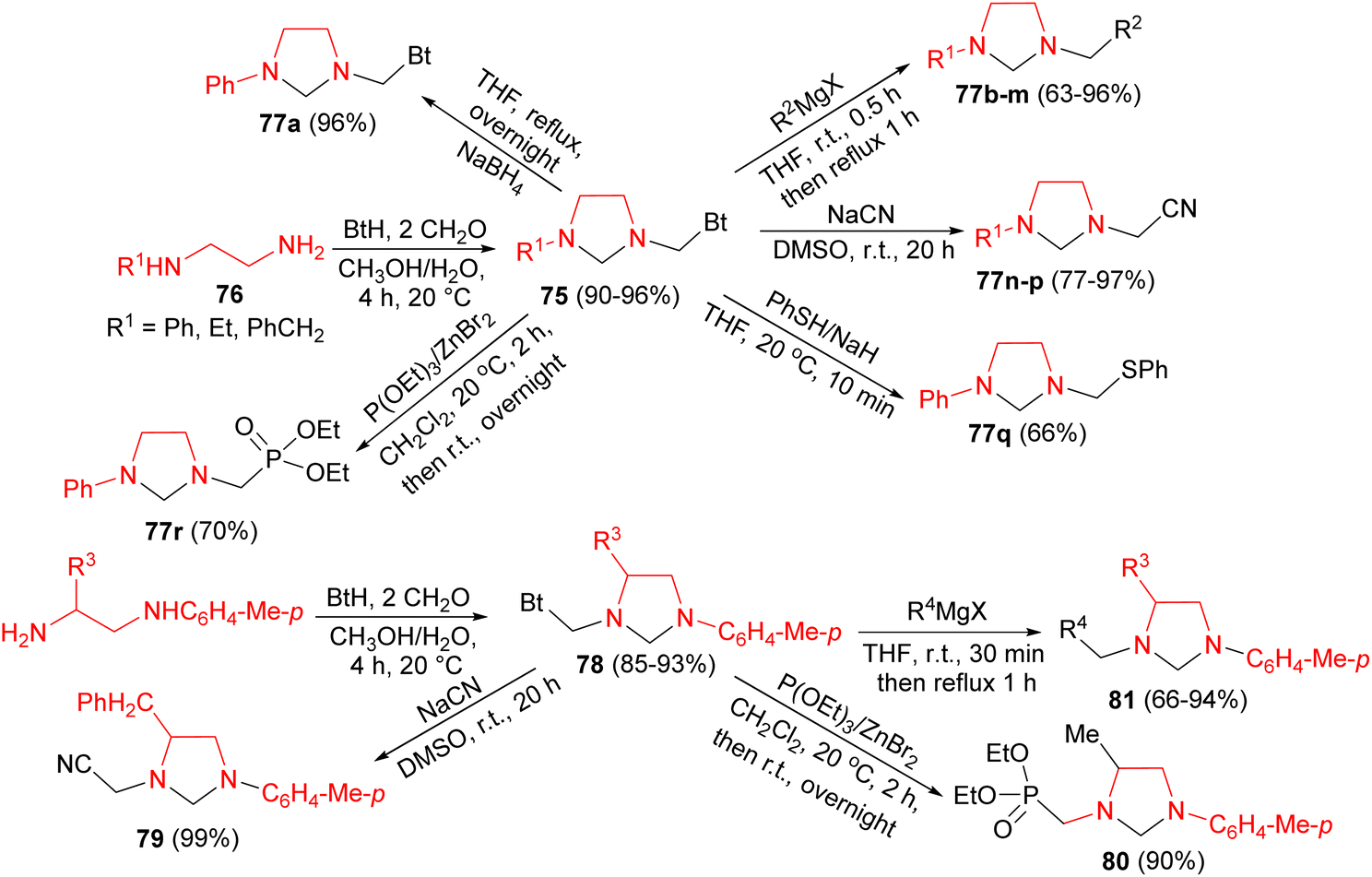 | ||
| Scheme 28 Synthesis of unsymmetrical imidazolidines 75, 77, 78–81 by Mannich reaction. | ||
In addition, several 1,2,3-trisubstituted tetrahydroimidazoles 82 in 25–79% yields were synthesized by Khan and Chawla. Synthesis of these compounds is based on formation of di-Schiff bases 83 formed by condensing two moles of aromatic aldehydes with ethylene diamine in dry benzene under reflux conditions. These Schiff bases on reduction with NaBH4 in methanol and methylene chloride under ice-cold condition for 7 hours gave substituted N,N′-dibenzyl ethylene diamines 84 which on subsequent condensation with various aromatic aldehydes in ethanol for 5–10 hours at room temperature gave tetrahydroimidazoles 82 (Scheme 29). These compounds showed excellent and far superior anti-inflammatory activity as compared to indomethacin.46
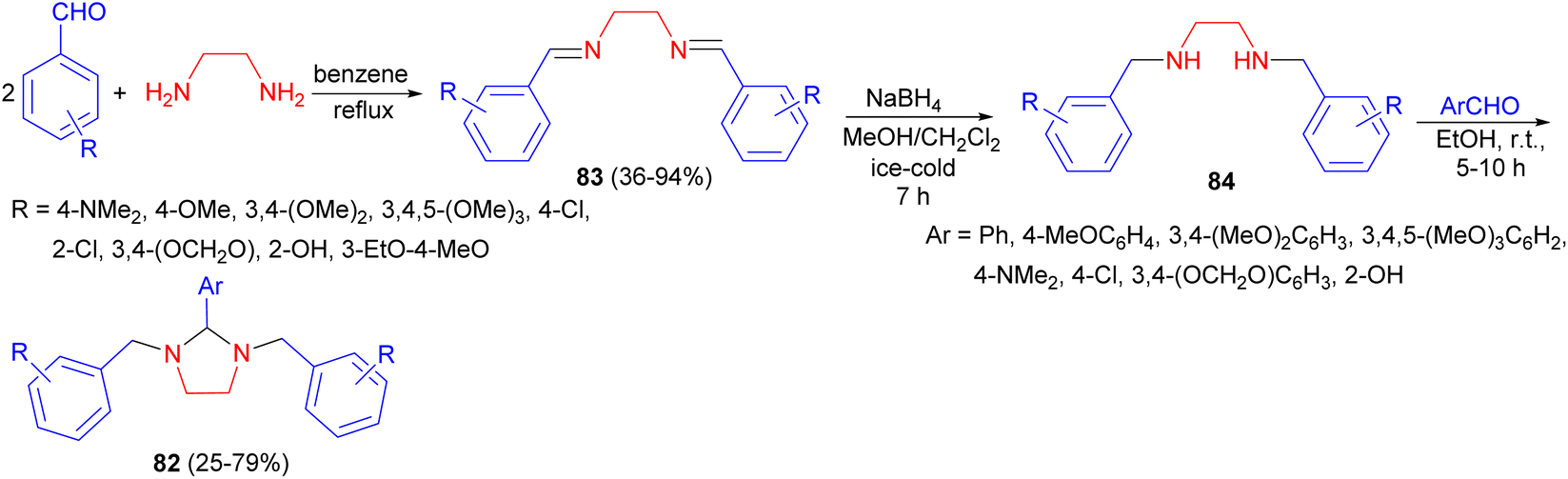 | ||
| Scheme 29 Synthesis of 1,2,3-trisubstituted tetrahydroimidazoles 82. | ||
Next, Katritzky group described synthesis of unsymmetrical imidazolidines 85 in 85–96% yields by Mannich reactions of 1,2-ethanediamines 86 with benzotriazole 87 and formaldehyde in MeOH/H2O at room temperature for 4 hours. Nucleophilic substitutions of 85 with NaBH4, Grignard reagents, sodium cyanide, benzenethiol, and triethyl phosphite afforded unsymmetrical imidazolidines 88–92 in 63–97% yields. Also, the reaction of diamines 93 with benzotriazole and formaldehyde generated benzotriazol-1-yl intermediates 94 in 85–93% yields. Nucleophilic substitutions of 94 by Grignard reagents, triethyl phosphite, or sodium cyanide gave optically active imidazolidines 95–97 in 66–99% yields (Scheme 30).47
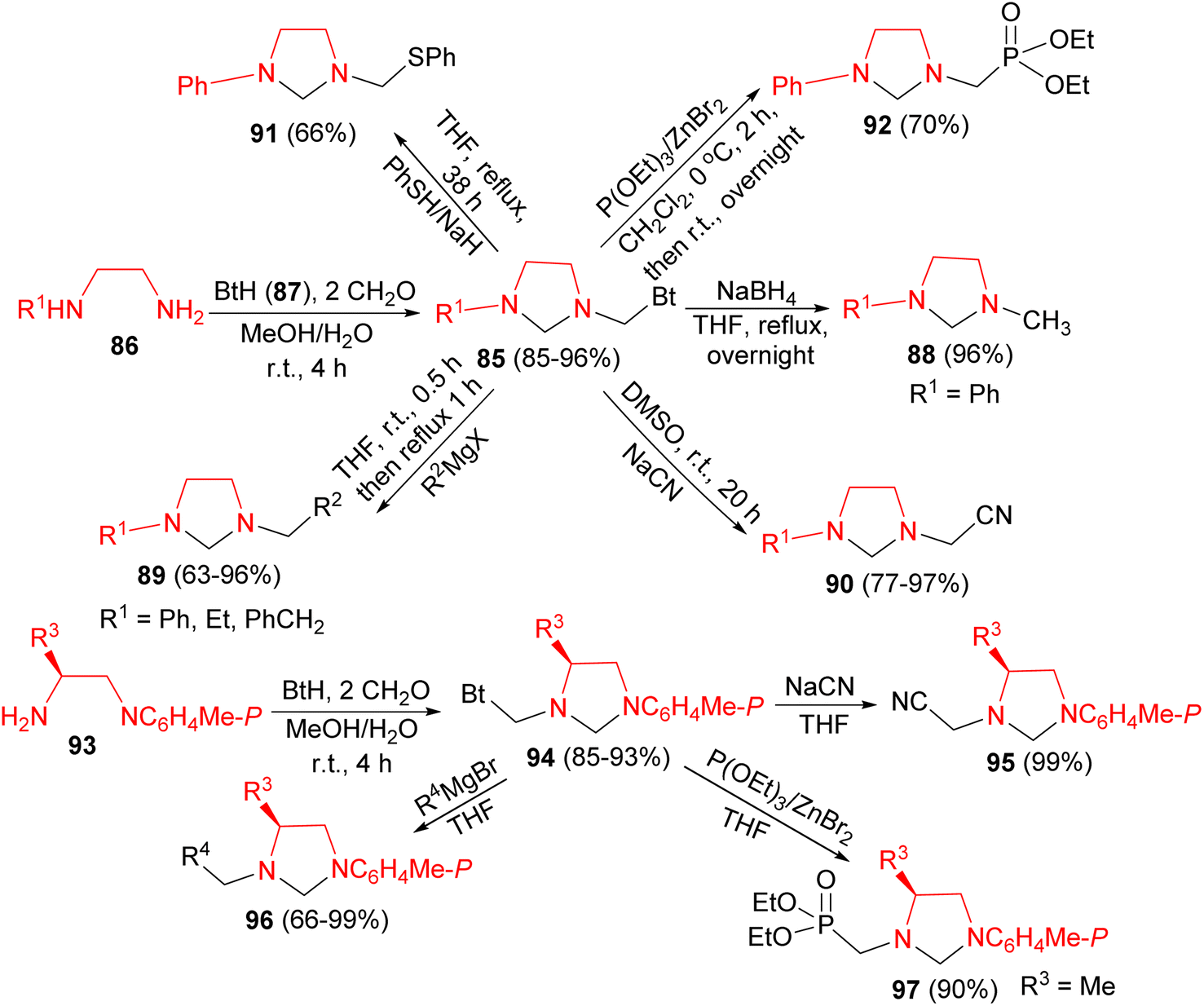 | ||
| Scheme 30 Convenient syntheses of unsymmetrical imidazolidines 85, 88–92 and 94–97. | ||
In addition, the Pearson group reported deprotonation of the imine 98 in THF or benzene, with LDA in cyclohexane which cannot lead to cycloaddition owing to the absence of an alkene, resulted in the formation of the imidazolidines 99 and 100 in 83% yield after 5 hours. Interestingly, the regioselectivity of the dimerization was found to depend on the solvent, providing complementary results. Also, the reaction of (D,L-)-stilbenediamine 101 with acetone in THF at room temperature for overnight afforded imidazolidine 102. The crude compound 102 was dissolved in absolute ethanol and treated with NaBH4 in EtOH for overnight resulted (1R*, 2S*)-N-(1-methylethyl)-1,2-diphenyl-1,2-ethanediamine 103 in 89% yield.
Then, compound 103 was treated with 5-hexenal or with 5-phenyl-5-hexenal in ether for overnight afforded imidazoldines 104 (88%) and 105 (86%), respectively (Scheme 31).48
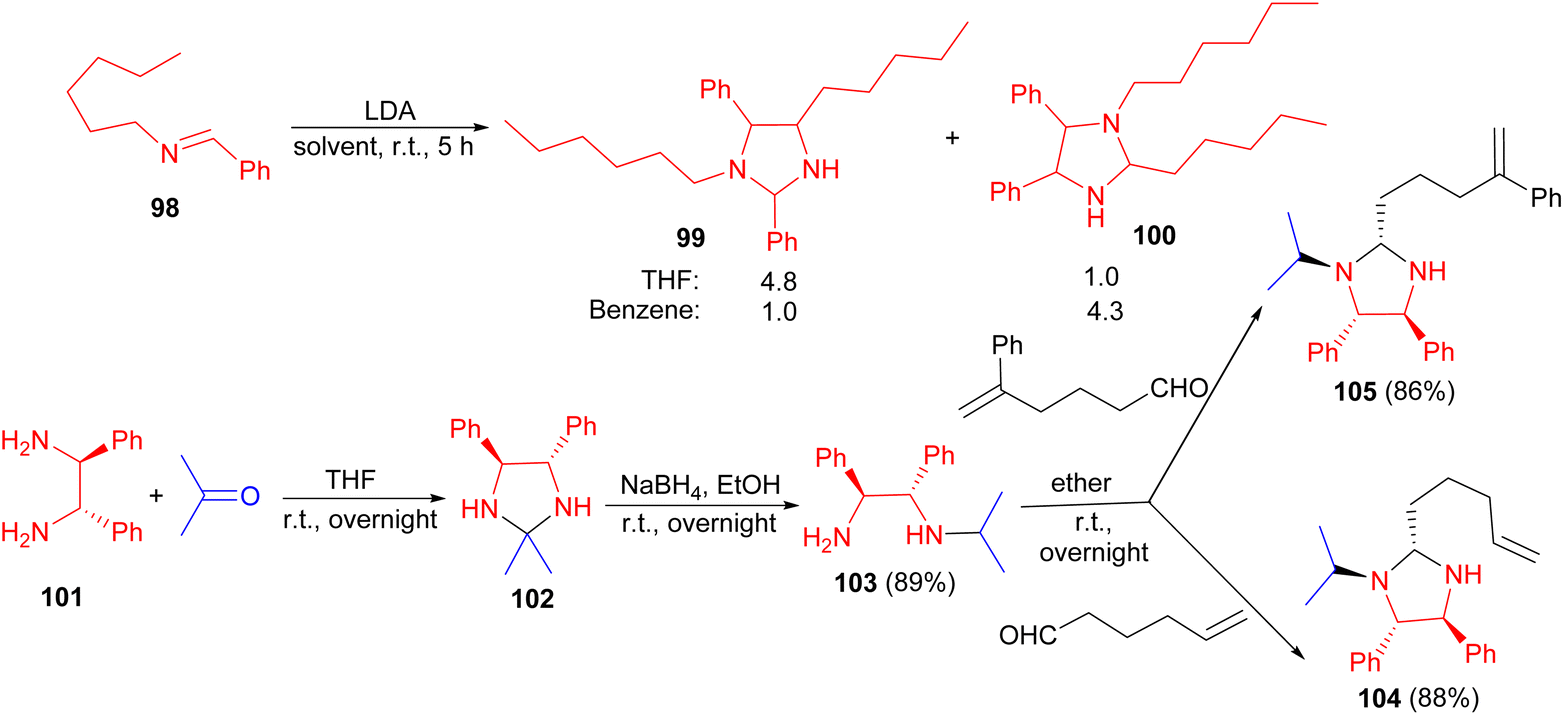 | ||
| Scheme 31 Synthesis of imidazolidine derivatives 99, 100, 102, 104 and 105. | ||
In 2003, Kelarev and co-workers revealed that by acylating bisazomethine 106 with aromatic and heteroaromatic acid chlorides in acetonitrile (boiling, 2–3 hours) in the presence of triethylamine, 1,3-diacyl-2-[3,5-di(tert-butyl)-4-hydroxyphenyl]imidazolidines 107 were formed in good yields (Scheme 32).49
 | ||
| Scheme 32 Synthesis of 1,3-diacyl-2-[3,5-di(tert-butyl)-4-hydroxyphenyl]imidazolidines 107. | ||
Next, Khan and Gupta reported synthesis and evaluation of anti-inflammatory and analgesic activity of some 1,3-diphenyl-2-aryltetrahydroimidazoles 108 in 54–60% yields via condensation of 1,2-dianilinoethane (109) with various aldehydes in EtOH at room temperature for 6–8 hours (Scheme 33). These compounds demonstrated significant anti-inflammatory effects compared to animals treated with normal saline. Furthermore, their analgesic activity was assessed. Compounds 108a–b exhibited similar effectiveness to aspirin. The Maximum Tolerated Dose (MTD) for all compounds was determined to be greater than 1800 mg kg−1.50
 | ||
| Scheme 33 Synthesis of 1,3-diphenyl-2-aryltetrahydroimidazoles 108. | ||
After that, the Hu group demonstrated unsymmetrical imidazolidines 110 were obtained in 75–91% yields by treating monoalkoxycarbonyl vicinal diamines 111 at room temperature with aqueous 37% formaldehyde in the presence of montmorillonite KSF as a solid catalyst in THF at room temperature for 1 hour (Scheme 34). The imidazolidines were shown to be useful intermediates in a novel protection strategy for the synthesis of peptide analogues containing a reduced glycine amide bioisostere. Moreover, the imidazolidine intermediate was cleaved conveniently and efficiently by 50% TFA in methylene chloride.51
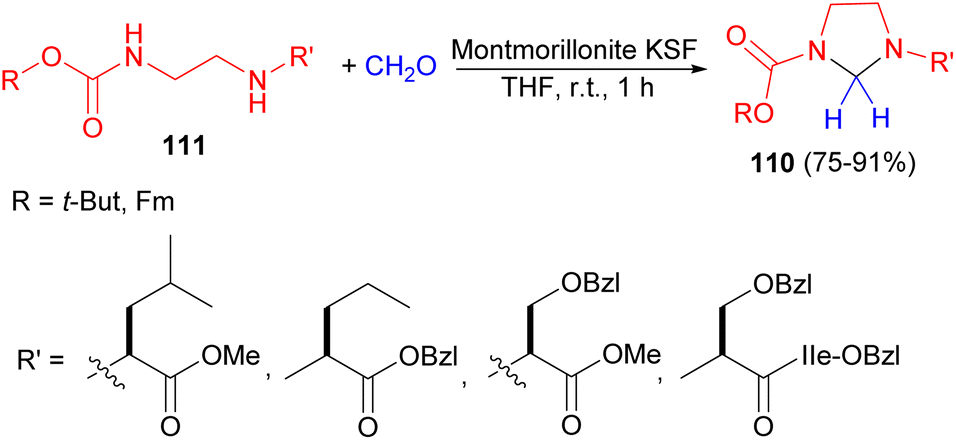 | ||
| Scheme 34 Montmorillonite KSF catalyzed synthesis of unsymmetrical imidazolidines 110. | ||
In addition, a procedure for the asymmetric synthesis of imidazolidines 112–113 in 37–80% yields have been reported. The 1,3-dipolar cycloaddition between nonracemic p-tolylsulfinimines 114 and azomethine ylides 115 generated in situ from iminoesters and LDA produces N-sulfinylimidazolidines 112–113 in THF at −78 to 4 °C for 20 hours with a high degree of stereo control (Scheme 35). In contrast, the presence of Lewis acids promotes formation of the cycloadducts through a highly diastereoselective process with opposite stereochemistry.52
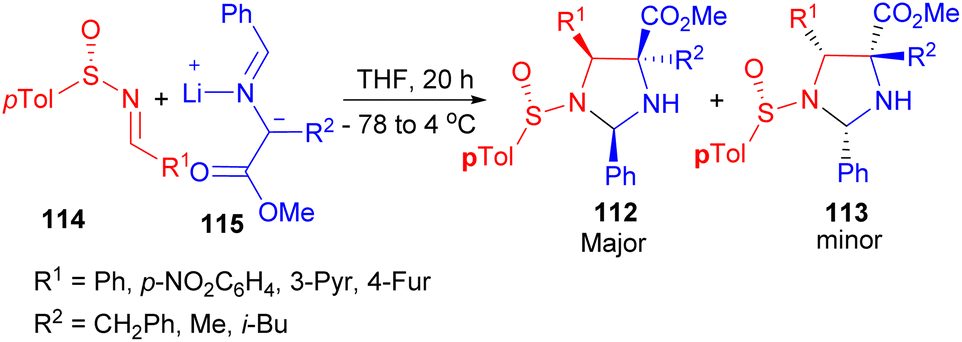 | ||
| Scheme 35 Asymmetric synthesis of imidazolidines 112–113. | ||
In 2004, Ray and co-workers described synthesis of tetraaza m-bis(bidentate) acyclic ligand 116 in 60% yield through the condensation reaction of one equivalent of trien 117 and 3 equivalents of benzaldehyde in MeOH at low temperature for 1 hour (Scheme 36). This ligand yielded new cationic dicopper(I/I) and dicopper(II/II) complexes in good yield.53
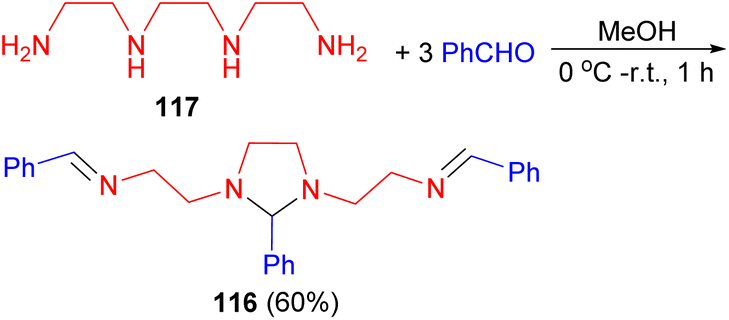 | ||
| Scheme 36 Synthesis of tetraaza m-bis(bidentate) acyclic ligand 116. | ||
Next, the Hedrick group reported synthesis of imidazolidine derivatives 118 in 68–96% yields by the reaction of ethylene diamines 119 with benzaldehyde derivatives in glacial acetic acid/CH2Cl2/toluene/Et2O in the presence of p-toluenesulfonic acid/anhydrous magnesium sulfate/Na2SO4 or absence of these catalyst at room temperature or warm conditions from 30 minutes to 24 hours (Scheme 37). Moreover, the pentafluorobenzene-based adducts are stable at room temperature. Thermolysis of these adducts generates the carbenes in solution, which they have shown are effective organic catalysts for transesterification reactions and ring-opening polymerization reactions. These adducts also provide convenient synthons for the generation of transition-metal complexes.54
 | ||
| Scheme 37 Synthesis of imidazolidine derivatives 118 in various conditions. | ||
In 2005, the Erkizia group reported that substituted imidazolidines 120 are the unexpected cycloadducts obtained in 30–50% yields through the reaction between imines 121 and isocyanates 122 in the presence of AgClO4 and NEt3 in CH3CN at room temperature (Scheme 38). The reaction is shown to take place via stepwise [3 + 2] cycloaddition between the N-metallated azomethine ylide formed in situ and the starting imine, followed by nucleophilic addition of the resulting imidazolidine on the sp hybridized carbon atom of the isocyanate.55
 | ||
| Scheme 38 AgClO4 catalyzed preparation of substituted imidazolidines 120. | ||
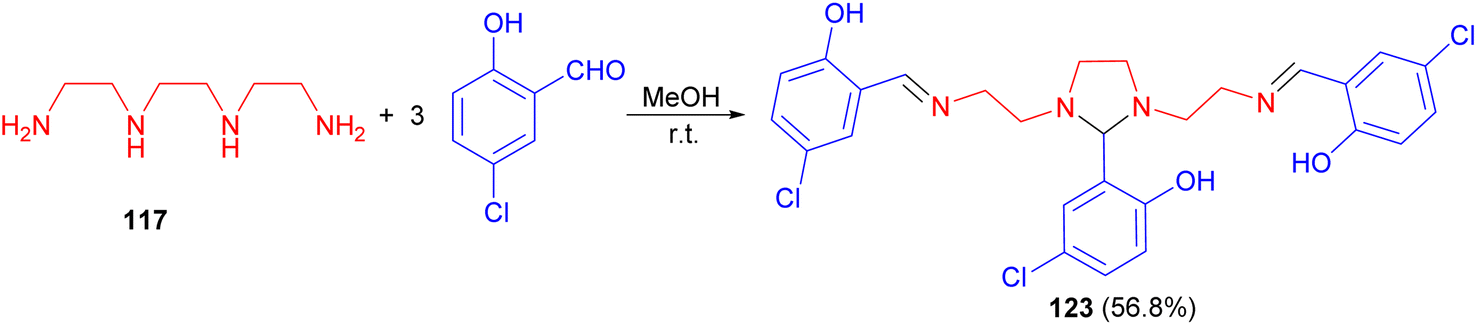
Further, Elmali et al. described synthesis of imidazolidine 123 in 56.8% yield via the reaction of triethylenetetramine 117 with 5-chlorosalicylaldehyde in methanol at room temperature (Scheme 39). Then, 123 reacts with Fe(ClO4)2·6H2O in aqueous methanol to form the mononuclear [Fe(L)](ClO4) complex with the imidazolidine ring cleaved by hydrolysis.56
 | ||
| Scheme 39 Synthesis of imidazolidine 123. | ||
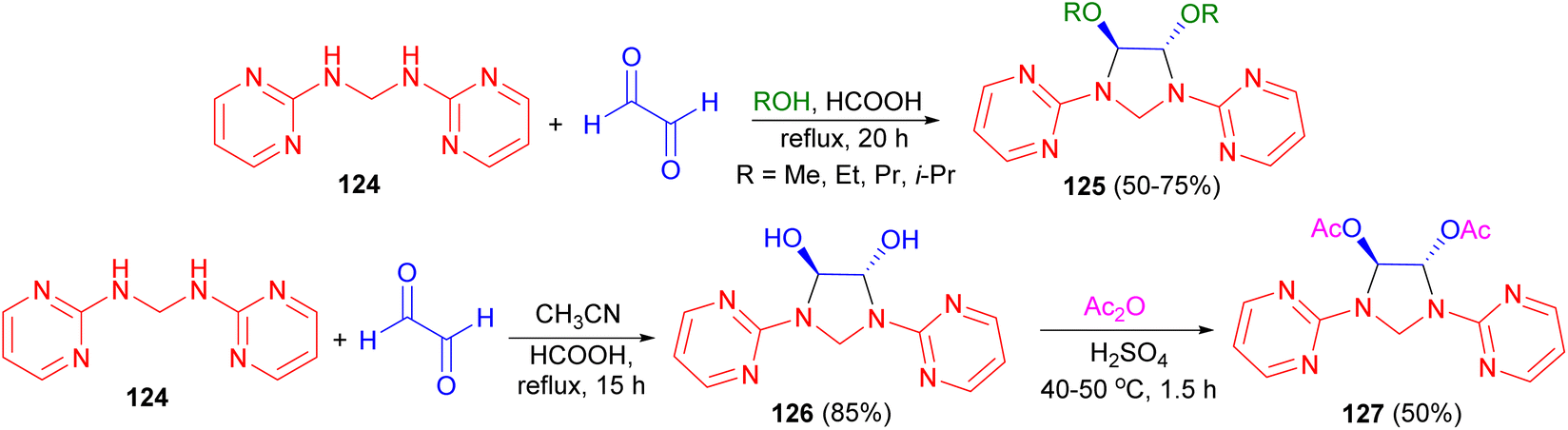
In 2006, Ghandi and co-workers demonstrated that cyclocondensation of N,N′-bis(2-pyrimidinyl)methanediamine 124 with glyoxal in alcohols (MeOH, EtOH, PrOH and i-PrOH) using formic acid as catalyst under reflux conditions for 20 hours led to the formation of the corresponding 4,5-dialkoxy-1,3-bis(2-pyrimidinyl)imidazolidines 125 in 50–75% yields. 4,5-Dihydroxy-1,3-bis(2-pyrimidinyl) imidazolidine 126 was obtained after 15 hours in 85% yield when the reaction was carried out in refluxing acetonitrile in the presence of formic acid. Moreover, the reaction of compound 126 with acetic anhydride in the presence of H2SO4 at 40–50 °C for 1.5 hours resulted in the formation of the corresponding trans-4,5-diacetoxy-1,3-bis(2-pyrimidinyl)imidazolidine 127 in 50% yield. Based on 1H-NMR analysis, it was found that the trans-isomers were selectively obtained in these cyclocondensation reactions (Scheme 40).57
 | ||
| Scheme 40 Formic acid catalyzed synthesis of imidazolidine derivatives 125–127. | ||
In addition, reactions of N-tosylimidoyl chlorides 128 with the Schiff bases of the general formula TsNH(CH2)2NCHR 129 using Et3N in CH3CN at 20 °C for 1–4 hours afforded 2-substituted 1-tosyl-3-(1-tosyliminoalkyl)imidazolidines 130 in 30–83% yields (Scheme 41).58
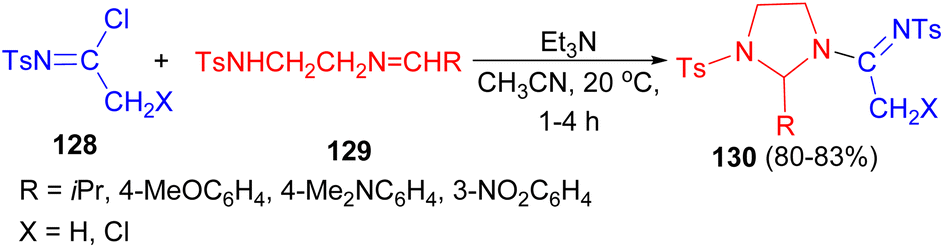 | ||
| Scheme 41 Synthesis of 2-substituted 1-tosyl-3-(1-tosyliminoalkyl)imidazolidines 130. | ||
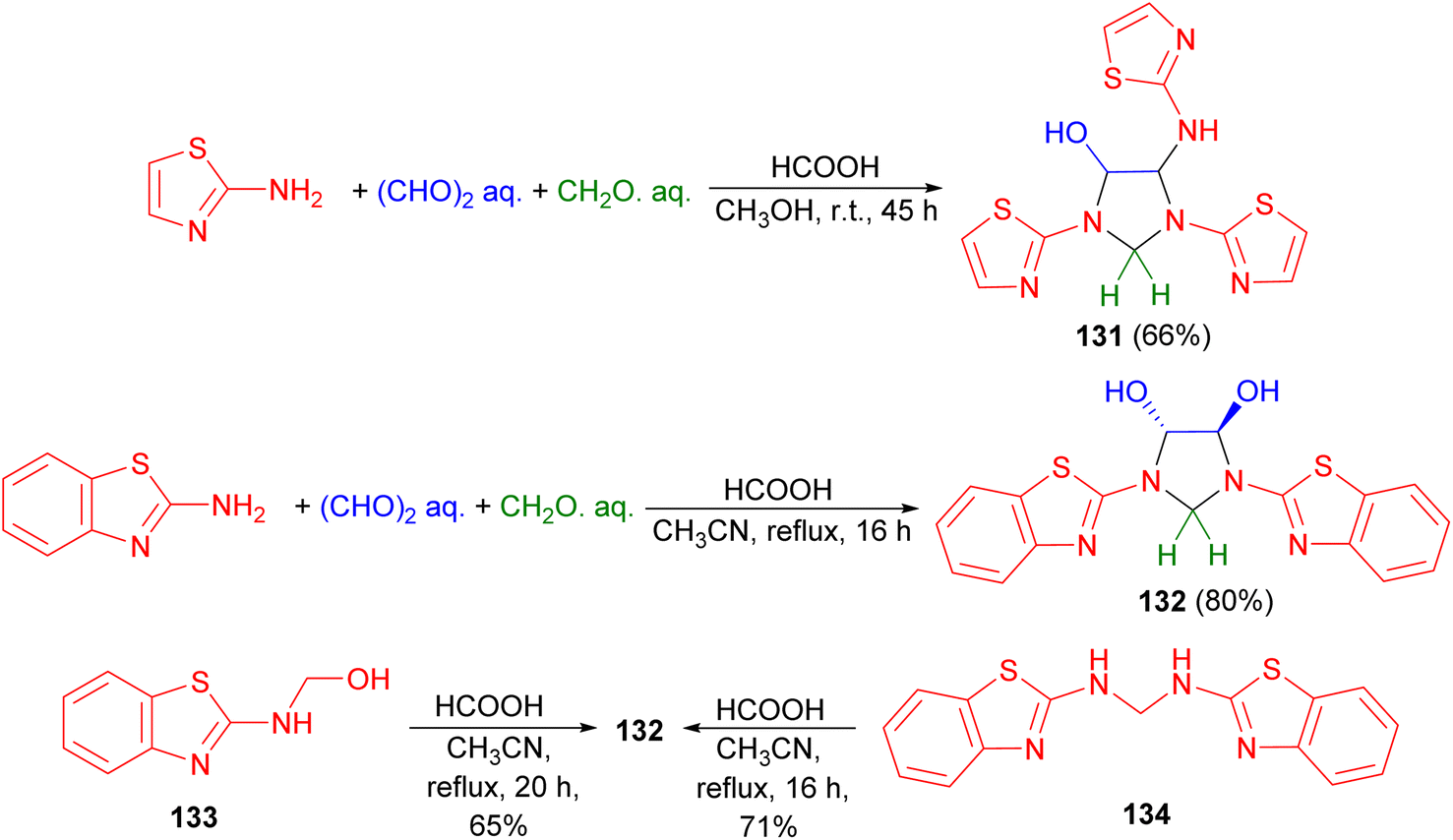
In 2007, Ghandi and Olyaei revealed that the reaction of 2-aminothiazole with aqueous glyoxal and aqueous formaldehyde in MeOH using HCOOH as catalyst at room temperature for 45 hours produced imidazolidine 131 in 66% yield. On the other hand, acid catalyzed one-pot three-component reaction of 2-aminobenzothiazole, aqueous glyoxal and aqueous formaldehyde in CH3CN under reflux conditions for 16 hours afforded trans-4,5-dihydroxy-1,3-bis(2-benzothiazolyl)imidazolidine 132 in 80% yield. Finally, the reaction of compound 133 or 134 with aqueous glyoxal in refluxing CH3CN using HCOOH as catalyst for 16–20 hours produced 132 (Scheme 42).59
 | ||
| Scheme 42 Preparation of imidazolidines 131 and 132 starting from heteroaryl amines. | ||
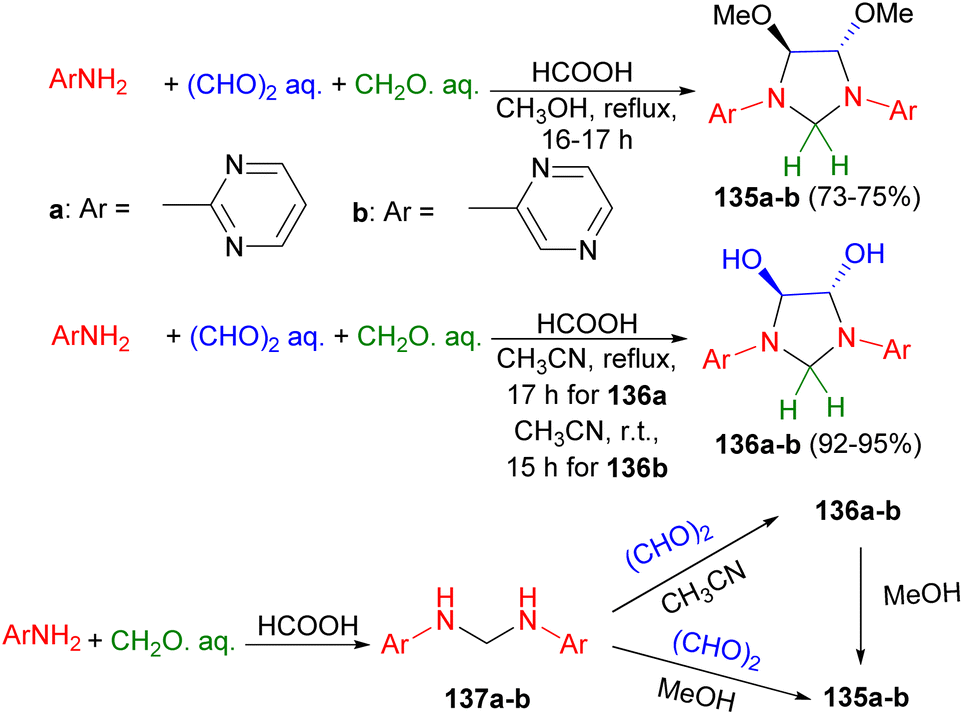
Next, four-component reaction of aminodiazines (2-aminopyrimidine and 2-aminopyrazine), glyoxal and formaldehyde in methanol under reflux conditions for 16–17 hours afforded trans-4,5-dimetoxy-1,3-bis(2-pyrimidinyl)imidazolidine (135a) in 75% yield and trans-4,5-dimetoxy-1,3-bis(2-pyrazinyl)imidazolidine (135b) in 73% yield, respectively. Changing methanol to acetonitrile resulted the corresponding 1,3-bis(2-pyrimidinyl) and-1,3-bis(2-pyrazinyl)-derivatives of trans-4,5-dihydroxyimidazolidine (136a–b) in 92–95% yields. The proposed mechanism is illustrated in Scheme 43. The condensation of aminodiazines with formaldehyde produces the intermediates 137a–b, which then undergo reaction with glyoxal to form 136a–b. Subsequent the reaction of 136a–b with methanol leads to the formation of 135a–b, respectively.60
 | ||
| Scheme 43 Formic acid catalyzed synthesis of imidazolidine derivatives 135 and 136 starting from aminodiazines. | ||
