
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
TiF4-mediated, one-pot, reductive amination of carboxylic acids with borane–ammonia†
Madison J. Snyder,
Abdulkhaliq A. Alawaed,
Chunge Li,
Samantha Pacentine,
Henry J. Hamann and
P. Veeraraghavan Ramachandran*
and
P. Veeraraghavan Ramachandran*
Herbert C. Brown Center for Borane Research, Department of Chemistry, Purdue University, West Lafayette, Indiana 47907, USA. E-mail: chandran@purdue.edu
First published on 30th September 2024
Abstract
A facile one-pot, two-step, reductive alkylation of amines with carboxylic acids has been achieved with BH3–NH3 as an air- and moisture-stable reductant in the presence of TiF4. The catalyst is effective for both amidation and reduction steps, and the product amines are isolated in high yields as either the free amines, for those products containing an arylamine, or the borane-complexes. The free amine can be separated from these complexes using BF3–Et2O, followed by hydrolysis. The amide reduction has been demonstrated for primary, secondary, and tertiary amides, as well as lactams, and the reductive amination is applicable to a wide variety of aromatic and aliphatic acids as well as amines.
Introduction
Amines constitute an integral part of natural and synthetic molecules, with wide-ranging applications, particularly in agrochemical and pharmaceutical industry.1 Nucleophilic substitutions and transition-metal catalyzed aminations are common procedures to prepare alkyl or aryl amines.2–5 Reduction of nitriles,6,7 amides,8,9 or imines, and reductive amination of carbonyls (or reductive alkylation of amines) are also well-established protocols to access amines. Borohydrides and borane–Lewis bases, particularly borane–amines, are well studied for both classes of reactions.10–12While the reductive amination of aldehydes and ketones has been well studied,13–16 a similar reaction of carboxylic acids is undergoing a renewed interest as a field of research. Recent reports on the reductive amination of carboxylic acids involve conversion of acids to amides, followed by reduction with silanes17–19 or selective reduction of acids to aldehydes or silyl acetals,20–25 followed by reductive amination. The silane reduction protocols suffer from several drawbacks including stoichiometric waste, moisture sensitivity, reagent cost, and the cumbersome separation of the amine from the siloxane byproduct. Hydrogenation of a mixture of acid and amine in the presence of homogeneous and heterogeneous catalysts has been studied with a variety of metals and catalysts.26–33 Intriguingly, there are no reported practical procedures for the reductive amination of carboxylic acids utilizing versatile borane–amines.
The first report of a reductive alkylation of amines with acids as the electrophile was described, independently, by Gribble34 and Marchini35 and their co-workers five decades ago (Scheme 1(i) and (ii)). They described the use of sodium borohydride as the reductant in carboxylic acid or benzene solvent, although the reaction, presumably, proceeded via the acyloxyborohydride. However, these protocols are not practical and, accordingly, have not received much attention. A similar reductive alkylation via the intermediacy of amides, in varying yields, with borane–trimethylamine was described, nearly a decade later, by Trapani36 and co-workers (Scheme 1(iii)). Given the popularity of borane–amines for reductive amination,37–39 it is surprising that this work has not received much attention.