In 2008, Perillo et al. described synthesis of a series of imidazolidines 138 in 70–90% yields by the reaction of the corresponding N,N′-disubstituted ethylenediamines and aldehydes or aqueous formaldehyde (37%, excess) in ethanol under reflux during 1.5 hours (Scheme 44). Some of the derivatives were found to have high and selective activity as anti-trypanosoma cruzi agents.61
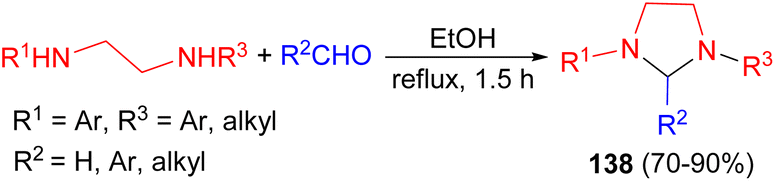 | ||
| Scheme 44 Synthesis of imidazolidines 138 as anti-trypanosoma cruzi agents. | ||
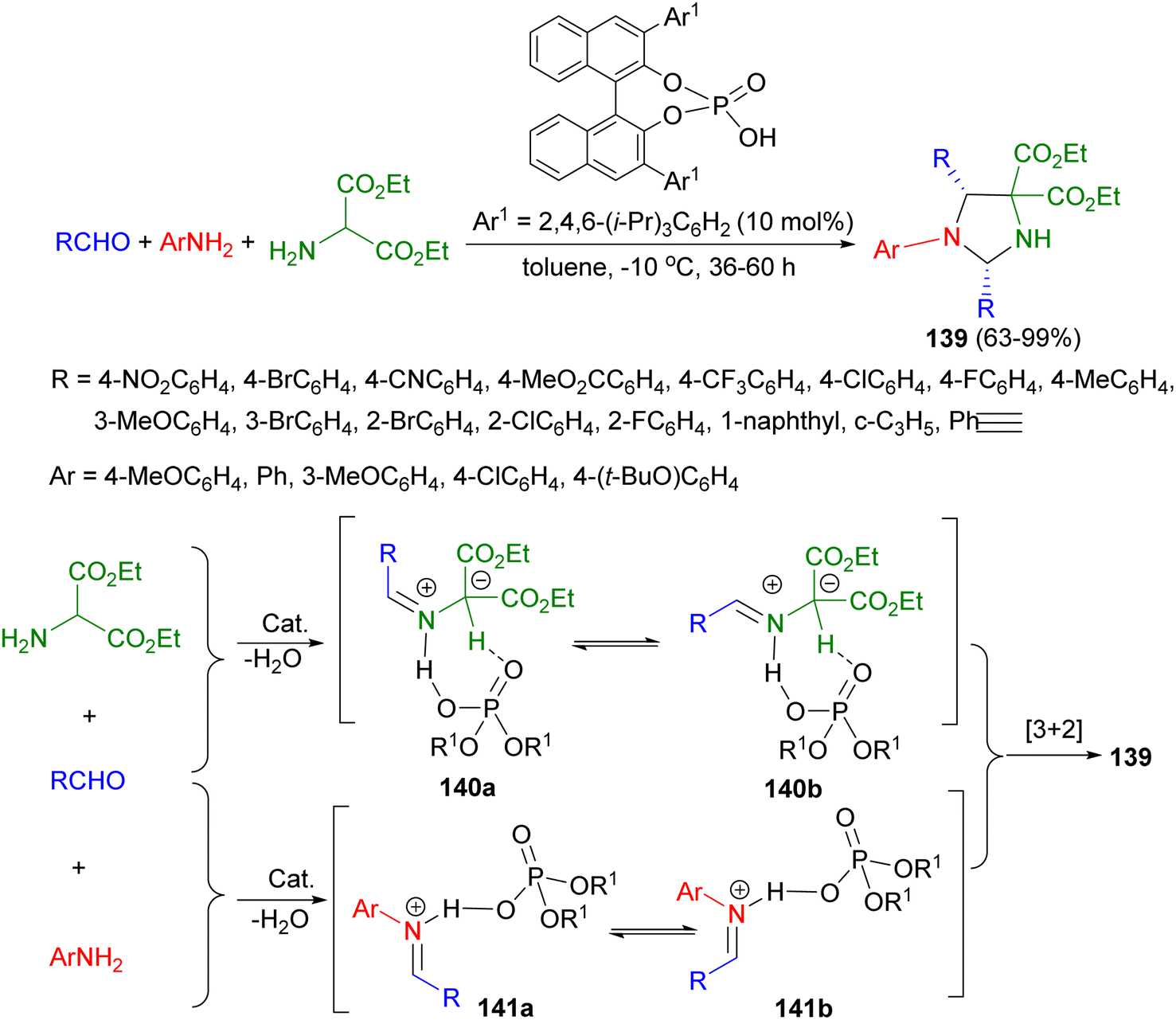
After that, a chiral Brønsted acid as catalyst applied for the synthesis of chiral imidazolidines 139 in 63–99% yields with high levels of stereoselectivity (up to 91/9 dr and 98% ee) via 1,3-dipolar cycloaddition reaction of aldehydes, amino esters, and anilines in toluene at −10 °C for 36–60 hours. The proposed mechanism is outlined in Scheme 45. Intermediates 140a or 140b as a chiral dipole obtained by the reaction of aldehyde with amino ester in the presence of catalyst. An imine generated in situ from an aldehyde and an amine could be activated by formation of an iminium species, either 141a or 141b, with a Brønsted acid and showed high reactivity toward nucleophiles. The iminium intermediates would be captured by the chiral Brønsted acid activated dipole 140a or 140b to thereby undergo an enantioselective [3 + 2] cycloaddition.62
 | ||
| Scheme 45 Chiral Brønsted acid catalyzed synthesis of chiral imidazolidines 139. | ||
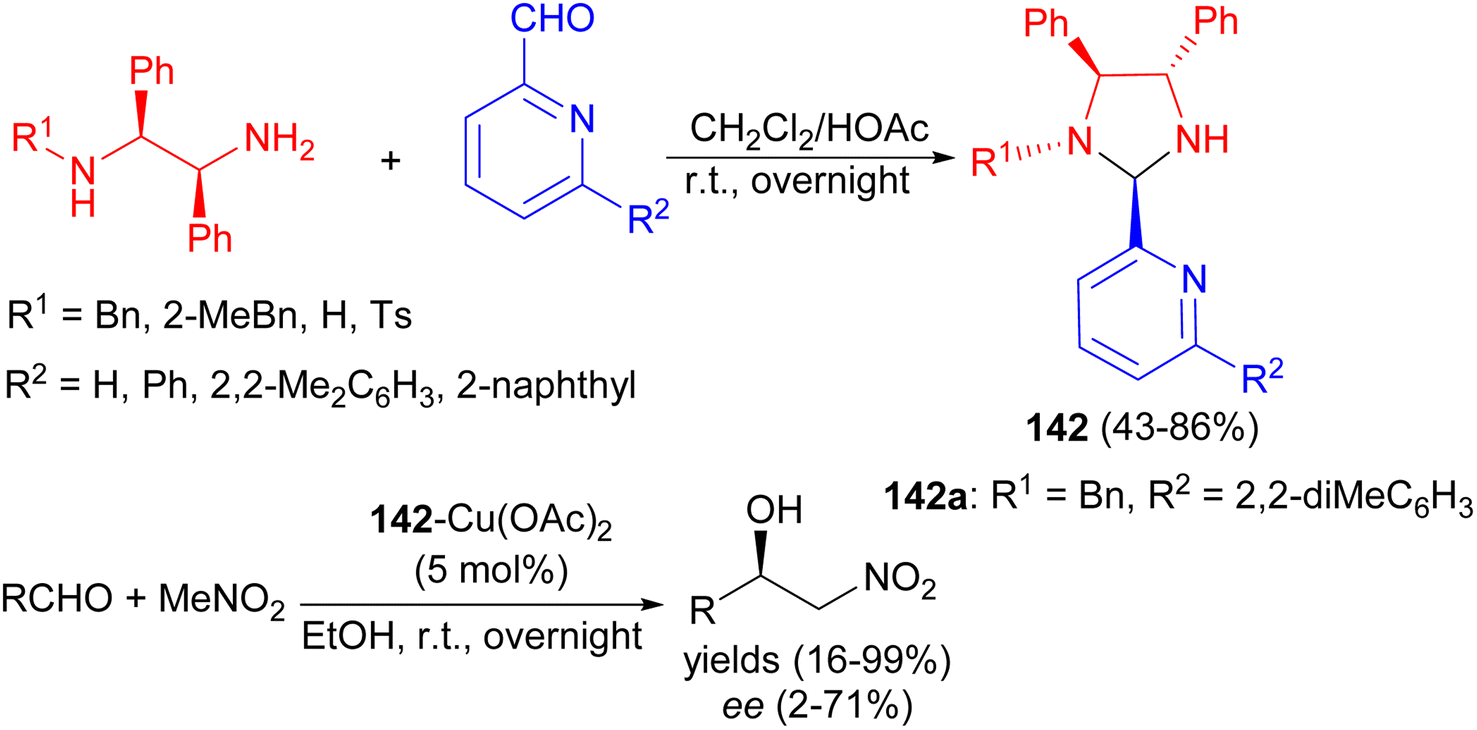
In 2009, Arai et al. reported condensation of chiral diamines and aldehydes in CH2Cl2/HOAc at room temperature for overnight gave a series of chiral imidazolidine-pyridines 142 in 43–86% yields with high diastereoselectivities. The ability of these compounds to act as chiral ligands was examined in the catalytic Henry reaction. Compound 142a showed better selectivity to provide the adduct with 71% ee (Scheme 46).63
 | ||
| Scheme 46 Synthesis of chiral imidazolidine-pyridines 142. | ||
Next, the synthesis of 1-acyl-3-arylimidazolidines 143 in 77–98% yields were performed by the reaction of N-acyl-N′-arylethylenediamines 144 with formaldehyde in THF in the presence of montmorillonite clay K-10 as a catalyst under microwave irradiation (180 W) for 2–5 minutes (Scheme 47).64
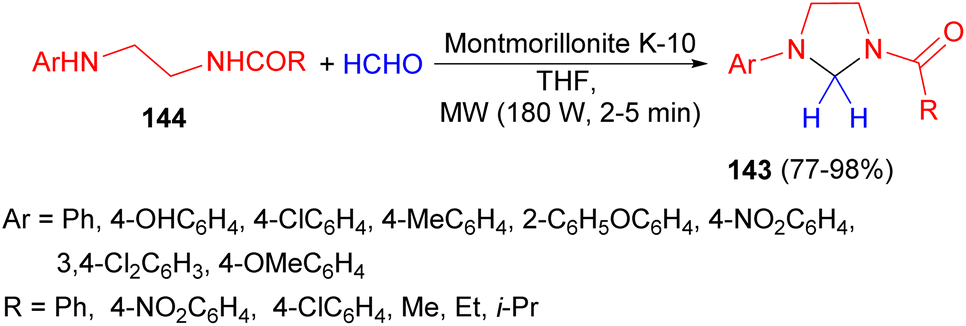 | ||
| Scheme 47 Synthesis of 1-acyl-3-arylimidazolidines 143 using montmorillonite clay K-10. | ||
In 2010, de Carvalho and his group disclosed synthesis of imidazolidine derivatives 145 in 70–79% yields by the classical method involving condensation between N,N′-disubstituted ethylenediamine with a variety of aromatic aldehydes in EtOH under reflux conditions for 1 hour (Scheme 48). The compounds showed a good activity against Leishmania without cytotoxicity on macrophages at the maximum concentration tested. 1,3-Bis(p-methoxybenzyl)imidazolidine showed the best activity on intracellular amastigotes, with IC50 value of 9.4 μg mL−1. In addition, none of compounds were cytotoxic against mammalian cells.65
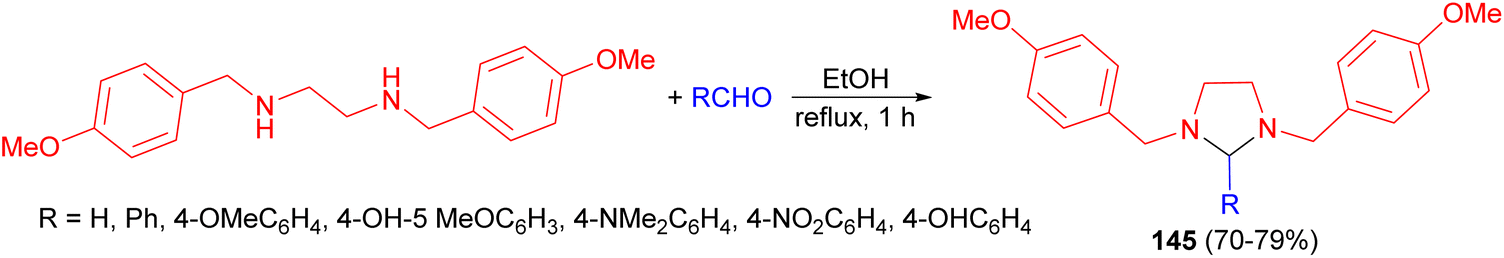 | ||
| Scheme 48 Synthesis of imidazolidine derivatives 145. | ||
Next, the Chen group explored an instance of diastereoselective silver-catalyzed 1,3-dipolar cycloaddition of azomethine ylides with imine compounds in THF at room temperature for 24 hours. This new method provided synthetically useful, highly substituted tetrahydroimidazole derivatives 146 with efficiency and high diastereoselectivity (Scheme 49).66
 | ||
| Scheme 49 AgOAc catalyzed synthesis of highly substituted tetrahydroimidazole derivatives 146. | ||
After that, Nenajdenko et al. reported synthesis of trifluoromethylated imidazolidines 147 in 35–97% yields by the reaction of β-halogeno-β-trifluoromethyl styrenes bearing an EWG at the aromatic ring with N,N′-binucleophile (ethylenediamine, N-methylethylenediamine, N,N′-dimethylethylenediamine) either in the refluxing THF for 7 hours or without solvent at room temperature from 1 hour to 7 days (Scheme 50).67
 | ||
| Scheme 50 Synthesis of trifluoromethylated imidazolidines 147. | ||
In 2011, the Helion group reported that the reductive coupling of imines in the presence of the lanthanide-originated zirconocene equivalent in THF for 1.5–12 hours afforded imidazolidines 148 in 70–85% yields under mild conditions in good yields with high diastereoselectivity. The proposed mechanism is shown in Scheme 51. Imines are partly converted (1.5 mmol of imine for 0.5 mmol of zirconocene) to diazazirconacyclopentanes 149. During hydrolysis, 149 gives corresponding diamines 150 and residual N-alkyl aldimines are hydrolysed to aldehydes, thus diamines 150 react slowly with aldehydes to give corresponding imidazolidines 148.68
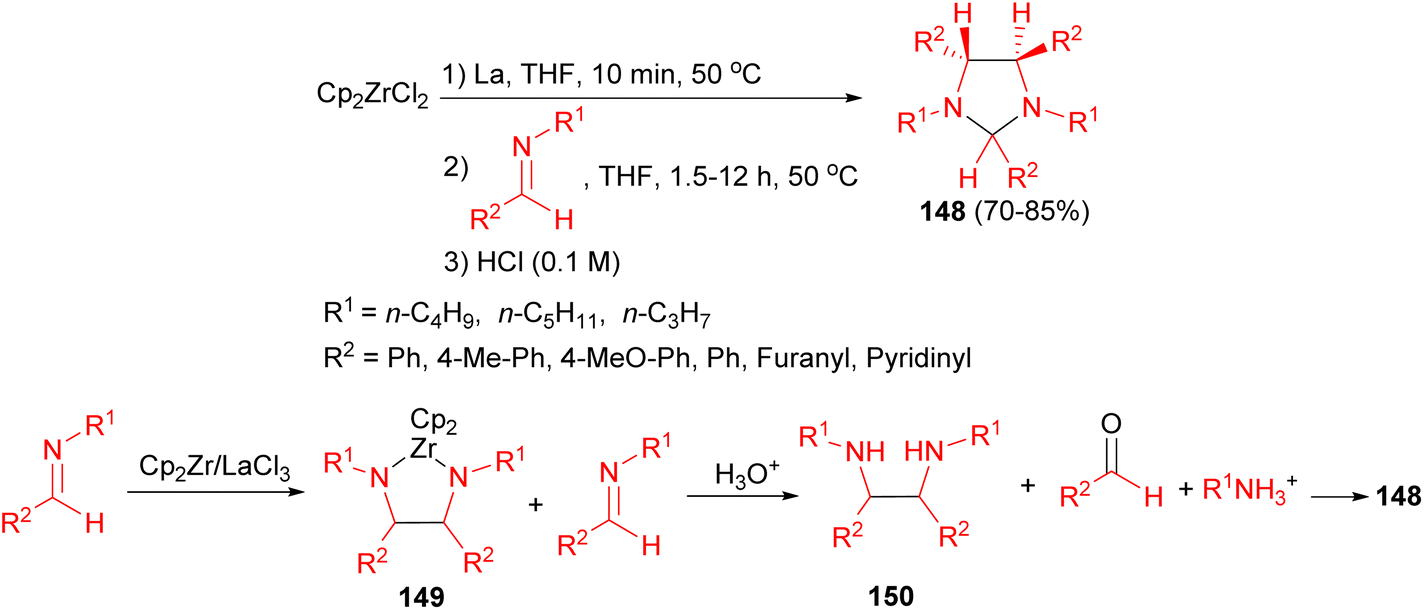 | ||
| Scheme 51 Synthesis of imidazolidines 148 in the presence of the lanthanide-originated zirconocene. | ||
The Lu group developed a diastereoselective synthesis of trans-2,5-disubstituted imidazolines 151 in 19–92% yields via the reaction of N-tosylaziridine 2,2-dicaboxylates 152 with trans-imines. The reaction involves a regioselective cleavage of the C–C bond of the aziridine ring and a diastereoselective [3 + 2] cycloaddition. AgOTf as catalyst in DCE was proved to be an effective Lewis acid catalyst for the formation of trans-2,5-imidazolidines at 50 °C after 12 hours. In the proposed mechanism, catalyzed by Lewis acid, the aziridine ring is opened to form azomethine ylide via a regioselective cleavage of the C–C bond. Then [3 + 2] cycloaddition reaction of the intermediate with trans-imine via the stable transition state afforded trans-2,5-imidazolidines (Scheme 52).69
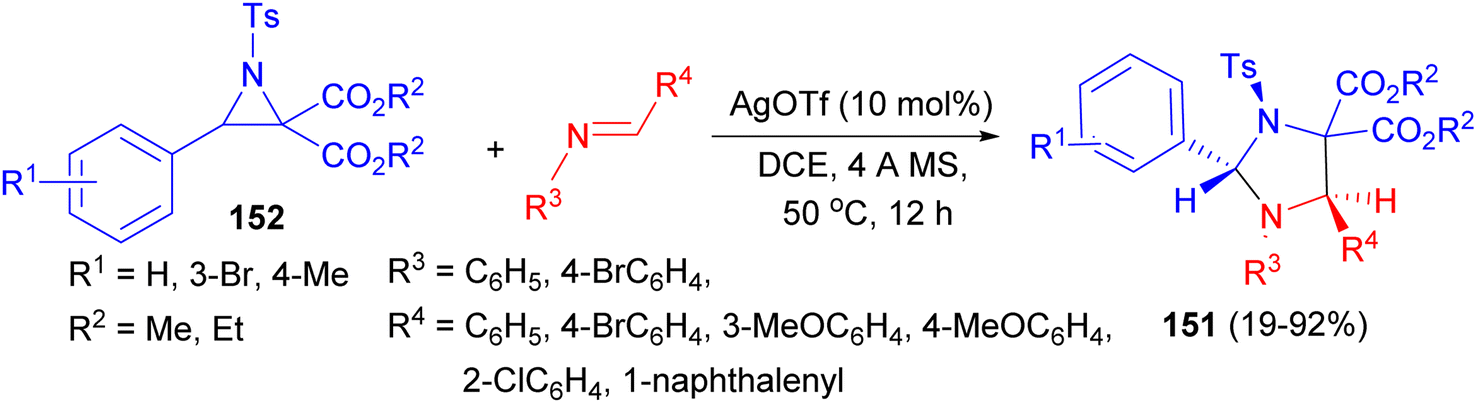 | ||
| Scheme 52 Synthesis of trans-2,5-disubstituted imidazolines 151 using AgOTf as catalyst. | ||
An efficient synthesis of highly substituted tetrahydroimidazole derivatives 153 45–94% yields by means of visible light-induced intramolecular cyclization reactions of 1,2-diamine derivatives described. This photoredox catalytic reaction in the presence of 1.0 mol% Ru(bpy)3Cl2 with O2 exhibited high diastereoselectivity and afforded the desired products at room temperature after 9–72 hours. A proposed mechanism and stereochemical course of this reaction depicted in Scheme 53. The addition of the nitrogen anion to the iminium ion from its Re is much more favorable than that to its Si face due to the steric repulsion. When the reaction time was prolonged, product epi-2 could be converted into the thermodynamically more stable cis form under the reaction conditions.70
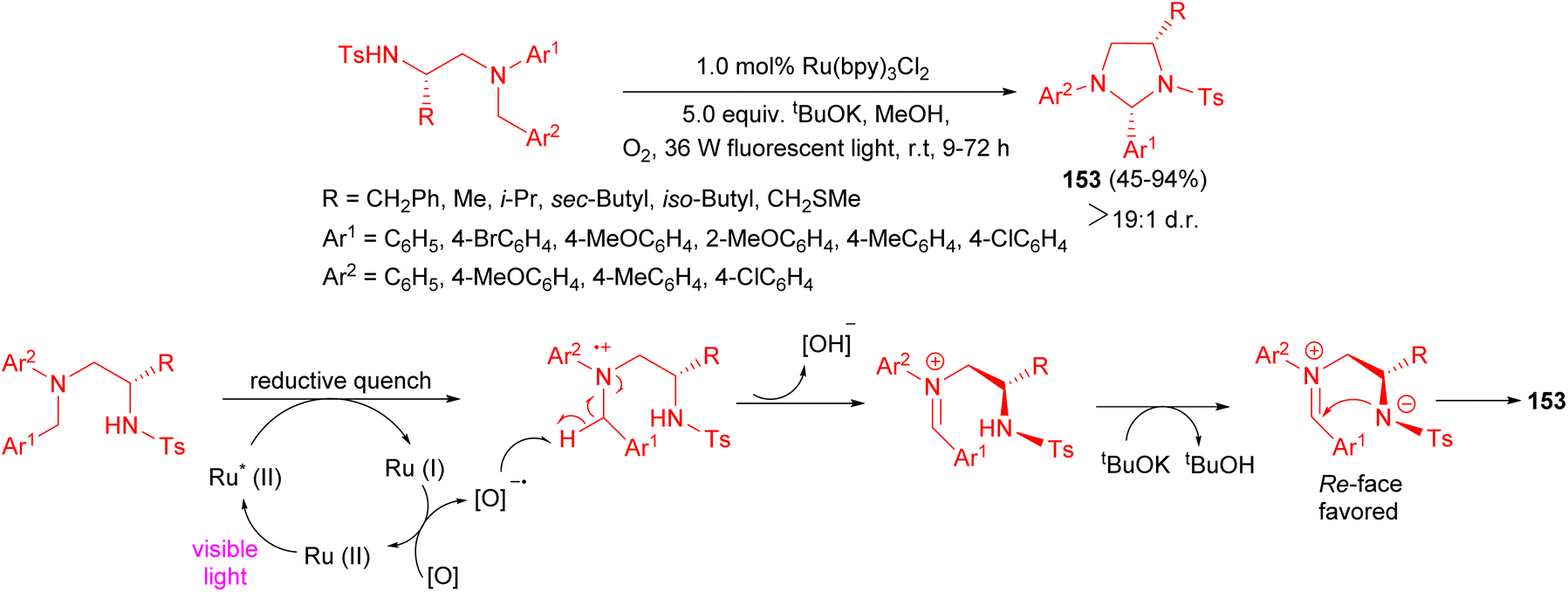 | ||
| Scheme 53 Ru(bpy)3Cl2 catalyzed diastereoselective synthesis of tetrahydroimidazole derivatives 153. | ||
The Lloyd-Jones group reported synthesis of imidazolidines 154 by the reaction of N-Ts and N-Boc derivatives of 1,2-diamines 155 with large range of electron-deficient alkenes via aza-Wacker reaction using Pd(II) as catalyst under the oxidative conditions (benzoquinone, DME, 40 °C) for 21 hours. In the proposed mechanism, as illustrated in Scheme 54, activation of the electron-deficient alkene by coordination to Pd(II) is followed by amino-palladation leading to the σ–Pd(II) species 156. β-Hydride elimination leads to the enamide 157 and Pd(0) which is then reoxidized by BQ to the active Pd(II) catalyst.71
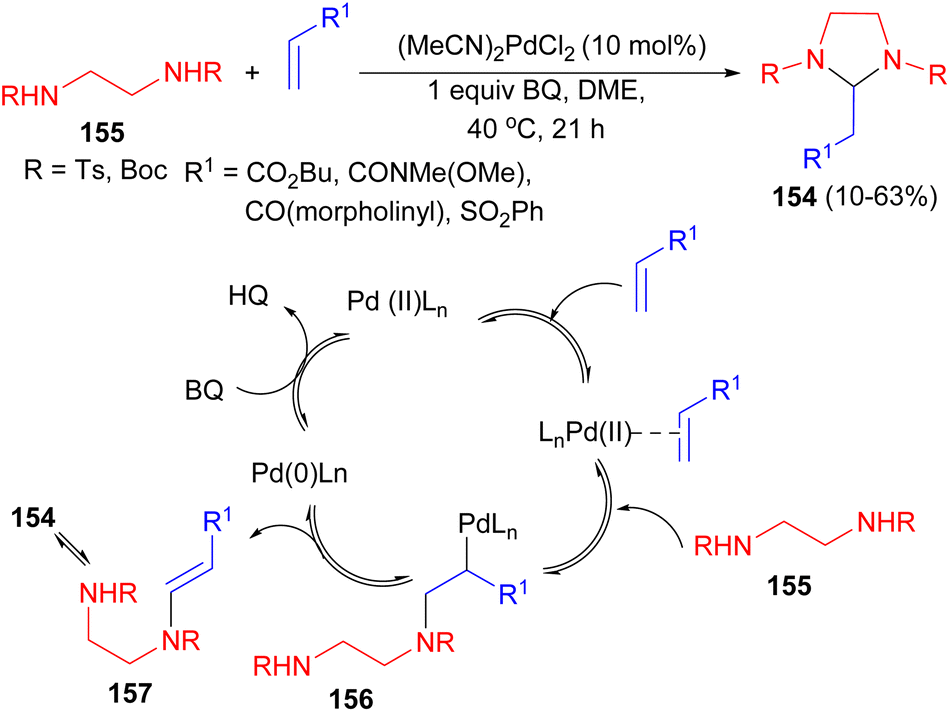 | ||
| Scheme 54 Synthesis of imidazolidines 154 in the presence of Pd(II) as catalyst. | ||
In 2012, Zhang et al. developed an efficient method for the diastereoselective synthesis of imidazolidines 158 through the reaction of aziridines with imines using Lewis acid catalyst (Y(OTf)3) in DCE at room temperature for 2–8 hours. In this procedure, the diastereoisomeric ratios are ranged from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to more than 50:1. Furthermore, both electron-deficient and electron-rich imines and aziridines exhibited excellent reactivity, yielding good to high yields (65–98%). A model that accounts for the trans selectivity observed in this cycloaddition is proposed in Scheme 55. A possible path is that the azomethine ylide 159 is first produced by C–C bond cleavage of 160, formed from aziridines through the selective coordination of Y(OTf)3 to the dicarboxylate groups. Subsequent diastereoselective addition of the imine would afford two zwitterionic intermediates, 161 and 162, which can be interconverted through iminium isomerization. Intermediate 162 is less stable than intermediate 161, owing to the steric hindrance of R and Ar, which are both in pseudo axial orientations within the envelope transition state, thus the trans isomer is produced preferentially.72
:1 to more than 50:1. Furthermore, both electron-deficient and electron-rich imines and aziridines exhibited excellent reactivity, yielding good to high yields (65–98%). A model that accounts for the trans selectivity observed in this cycloaddition is proposed in Scheme 55. A possible path is that the azomethine ylide 159 is first produced by C–C bond cleavage of 160, formed from aziridines through the selective coordination of Y(OTf)3 to the dicarboxylate groups. Subsequent diastereoselective addition of the imine would afford two zwitterionic intermediates, 161 and 162, which can be interconverted through iminium isomerization. Intermediate 162 is less stable than intermediate 161, owing to the steric hindrance of R and Ar, which are both in pseudo axial orientations within the envelope transition state, thus the trans isomer is produced preferentially.72
 | ||
| Scheme 55 Y(OTf)3 catalyzed synthesis of imidazolidines 158. | ||
After that, two series of 4-substituted-imidazolidines 163 and 164 in 53–72% yields were synthesized by reacting different tetrahydro-di-Schiff bases 165 and 166 with p-diethylaminobenzaldehyde/dimethylaminobenzaldehyde in EtOH at room temperature for 5 hours (Scheme 56). The title compounds were evaluated for their antibacterial and antifungal actions against some selected microbes. The results of microbiological evaluation revealed that two compounds (164) were good in their antibacterial as well as antifungal actions.73
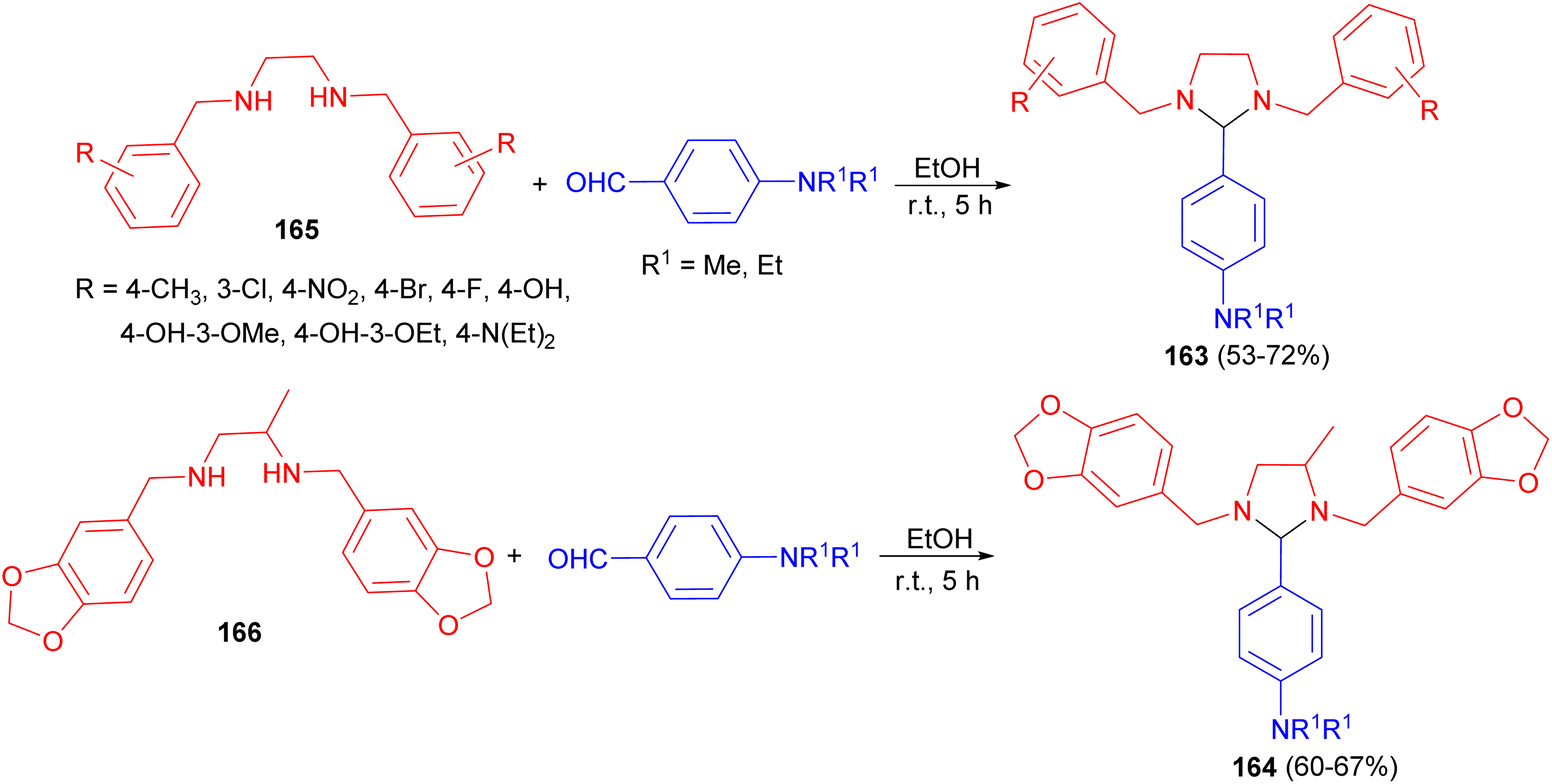 | ||
| Scheme 56 Preparation of 4-substituted-imidazolidines 163 and 164. | ||
In 2013, the Muthusubramanian group developed an atom-efficient, catalyst-free and environmentally friendly approach towards the synthesis of 1,3,4-trisubstituted imidazolidines 167 through a multicomponent reaction involving monophenacyl anilines 168, aromatic amines and formaldehyde. The reaction proceeds in refluxing ethanol for 3 hours providing higher yields (77–94% yields) of the imidazolidines 167. The mechanism for the formation of 167 is depicted in Scheme 57 in which two possible paths, route A and B, have been suggested. Imine 169 formed by the reaction of substituted aniline with formaldehyde could have undergone Mannich type reaction with the enolic form of monophenacyl aniline resulting in 170. 170 could have reacted with formaldehyde ultimately yielding 167 after dehydration. The initial formation of N-hydroxylmethyl derivative 171 has been proposed in route B. Compound 171 undergoes reaction with imine 169 resulting in the imidazolidine derivative 167.74
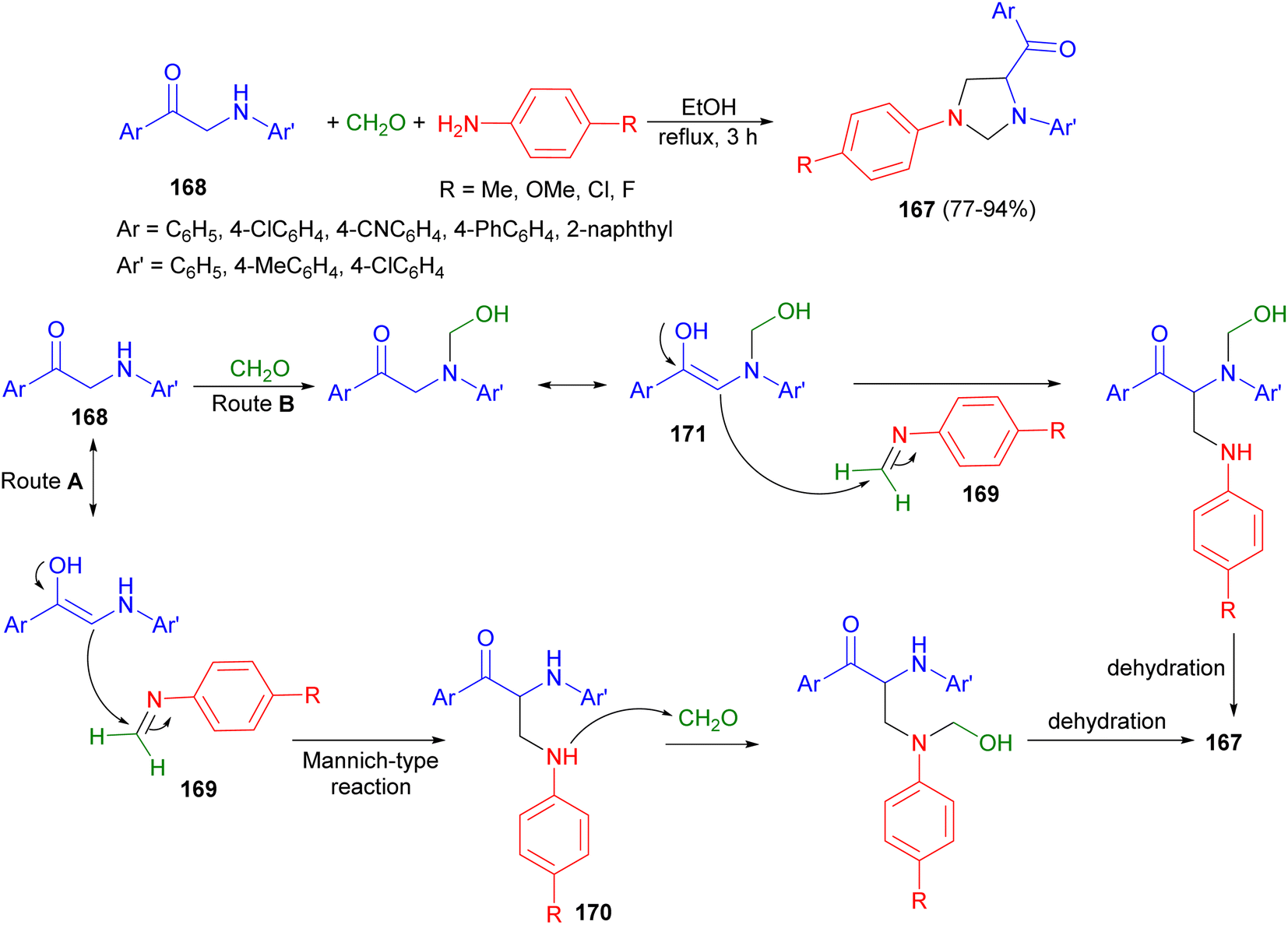 | ||
| Scheme 57 Catalyst-free synthesis of 1,3,4-trisubstituted imidazolidines 167. | ||
In addition, Olyaei and his group reported a facile, one-pot stereoselective synthesis of trans-4,5-dihydroxy-2-aryl-1,3-bis(heteroaryl)imidazolidines 172 in 75–88% yields by a cyclocondensation reactions of heteroarylamines, benzaldehydes and aqueous glyoxal in the presence of guanidinium chloride as a polyfunctional organocatalyst under solvent-free conditions for 23–76 minutes. The proposed mechanism is shown in Scheme 58. The catalyst initially acts as a hydrogen-bond donor to activate the aldehyde by formation of a six-membered ring. Subsequently, a Schiff base was formed by nucleophilic addition of the amine to the aldehyde and dehydration in the presence of the catalyst acting as an acid. Next, the Schiff base is further attacked by a second amine to give gem-diamine as intermediate 173. Finally, nucleophilic addition of 173 to the carbonyls of glyoxal gave the final product 172.75
 | ||
| Scheme 58 Guanidinium chloride catalyzed synthesis of trans-4,5-dihydroxy-2-aryl-1,3-bis(heteroaryl)imidazolidines 172. | ||
After that, a series of substituted-imidazolidine derivatives 174 synthesized in 53–71% yields by the reaction of N,N-bis(substituted-benzyl)ethane-1,2-diamines 175 with p-diethyl/dimethylaminobenzaldehyde in EtOH for 5 hours (Scheme 59). The results of biological evaluation of these compounds revealed that some of the compounds exhibited anti-inflammatory and analgesic activities. Additionally, these derivatives showed superior GI safety profile as compared to that of the standard drug in terms of low severity index.76
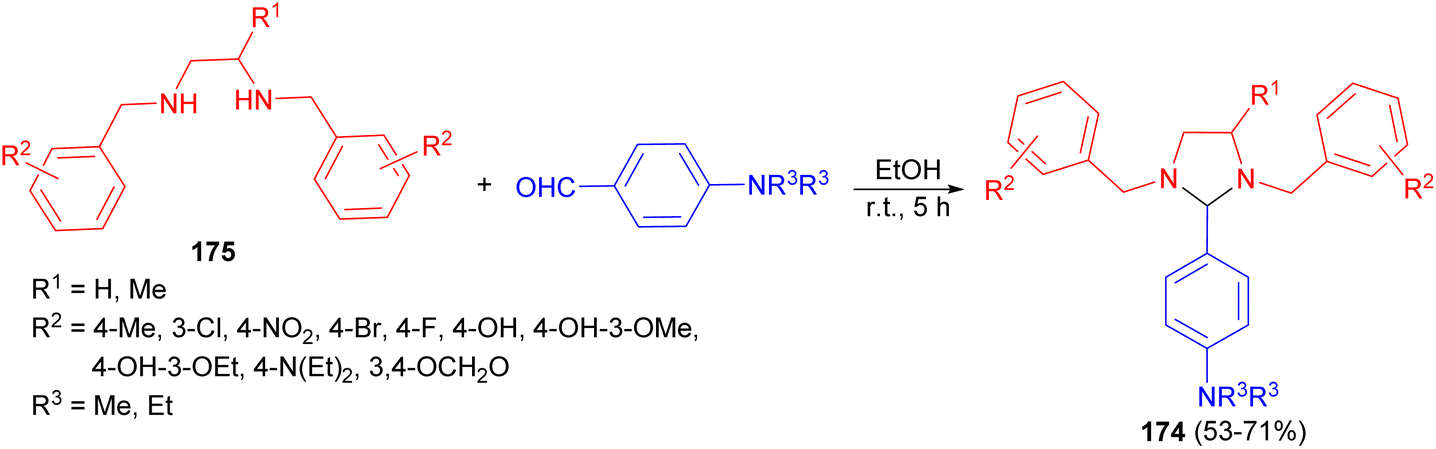 | ||
| Scheme 59 Preparation of substituted-imidazolidine derivatives 174. | ||
In 2013, the Wang group developed the first catalytic asymmetric synthesis of fluorinated 2,4-trans-imidazolidines 176 with excellent diastereoselectivity via Cu(I)/(S,Rp)-PPFOMe (3 mol%) catalyzed 1,3-dipolar cycloaddition of azomethine ylides 177 with various fluorinated imines 178 using Et3N in Et2O at −20 °C for 1–3 hours (Scheme 60).77
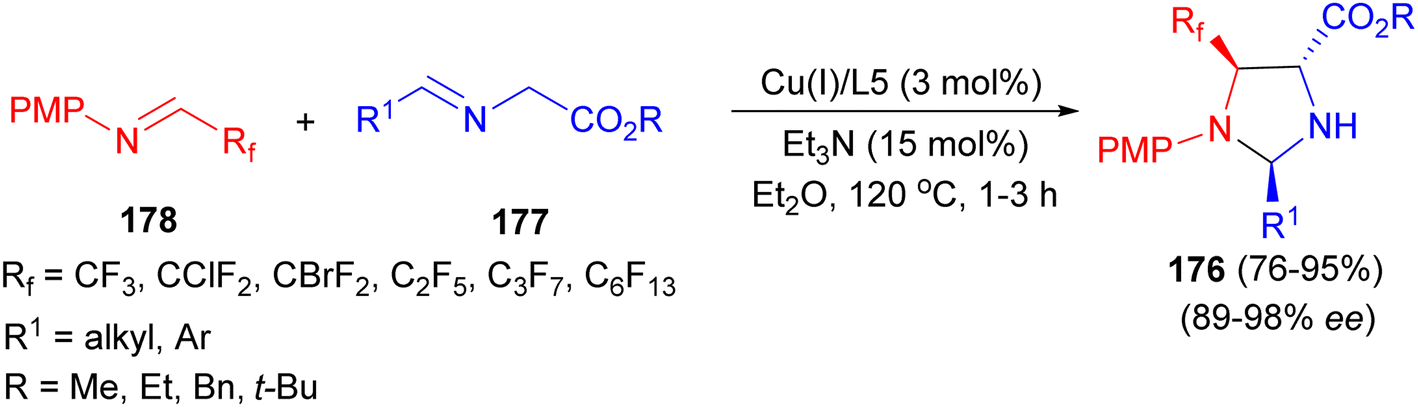 | ||
| Scheme 60 Asymmetric synthesis of fluorinated 2,4-trans-imidazolidines 176. | ||
After that, the Wang group reported synthesis of imidazolidine 179 in 89% yield via cascade reaction between N-phenyl glycine (180) and N-tosylimine (181) in the presence of 2 mol% fluorescein using an 11 W fluorescent bulb in MeOH at room temperature for 24 hours. A cascade process which contained both radical and ionic pathways was proposed for its formation (Scheme 61). The α-amino radical 182 was added to the 181 to afford a diamine intermediate 183. This adduct was added to iminium ion 184 to form ammonium intermediate 185. Losing an aniline led to iminium cation 186, which cyclized to form product 179.78
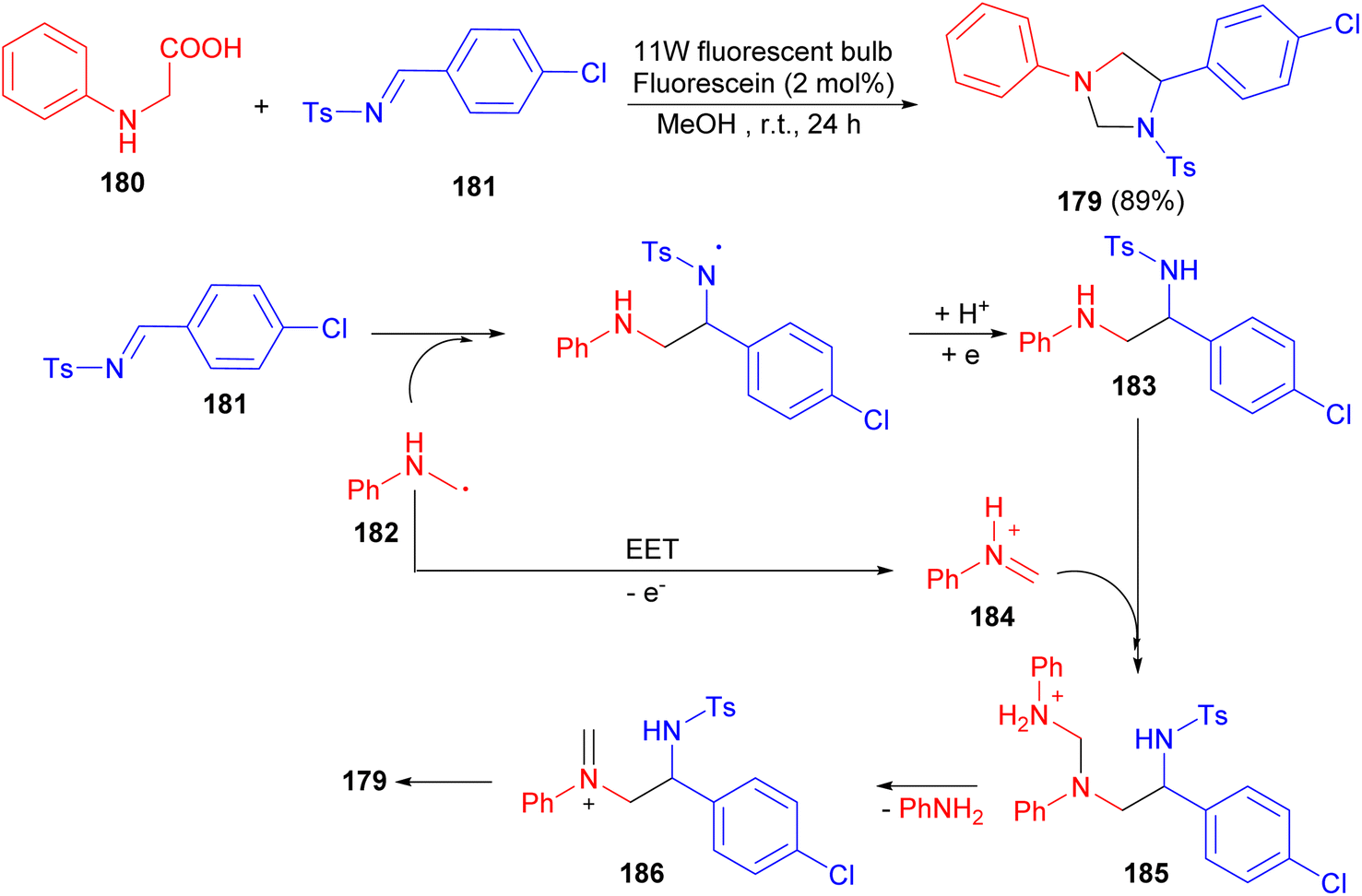 | ||
| Scheme 61 Fluorescein catalyzed synthesis of imidazolidines 179 using an 11 W fluorescent bulb. | ||
In 2014, a palladium complex bearing a chiral ammonium-phosphine hybrid ligand-catalyzed asymmetric [3 + 2] annulation reaction between racemic 5-vinyloxazolidinones 187 and N-sulfonyl imines 188 in toluene at 20 °C for 24 hours resulted imidazolidines 189 bearing α-amino quaternary stereocenters in 80–99% yields with excellent diastereo- and enantioselectivities (Scheme 62).79
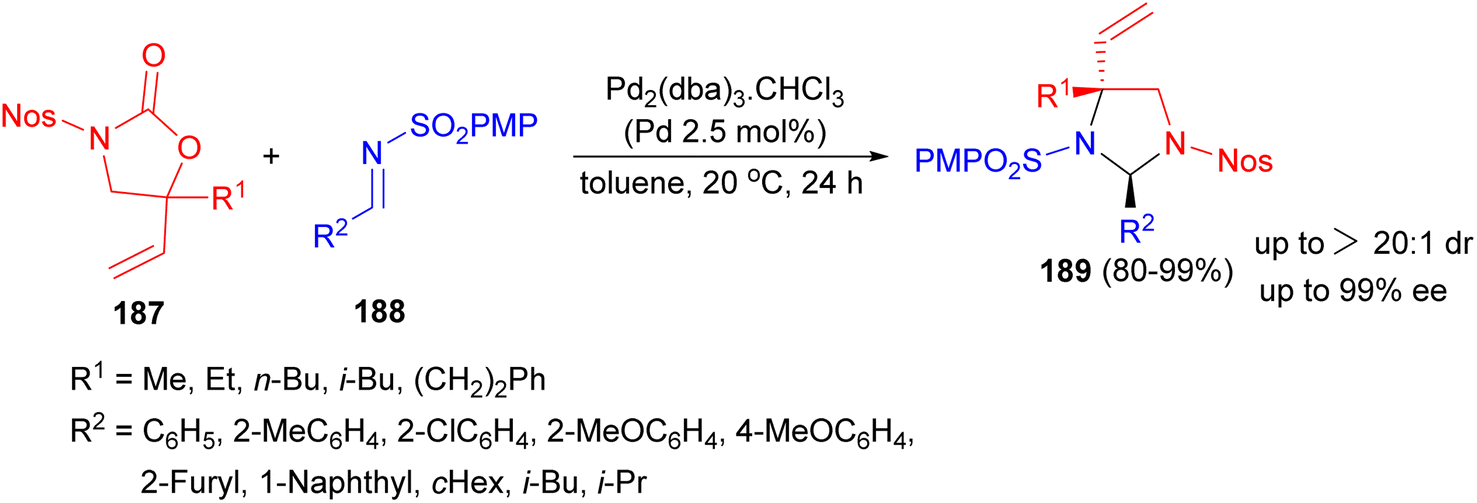 | ||
| Scheme 62 Palladium catalyzed synthesis of imidazolidines 189. | ||
Next, one convenient CdII-mediated C–C/C–N bond-forming strategy toward asymmetric tetra-(2-pyridine)-substituted imidazolidine 190 was reported by the Lin group. These compounds were formed from in situ solvothermal [3 + 2] asymmetric coupling dimerization of N-(2-pyridylmethyl)-pyridine-2-carbaldimine (191) with CdCl2/CdCl2 + NaSCN/CdCl2 + NaNO3 in the mixed solvents of methanol and pyridine at 70 °C or so for 3 days. Moreover, 190 could be obtained effectually from all three complexes, [Cd3L1Cl6]n (191a), [Cd2L1(SCN)Cl3]n (191b) and Cd2L1(NO3)4(MeOH) (191c), through the reactions of those compounds with Na2S (Scheme 63).80
 | ||
| Scheme 63 CdII catalyzed synthesis of tetra-(2-pyridine)-substituted imidazolidine 190. | ||
In addition, catalytic asymmetric homo-1,3-dipolar cycloadditions of azomethine ylides were established via SPINOL-derived chiral phosphoric acid-catalyzed pseudo four-component reactions of aldehydes and 2-aminomalonates in CHCl3 at 40 °C for 24 hours, resulted in the stereoselective construction of chiral imidazolidine scaffolds 192 with two stereogenic centers in generally high yields and with good stereoselectivities (36–81% yields, >20:1 dr, up to 93% ee). In the proposed mechanism as depicted in Scheme 64, the catalyst acted as a Brønsted acid/Lewis base bifunctional catalyst to simultaneously activate both the azomethine ylide and the aldimine via hydrogen bonding interactions, which facilitated subsequent [3 + 2] cycloadditions.81
 | ||
| Scheme 64 SPINOL-derived chiral phosphoric acid-catalyzed synthesis of chiral imidazolidine scaffolds 192. | ||
In 2015, the Hwu group an efficient method developed for the direct synthesis of various imidazolidines 193 in good to excellent yields (70–85%) and excellent diastereoselectivity from two equivalents of Schiff bases and one equivalent of 2-(trimethylsilyl)aryl triflates using CsF in CH3CN at room temperature for 10–12 hours. Scheme 65 illustrated a plausible mechanism by which arynes 194 can function as “initiators” of heterocyclic ring formation. After they are generated by the 1,2-elimination of silylphenyl triflates with CsF, arynes 194 first act as electrophiles to react with the Schiff bases. Then the nucleophilic aryl carbanionic center in the resultant betaines 195 abstracts an acidic proton at the g position to form ylides 196. Steric congestion between the two phenyl groups in ylides 196a with the cis configuration caused their isomerization to the trans isomers 196b. Then a regioselective [3 + 2] cycloaddition takes place between azomethine ylides 196b and the second equivalent of Schiff bases in situ. The remarkably high degree of endo stereocontrol and excellent diastereofacial discrimination of the transition state 197 cause the imidazolidines 193.82
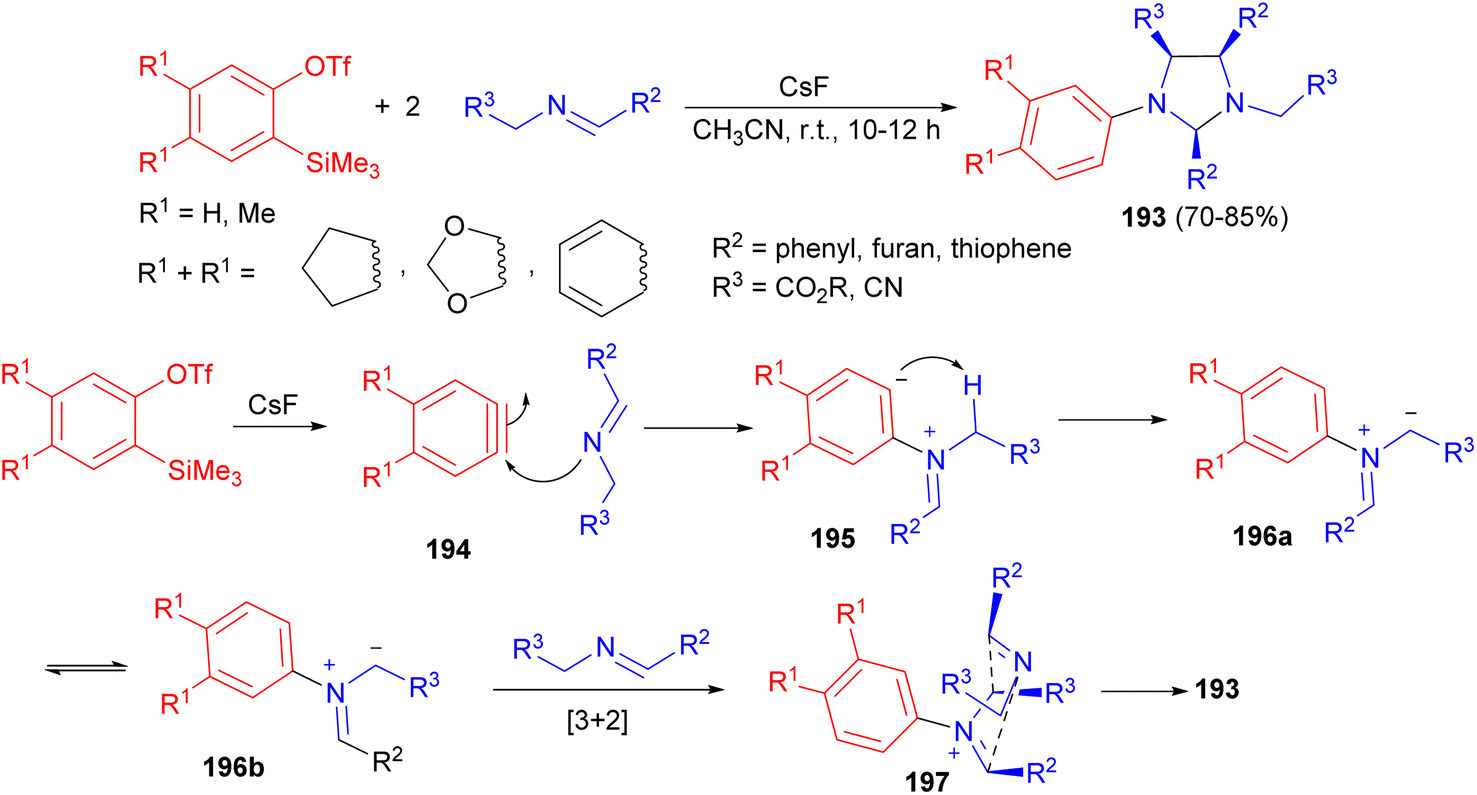 | ||
| Scheme 65 CsF catalyzed diastereoselective synthesis of imidazolidines 193. | ||
In 2016, Husain group synthesized a number of substituted-imidazolidine derivatives 198 in 51–70% yields starting from N,N′-bis(substituted-benzyl)ethane-1,2-diamines 199 and aromatic aldehydes in absolute ethanol. This reaction mixture was properly shaken for 5 hours using mechanical shaker and then kept in a refrigerator for whole night. The results of biological testing indicated that among the synthesized compounds only three imidazolidine derivatives 198a–c possess promising anti-inflammatory and analgesic actions. Additionally, these derivatives displayed superior GI safety profile (low severity index) with respect to the positive control, Indomethacin (Scheme 66).83
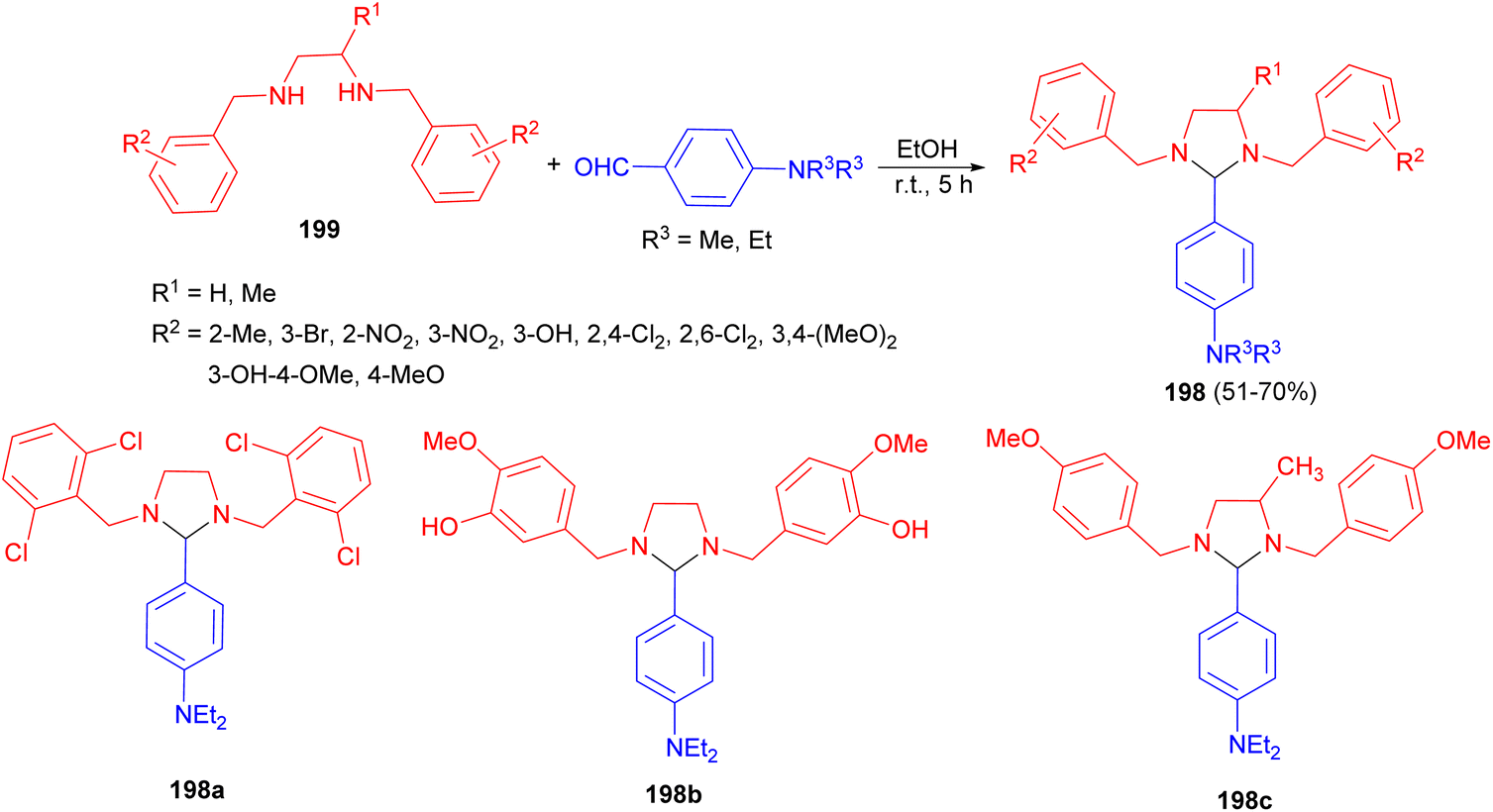 | ||
| Scheme 66 Synthesis of substituted-imidazolidine derivatives 198. | ||
Interaction of pyridoxal (3-hydroxy-5-hydroxymethyl-2-methylisonicotinaldehyde) (200) with amines, mono- and disubstituted diamines in EtOH at room temperature for 2 days or in benzene at 40–50 °C for 2–3 hours led to the formation of imidazolidines 201a–c in 44–82% yields (Scheme 67).84
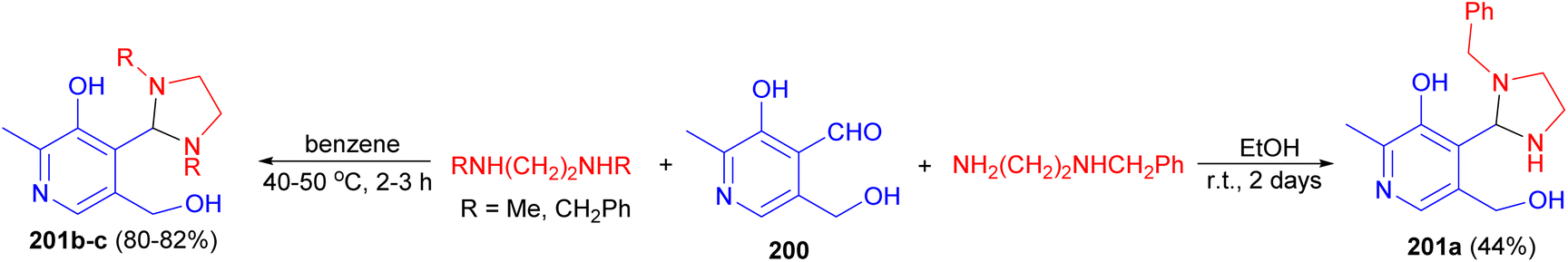 | ||
| Scheme 67 Preparation of imidazolidines 201. | ||
Copper(I) thiophene-2-carboxylate catalyzed regio- and chemoselective synthesis of indoloimidazolidines 202 in 73–84% yields by the reaction of the four-component reaction of diazoamides 203, electron-withdrawing imines 204, electron-donating aldehydes and amines 205 in 1,2-DCE under reflux conditions. The initially generated azomethine ylide from diazoamide and imine, formed from electron-donating aldehyde and amine, underwent [3 + 2]-cycloaddition with electron-withdrawing imine furnished indoloimidazolidine 202 in a chemo- and diastereoselective manner (Scheme 68).85
 | ||
| Scheme 68 Copper(I) thiophene-2-carboxylate catalyzed regio- and chemoselective synthesis of indoloimidazolidines 202. | ||
On water oxidative C(sp3)–H functionalization C–N bond formation using tetrabutylammonium iodide (TBAI) as the catalyst and tert-butyl hydroperoxide in water (T-Hydro) as the oxidant at 60 °C for 4–5 hours afforded a potential route for the construction of functionalized imidazolidines 206 and 207 in 61–84% yields. The proposed mechanism is depicted in Scheme 69. Thus, the oxidation of TBAI by T-Hydro may give iodine, tert-butoxyl radical, and hydroxyl ion (step (i)). Single electron transfer (SET) reduction of iodine may regenerate the catalyst with the formation of the radical cation (step (ii)). Homolysis of the methyl C–H bond induced by tert-butoxyl radical may give the iminium 208, which may convert into the target heterocycles 206 and 207 via the intermediate 209 (steps (iv) and (v)).86
 | ||
| Scheme 69 Construction of imidazolidines 206 and 207 using TBAI as the catalyst and tert-butyl hydroperoxide as the oxidant. | ||
The Sun group described gold-catalyzed synthesis of imidazolidines 210 in 38–85% yields by the reaction of donor/acceptor diazo esters 211, including aryl diazoacetates, alkyl diazoacetate, vinyl diazoacetates, cyclic diazo compounds and acceptor/acceptor diazoesters, with triazines 212 in the presence of tBuXPhosAuCl (5 mol%) in THF at 60 °C for 12 hours. The possible route by the formation of 210 illustrated in Scheme 70. First, the reaction of 212 with metal carbene 213 provides the intermediate 214 by ylide formation. The intramolecular electrophilic trapping associated with rearrangement, and subsequent reductive elimination affords the cycloaddition product 210.87
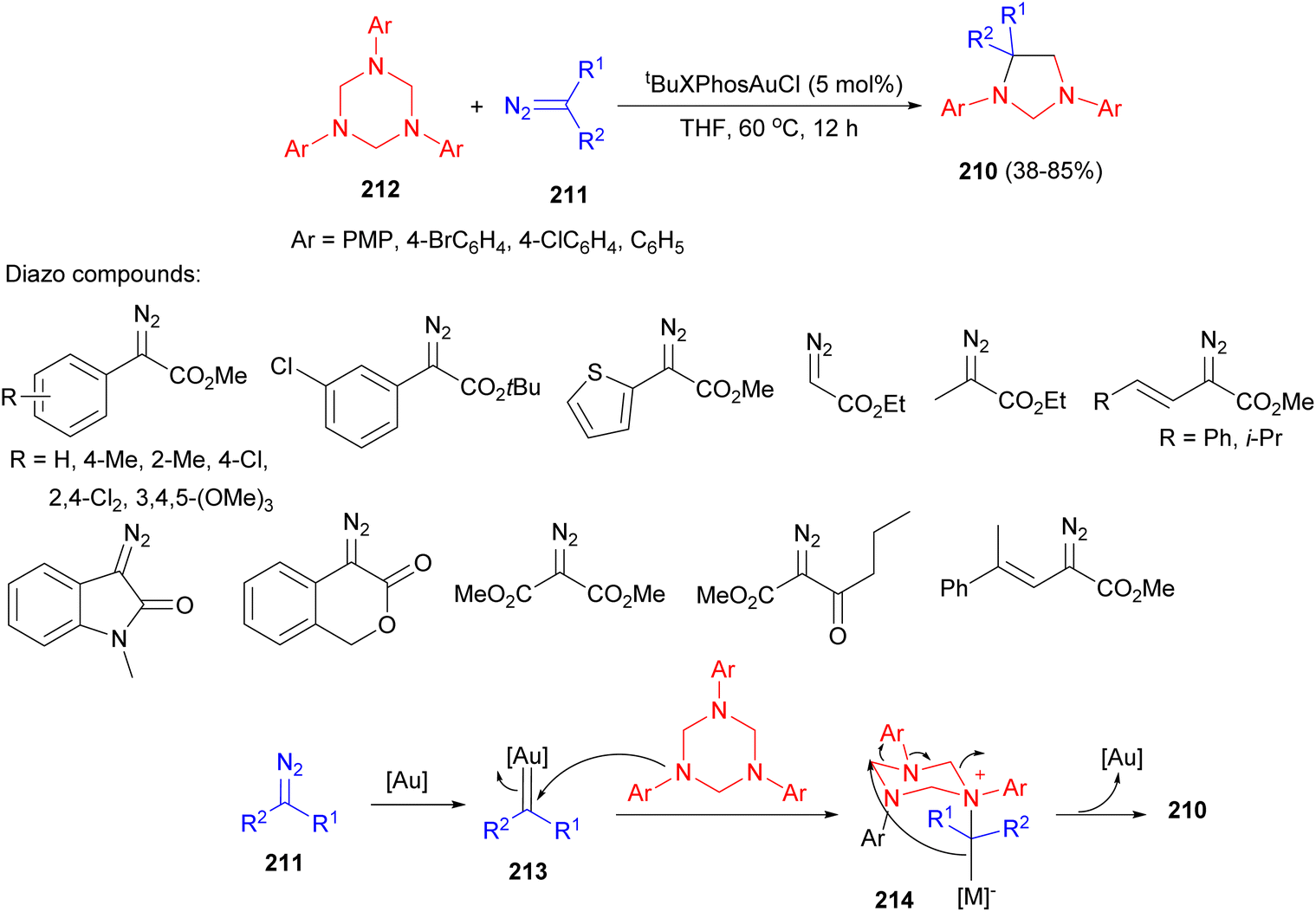 | ||
| Scheme 70 tBuXPhosAuCl catalyzed synthesis of imidazolidines 210. | ||
In 2017, an efficient synthesis of polysubstituted imidazolidines 215 in 42–81% yields using Pd(0)-catalyzed double-addition–cyclization of 2,3-allenyl amines 216 with aryl iodides and imines in the presence of K2CO3 in THF at 85 °C for 24 hours was reported by Gong and co-workers. A plausible mechanism for this cyclization reaction is proposed in Scheme 71. Oxidative addition of the aryl iodide to Pd0 affords aryl-palladium species 217. Coordination of one of the allene double bonds to electrophilic complex 217, and subsequent carbopalladation, affords π-allyl species 218, which reacts with imine and base to afford π-allyl species 219. Finally, intermediate 219 undergoes intramolecular nucleophilic attack on the inner π-allylic carbon atom, affording the imidazolidine structure, thereby releasing the active catalytic species.88