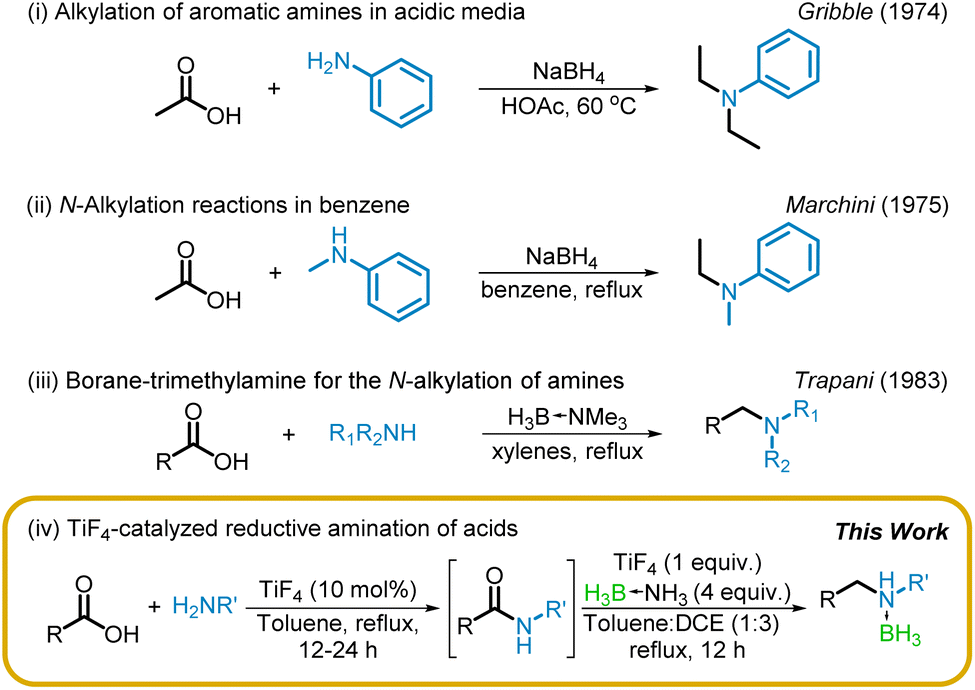 | ||
| Scheme 1 Reductive amination of acids via boranes. | ||
Our interest in the chemistry of borane–amines, particularly the reactions of borane–ammonia catalyzed by titanium tetrachloride40,41 has led to the successful reduction of carboxamides.42 We have also recently reported that TiF4 is an efficient catalyst for the direct amidation of acids.43 This prompted us to examine (i) the efficacy of TiF4 for the reduction of amides, so that (ii) a one-pot reductive amination of acids can be developed. Our success in both fronts resulting in a TiF4-mediated reductive amination of acids is detailed below (Scheme 1(iv)).
Results and discussion
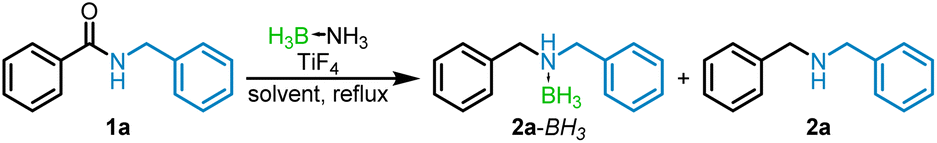
Our initial focus was to achieve the reduction of amides using borane–ammonia (BH3NH3). Other borane–amines were not examined for the reduction due to the menace of amine byproduct that will be difficult to separate. Prior TiCl4 catalyzed42,44 and tris(pentafluorophenyl)borane [(C6F5)3B] catalyzed45 amide reductions using BH3NH3 gave further motivation for the project.With N-benzylbenzamide (1a) as the representative amide, we began optimizing the reduction. The results are summarized in Table 1. Following our reported TiCl4 amide reduction using two equiv. of borane–ammonia and 20 mol% of the catalyst, the reaction was carried out using TiF4 in refluxing DCE for 24 h. Workup revealed, by 1H NMR spectroscopy, a mixture of 36% of N,N-dibenzylamine (2a) and 32% of a second product identified as the 2a–BH3 complex,46 along with 32% of the unreacted amide (Table 1, entry 1). Given the low conversion, the catalyst was increased to 100 mol%, which resulted in a 44% conversion to the amine–borane as the sole product (entry 2). It is noteworthy that the TiCl4 catalyzed reduction yields the amine or the amine–hydrochloride product in some cases,42,44 the halide being delivered from the catalyst. Noting that earlier reported45 borane reductions of amides have used either 3 or 4 equiv. of the reducing agent, the reaction was repeated with 3 and 4 equivalents of BH3NH3, resulting in a respective increase in yield of the borane–amine to 90% and 97% in 16 h (entries 3 and 4). Decreasing the reaction time to 12 h had no deleterious effect on the yield (96%, entry 5). Having obtained nearly quantitative transformation of the amide to the borane–amine, an attempt was made to decrease TiF4 load to 50 mol% when the yield decreased to 75% (entry 6).