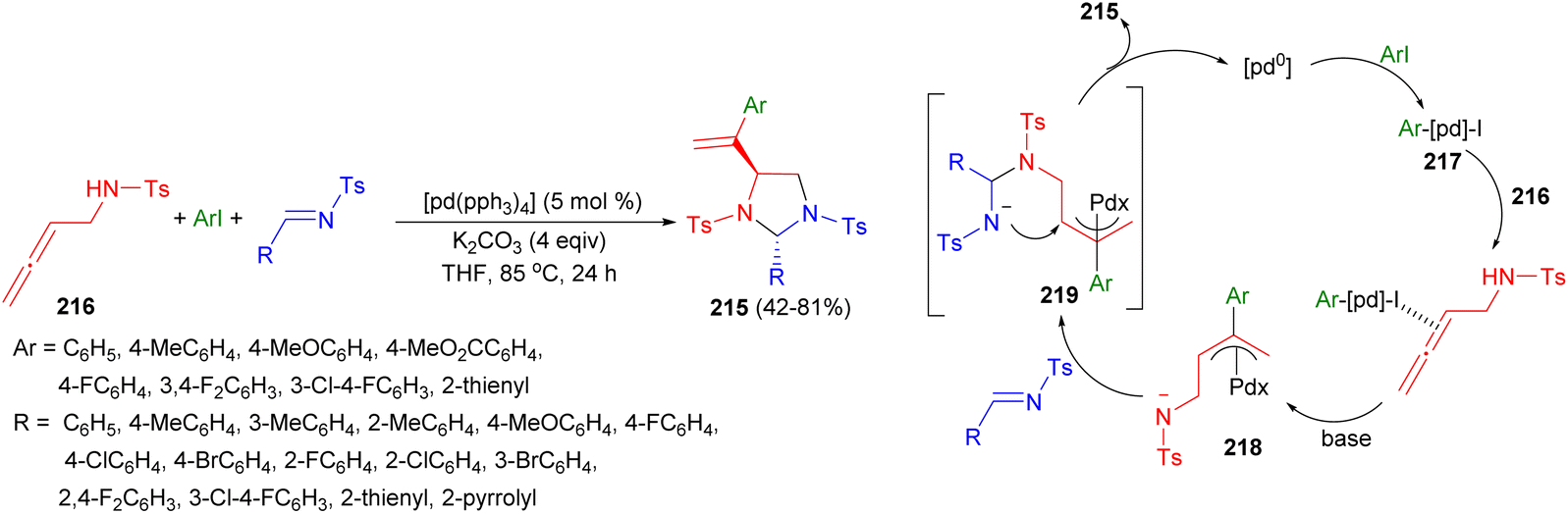 | ||
| Scheme 71 Pd(0)-catalyzed synthesis of polysubstituted imidazolidines 215. | ||
Next, stereocontrolled dimerization-type homo-1,3-dipolar [3 + 2] cycloaddition reaction of glycine aldimino esters 220 for creating new heterocycles bearing multiple stereogenic centers developed through the chiral phosphine ligand-involved silver catalysis. A variety of chiral imidazolidines 221 could be obtained with high yields and good diastereoselectivities as well as excellent enantioselectivities by employing Xing-Phos as chiral P-ligand in EtOAc at −20 °C for 24 hours. They believed that the homo-1,3-dipolar [3 + 2] cycloaddition of imino esters/azomethine ylides controlled by the silver/Xing-Phos catalyst aroused predominately from the steric repulsion and non-covalent interaction between the Xing-Phos ligand and glycine aldimino ester during the Mannich addition and subsequent intramolecular N-acetalization (cyclization) of the intermediate 222 (Scheme 72).89
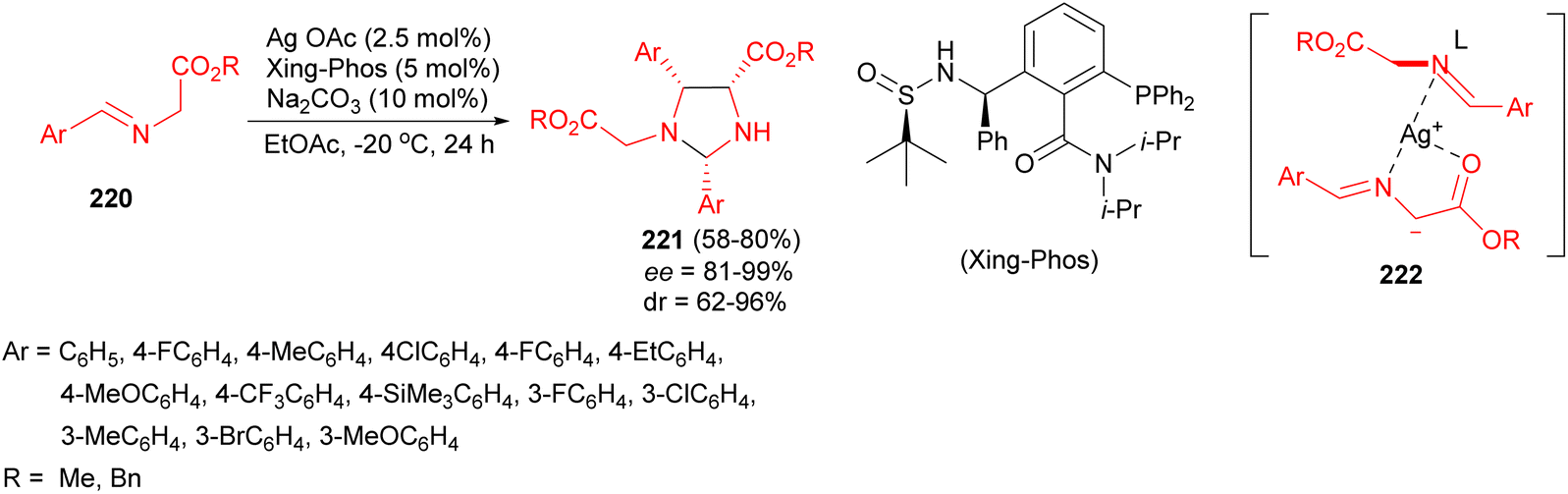 | ||
| Scheme 72 Silver/Xing-Phos-catalyzed synthesis of chiral imidazolidines 221. | ||
An enantioselective synthesis of biologically important imidazolidines 223 has been achieved via a tandem [3 + 2] cycloaddition/1,4-addition reaction of azomethine ylides 224 and aza-o-quinone methides 225 using Ag(I) salts as the precatalyst and ferrocenylphosphine P–N ligand 226 as the chiral ligand in the presence of KOH and 18-crown-6 in DCM at −30 °C for 4 hours. With the use of this tool, various imidazolidine derivatives were obtained in 28–90% yields with excellent diastereoselectivities and enantioselectivities (62–99%). A plausible mechanism is proposed in Scheme 73. Treatment of 224 with a base in the presence of the in situ generated silver complex would lead to the formation of the metalloazomethine ylide 227 as an active species. A regioselective [3 + 2] cycloaddition of azomethine ylides 227 and the second equivalent of Schiff base 224 then occurs. The high degree of endo stereocontrol and excellent diastereofacial discrimination of the transition state 228 generates the imidazolidine complex 229 as the exclusive intermediate. The intermediate 229 was protonated to form the intermediate 230, which was then captured by a o-QM generated in situ from 225 to accomplish the final product 223.90
 | ||
| Scheme 73 Enantioselective synthesis of imidazolidines 223 using AgOTs and ferrocenylphosphine P–N ligand. | ||
The Punniyamurthy group reported stereospecific copper catalyzed nucleophilic ring opening in the presence of tert-butyl hydroperoxide to afford functionalized imidazolidines 231 starting from N-sulfonylaziridines 232 and N-alkylanilines 233 in DCE at 60 °C. The products were obtained after 1.5–10 hours in 42–89% yields and high optical purities (95–99% ee) with excellent functional group tolerance. In the proposed mechanism as illustrated in Scheme 74, single-electron transfer (SET) reduction of Cu(OTf)2 using the nitrogen lone pair of 234 may lead to the formation of an intermediate 235. Homolysis of the N-methyl C–H bond using tert-butoxy radical can generate imine derivative 236, which may lead to cyclization to furnish the target heterocycles. Oxidation of Cu(OTf)2− using TBHP may regenerate Cu(OTf)2 to complete the catalytic cycle.91
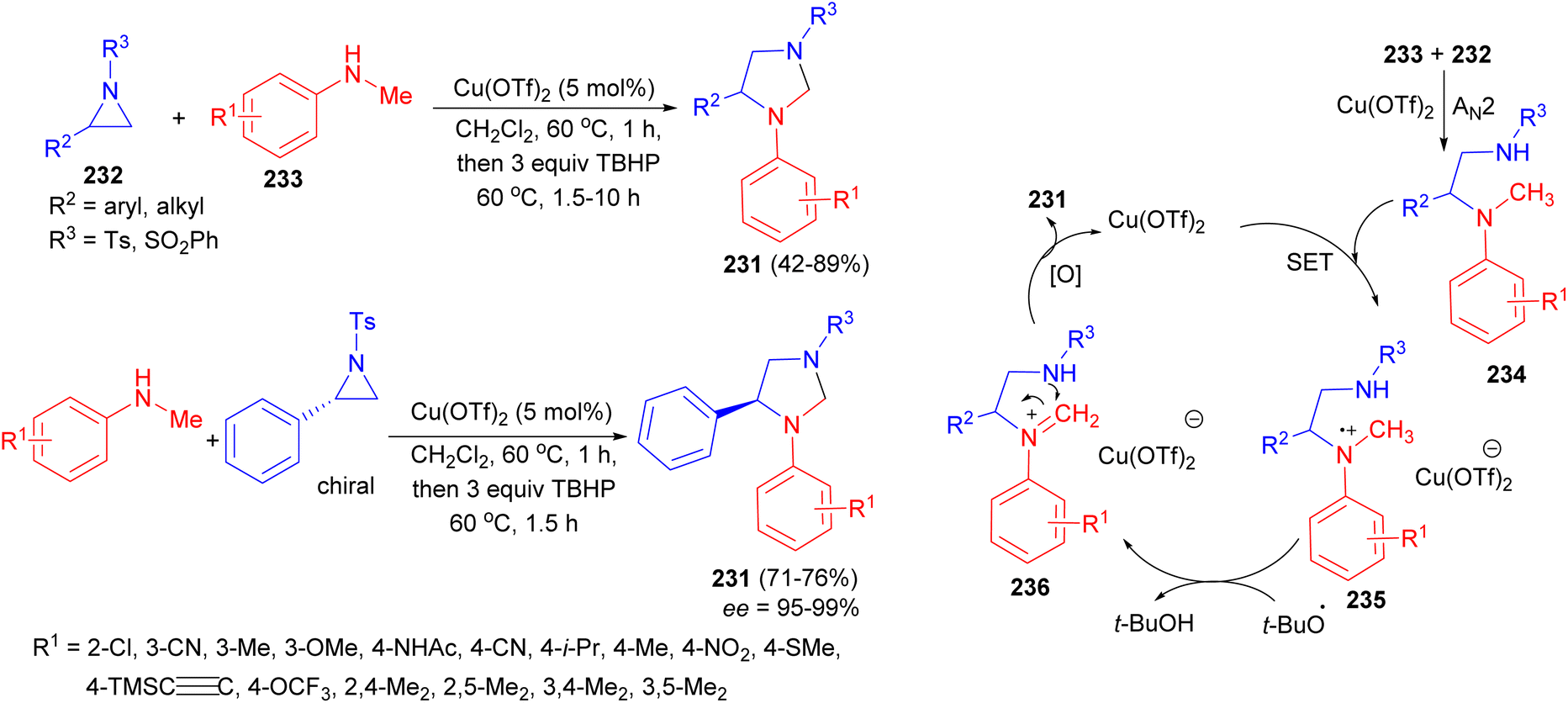 | ||
| Scheme 74 Cu(OTf)2 catalyzed synthesis of functionalized imidazolidines 231. | ||
The Sun group further explored a protocol toward imidazolidines 237 through a stepwise [2 + 1 + 2] process using tosylhydrazones 238 and hexahydro-1,3,5-triazines 239 as the substrates under metal-free reaction conditions in the presence of tBuOLi in toluene at 60 °C for 15 hours. Importantly, the role of tBuOLi confirmed not only to release the diazo but also to promote the cycloaddition. Mechanistically, in this process, diazo 240 is slowly released from tosylhydrazone 238 by tBuOLi. The cycloaddition would be initiated by the first nucleophilic addition between diazo and formaldimine 241, generating aziridine intermediate 242 through transition state 243 or 244. Then, a base-promoted ring opening reaction of aziridine by another molecule of 241 occurs and delivers the final product 237 (38–80% yields) via intermediate 245 (Scheme 75).92
 | ||
| Scheme 75 Metal-free synthesis of imidazolidines 237 in the presence of tBuOLi. | ||
In addition, Laha and co-workers reported synthesis of N-sulfonyl imidazolidines 246 in 66–89% yields via 1,3-dipolar cycloaddition reaction of nonstabilized azomethine ylides 247 and N-sulfonyl aldimine or ketimines 248 using AgF in THF at ice-bath to room temperature for 1 hour. The strategy could complement the preparation of N-sulfonyl imidazolidines via selective N-sulfonylation. Furthermore, novel ring cleavage reactions of N-sulfonyl imidazolidines yielded synthetically useful 1,2-diamines that are otherwise difficult to prepare. Moreover, reaction of 247 with N-sulfonyl ketimine 248 gave sulfamidate fused imidazolidine 249 bearing a quaternary center (Scheme 76).93
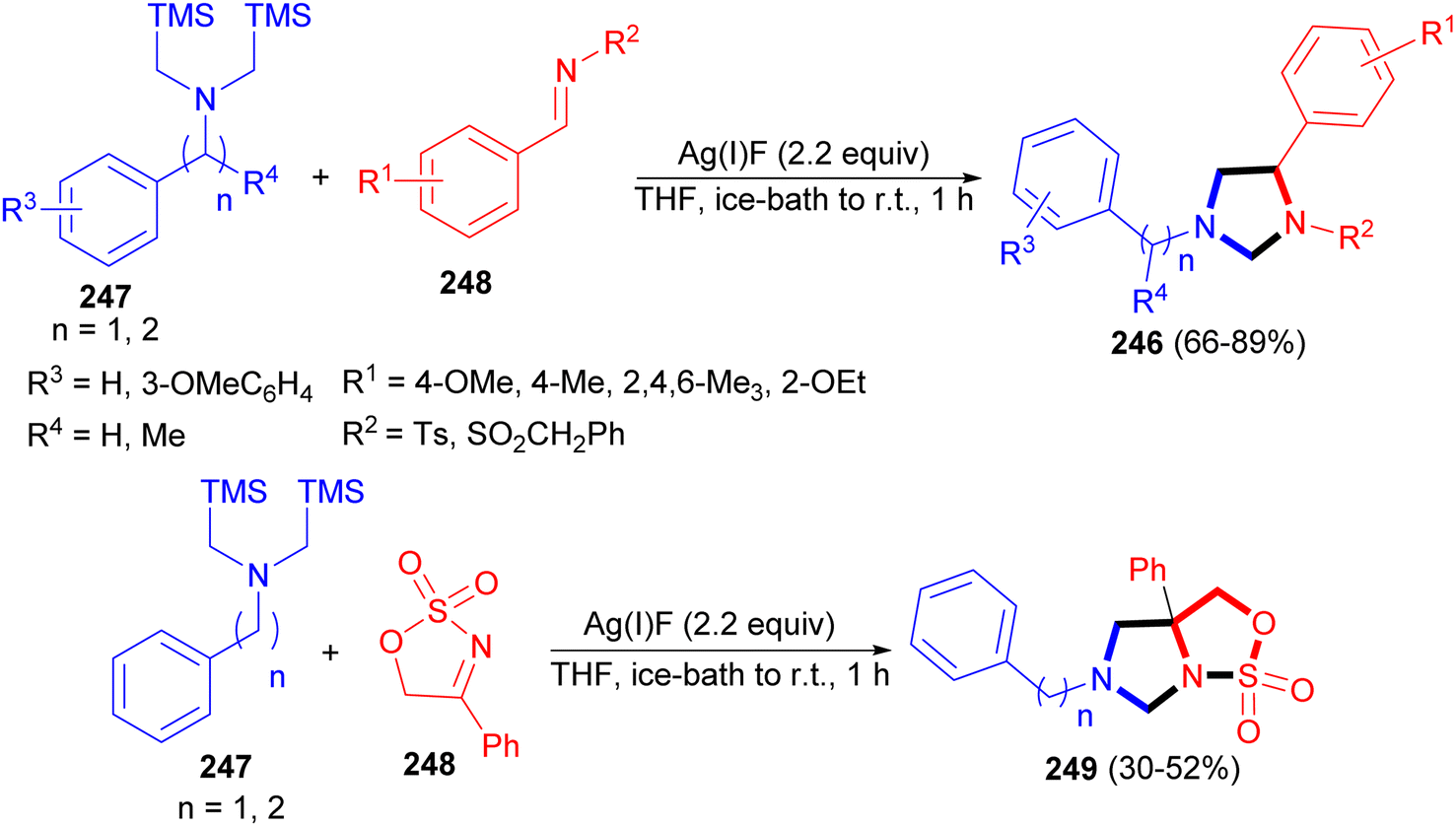 | ||
| Scheme 76 Synthesis of N-sulfonyl imidazolidines 246 and sulfamidate fused imidazolidine 249. | ||
Further, the Sun group demonstrated iron-catalyzed cycloaddition reaction of diazo surrogates 250 with hexahydro-1,3,5-triazines using a mixture of MnO2 and MgSO4 in CH2Cl2 at 0 °C to room temperature for 7 hours afforded imidazolidine derivatives 251 in 41–84% yields. Next, the reaction of substituted phenyl diazoacetates 252 with hexahydro-1,3,5-triazines 253 in the presence of 5 mol% of Fe(acac)3, resulted the corresponding products 254 in 52–80% yields. In addition, cycloaddition of a tosyl hydrazine 255 with hexahydro-1,3,5-triazines led to the formation of imidazolidine 256 in 76% yield. The plausible reaction mechanism is proposed in Scheme 77. Firstly, the reaction of the diazo compound with the iron catalyst generates iron-carbene 257. Then, N-methyleneamine 258 (formed in situ from hexahydro-1,3,5-triazines) reacts rapidly with 257 to afford 259, which undergoes nucleophilic attack by another molecule of 258, followed by ring closing to give the final cycloaddition product and the iron catalyst regenerates.94
 | ||
| Scheme 77 Iron-catalyzed synthesis of imidazolidine derivatives 251, 254 and 256. | ||
In 2018, the highly diastereo- and enantioselective synthesis of 2,4-disubstituted imidazolidines 260 in 60–82% yields developed via a formal [3 + 2] cyclization reaction of bidentate aminomethyl enones 261 and N-tosyl imines 262 in the presence of bifunctional squaramide catalyst 263 in toluene at room temperature for 48 hours. A plausible TS depicted in Scheme 78 which dictates a bifunctional mode of activation by the catalyst. Since the CN of 262 is activated by hydrogen bonding of the squaramide motif, Re face is blocked. Thus, the addition of deprotonated 261 will take place only from the Si face and thus intermediate 264 is formed. Intermediate 264 then undergoes Michael addition from the Re face of enone moiety to provide product 260.95
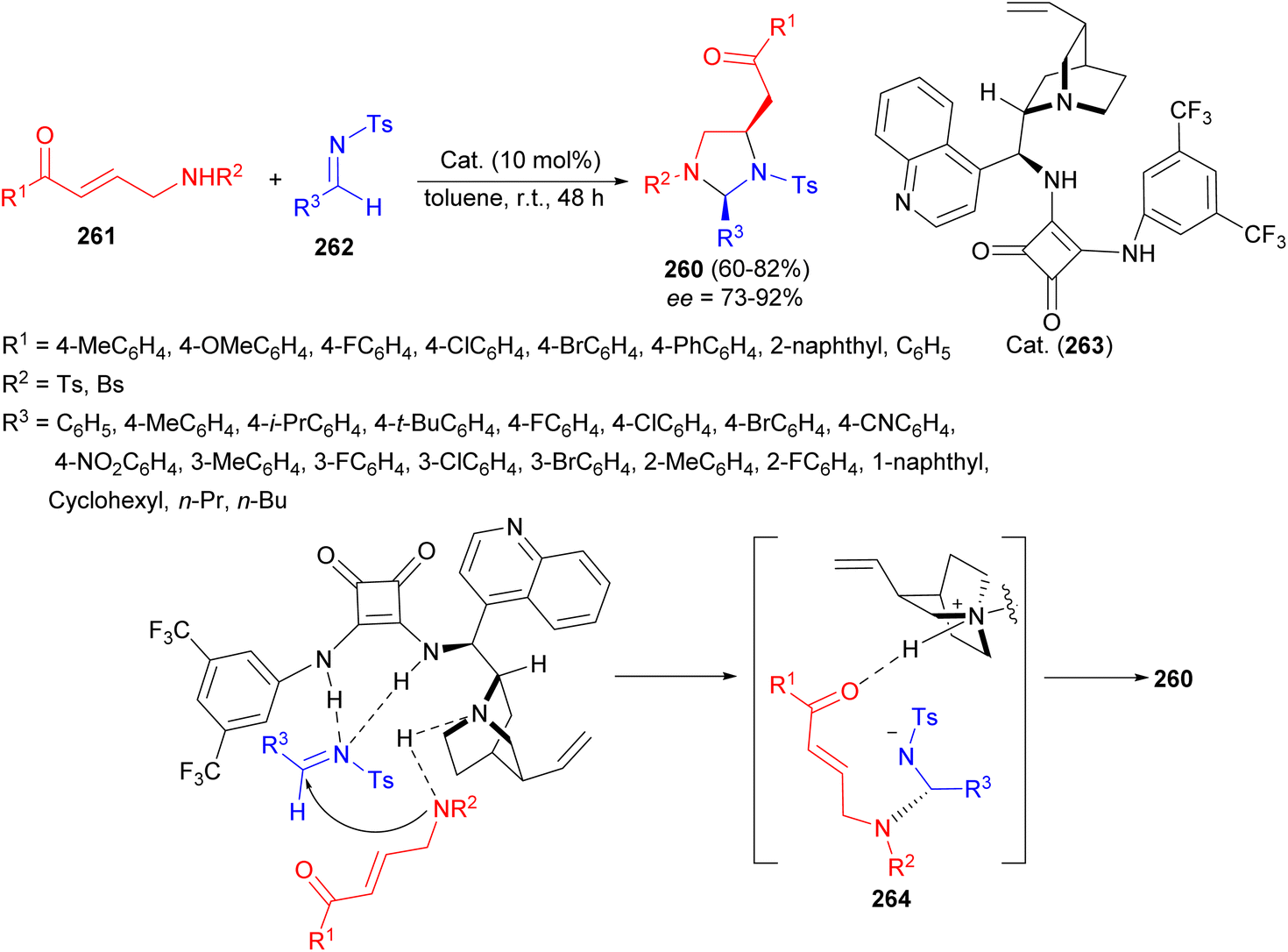 | ||
| Scheme 78 Organocatalytic asymmetric synthesis of 2,4-disubstituted imidazolidines 260. | ||
Further, the Huo group revealed a straightforward and efficient aerobic oxidative dehydrogenative formal [2 + 3]-cyclization of glycine derivatives 265 with aziridines 266. The reaction provides facile access to a series of highly functionalized imidazolidine derivatives 267 in 33–81% yields and diastereomeric ratios up to 3.3:1 using Cu(OTf)2 and TFA in toluene at 100 °C for 3–10 hours. In this process, aziridine 266 is initially attacked by glycine derivative 265 to form ring-opened intermediate 268 (SN2). Intermediate 268 is then auto-oxidized to give hydroperoxide intermediate 269. Subsequently, iminium ion intermediate 270 is then formed from 269 through an acid catalyzed SN1-type procedure. Finally, intramolecular C–N bond formation results in the desired product 267 (Scheme 79).96
 | ||
| Scheme 79 Synthesis of functionalized imidazolidine derivatives 267 using Cu(OTf)2 and TFA. | ||
The tandem nucleophilic addition–cycloaddition reaction developed for the synthesis of functionalized imidazolidine derivatives 271 by the reaction of a variety of α-iminoesters 272 with silylaryl triflates 273 at −10 °C in the presence of Ag(Tf2N), chiral ligand 274, CsF and 18-crown-6 in acetonitrile for overnight. This asymmetric cycloaddition afforded imidazolidine derivatives with high yields (up to 99%), complete regioselectivities, and excellent diastereo (>20:1)- and enantioselectivities (up to 97% ee) (Scheme 80). In this process, aryne-induced ylides working as 1,3-dipoles for asymmetric cycloaddition is the notable feature of the present reaction. In the tandem reaction, the [3 + 2] cycloaddition of aryne induced ylides with metallized α-iminoesters and metal-catalyzed [3 + 2] cycloaddition of azomethine ylide with α-iminoesters are two concurrent pathways to imidazolidines.97
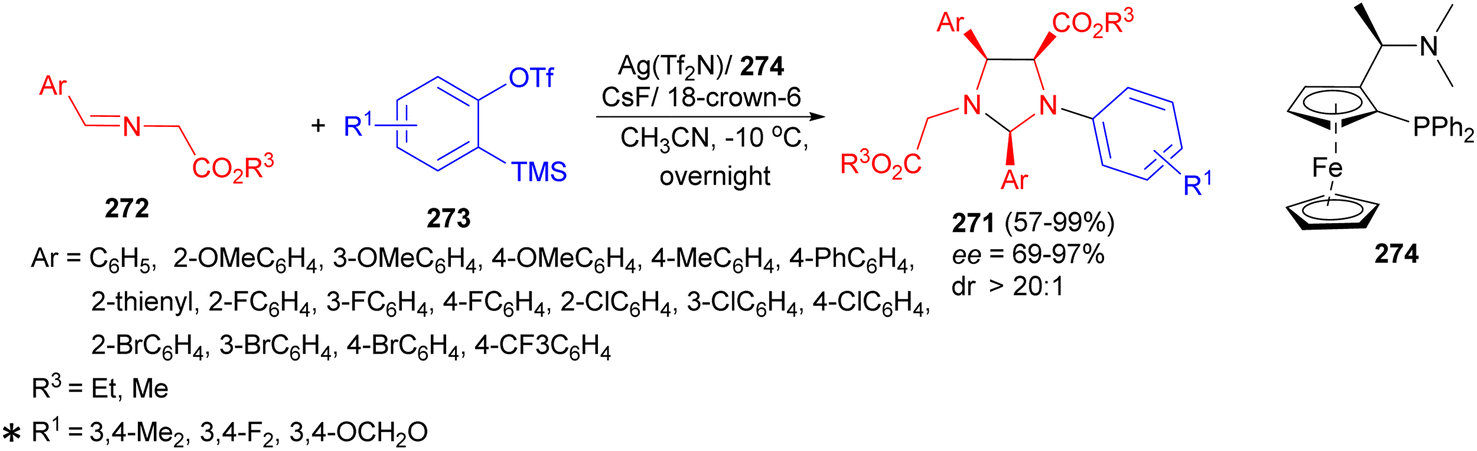 | ||
| Scheme 80 Synthesis of functionalized imidazolidine derivatives 271 in the presence of Ag(Tf2N). | ||
The Feng group reported a highly efficient and stereoselective synthesis of enantioenriched imidazolidines 275 by rhodium-catalyzed intermolecular [3 + 2] cycloaddition reaction of chiral vinyl aziridines 276 and oxime ethers 277 using AgSbF6 in DCE at −5 °C for 12 hours. This method delivers enantioenriched imidazolidines in up to 99% yield and up to 99% ee by a chirality-transfer strategy. A plausible mechanism is proposed in Scheme 81. Both the olefin and the nitrogen atom in vinylaziridine 276 could coordinate to the rhodium catalyst to give complex 278, which led to enyl (σ + π) rhodium species 279 with the retention of configuration formed by oxidative addition. Subsequently, nucleophilic attack of the oxime ether 277 onto the rhodium complex 279 from the back face would produce the intermediate 280 with a net inversion of absolute configuration and regenerate the rhodium catalyst. Finally, intermediate 280 underwent intramolecular cyclization to afford the less sterically hindered [3 + 2] cycloadducts 275.98
 | ||
| Scheme 81 Rhodium-catalyzed synthesis of enantioenriched imidazolidines 275. | ||
A palladium-catalyzed controllable cyclization of vinyl ethylene carbonates 281 with 1,3,5-triaryl-1,3,5-triazinanes 282 has been reported by Yang group. The reaction proceeds through formal migration [2 + 3] cycloaddition in MeOH/H2O at 80 °C for 2 hours. The transformation affords imidazolidine derivatives 283 in 26–96% yields. A plausible mechanism is proposed in Scheme 82. The reaction begins with the oxidative addition of Pd(0) to vinyl ethylene carbonate 281, generating the zwitterionic p-allyl palladium intermediate 284 by releasing carbon dioxide. Meanwhile, three imines are generated in situ from triazinane 282 via C–N bond cleavage, which would attack 284 followed by reductive elimination to produce the formal [5 + 2] cycloaddition product 285 and regenerate Pd(0) species. Under controllable conditions, the 7-membered ring product 285 could further undergo oxidative addition with Pd(0) species to give intermediate 286, which is then captured by imine and delivers the ten-membered palladacycle 287. Subsequently, reductive elimination and intramolecular aza [3,3]-sigmatropic rearrangement events take place to furnish the formal migration [2 + 3] cycloaddition product 283 with extrusion of formaldehyde.99
 | ||
| Scheme 82 Palladium-catalyzed synthesis of imidazolidine derivatives 283. | ||
In 2019, Mani et al. reported the Mannich reaction of pyrrole (288) with a mixture of ethylenediamine dihydrochloride (289) and formaldehyde using K2CO3 in aqueous methanol at 0 °C gave a mixture of products N1,N1,N2,N2-tetrakis(pyrrol-2-ylmethyl)ethane-1,2-diamine 290 and 1,3-bis(pyrrol-2-ylmethyl)imidazolidine 291 which were isolated in 14% and 1% yield, respectively, after basic alumina column chromatography (Scheme 83). The X-ray structure of 291 along with intermolecular hydrogen bonding in its crystal lattice.100
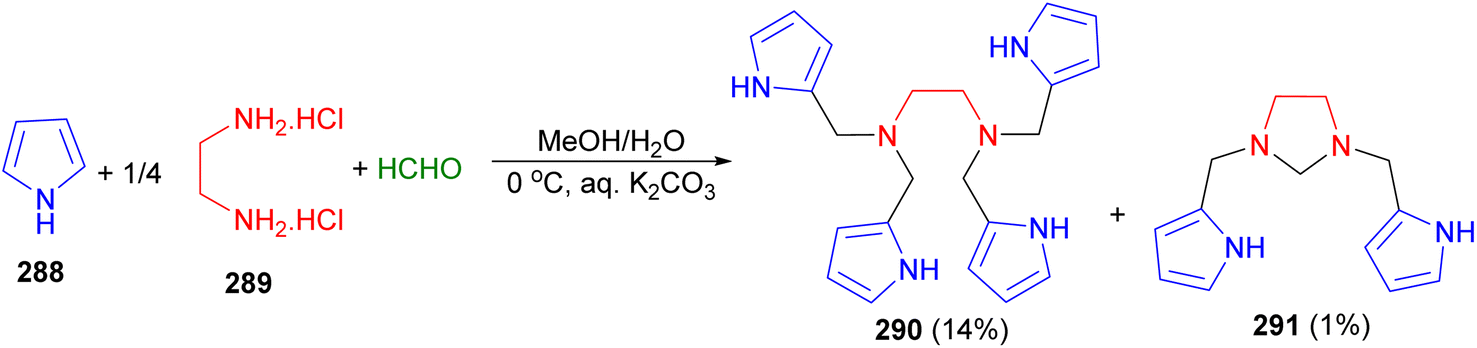 | ||
| Scheme 83 Synthesis of 1,3-bis(pyrrol-2-ylmethyl)imidazolidine 291. | ||
The Tu group described synthesis of functionalized imidazolidines 292 in 36–92% yields via unprecedented 1,3-dipolar cycloadditions of 1,3,5-triazinanes 293 with aziridines 294 in the presence of ZnBr2 in toluene at 80 °C for 36 hours. A plausible mechanism is depicted in Scheme 84. The formaldimine was first generated in the presence of Lewis acid. Next, formaldimine attacked the activated aziridine through a second-order nucleophilic substitution (SN2)-like pathway, leading to the inversion of configuration and the ring open of the aziridine. Meanwhile, the racemic product was generated through the ring opened zwitterion, which served as both a nucleophile and an electrophile to react with N-phenyl formaldimine. The ee value of 292 suggested that the ring opened zwitterionic pathway product dominated over the SN2-like product.101
 | ||
| Scheme 84 ZnBr2 catalyzed synthesis of functionalized imidazolidines 292. | ||
In 2020, Ghorai and co-workers developed a mild one-pot stereospecific synthetic route to highly functionalized imidazolidines 295 via SN2-type ring-opening of the corresponding activated aziridines 296 with amines followed by p-toluenesulfonic acid catalyzed intramolecular cyclization with aldehydes using MgSO4 in DCE at 65 °C for 6 hours. The methodology tolerates a variety of functional groups and furnishes the desired products in high yields (up to 92%) with excellent stereoselectivities (ee > 99%). Interestingly, imidazolidines were formed as the cis-isomers. A plausible mechanism is depicted in Scheme 85. The ring-opening reaction of activated aziridines proceed via a regioselective SN2-type pathway. Amine nucleophile attacks the aziridine at the benzylic position to produce the corresponding ring-opening product 297, which in the presence of acid catalyst and additive MgSO4 forms the corresponding iminium ion 298 when reacted with aldehyde. Subsequently, the intramolecular nucleophilic attack by the tosyl amide on the iminium ion probably through the cationic intermediate 299 occurs in such a way that it leads to the more favorable TS 300 where the electronic 1,4-π–π stacking interaction outweighs the steric repulsion arising from the interaction between the ortho-hydrogens of the aromatic ring at C-2 and methylene hydrogen at C-5 of the ring to produce the 2,4-cis diastereomer 295 of imidazolidine derivatives as the only product.102
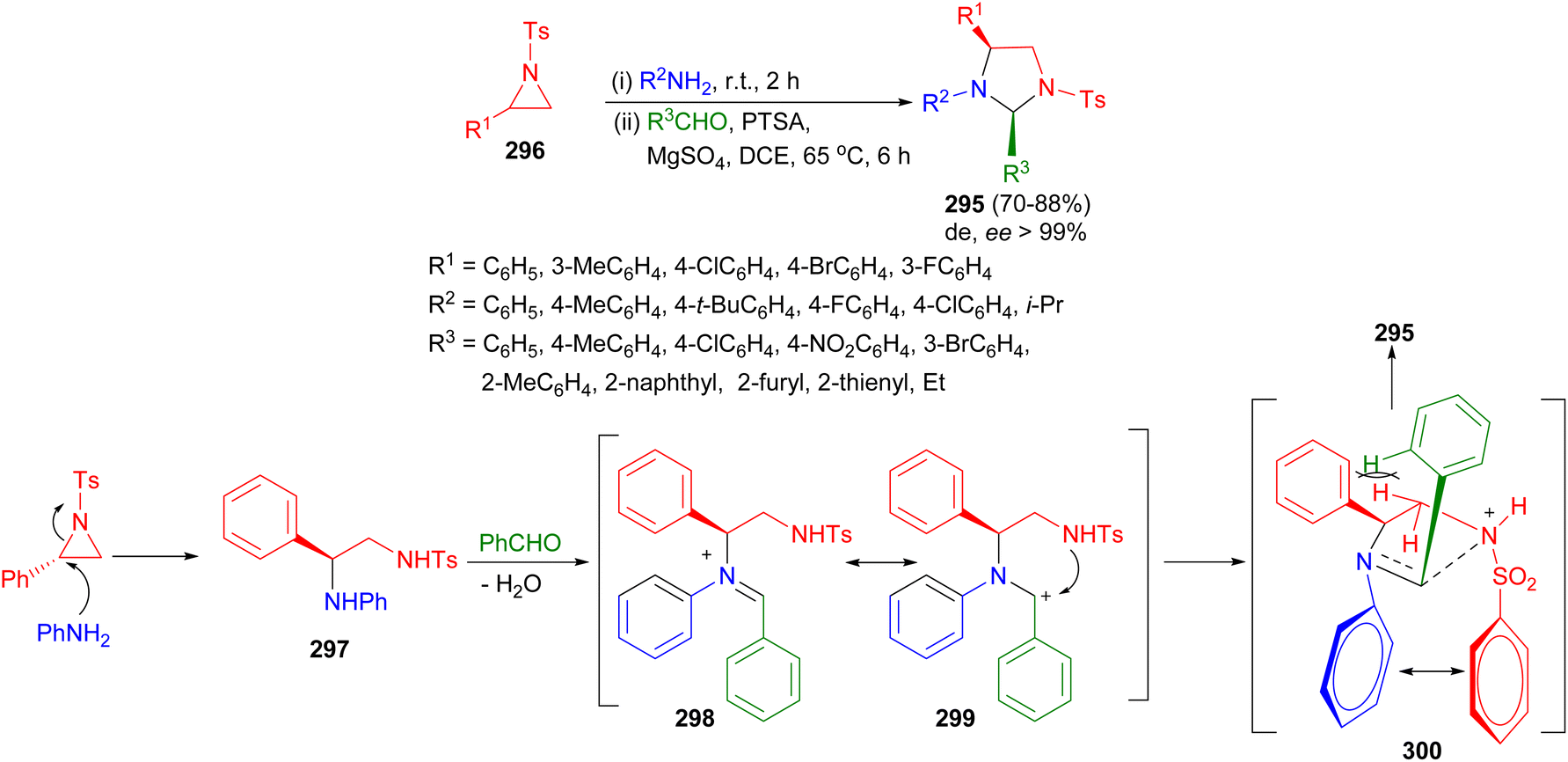 | ||
| Scheme 85 Synthetic of highly functionalized imidazolidines 295 via SN2-type ring-opening of aziridines. | ||
The Hu group described the effectiveness of trifluoromethylated N-acylhydrazones 301 as dipolarophiles in conducting 1,3-dipolar cycloaddition with azomethine ylides 302. This reaction occurs in the presence of AgNO3 and Et3N in toluene at room temperature, yielding trifluoromethylated imidazolidines 303 in 76–91% yields after 24 hours. A proposed mechanism is depicted in Scheme 86. The in situ-formed azomethine ylides 302 was coordinated to the Ag(I) to give complex 304, which performed addition reaction to the CN of 301 to generate the zwitterionic intermediate 305. The silver atom switched from the nitrogen atom of imine ester to the N′-nitrogen atom of trifluoromethylated N-acylhydrazone to form the species 306. Rotation of C–N σ-bond formed intermediate 307, which performed cyclization to give the final product 303.103
 | ||
| Scheme 86 AgNO3 catalyzed synthesis of trifluoromethylated imidazolidines 303. | ||
In 2021, the Wang and Xuan group demonstrated a visible light-promoted divergent cycloaddition of α-diazo esters 308 with hexahydro-1,3,5-triazines 309 in DCM at room temperature for 12 hours. This reaction yielded a series of imidazolidine frameworks 310 in 41–79% yields (Scheme 87). It is noteworthy that the reaction occurs under sole visible light irradiation without the need for exogenous photo redox catalysts. Mechanistic studies based on control experiment results and DFT calculations revealed that both 1,3,5-triazines and the in situ formation of formaldimines could serve as carbene trapping reagents to form key nitrogen ylide intermediates.104
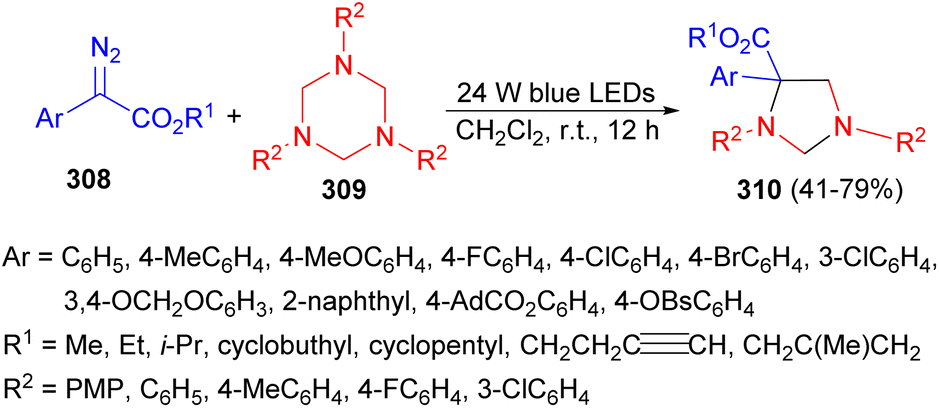 | ||
| Scheme 87 Synthesis of a series of imidazolidine frameworks 310 under sole visible light irradiation. | ||
The synthesis of 1,3-disubstituted imidazolidines 311 in 34–80% yields was reported by the Ye and Fu group. To begin, diamine 312 reacted with formaldehyde in water at 100 °C for 0.5–2 hours, yielding intermediate imidazolidines 313. Subsequently, aromatic acyl chloride in acetone was added to imidazolidines 313 under pH = 8–10, resulting in the formation of imidazolidines 311 after 2 hours. The bioassay results indicated that the majority of the target compounds exhibited softening activity against nicosulfuron in sensitive Kennian 1 maize (Scheme 88).105
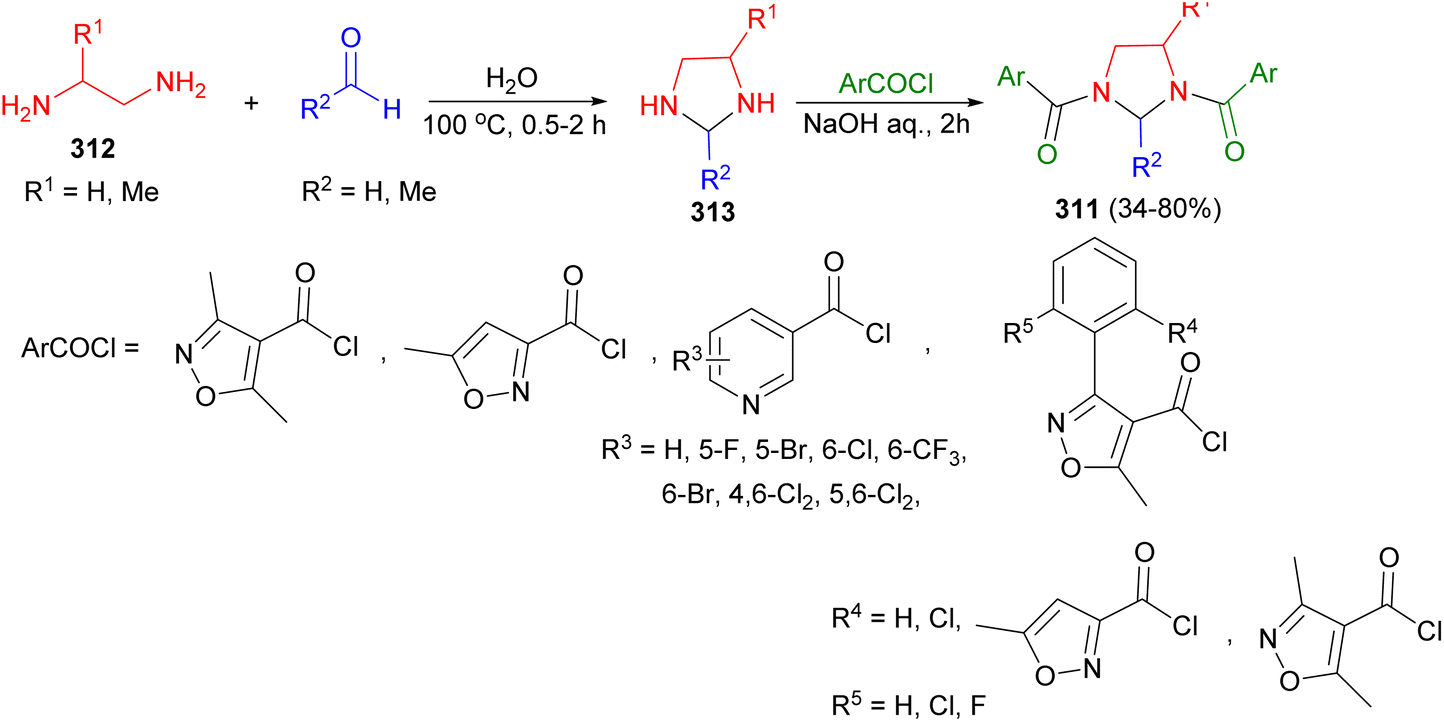 | ||
| Scheme 88 Synthesis of 1,3-disubstituted imidazolidines 311. | ||
The reaction of equimolar amounts of pyridoxal 314 and 1,2-propylenediamine 315 in EtOH at 0 °C afforded the formation of monoamine 316 after 12 hours, which affords as a cyclic 5-(hydroxymethyl)-2-methyl-4-(4-methylimidazolidin-2-yl)pyridin-3-ol tautomer 317 in 100% yield (Scheme 89).106
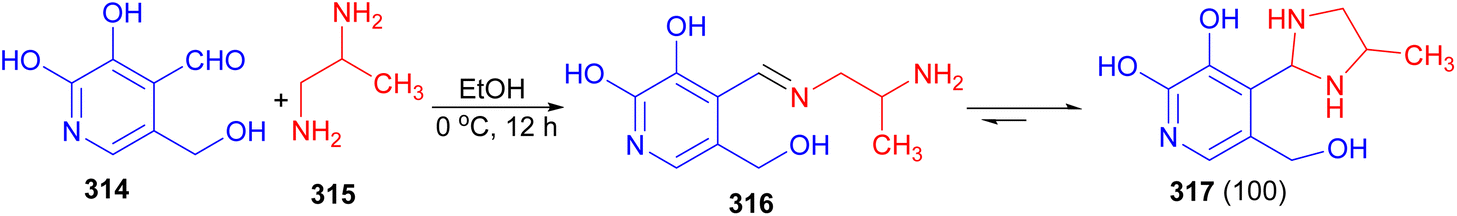 | ||
| Scheme 89 Synthesis of cyclic 5-(hydroxymethyl)-2-methyl-4-(4-methylimidazolidin-2-yl)pyridin-3-ol tautomer 317. | ||
Synthesis of methyl (2E)-3-[3-benzyl-2-(3-methoxy-3-oxoprop-1-yn-1-yl)-2-(1-naphthyl)imidazolidine-1-yl]acrylate 318, has been reported via domino-reaction, employing easily available 1-benzyl-2-(1-naphthyl)-4,5-dihydro-1H-imidazole 319 and methyl propiolate (320) in dry ether at room temperature for 3 hours in 92% yield. The mechanism of imidazolidine 320 formation includes the conjugated addition of 2-imidazoline 318 at the triple bond of the first alkyne molecule, leading to the zwitterion 321, which then deprotonates the second methylpropiolate molecule to form an acetylenide ion. At the final stage, nucleophilic addition of the obtained acetylenide ion occurs at the position 2 of the 2-imidazolinium ion (Scheme 90).107
 | ||
| Scheme 90 Synthesis of imidazolidine derivative 318 via domino-reaction. | ||
The Jia group disclosed a facile BF3·Et2O-catalysed [3 + 2] annulation between readily available 1,3,5-triazinanes 322 and 3-aminooxetanes 323 in THF at 50 °C for 10 hours furnishing a wide range of 4-hydroxymethyl imidazolidines 324 in useful to good yields. A plausible [3 + 2] annulation reaction mechanism is proposed in Scheme 91. With the assistance of BF3·Et2O, the nucleophilic addition of 323 to 322 affords a usually unstable intermediate 325 under basic conditions. The intermediate 325 further undergoes an intramolecular ring-opening produce intermediate 326. The intermediate 326 generate the 4-hydroxymethyl imidazolidine product 324. It should be noted that the ring-opening step should be fast enough to outcompete the decomposition of intermediate 325.108
 | ||
| Scheme 91 A facile BF3·Et2O-catalysed synthesis of 4-hydroxymethyl imidazolidines 324. | ||
A strategy of asymmetric carbonyl catalysis via a chiral Lewis acid-bonded aldehyde has been developed for the direct Mannich/condensation cascade reaction of glycine ester 327 with aromatic aldimines 328. The co-catalytic system of 2-picolinaldehyde and chiral YbIII-N,N′-dioxides as the Lewis acid catalyst was identified to be efficient in EtOH at 35 °C for 48 hours, providing a series of trisubstituted imidazolidines 329 in moderate to good yields (up to 66%), and excellent diastereo- and enantioselectivities with high diastereo- and enantioselectivities (up to 95:5 dr, 97% ee). Enantiodivergent synthesis was achieved via changing the sub-structures of the chiral ligands. The reaction could be carried out in a three-component version involving glycine ester, aldehydes, and anilines with equally good results. The chiral N,N′-dioxide/Yb(OTf)3 complex bonded aldehyde enabled carbonyl activation of glycine ester for α-addition transformation (Scheme 92).109
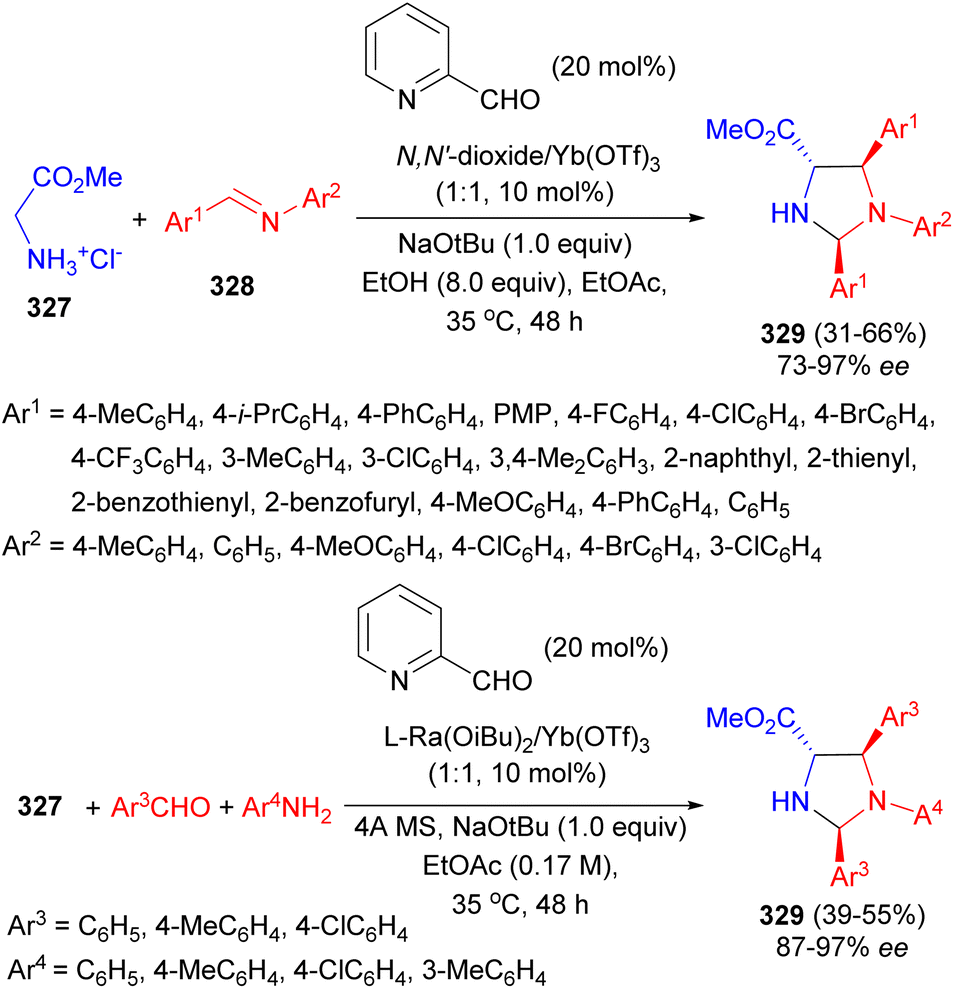 | ||
| Scheme 92 2-Picolinaldehyde and chiral YbIII-N,N′-dioxides catalyzed synthesis of a series of trisubstituted imidazolidines 329. | ||
A Y(OTf)3-catalyzed [3 + 2] cycloaddition of 1,3,5-triazinanes 330 with donor–acceptor aziridines 331 has been developed by Lin and co-workers. The reaction conducted in DCM at 30 °C for 12 hours yielded substituted imidazolidines 332 in 39–98% yields. A plausible mechanism is depicted in Scheme 93. In the presence of Y(OTf)3, the formaldimine 333 was first generated and the 1,3-dipole 334 was produced in situ from the ring-opening reaction of the donor acceptor aziridine, followed by a [3 + 2] cycloaddition between the formaldimine 333 and 1,3-dipole 334, furnishing the desired product 332 through an SN1-like pathway. Moreover, the compound 332A exhibited promising anti-proliferative activity against a number of human cancer cell lines, which could serve as a hit compound for the anti-tumor research.110
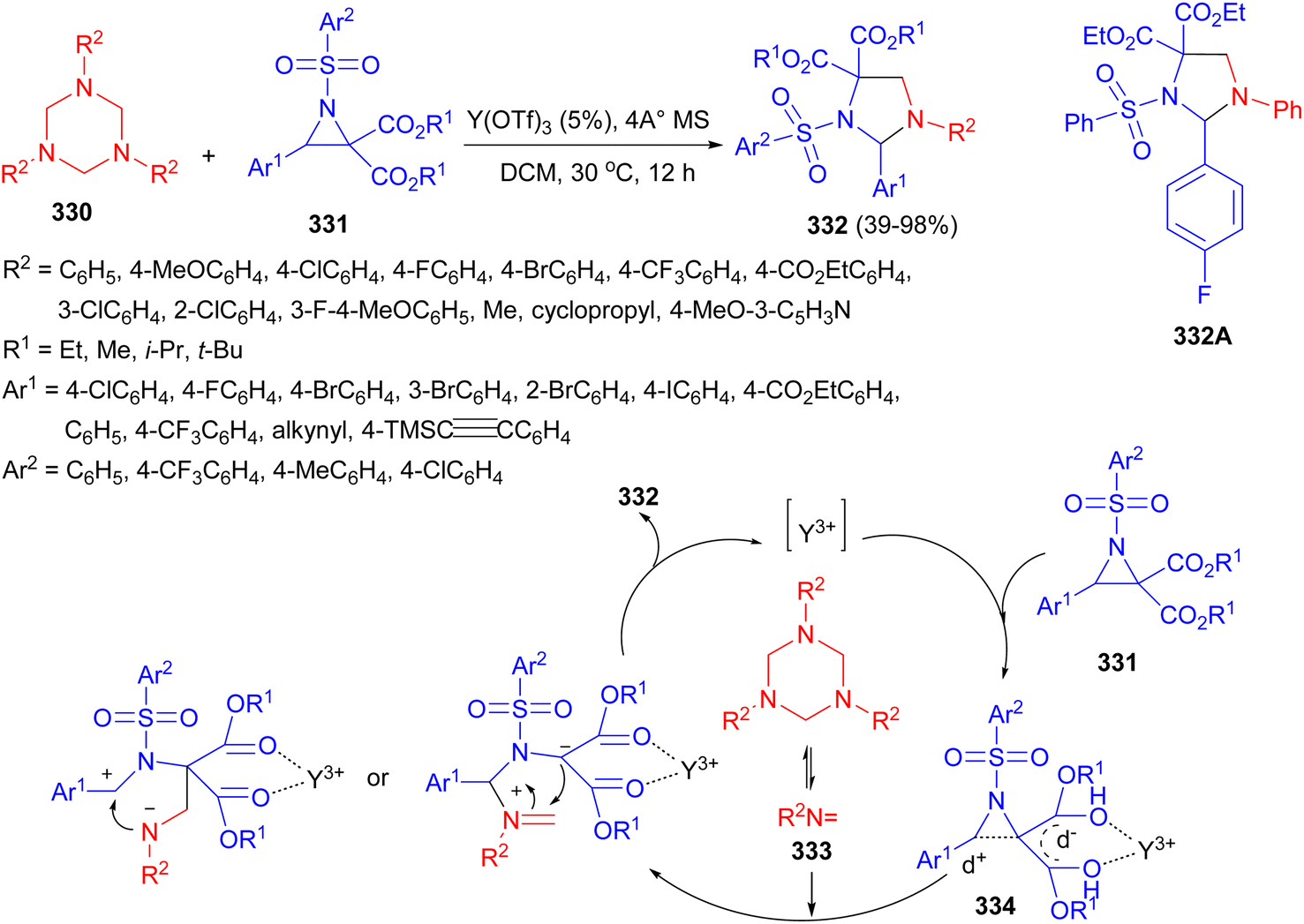 | ||
| Scheme 93 Y(OTf)3-catalyzed synthesis of substituted imidazolidines 332. | ||