|
|||||
|---|---|---|---|---|---|
| Entry | BH3NH3 (equiv.) | TiF4 (equiv.) | Solvent | Time (h) | Conversiona 2a–BH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a:1a :2a:1a |
| a Ratio determined by 1H NMR spectroscopy.b Isolated yield of 2a–BH3.c Combined value of benzyl alcohol and 1a.d Isolated yield of 2a. | |||||
| 1 | 2 | 0.2 | DCE | 24 | 32:36:32 |
| 2 | 2 | 1 | DCE | 24 | 44:0:56 |
| 3 | 3 | 1 | DCE | 24 | (90)b |
| 4 | 4 | 1 | DCE | 16 | (97)b |
| 5 | 4 | 1 | DCE | 12 | (96)b |
| 6 | 4 | 0.5 | DCE | 24 | (75)b |
| 7 | 2 | 0.2 | Toluene | 24 | 4:32:64c |
| 8 | 2.5 | 1 | Toluene | 24 | 13:59:28c |
| 9 | 3 | 1 | Toluene | 24 | 4:71:25c |
| 10 | 4 | 1 | Xylenes | 12 | (67)d |
| 11 | 2 (HBpin) | 0.1 | Toluene | 24 | 0:71:29 |
| 12 | 3 (HBpin) | 0.1 | Toluene | 24 | 0:100:0 (88)d |
Since the TiF4-mediated amidation is more efficient in toluene as the solvent,43 the reduction was now optimized in toluene, to develop a one-pot reductive amination of acids. Although it was noticed that the use of toluene greatly encouraged conversion to the free amine (32%) over the borane–amine (4%), very low combined yields were obtained (entry 7). Increasing the catalyst or reductant, or switching the solvent to xylenes did not improve the results considerably (entries 8–10). In addition, competing reduction to the alcohol was also observed! As can be seen from entries 11 and 12, satisfactory conversions to 2a were achieved using pinacolborane (HBpin) as an alternate reductant, however, isolation of the product was hindered by the presence of byproduct pinacol. Thus, a reaction in refluxing DCE with four equiv. of BH3NH3 and 100 mol% TiF4 was chosen as the optimal conditions for subsequent studies. The scope of the TiF4-catalyzed BH3NH3 reduction of amides was examined for a variety of amides derived from both aromatic and aliphatic acids and amines. The borane–amines synthesized using this reduction protocol and their respective yields are summarized in Scheme 2. For the reduction of amides derived from aromatic acids, electron donating groups in the para position (–CH3 and –OMe) of the aryl moiety were well tolerated, producing excellent yields of 2b–BH3 and 2c–BH3 (96% and 97% respectively). The electron withdrawing –CF3 group performed similarly (95% yield of 2d–BH3) showing tolerance for electronic substitution. Amides derived from aliphatic acids (1e–1q) were also reduced in excellent yields (89–98%). X-ray crystallographic analysis of 2k–BH3, crystallized from hexane, served to confirm the formation of the borane–amine (Scheme 2, 2k–BH3). As a representative primary amine, N-hexanamide (1e) was reduced, providing a 92% yield of the corresponding borane–amine (2e–BH3). N-Benzylformamide (1f) and N-phenylacetamide (1g) were reduced in 92% and 98% yields respectively. The method also proved effective for the reduction of a cyclic amide, caprolactam (1p), to borane–azepane (2p–BH3) in 89% yield.
 | ||
| Scheme 2 TiF4-catalyzed reduction of amides with BH3NH3. Isolated yields shown. | ||
Notably, the reduction of amides (anilides) derived from aromatic amines (1s–1x) resulted in none of the borane–amine, and the free amines (2s–2x) were isolated in 90–99% yields. This can be rationalized by the weak nucleophilicity of anilines resulting in poor coordination with borane. Thus, N-phenylbenzamide (1s) was reduced to N-benzylaniline (2s) in 94% yield. When the acyl moiety of the amide contained an electron withdrawing fluoride in the meta position (1t) the reduction resulted in excellent yields (97%) of the N-benzylaniline (2t). As with the acyl moiety, the aniline moiety tolerated both electron withdrawing and donating groups. Both N-(4-methoxyphenyl)benzamide (1u) and N-(4-methoxyphenyl)-2-phenylacetamide (1v) were reduced to their corresponding amines (2u and 2v) in excellent yields (97% and 90%) and 3-bromo-N-phenethylaniline (1w) yielded 99% of 2w. Curiously, the reduction of N-benzyl-2,2,2-trichloroacetamide (1x) also resulted in producing the corresponding free amine (2x), probably due to the strong inductive effect of the –CCl3 group weakening the borane coordination (Scheme 3).