An unprecedented 1,3-dipole cycloaddition between acyclic CF3-ketimines 335 and N-benzyl amine 336 has been allowed by tungsten (W) catalysis in o-xylene at 150 °C for 48 hours, resulting in a variety of imidazolidines 337 in 32–99% yields bearing a trifluoromethylated tetrasubstituted carbon center. A proposed mechanism is illustrated in Scheme 94. The coordination of reagent 336 with W catalyst occurred to form intermediate 338 through CO dissociation, which further generated azomethine 339 by releasing W(CO)5 and TMSOMe. The active intermediate 339 could undertake a concerted 1,3-dipolar cycloaddition reaction with imine 340 to furnish 337 or might undergo a stepwise cyclization to give product 337 under the assistance of W catalyst.111
 | ||
| Scheme 94 Tungsten catalyzed synthesis of imidazolidines 337. | ||
The Ma Group described the TiO2 photocatalytic synthesis of five-membered N-heterocyclic imidazolidines 341 in 32–98% yields from a common imine (N-benzylidenebenzylamine) 342 and alcohols 343 via a 1,3-dipolar azomethine ylide intermediate, notably without pre-installed electron-withdrawing groups (EWG) on the substrates (Scheme 95).112
 | ||
| Scheme 95 TiO2 photocatalytic synthesis of imidazolidines 341. | ||
In 2022, Wang and co-workers reported a simple and practical method for the construction of 1,3,5-trisubstituted imidazolidine derivatives 344 via [3 + 2] cycloaddition reaction. This reaction could smoothly proceed between nonstabilized azomethine ylide 345, generated in situ from N-(methoxymethyl)-N-(trimethylsilyl-ethyl)-benzyl amine (346), and 2-benzothiazolamines 347 in the presence of TFA in DCM at room temperature for 1 hour to deliver a wide scope of differently substituted imidazolidines in high yields (up to 98%). A possible mechanism for this transformation is proposed in Scheme 96. First, the nonstabilized azomethine ylide 345 from 346 is generated in the presence of TFA. Then, this nonstabilizedazomethine ylide could react with 347 to obtain the desired product 344 via [3 + 2] cycloaddition reaction with high regioselectivity.113
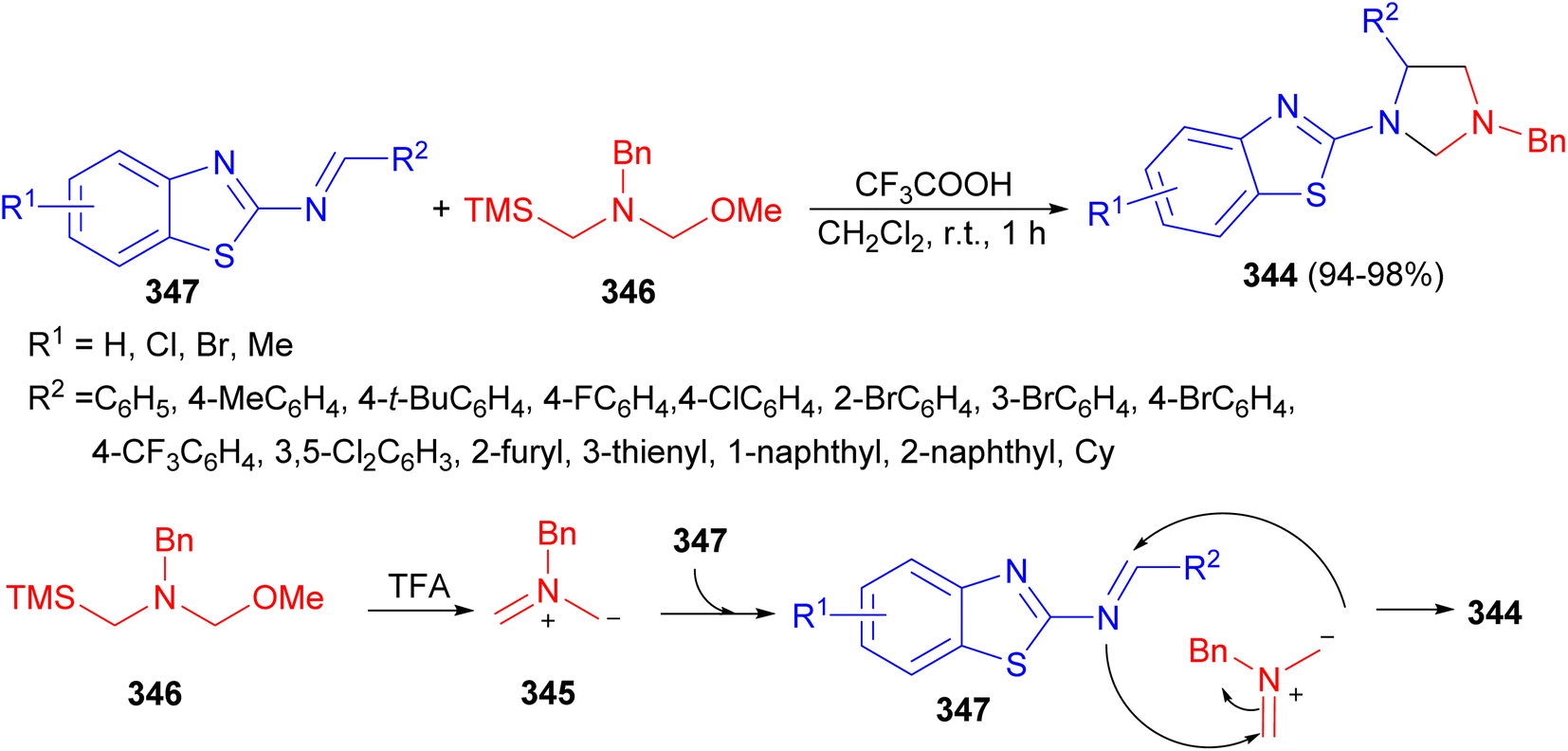 | ||
| Scheme 96 TFA catalyzed synthesis of 1,3,5-trisubstituted imidazolidine derivatives 344. | ||
In addition, the Tamang group described the synthesis of highly stable CsPbBr3 QD based photocatalysts using dibromoisocyanuric acid (DBI) as a benign non-toxic bromide precursor. The QDs were applied as a visible light photocatalyst for intramolecular cyclisation of diamines 348 and 349 to obtain a range of enantiopure bioactive heterocycles such as imidazolidines 350 and fused-imidazolidines 351 in high yields (isolated yield up to 82%, ee > 99%). The reaction carried out in open air at room temperature in DCM for 3–14 hours. The proposed mechanism is depicted in Scheme 97.114
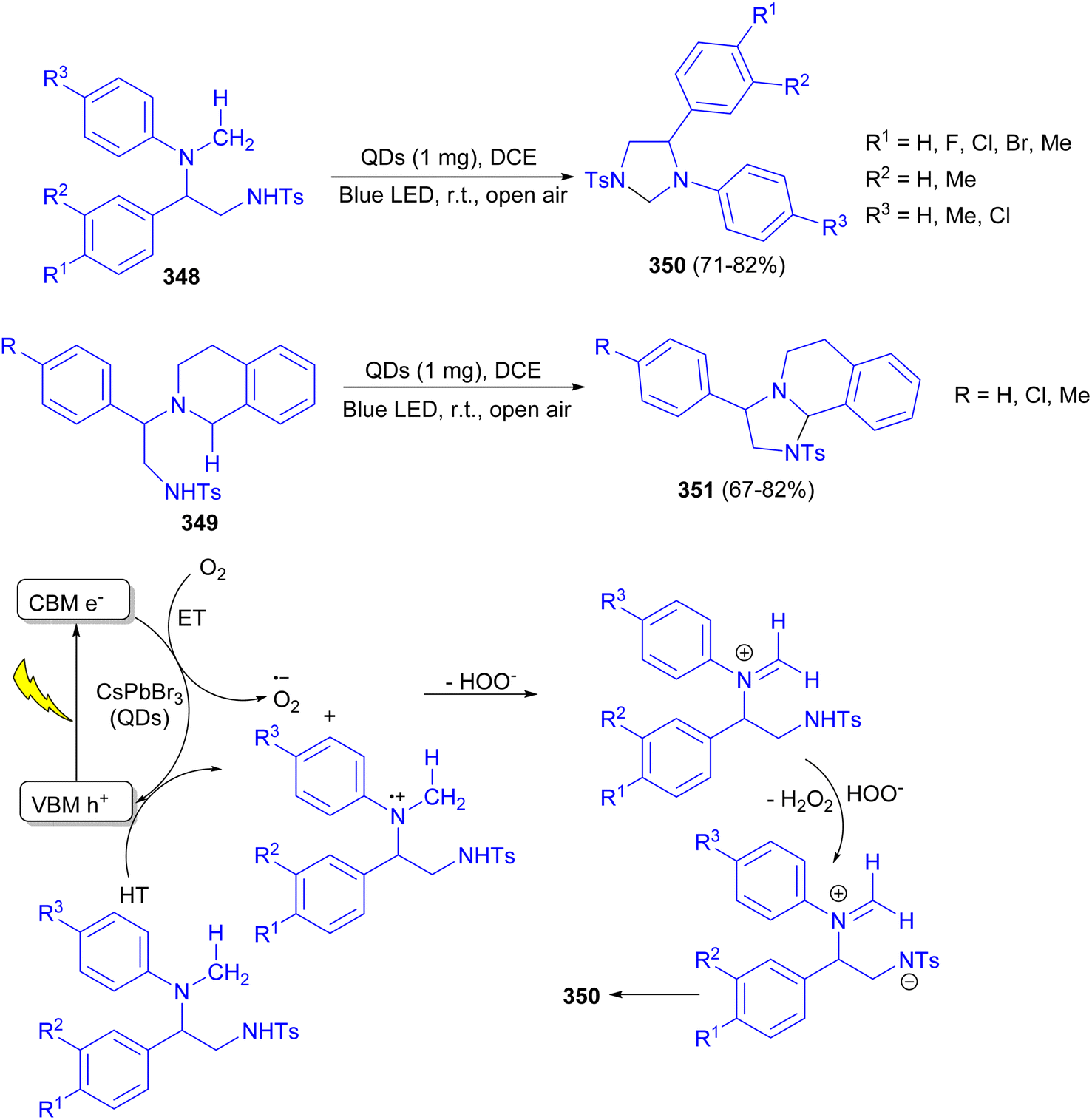 | ||
| Scheme 97 Synthesis of imidazolidines 350 and 351 using QDs as a visible light photocatalyst. | ||
In 2023, Moloney et al. described the reaction of diethyl aminomalonate hydrochloride 352, along with 2 eq. of substituted benzaldehydes and benzylamine under basic conditions and heated to more than 100 °C in a Dean–Stark trap afforded imidazolidines 353 in 58%-quant. Yields, as either a single diastereomer or as a mixture of diastereomers, by a one-pot, three component 1,3-dipolar cycloaddition. Also, imidazolidines 353 were then N-acylated with ethyl malonyl chloride in DCM under reflux conditions for overnight to form imidazolidines 354 as single diastereomers in 56%-quant. Yields (Scheme 98).115
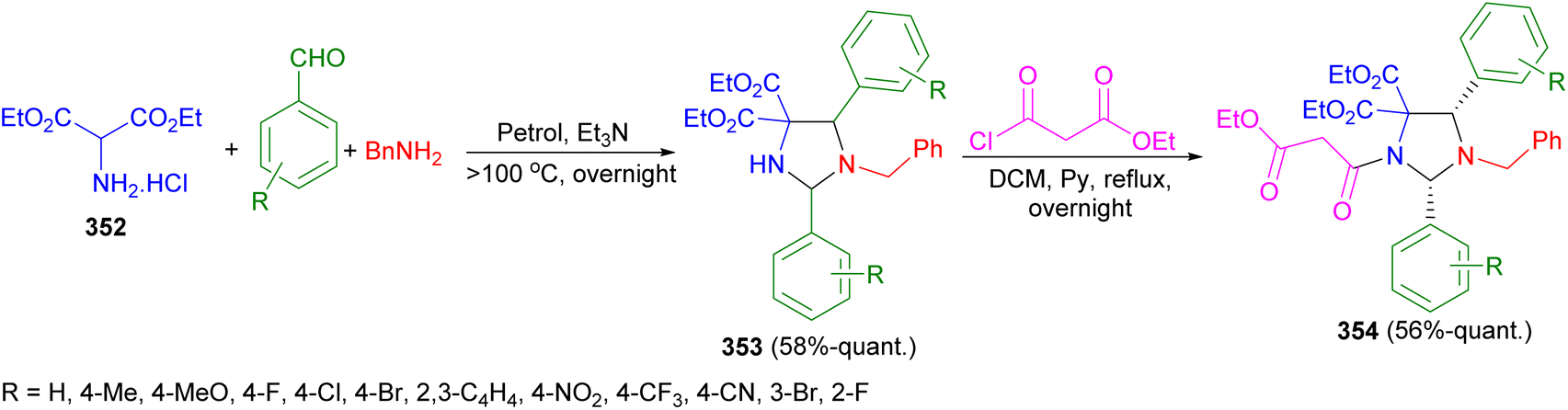 | ||
| Scheme 98 Preparation of imidazolidines 353 and 354 under basic conditions. | ||
The Vchislo group developed synthesis of 1,3-imidazolidine 355 in 93% yield from 2,5-bis-(butylsulfanyl)-2,3-dihydro-4H-pyran-2-carbaldehyde 356 and N,N′-diphenylethylenediamine 357 in EtOH at reflux for 8 hours. At room temperature, this reaction takes 12 days, while, upon heating, the duration to reaction completion is reduced to 8 hours (Scheme 99).116
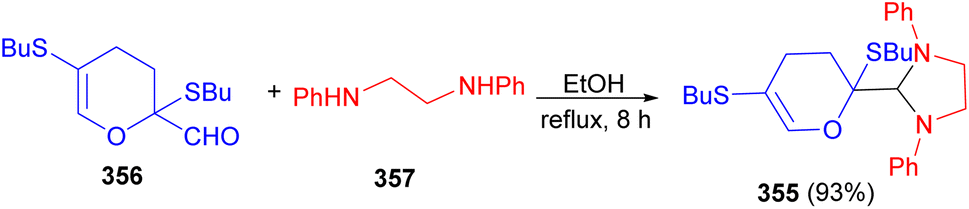 | ||
| Scheme 99 Synthesis of 1,3-imidazolidine 355. | ||
The Itoh group developed an unprecedented formal [3 + 2] photocycloaddition reaction of aromatic imines 358 with N,N,N′,N′-tetramethyldiaminomethane 359 to synthesize imidazolidines 360 in 30–48% yields using visible-light photo redox catalysis in CH3CN at room temperature for 24 hours (Scheme 100).117
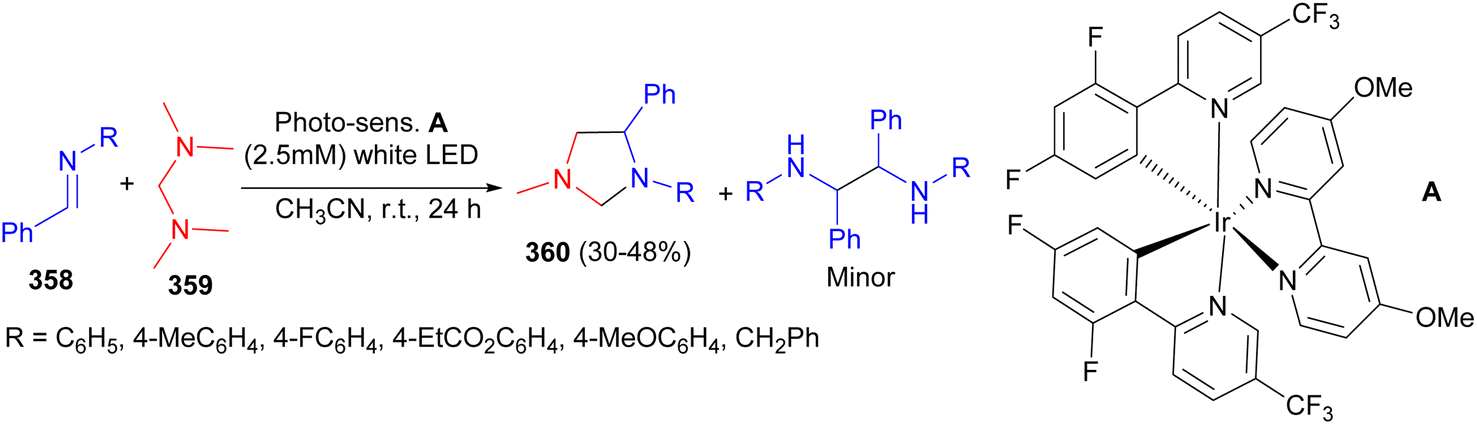 | ||
| Scheme 100 Visible-light photoredox catalyzed synthesis of imidazolidines 360. | ||
The Pariyar group utilized metal–organic framework (MOF), [Cu(BTC)(Mim)]n Cu-SKU-3, for the synthesis of biologically valued chiral imidazolidine motifs 361 in a one-pot fashion starting from aziridines 362 and secondary amines 363. The chiral imidazolidines are synthesized in good yield (up to 89%) and with high optical purity (ee > 98–99%). The proposed mechanism is depicted in Scheme 101. Initially, the ring-opening product 364 gets oxidized by Cu(II) metal center via single electron transfer (SET), forming a radical cation intermediate 365 stabilized by the framework's microenvironment. In the second step, the oxidation of the catalyst [Cu(I) to Cu(II)] using tBuOOH generates a tertiary butoxide radical (tBuȮ) that abstracts a hydrogen atom homolytically via sp3 C–H bond cleavage forming an iminium ion intermediate 366. Lastly, the consecutive intramolecular cyclization of intermediate 366 results in the formation of the desired product 361.118
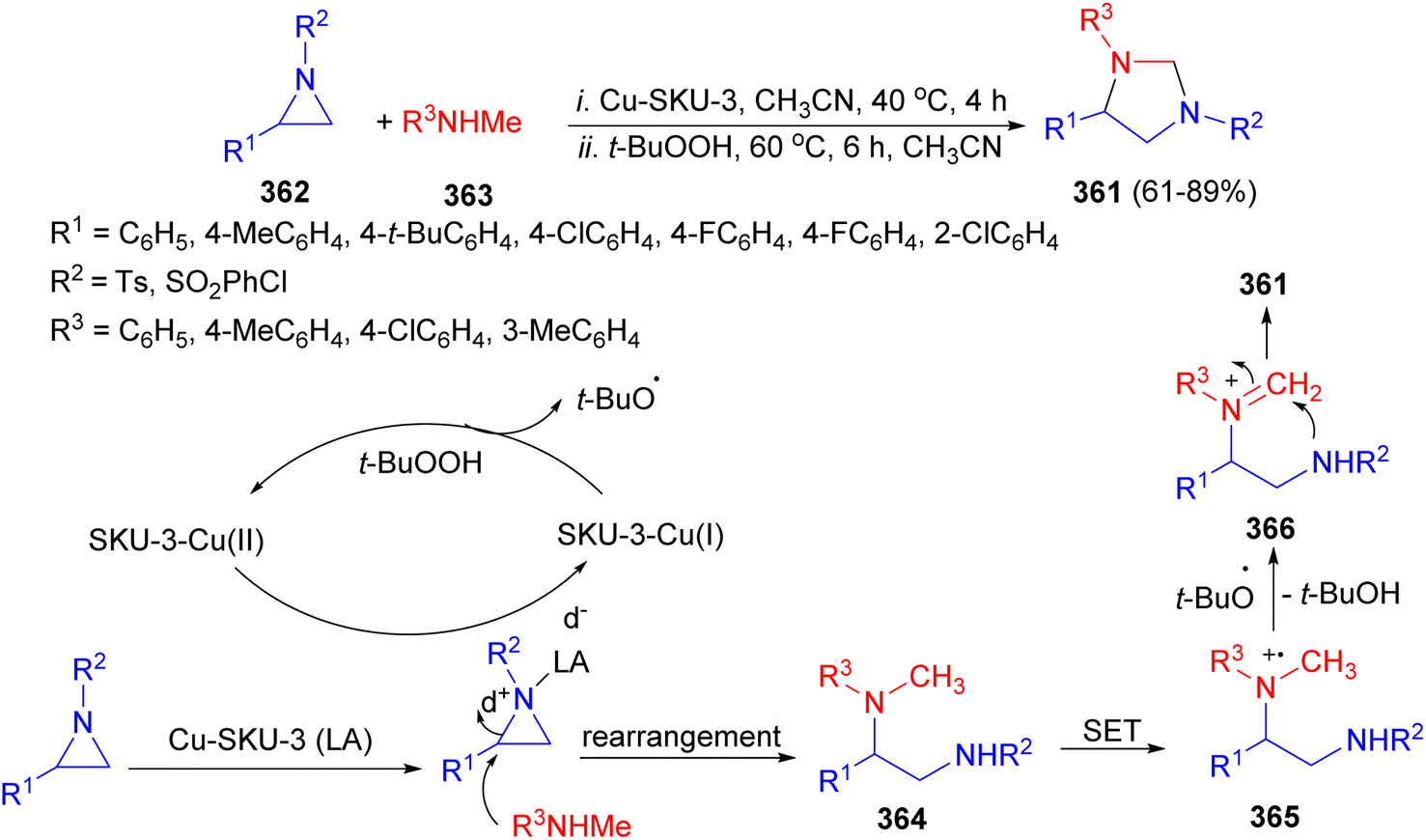 | ||
| Scheme 101 Cu-SKU-3-catalyzed synthesis of substituted imidazolidines 361. | ||
Cu-catalyzed synthesis of imidazolidines 367 in 30–76% yields through heterocyclic recombination between aziridines 368 and diazetidine 369 in toluene at 120 °C for 20 hours was reported by Murakami and co-workers. The proposed mechanism is illustrated in Scheme 102. The reaction involves two catalytic cycles: (1) the formation of imines from diazetidines 369. The reaction of aziridine with the resulting imine. In cycle 1, Cu catalyst L reacts with diazetidine 369 to provide intermediate 370. Reductive C–C bond cleavage provides the Cu intermediate 371, which releases imine 372. In cycle 2, copper-catalyst L activates aziridine 368 through coordination to give 373 (or 374). Imine 372 then attacks 373 to furnish intermediate 375 (or 376). Finally, ring-closing cyclization gives product 367.119
 | ||
| Scheme 102 Cu-catalyzed synthesis of imidazolidines 367. | ||
Liu and co-workers developed an enantioselective [3 + 2] cycloaddition of donor acceptor aziridines 377 with N-aryl protected imines 378 with a Ni(ClO4)2·6H2O/N,N′-dioxide catalyst system under nitrogen atmosphere in DCM at 35 °C for 36 hours, providing a broad range of chiral trans-substituted imidazolidine compounds 379 with good yields and excellent enantioselectivities (up to 99% yield, up to 98% ee) (Scheme 103).120
 | ||
| Scheme 103 Ni-catalyzed chiral trans-substituted imidazolidines 379. | ||
Murakami and co-workers described a copper-catalyzed reaction between aziridine 380 and imine 381, utilizing ligand L in toluene at 120 °C for 20 hours, resulting in imidazolidine derivatives 382 in 25–99% yields. These developed reactions exhibited broad functional group compatibility to access a diverse array of potential bioactive 5-membered azaheterocycles. A proposed mechanism is shown in Scheme 104. The reaction initiates with the coordination of aziridine 380 to copper catalyst A to give the corresponding intermediate 383 (or 384). Subsequently, imine 381 attacks to open the aziridine ring to give intermediate 385 (or 386). Finally, a cyclization reaction takes place from 385 to give product 382.121
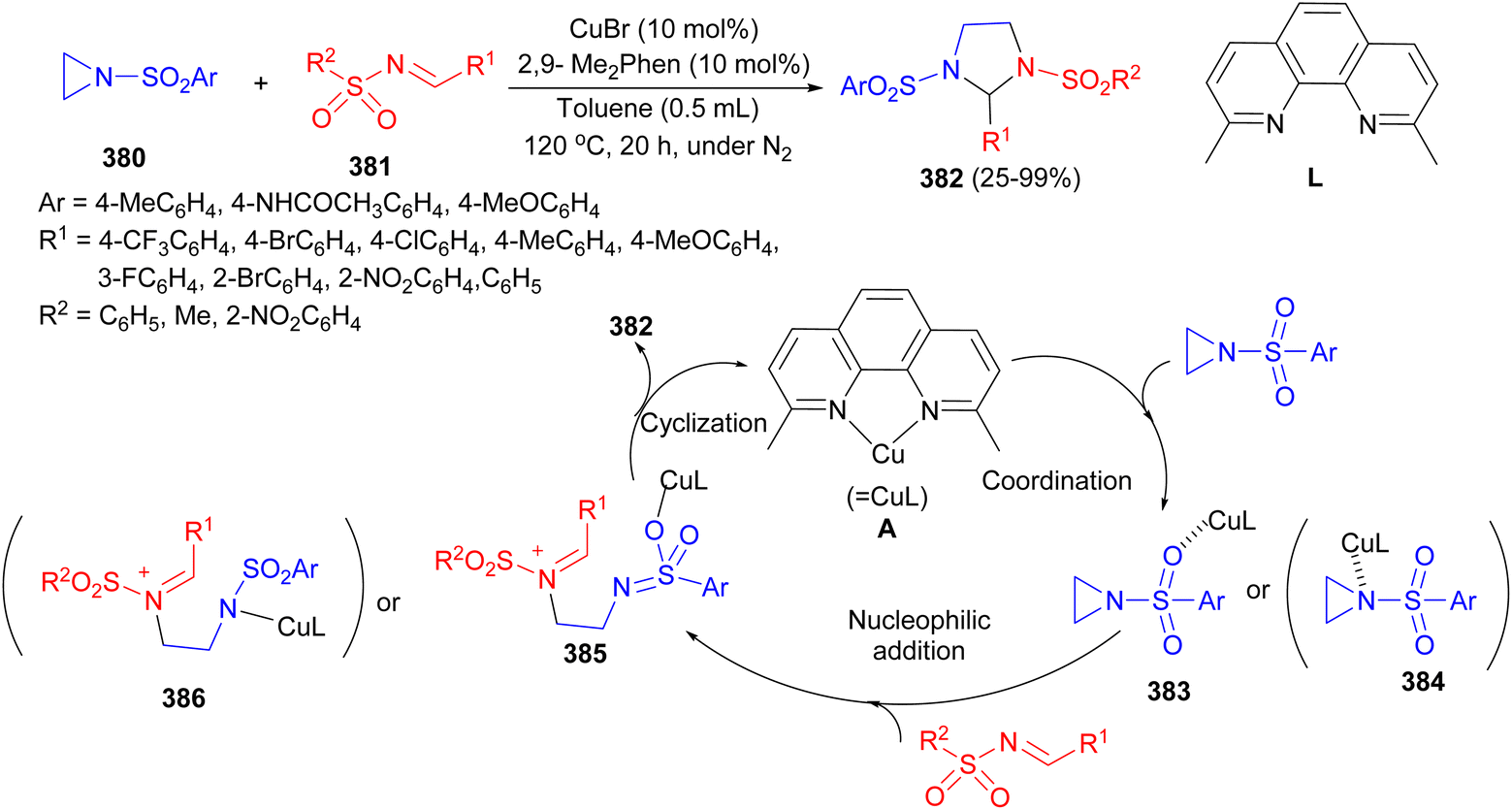 | ||
| Scheme 104 Copper-catalyzed synthesis of imidazolidine derivatives 382. | ||
The Shivachev group reported the synthesis of imidazolidine 387 in 47% yield by three-component Mannich-type condensation reaction of naphthalene-2-ol, paraformaldehyde, and R,R-cyclohexane-1,2-diamine in methanol under reflux for 2 hours. Additionally, they synthesized imidazolidine 388 in 31% yield by using S,S-cyclohexane-1,2-diamine in an ethanol/water mixture in the presence of K2CO3 at 50 °C for 24 hours (Scheme 105).122
 | ||
| Scheme 105 Synthesis of imidazolidines 387 and 388. | ||
2.2. Synthesis of spiro-imidazolidines
In 1987, Gruseck and Heuschmann reported the reaction of 2-cyclopropylidene imidazolidine 389 with 2,3-naphthacene 390 or pyridazine 391 as dienophiles afforded spiro-imidazolidines 392 and 393 in 94 and 96% yields, respectively. At 0 °C nitrogen evolution was complete within minutes with 391 and within seconds using 390. The mechanism is outlined in Scheme 106.123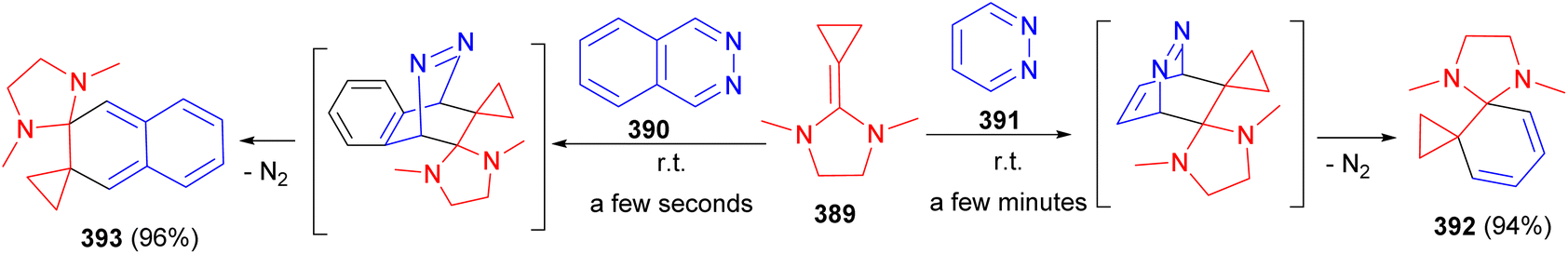 | ||
| Scheme 106 Synthesis of spiro-imidazolidines 392 and 393. | ||
In 2006, Ishikawa et al. successfully isolated a spiro imidazolidine-oxazolidine intermediate 394 in the reaction of diphenylguanidinium salt 395 with (Z)-R-bromocinnamaldehyde 396 in the presence of tetramethylguanidine (TMG) in THF at −40 to −10 °C for 6 hours. In this reaction, compound 394 was obtained in a yield of 73%, along with trans-aziridine 397, which was obtained in a yield of 9%. X-ray crystallographic analysis unambiguously revealed that the stereogenic centers of the spiro intermediate were in a trans configuration. The proposed mechanism is illustrated in Scheme 107.124
 | ||
| Scheme 107 Tetramethylguanidine (TMG) catalyzed synthesis of spiro imidazolidine-oxazolidine 394. | ||
In 2011, an array of spiro imidazolidine derivatives 398 was synthesized in 88–98% yields by the reaction of 6-carbethoxy-3,5-diarylcyclohex-2-enones 399 with ethylene diamine using catalytic amount of activated fly ash under microwave irradiation at 90 °C for 4–8 min at 2 bar pressure and was screened for their antibacterial and antifungal activities (Scheme 108). The results revealed that some of these compounds against P. aeruginosa, S. aureus, K. pneumonia, β-H. streptococcus, M. luteus, P. vulgaris, M. gypseum and C. albicans exhibited excellent antibacterial activity at a minimum inhibitory concentration (MIC) value of 6.25 μg mL−1.125
 | ||
| Scheme 108 Synthesis of spiro-imidazolidine derivatives 398 using catalytic amount of fly ash under microwave irradiation. | ||
In 2015, Zhao and co-workers reported preparation of imidazolidine-dispirooxindoles 400 in 61–84% yields with up to >99:1 diastereoselectivity through the [3 + 2] cycloaddition of isatins, 2-(aminomethyl)pyridine and isatin-based imines in the presence of Et3N in anhydrous CH2Cl2 at room temperature for 72 hours. The proposed mechanism is depicted in Scheme 109. Initially, under catalysis of Et3N, isatin condenses easily with 2-(aminomethyl)pyridine to afford imine 401. Subsequently, the deprotonation of imine 401 with Et3N give rise to enolate 402. Finally, the cyclization of the resulted enolate 402 with imine formed diastereoisomer 400 via the transition state 403.126
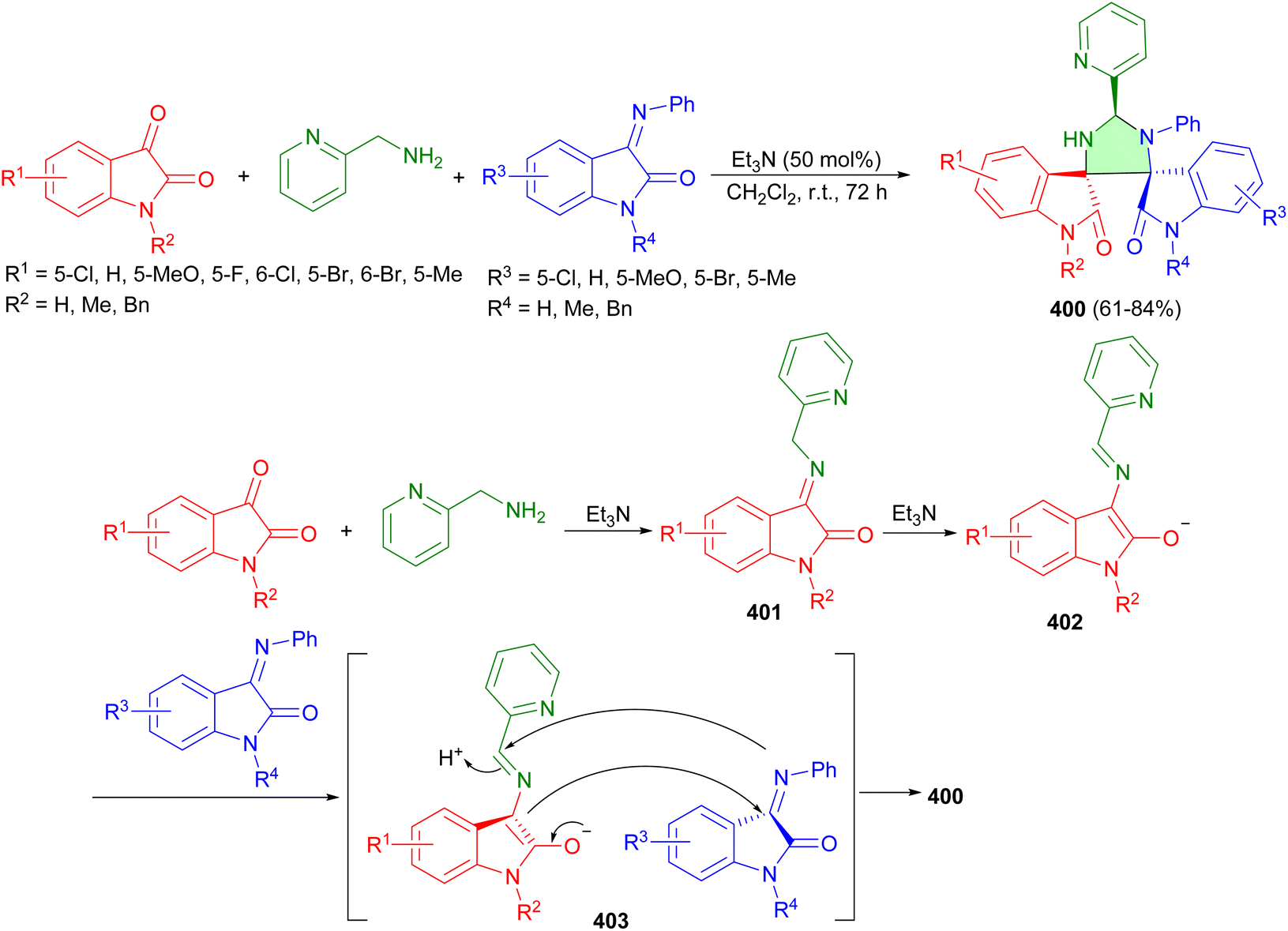 | ||
| Scheme 109 Preparation of imidazolidine-dispirooxindoles 400 in the presence of Et3N. | ||
In addition, an acid-promoted (3,5-dinitrobenzoic acid (3,5-DNBA)) self-1,3-dipolar cycloaddition of ketimines derived from isatins and benzylamines in THF at room temperature for 12 hours developed to assemble unprecedented dispirooxindole-imidazolidine derivatives 404. Generally, excellent diastereoselectivities (only single stereoisomer formed) and good yields (up to 94%) were obtained. A plausible reaction pathway illustrated in Scheme 110. Imine 405 was firstly generated from the condensation between isatin and amine in the presence of acid. A subsequent 1,2-prototropy of 405 led to the formation of azomethine ylide 406. When these two reaction partners approaching each other, transitions state 407 could be generated. Presumably, it would be favored. The subsequent cycloaddition resulted in the formation of dispirooxindole-imidazolidine 404.127
 | ||
| Scheme 110 3,5-Dinitrobenzoic acid promoted synthesis of dispirooxindole-imidazolidine derivatives 404. | ||
In 2016, a catalytic asymmetric chemoselective 1,3-dipolar cycloaddition of azomethine ylide with imines established via a three-component reaction of isatin derived imines, aldehydes and amino-ester in the presence of chiral phosphoric acid in toluene at 0 °C for 48 hours, which efficiently constructed biologically important spiro[imidazolidine-2,3′-oxindole] 408 frameworks in good yields, high diastereo- and enantioselectivities (43–72% yields, 97:5 ee, all >95:5 dr). In the proposed mechanism, as illustrated in Scheme 111, initially, in the presence of catalyst, a homo-1,3-DC occurred, which generated the key intermediate 409. Then, again promoted by catalyst via dual hydrogen bonding activation, this key intermediate 409 performed an enantioselective cascade reaction with isatin-derived imine, which ultimately gave the experimentally observed product 408 with concomitant regeneration of the azomethine ylide.128
 | ||
| Scheme 111 Preparation of spiro[imidazolidine-2,3′-oxindole] 408 in the presence of chiral phosphoric acid. | ||
Next, regioselective 1,3-dipolar cycloaddition reaction of appropriate diazoamide 410, anilines, aldehydes and ketimine derived from boc-protected isatin 411 in the presence of copper(I) thiophene-2-carboxylate as catalyst in DCE refluxing under nitrogen atmosphere at room temperature afforded dispiroimidazolidine 412 in 71–84% yields in a chemo- and diastereoselective manner (Scheme 112). Mechanistically, the electron-donating imines, generated from amines and aromatic aldehydes, chemoselectively react with electron deficient copper carbenoid, generated from cyclic diazoamide, affording the respective intermolecular azomethine ylides. The S-shaped conformation of azomethine ylide intermediate in providing the stereoselective products.129
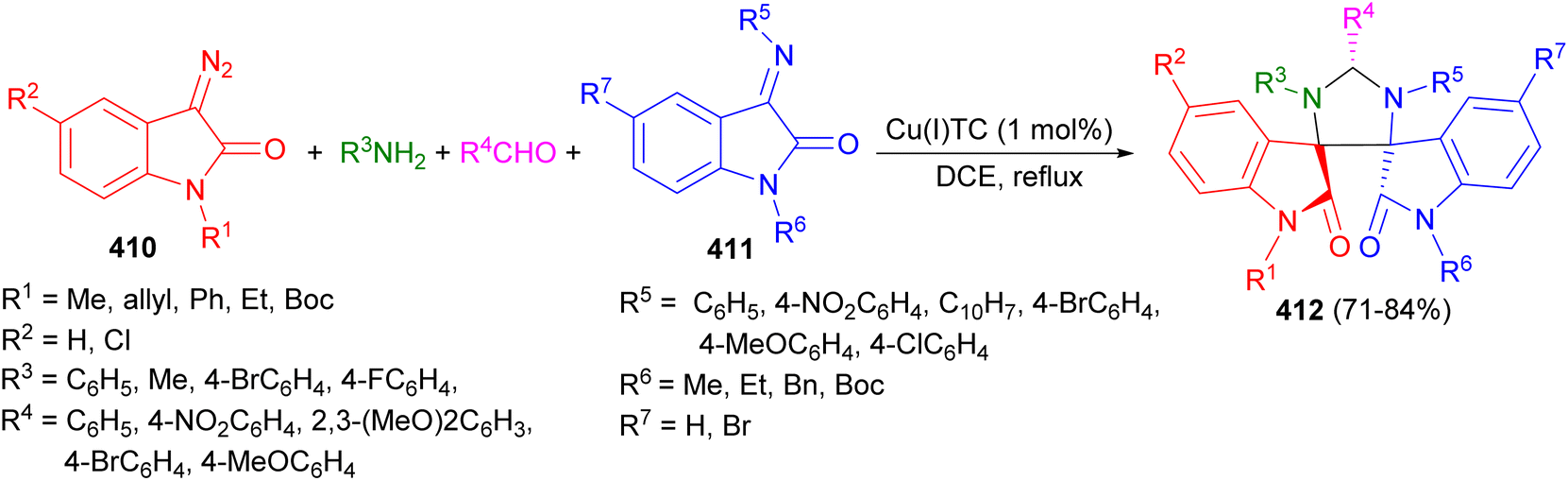 | ||
| Scheme 112 Copper(I) thiophene-2-carboxylate catalyzed synthesis of dispiroimidazolidine 412. | ||
In 2018, the Wazzan group reported synthesis of imidazolidine derivatives 413a–c by the reaction of 1,2-ethylenediamine with the appropriate cyclic ketones in dry benzene under reflux condition using dean-stark trap until no more water was collected (about 2–4 hours). The three imidazolidine derivatives tested were good corrosion inhibitor for X60 steel in 1 M HCl solution and they functioned as mixed-type inhibitors during the electrochemical acid corrosion of the steel in 1 M HCl. Moreover, experimentally determined inhibition efficiency increases in the order: 413c > 413b > 413a (Scheme 113).130
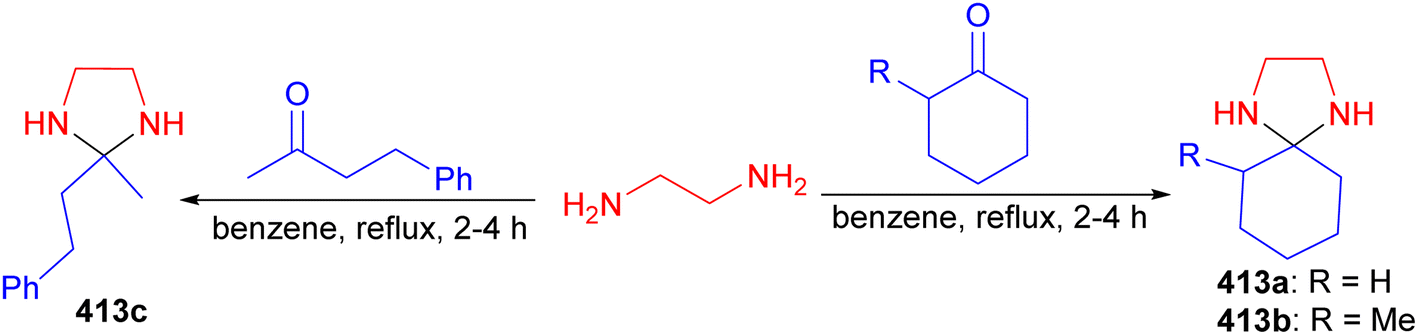 | ||
| Scheme 113 Synthesis of imidazolidine derivatives 413a–c. | ||
A facile and efficient synthesis of spiro[imidazolidine-4,3′-indolin]-2′-imines 414 in 36–89% yields via a copper(I)-catalyzed cascade reaction of 3-diazoindolin-2-imines 415 with 1,3,5-triazines 416 in DCE at room temperature for 12 hours. The cascade process involves the formation of a copper–carbene intermediate and a formal [2 + 2 + 1] cycloaddition. They proposed two plausible reaction mechanisms, as shown in Scheme 114. First, the copper-carbene intermediate 417 is generated from 415. In the next step, there are two possible pathways to obtain product 414. In pathway a, the nucleophilic addition of 1,3,5-triazine derived formaldimine 418 to copper-carbene 417 generates ylide 419, which releases Cu(I) to form ylide 420. Subsequent addition with another formaldimine gives intermediate 421. Finally, 421 undergoes an intramolecular nucleophilic addition to furnish the final product 414.131
 | ||
| Scheme 114 Copper(I)-catalyzed synthesis of spiro[imidazolidine-4,3′-indolin]-2′-imines 414. | ||
In 2020, Habarurema and co-workers reported synthesis of imidazolidine 422 in 69% yield from the reaction of the bridging pyridyl; 2,2′-dipyridylketone (423) with 1,2-diaminoethane in methanol at reflux temperature under nitrogen for 4 hours. Then, rhenium(I) complex 424 of this compound synthesized in 73% yield in toluene at reflux for 3 hours under nitrogen (Scheme 115).132
 | ||
| Scheme 115 Synthesis of imidazolidine 422 and spiroimidazolidine 424. | ||
Recently, the Guo group developed heterogeneous carbon nitride photocatalyst for the four-component synthesis of spiroimidazolidines 425 in 21–93% yields under visible light irradiation, starting from simple amines, cyclic ketones, amino acids, and aldehydes in DCM at room temperature for 6 hours. The heterogeneous nature of the catalytic system enables the recovery and reuse of the photocatalyst without loss of reactivity, and the multicomponent reaction can be carried out in a continuous flow fashion. A plausible mechanism is proposed in Scheme 116. Initially, the oxidation of α-amino acid by the photogenerated holes at the VB of 1.0 Ci-C3N4 leads to rapid decarboxylation, producing α-amino radical intermediate 426 with the release of CO2 and proton. In the meantime, primary amine and ketone undergo a condensation process to in situ generate iminium ion species 427 in the presence of protons. Then the α-amino radical 426 undergoes a free radical addition to the iminium ion species 427 to give radical adducts 428, which is subsequently reduced by visible-light-induced electrons via a single electron transfer process. The generated diamine species 429 reacts with paraformaldehyde to form the final spiro-imidazolidine product 425.133
 | ||
| Scheme 116 Carbon nitride photocatalyzed synthesis of spiro-imidazolidines 425. | ||
2.3. Synthesis of bis-imidazolidines
In 1999, the Boca group reported synthesis of bis-imidazolidine 430 in 53% yield by the condensation of triethylenetetramine with 2-pyridinecarboxaldehyde N-oxide in CH3OH under heating on the water bath for 15 min (Scheme 117).134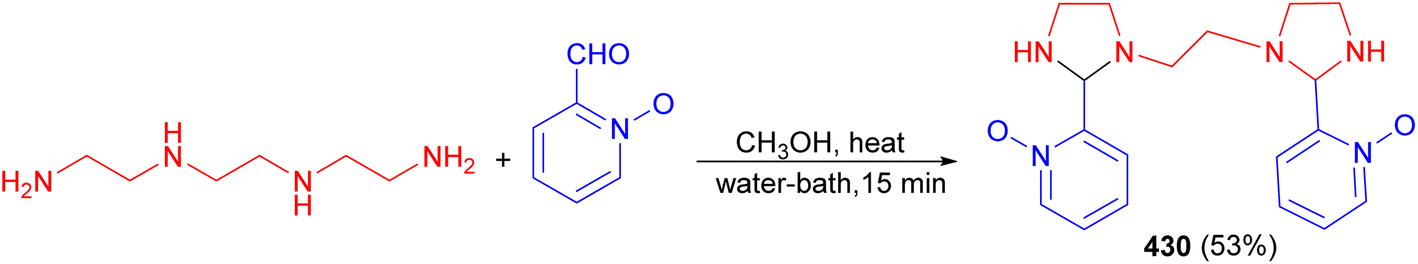 | ||
| Scheme 117 Synthesis of bis-imidazolidine 430. | ||
After that, bis(3-arylimidazolidinyl-1)methanes 431 synthesized in 75–89% yields by condensation reaction between N-arylethylenediamines and an excess aqueous formaldehyde (37%) in ethanol under reflux conditions (Scheme 118). All synthesized compounds showed antibacterial activity against Escherichia coli, Micrococcus luteus, Bacillus subtilis, Listeria monocytogenes, Pseudomonas aeruginosa and Staphylococcus aureus.135
 | ||
| Scheme 118 Preparation of bis(3-arylimidazolidinyl-1)methanes 431. | ||
Next, Ghandi and his group demonstrated synthesis of 2,4,6,8-tetraphenyl-2,4,6,8-tetraazabicyclo[3.3.0]octane 432 in 78% yield by the reaction of N,N′-bisphenylmethanediamine 433 (2.0 mmol) with glyoxal (1.0 mmol, 40% aq.) in the presence of formic acid as catalyst in acetonitrile at room temperature for 5 hours (Scheme 119).136,137
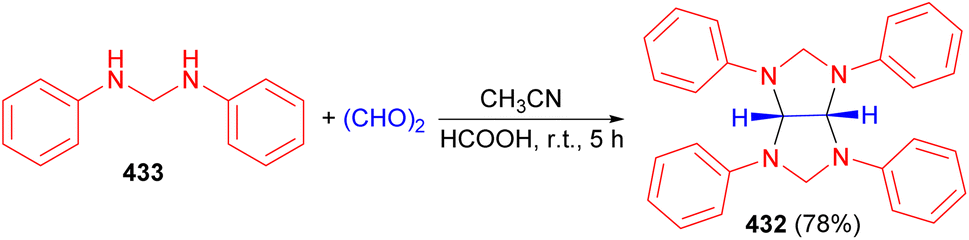 | ||
| Scheme 119 Synthesis of 2,4,6,8-tetraphenyl-2,4,6,8-tetraazabicyclo[3.3.0]octane 432. | ||
Recently, the condensation reactions between N-alkylethylenediamines 434 and aqueous formaldehyde in THF at 5 °C for 10 minutes and maintained at reflux for 6 hours afforded bis(3-alkyl-imidazolidin-1-yl)methanes 435 in 82–92% yields (Scheme 120).138
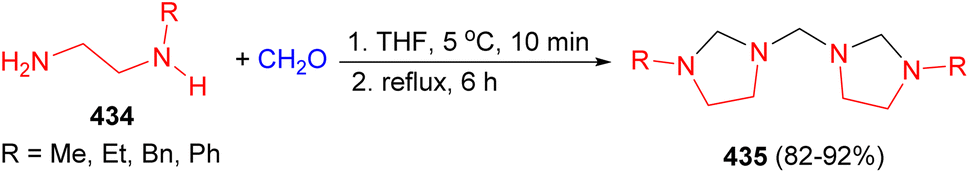 | ||
| Scheme 120 Synthesis of bis(3-alkyl-imidazolidin-1-yl)methanes 435. | ||
2.4. Synthesis of tris-imidazolidines
In 2013, synthesis of imidazolidines 436 in 37–94% yields reported by the reaction of a series of phenol derivatives with macrocyclic aminal 1,3,6,8-tetraazatricyclo[4.4.1.13,8]dodecane (TATD) (437) under solvent-free conditions at 150 °C for 20 minutes. The reaction proceeds through intramolecular cyclization via intermediate 438. Moreover, the formation of 439 depend on the stoichiometric ratio. When employing a 1:2 (TATD 1:imidazolidine 436a) ratio, the major product is a trimer of benzylimidazolidine 439 (Scheme 121).139
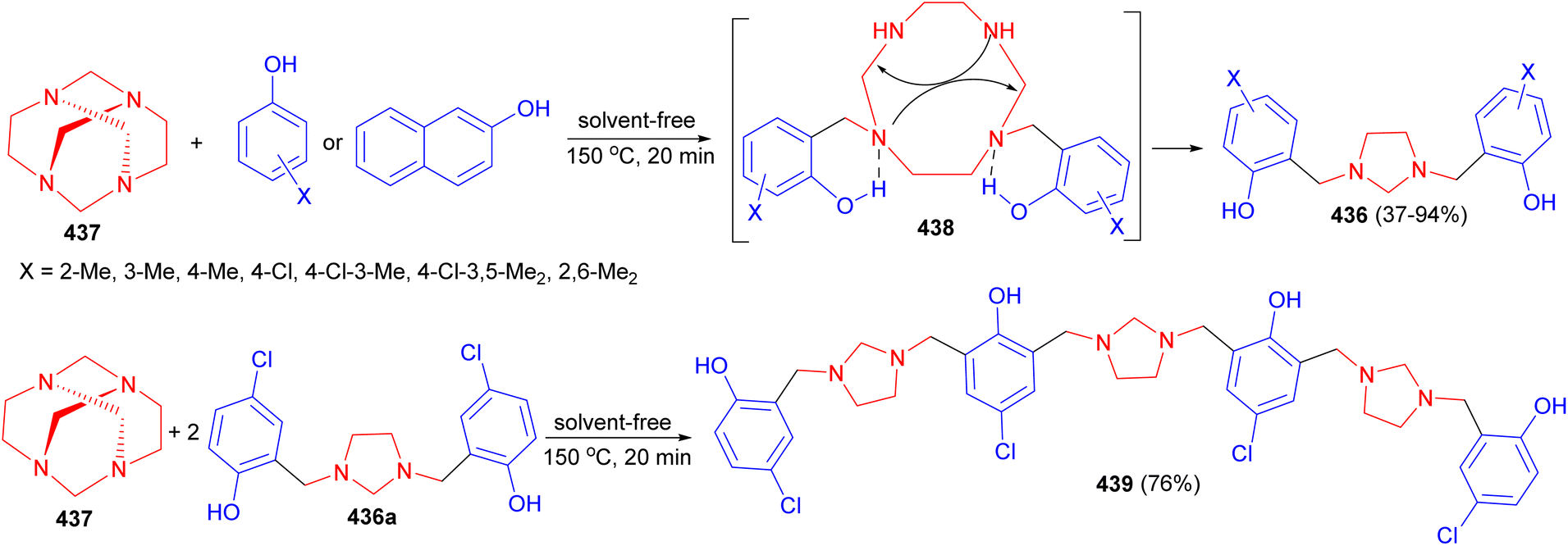 | ||
| Scheme 121 Preparation of imidazolidines 436 and trimer of benzylimidazolidine 439. | ||
2.5. Synthesis of macrocyclic imidazolidines
In 2009, Raghunathan and co-workers described one-pot three-component synthesis of macrocyclic imidazolidines 440a–f in 60–69% yields via a facile [3 + 2] cycloaddition reaction of azomethine ylide, derived from paraformaldehyde and sarcosine, with various macrocyclic imines 441a–f as dipolarophiles in toluene under reflux conditions for 22–26 hours as illustrated in Scheme 122.140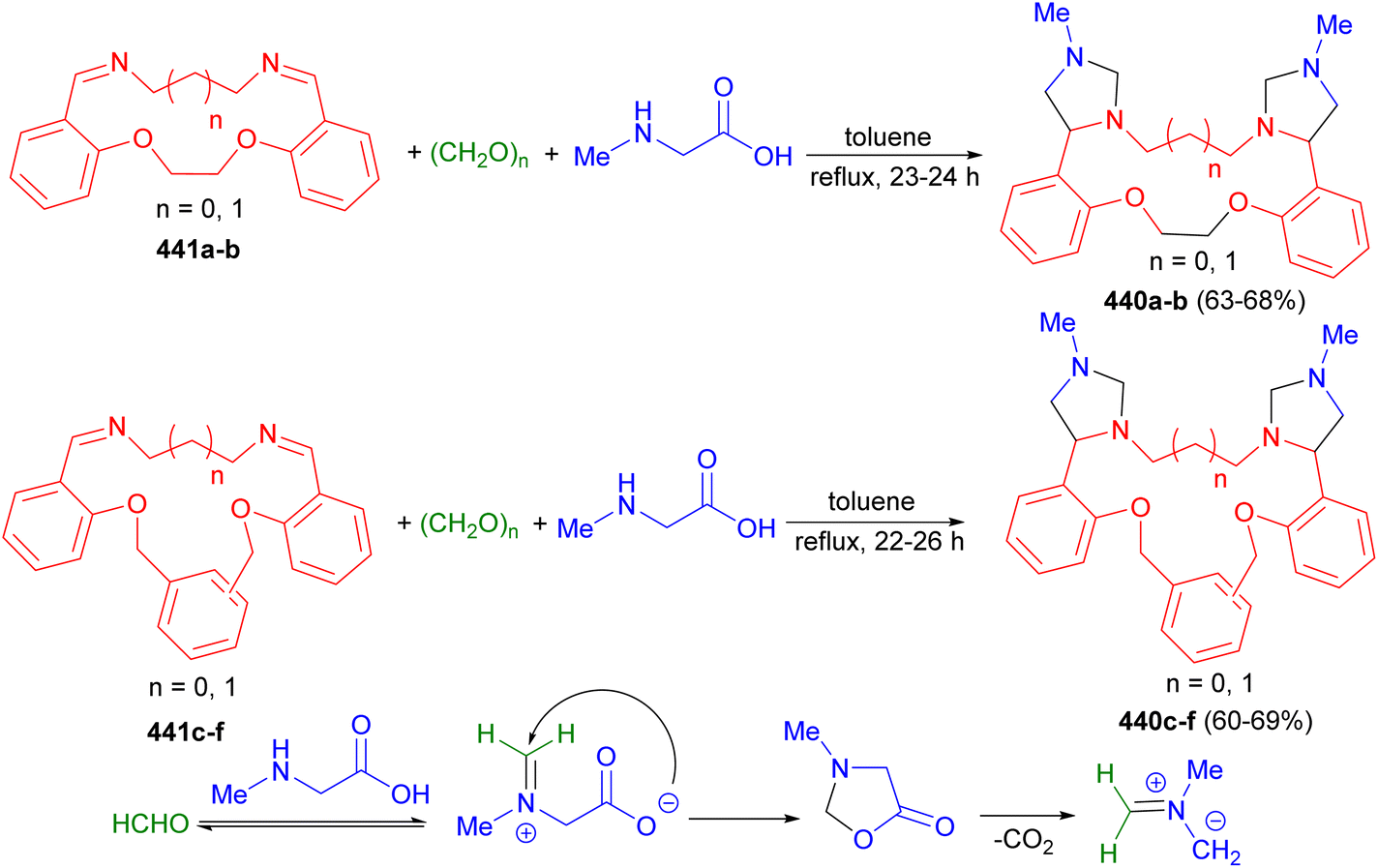 | ||
| Scheme 122 Synthesis of macrocyclic imidazolidines 440a–f. | ||
3 Conclusions
This review highlights various synthetic strategies to the imidazolidines, chiral imidazolidines with high diastereoselectivities and enantioselectivities, bis-imidazolidines, and spiro-imidazolidines by approaching different methodologies. The general synthetic strategy for preparing imidazolidine derivatives involves the condensation of aldehydes or ketones with 1,2-diamines under various conditions. Additionally, other synthetic methodologies for these scaffolds include intermolecular amination reactions, Mannich cyclization, reactions of aziridines with imines, amines, or diazetidines, condensation reactions of methane diamines with glyoxal or imines, and reactions of 1,3,5-triazines with diazoesters, tosylhydrazones, vinyl ethylene carbonates, aziridines, or 3-aminooxetanes via [3 + 2] formal cycloadditions and 1,3-dipolar cycloadditions. Moreover, imidazolidine derivatives have reported to possess a wide range of biological and pharmaceutical applications such as anti-inflammatory, anti-bacterial, anti-trypanosoma cruzi agents, anti-fungal, anti-proliferative and analgesic activity.Data availability
No new data were generated for this article.Author contributions
All authors discussed the concept of this article, and contributed to the scientific writing of the original manuscript. All authors have read and approved the final manuscript.Conflicts of interest
We have no conflicts of interest to disclose.Acknowledgements
The authors thank the Research Council of Imam Khomeini International University and Qazvin Islamic Azad University for their support.References
- R. H. Jiao, S. Xu, J. Y. Liu, H. M. Ge, H. Ding, C. Xu, H. L. Zhu and R. X. Tan, Org. Lett., 2006, 8, 5709–5712 CrossRef CAS PubMed.
- K. Macharoen, Q. Li, A. M. Veronica, J. M. Corbin, C. B. Lebrilla, S. Nandi and K. A. Mcdonald, Int. J. Mol. Sci., 2020, 21, 6896 CrossRef CAS PubMed.
- B. Malgesini, B. Forte, D. Borghi, F. Quartieri, C. Gennari and G. Papeo, Chem.–Eur. J., 2009, 15, 7922–7929 CrossRef CAS PubMed.
- Y. Nakao, J. Kuo, W. Y. Yoshida, M. Kelly and P. Scheuer, J. Org. Lett., 2003, 5, 1387–1390 CrossRef CAS PubMed.
- Z. Y. Mao, H. Geng, T. T. Zhang, Y. P. Ruan, J. L. Ye and P. Q. Huang, Org. Chem. Front., 2016, 3, 24–37 RSC.
- G. Yu, G. Zhou, M. Zhu, W. Wang, T. Zhu, Q. Gu and D. Li, Org. Lett., 2016, 18, 244–247 CrossRef CAS PubMed.
- J. Clardy, J. P. Springer, G. Béchi, K. Matsuo and R. Wightman, J. Am. Chem. Soc., 1975, 97, 663–665 CrossRef CAS PubMed.
- A. Erkkila, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416–5470 CrossRef PubMed.
- T. Arai, A. Mishiro, N. Yokoyama, K. Suzuki and H. Sato, J. Am. Chem. Soc., 2010, 132, 5338–5339 CrossRef CAS PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- A. E. Allen and D. W. C. MacMillan, J. Am. Chem. Soc., 2011, 133, 4260–4263 CrossRef CAS PubMed.
- I. R. Sadarangani, S. Bhatia, D. Amarante, I. Lengyel and R. A. Stephani, Bioorg. Med. Chem. Lett., 2012, 22, 2507–2509 CrossRef CAS PubMed.
- X. X. Jiang, Y. Q. Wang, G. Zhang, D. Fu, F. T. Zhang, M. Kai and R. Wang, Adv. Synth. Catal., 2011, 353, 1787–1796 CrossRef CAS.
- M. van der Stelt, J. Cals, S. Broeders-Josten, J. Cottney, A. A. van der Doelen, M. Hermkens, V. de Kimpe, A. King, J. Klomp, J. Oosterom, I. P. Rooij, J. de Roos, M. van Tilborg, S. Boyce and B. James, J. Med. Chem., 2011, 54, 7350–7362 CrossRef CAS PubMed.
- M. C. Caterina, I. A. Perillo, L. Boiani, H. Pezaroglo, H. Cerecetto, M. González and A. Salerno, Bioorg. Med. Chem., 2008, 16, 2226–2234 CrossRef CAS PubMed.
- V. Sharma and M. S. Khan, Eur. J. Med. Chem., 2001, 36, 651–658 CrossRef CAS PubMed.
- R. J. Ferw and J. L. Riebsomer, Chem. Rev., 1954, 54, 593–613 CrossRef.
- R. A. Donia, J. A. Shotton, L. O. Bentz and G. E. P. Smith, J. Org. Chem., 1949, 14, 952–961 CrossRef CAS.
- J. H. Billman, J.-Y. Chen Ho and L. R. Caswell, J. Org. Chem., 1957, 22, 538–539 CrossRef CAS.
- L. V. Jaenlcke and E. Erode, Justus Liebigs Ann. Chem, 1959, 624, 120–136 CrossRef.
- M. M. Joullie, G. M. J. Slusarczuk, A. Dey, P. B. Venuto and R. H. Yocum, J. Org. Chem., 1967, 32, 4103–4105 CrossRef CAS.
- J. W. Lown, J. P. Moser and R. Westwo, Can. J. Chem., 1969, 47, 4335–4345 CrossRef CAS.
- A. J. Birch and K. P. Dastu, Aust. J. Chem., 1973, 26, 1363–1364 CrossRef CAS.
- J. Hine and K. W. Narducy, J. Am. Chem. Soc., 1973, 95, 3362–3368 CrossRef CAS.
- H. Suzuki, M. Ohashi, K. Itoh, I. Matsuda and Y. Ishii, Bull. Chem. Soc. Jpn., 1975, 48, 1922–1924 CrossRef CAS.
- G. P. Tuszynski and R. G. Kallen, J. Am. Chem. Soc., 1975, 97, 2860–2875 CrossRef CAS PubMed.
- J. F. W. Keana, R. S. Norton, M. Morello, D. V. Engen and J. Clardy, J. Am. Chem. Soc., 1978, 100, 934–937 CrossRef CAS.
- K. Amornraksa and R. Grigg, Tetrahedron Lett., 1980, 21, 2197–2200 CrossRef CAS.
- A. J. Carpenter and D. J. Chadwick, Tetrahedron, 1985, 41, 3803–3812 CrossRef CAS.
- A. Rivera, G. I. Gallo and M. E. Gaybn, Synth. Commun., 1993, 23, 2921–2929 CrossRef CAS.
- M. Perisamy, M. R. Reddy and J. V. Bhaskar Kanth, Tetrahedron Lett., 1996, 37, 4767–4770 CrossRef.
- I. Coldham, P. M. A. Houdayer, R. A. Judkins and D. R. Witty, Synlett, 1996, 1109–1111 CAS.
- J. Li, S. Wang, J. Hu and W. Chen, Tetrahedron Lett., 1999, 40, 1961–1962 CrossRef CAS.
- P. K. S. Chowdhury, U. Mokhopadhyay and D. Ray, Indian J. Chem., 1999, 38, 1159–1163 Search PubMed.
- H. A. Craig, J. R. Goerlich, W. J. Marshall and M. Unverzagt, Tetrahedron, 1999, 55, 14523–14534 CrossRef.
- V. Y. Sosnovskikh and P. A. Kutsenko, Russ. Chem. Bull., 1999, 48, 540–551 CrossRef CAS.
- K. Tanaka and R. Shiraishi, Green Chem., 2000, 2, 272–273 RSC.
- I. K. Kavrakova and M. J. Lyapova, Collect. Czech. Chem. Commun., 2000, 65, 1580–1586 CrossRef CAS.
- A. Rivera, J. F. León, J. Rivera, E. C. Parra, J. Purmova, E. Burgueno-Tapia and P. Joseph-Nathan, Synth. Commun., 2000, 30, 2029–2040 CrossRef CAS.
- G. V. Pokhvisneva and O. A. Lukyanov, Russ. Chem. Bull., 2000, 49, 894–898 CrossRef CAS.
- V. Sharma and M. S. Y. Khan, Eur. J. Med. Chem., 2001, 36, 651–658 CrossRef CAS PubMed.
- I. Coldham, R. C. B. Copley, T. F. N. Haxell and S. Howard, Org. Lett., 2001, 3, 3799–3801 CrossRef CAS PubMed.
- Z. Liz and Y. Zhang, Org. Prep. Proced. Int., 2001, 33, 185–187 CrossRef.
- V. Dryanska, I. Pashkuleva, S. Simova and S. Angelova, J. Chem. Res., 2001, 457–459 CrossRef CAS.
- A. R. Katritzky, K. Suzuki and H.-Y. He, J. Org. Chem., 2002, 67, 3109–3114 CrossRef CAS PubMed.
- M. S. Y. Khan and G. Chawla, Indian J. Chem., 2002, 41, 653–663 Search PubMed.
- A. R. Katritzky, K. Suzuki and H.-Y. He, J. Org. Chem., 2002, 67, 3109–3114 CrossRef CAS PubMed.
- W. H. Pearson, M. A. Walters, M. K. Rosen and W. G. Harter, Arkivoc, 2002, 91–111 Search PubMed.
- V. I. Kelarev, M. A. Silin and O. A. Borisova, Chem. Heterocycl. Compd., 2003, 39, 729–735 CrossRef CAS.
- M. S. Y. Khan and M. Gupta, Indian J. Chem., 2003, 42, 2086–2090 Search PubMed.
- J. Zhao, V. Pattaropong, Y. Jiang and L. Hu, Tetrahedron Lett., 2003, 44, 229–232 CrossRef CAS.
- A. Viso, R. Fernandez de la Pradilla, A. Garcia, C. Guerrero-Strachan, M. Alonso, M. Tortosa, A. Flores, M. Martinez-Ripoll, I. Fonseca, I. Andre and A. Rodriguez, Chem.–Eur. J., 2003, 9, 2867–2876 CrossRef CAS PubMed.
- M. Bera, P. K. Nanda, U. Mukhopadhyay and D. Ray, J. Chem. Sci., 2004, 116, 151–158 CrossRef CAS.
- G. W. Nyce, S. Csihony, R. M. Waymouth and J. L. Hedrick, Chem.–Eur. J., 2004, 10, 4073–4079 CrossRef CAS PubMed.
- E. d. Erkizia, E. Aldaba, Y. Vara, A. Arrieta, H. Gornitzka and F. P. Cossio, Arkivoc, 2005, 189–199 Search PubMed.
- C. T. Zeyrek, A. Elmali and Y. Elerman, Z. Naturforsch., 2005, 60, 520–526 Search PubMed.
- M. Ghandi, F. Salimi and A. Olyaei, J. Heterocycl. Chem., 2006, 43, 791–794 CrossRef CAS.
- G. V. Pokhvisneva and O. A. Lukyanov, Russ. Chem. Bull., 2006, 55, 903–906 CrossRef CAS.
- M. Ghandi and A. Olyaei, J. Heterocycl. Chem., 2007, 44, 323–327 CrossRef CAS.
- M. Ghandi, A. Olyaei and F. Salimi, Synth. Commun., 2007, 37, 247–256 CrossRef CAS.
- M. C. Caterina, I. A. Perillo, L. Boiani, H. Pezaroglo, H. Cerecetto, M. Gonzalez and A. Salerno, Bioorg. Med. Chem., 2008, 16, 2226–2234 CrossRef CAS PubMed.
- W.-J. Liu, X.-H. Chen and L.-Z. Gong, Org. Lett., 2008, 10, 5357–5360 CrossRef CAS PubMed.
- T. Arai and K. Suzuki, Synlett, 2009, 3167–3170 CrossRef CAS.
- M. C. Caterina, M. V. Corona, I. Perillo and A. Salerno, Heterocycles, 2009, 78, 771–781 CrossRef CAS.
- G. S. G. de Carvalho, P. A. Machado, D. T. S. de Paula, E. S. Coimbra and A. D. da Silva, Sci. World J., 2010, 10, 1723–1730 CrossRef CAS PubMed.
- H. Xie, J. Zhu, Z. Chen, S. Li and Y. Wu, J. Org. Chem., 2010, 75, 7468–7471 CrossRef CAS PubMed.
- V. G. Nenajdenko, V. M. Muzalevskiy, A. V. Shastin, E. S. Balenkova, E. V. Kondrashov, I. A. Ushakov and A. Yu. Rulev, J. Org. Chem., 2010, 75, 5679–5688 CrossRef CAS PubMed.
- M. Soueidan, F. Helion, J.-L. Namy and J. Szymoniak, Tetrahedron Lett., 2011, 52, 1348–1350 CrossRef CAS.
- Z. Jiang, J. Wang, P. Lu and Y. Wang, Tetrahedron, 2011, 67, 9609–9617 CrossRef CAS.
- J. Xuan, Y. Cheng, J. An, L.-Q. Lu, X.-X. Zhang and W.-J. Xiao, Chem. Commun., 2011, 47, 8337–8339 RSC.
- L. D. Elliott, J. W. Wrigglesworth, B. Cox, G. C. Lloyd-Jones and K. I. Booker-Milburn, Org. Lett., 2011, 13, 728–731 CrossRef CAS PubMed.
- X. Wu and J. Zhang, Synthesis, 2012, 44, 2147–2154 CrossRef CAS.
- M. S. Y. Khan, A. Husain, S. Sharma and M. Rashid, Indian J. Pharm. Sci., 2012, 74, 80–83 CrossRef CAS PubMed.
- S. Kaladevi, N. Paul, S. Muthusubramanian and S. Sivakolunthu, Tetrahedron Lett., 2013, 54, 3702–3705 CrossRef CAS.
- A. Olyaei, M. Karbalaei Karimi and R. Razeghi, Tetrahedron Lett., 2013, 54, 5730–5733 CrossRef CAS.
- A. Husain, R. Bhutani, D. Kumar and D.-S. Shin, J. Korean Chem. Soc., 2013, 57, 227–233 CrossRef CAS.
- Q.-H. Li, L. Wei, X. Chen and C.-J. Wang, Chem. Commun., 2013, 49, 6277–6279 RSC.
- L. Chen, C. S. Chao, Y. Pan, S. Dong, Y. C. Teo, J. Wang and C.-H. Tan, Org. Biomol. Chem., 2013, 11, 5922–5925 RSC.
- K. Ohmatsu, S. Kawai, N. Imagawa and T. Ooi, ACS Catal., 2014, 4, 4304–4306 CrossRef CAS.
- Y.-J. Ou, Z.-P. Zheng, X.-J. Hong, L.-T. Wan, L.-M. Wei, X.-M. Lin and Y.-P. Cai, Cryst. Growth Des., 2014, 14, 5339–5343 CrossRef CAS.
- R.-Y. Zhu, C.-S. Wang, F. Jiang, F. Shi and S.-J. Tu, Tetrahedron: Asymmetry, 2014, 25, 617–624 CrossRef CAS.
- S. P. Swain, Y.-C. Shih, S.-C. Tsay, J. Jacob, C.-C. Lin, K. C. Hwang, J.-C. Horng and J. R. Hwu, Angew. Chem., 2015, 127, 10064–10068 CrossRef.
- A. Husain, A. Ahmad, S. A. Khan, M. Asif, R. Bhutani and F. A. Al-Abbasi, Saudi Pharm. J., 2016, 24, 104–114 CrossRef PubMed.
- L. K. Kibardina, A. V. Trifonov, R. H. Bagautdinova, A. B. Dobrynin, E. M. Pudovik, A. R. Burilov and M. A. Pudovik, Russ. J. Gen. Chem., 2016, 86, 607–612 CrossRef CAS.
- S. Muthusamy and S. G. Kumar, Org. Biomol. Chem., 2016, 14, 2228–2240 RSC.
- V. Satheesh, M. Sengoden and T. Punniyamurthy, J. Org. Chem., 2016, 81, 9792–9801 CrossRef CAS PubMed.
- C. Zhu, G. Xu and J. Sun, Angew. Chem., 2016, 55, 11867–11871 CrossRef CAS PubMed.
- J. Hu, B. Kong, Y. Liu, B. Xu, Y. Zhao and P. Gong, ChemCatChem, 2017, 9, 403–406 CrossRef CAS.
- B. Yu, X.-F. Bai, J.-Y. Lv, Y. Yuan, J. Cao, Z.-J. Zheng, Z. Xu, Y.-M. Cui, K.-F. Yang and L.-W. Xu, Adv. Synth. Catal., 2017, 359, 3577–3584 CrossRef CAS.
- H. Jia, H. Liu, Z. Guo, J. Huang and H. Guo, Org. Lett., 2017, 19, 5236–5239 CrossRef CAS PubMed.
- M. Sengoden, A. Bhowmick and T. Punniyamurthy, Org. Lett., 2017, 19, 158–161 CrossRef CAS PubMed.
- P. Liu, G. Xu and J. Sun, Org. Lett., 2017, 19, 1858–1861 CrossRef CAS PubMed.
- J. K. Laha, K. P. Jethava, K. S. Satyanarayana Tummalapalli and S. Sharma, Eur. J. Org Chem., 2017, 4617–4624 CrossRef CAS.
- P. Liu, C. Zhu, G. Xu and J. Sun, Org. Biomol. Chem., 2017, 15, 7743–7746 RSC.
- S. Mukhopadhyay and S. C. Pan, Chem. Commun., 2018, 54, 964–967 RSC.
- H. Li, S. Huang, Y. Wang and C. Huo, Org. Lett., 2018, 20, 92–95 CrossRef CAS PubMed.
- H. Jia, Z. Guo, H. Liu, B. Mao, X. Shi and H. Guo, Chem. Commun., 2018, 54, 7050–7053 RSC.
- T.-Y. Lin, H.-H. Wu, J.-J. Feng and J. Zhang, Org. Lett., 2018, 20, 3587–3590 CrossRef CAS PubMed.
- Y. Yang and W. Yang, Chem. Commun., 2018, 54, 12182–12185 RSC.
- D. Jana, T. Guchhait, V. Subramaniyan, A. Kumar and G. Mani, Tetrahedron Lett., 2019, 60, 151247 CrossRef CAS.
- L. Tu, Z. Li, T. Feng, S. Yu, R. Huang, J. Li, W. Wang, Y. Zheng and J. Liu, J. Org. Chem., 2019, 84, 11161–11169 CrossRef CAS PubMed.
- S. Tarannum, S. Sk, S. Das, I. A. Wani and M. K. Ghorai, J. Org. Chem., 2020, 85, 367–379 CrossRef CAS PubMed.
- F. Zhao, K.-H. Wang, L. Wen, Z. Zhao, Y. Hu, W. Xu, D. Huang, Y. Su, J. Wang and Y. Hu, Asian J. Org. Chem., 2020, 9, 1036–1039 CrossRef CAS.
- X. Cheng, B.-G. Cai, H. Mao, J. Lu, L. Li, K. Wang and J. Xuan, Org. Lett., 2021, 23, 4109–4114 CrossRef CAS PubMed.
- T. Kang, S. Gao, L.-X. Zhao, Y. Zhai, F. Ye and Y. Fu, J. Agric. Food Chem., 2021, 69, 45–54 CrossRef CAS PubMed.
- R. H. Bagautdinova, L. K. Kibardina, A. R. Burilov and M. A. Pudovik, Russ. J. Gen. Chem., 2021, 91, 1265–1270 CrossRef CAS.
- A. S. Golubenkova, N. E. Golantsov and L. G. Voskressensky, Molbank, 2021, 2021, M1176 CrossRef.
- J. Zhang, Y.-F. Li, F.-C. Jia, Y. Gao and X.-Q. Hu, Org. Chem. Front., 2021, 8, 6616–6621 RSC.
- X. Zhong, Z. Zhong, Z. Wu, Z. Ye, Y. Feng, S. Dong, X. Liu, Q. Peng and X. Feng, Chem. Sci., 2021, 12, 4353–4360 RSC.
- Z. Shi, T. Fan, X. Zhang, F. Zhan, Z. Wang, L. Zhao, J.-S. Lin and Y. Jiang, Adv. Synth. Catal., 2021, 363, 2619–2624 CrossRef CAS.
- Z. Chen, Y. Zhou, T. Hu, H.-Y. Xiong and G. Zhang, J. Org. Chem., 2021, 86, 7714–7724 CrossRef CAS PubMed.
- A. Liu, D. Ma, Y. Qian, J. Li, S. Zhai, Y. Wang and C. Chen, Org. Biomol. Chem., 2021, 19, 2192–2197 RSC.
- K.-K. Wang, Y.-L. Li, M.-Y. Wang, J. Jing, Z.-Y. Wang and R. Chen, RSC Adv., 2022, 12, 28295–28298 RSC.
- B. Gurung, S. Pradhan, D. Sharma, D. Bhujel, S. Basel, S. Chettri, S. Rasaily, A. Pariyar and S. Tamang, Catal. Sci. Technol., 2022, 12, 5891–5898 RSC.
- L. Saney, T. Panduwawala, X. Li, K. E. Christensen, M. Genov, A. Pretsch, D. Pretsch and M. G. Moloney, Org. Biomol. Chem., 2023, 21, 4801–4809 RSC.
- E. A. Verochkina, V. G. Fedoseeva, L. I. Larina and N. V. Vchislo, Arkivoc, 2023, 202311977 Search PubMed.
- K. Itoh, N. Ishii, A. Takashino, A. Hara, S. Kon, T. Mizuguchi, F. Karaki, S. Hirayama, Y. Shibagaki, K. Nagai, N. Sato, K. Tokunaga, M. Suzuki, M. Hashimoto and H. Fujii, J. Photochem. Photobiol., A, 2023, 434, 114239 CrossRef CAS.
- D. Sharma, S. Rasaily, S. Chettri, D. Sureka, S. Tamang and A. Pariyar, Inorg. Chem., 2023, 62, 4540–4549 CrossRef CAS PubMed.
- D. Higuchi, S. Matsubara, H. Kadowaki, D. Tanaka and K. Murakami, Chem.–Eur. J., 2023, e202301071 CrossRef CAS PubMed.
- J. Qiao, S. Wang, X. Liu and X. Feng, Enantioselective [3+2] Cycloaddition of Donor-Acceptor Aziridines and Imines to Construct 2,5-trans-Imidazolidines, Chem.–Eur. J., 2023, 29, e202203757 CrossRef CAS PubMed.
- K. Hashimoto, D. Higuchi, S. Matsubara and K. Murakami, Front. Chem., 2023, 11, 1272034 CrossRef CAS PubMed.
- M. Tavlinova-Kirilova, K. Dikova, M. K. Marinova, M. Kamenova-Nacheva, R. Rusew, H. Sbirkova-Dimitrova, B. Shivachev, K. Kostova and V. Dimitrov, Crystals, 2023, 13, 1495 CrossRef CAS.
- U. Gruseck and M. Heuschmann, Tetrahedron Lett., 1987, 28, 6027–6030 CrossRef CAS.
- W. Disadee, T. Ishikawa, M. Kawahata and K. Yamaguchi, J. Org. Chem., 2006, 71, 6600–6603 CrossRef CAS PubMed.
- V. Kanagarajan, J. Thanusu and M. Gopalakrishnan, J. Enzyme Inhib. Med. Chem., 2011, 26, 280–287 CrossRef CAS PubMed.
- H.-W. Zhao, X.-Q. Chen, Z. Yang, T. Tian, B. Li, W. Meng, X.-Q. Song and H.-L. Pang, RSC Adv., 2015, 5, 103116–103122 RSC.
- Y.-H. Sun, Y. Xiong, C.-Q. Peng, W. Li, J.-A. Xiao and H. Yang, Org. Biomol. Chem., 2015, 13, 7907–7910 RSC.
- Y.-M. Wang, H.-H. Zhang, C. Li, T. Fan and F. Shi, Chem. Commun., 2016, 52, 1804–1807 RSC.
- S. Muthusamy and S. G. Kuma, Tetrahedron, 2016, 72, 2392–2401 CrossRef CAS.
- N. Wazzan, I. B. Obot, H. Faidallah and J. Adhes, Sci. Technol., 2018, 32, 2569–2589 CAS.
- Y. Zhou, F. Ma, P. Lu and Y. Wang, Org. Biomol. Chem., 2019, 17, 8849–8852 RSC.
- G. Habarurema, J. Mukiza, T. I. A. Gerber, T. Mukabagorora, E. C. Hosten and R. Betz, J. Organomet. Chem., 2020, 906, 121033 CrossRef CAS.
- J. Tian, L. Zhao, C. Yang, C. Yang, L. Guo and W. Xia, ACS Catal., 2023, 13, 866–876 CrossRef CAS.
- M. Boca, P. Baran, R. Boca, G. Kickelbick, F. Renz and W. Linert, Inorg. Chem. Commun., 1999, 2, 188–190 CrossRef CAS.
- I. Perillo, E. Repetto, M. C. Caterina, R. Massa, G. Gutkind and A. Salerno, Eur. J. Med. Chem., 2005, 40, 811–815 CrossRef CAS PubMed.
- M. Ghandi, F. Salimi and A. Olyaei, Molecules, 2006, 11, 556–563 CrossRef CAS PubMed.
- A. Kakanejadifard and S. M. F. Farnia, Tetrahedron, 1997, 53, 2551–2556 CrossRef CAS.
- R. Colorado-Peralta, S. A. Sánchez-Ruiz and A. Flores-Parra, Organics, 2023, 4, 297–312 CrossRef CAS.
- A. Rivera and R. Quevedo, Tetrahedron Lett., 2013, 54, 1416–1420 CrossRef CAS.
- S. Kathiravan and R. Raghunathan, Synlett, 2009, 1126–1130 CAS.
| This journal is © The Royal Society of Chemistry 2024 |