 | ||
| Scheme 3 TiF4-catalyzed reduction of anilides with BH3NH3. Isolated yields shown. | ||
The amide reduction methodology was then applied to a competitive reaction between a secondary (1h) and tertiary amide (1n). Using the standard condition (4 equiv. BH3NH3 and 1 equiv. TiF4 in refluxing DCE), the reaction mixture was analyzed using 1H NMR after 12 h. The expected borane–amine products 2h–BH3 and 2n–BH3 were present in a ratio 37% to 63% respectively, along with the corresponding quantities of unreacted amide. These results indicate a nearly 2 to 1 preference for the reduction of the tertiary vs. the secondary amide (Scheme 4).
 | ||
| Scheme 4 Competitive reduction of 2° vs. 3° amide. | ||
After achieving the TiF4-mediated BH3NH3 reduction of a variety of amides to the corresponding amines or borane–amines in good to excellent yields, optimization of a one-pot tandem reductive amination procedure was performed. First, the amidation was carried out with the previously reported conditions,43 in refluxing toluene for 12 h for aliphatic and 24 h for aromatic acids. The challenge was overcoming the change of solvent from toluene to DCE for the reduction. As discussed above, the reduction in toluene is not satisfactory, but removing toluene to substitute DCE will make the process tedious. Accordingly, the use of mixed solvent system for the reduction step was envisioned. After completing the amidation in toluene in the presence of 10 mol% TiF4, three reduction experiments were performed by adding DCE so that the overall ratios of toluene and DCE were 1:3, 1:1, and 3:1. An equiv. of TiF4 and 4 equiv. of BH3NH3 were also added and the reductions completed. Among these, the 1:3 ratio of toluene:DCE was found to provide the best yields of dibenzylamine–borane (89%) with the least amount of the benzyl alcohol side-product (vide supra: optimization). This solvent mixture was used for all subsequent one-pot reductive aminations. An attempt to carry out the amidation of benzoic acid with benzylamine in this solvent mixture was not satisfactory.
The tandem conversion of the carboxylic acid to the borane–amine proved in many cases to be equally effective as the stepwise process, resulting in good to excellent yields of the borane–amine or free-amine products. The reaction of benzoic, phenylacetic, cyclohexanecarboxylic, and trifluoropropanoic acids with benzylamine yielded the borane–amines (3a–BH3, 3h–BH3, 3k–BH3, and 3aa–BH3) without difficulty (79–98%). In cases where trace amounts of alcohol formed, it was simply washed away with hexane, leaving pure borane–amine. Decorating the carboxylic acid with an electron donating group (–OMe) in the para position, resulted in a near quantitative conversion to the borane–amine 3c–BH3 (98%). However, 4-nitrophenylacetic acid had moderate conversion to 3q–BH3 after column chromatography (41%) due to the reduction to the alcohol in place of the amine. Similarly, the use of both 4-chlorobenzylamine and 4-methoxybenzylamine resulted in significant formation of the alcohol product. After separation by column chromatography, they yielded their respective borane–amines (3r–BH3 and 3y–BH3) in 51% and 59%. The conversion to the free amine by use of aniline proceeded in good yields when paired with benzoic and phenylacetic acids (87 and 68% respectively for 3s and 3z) with the lower yield of N-phenylethylaniline (3z) due to alcohol formation. These results are summarized in Scheme 5.
 | ||
| Scheme 5 One pot TiF4-catalyzed reductive amination of acids with BH3NH3. Isolated yields shown. | ||
While the reaction products containing an aromatic amine (2s–2w, 3s, 3z) or a strongly withdrawing group at the alpha position (2x) were obtained as the free amine, the majority of the products of the amide reduction and reductive amination were isolated as the corresponding complexes with borane (BH3). The simple conversion of BH3 complex to free amine was demonstrated by reacting product 2h–BH3 with BF3–OEt2, where, after acidic aqueous workup, the free amine (2h) was obtained in 91% isolated yield (Scheme 6). A one-pot reaction to prepare 2h, without isolation of 2h–BH3 was also examined. Starting from phenylacetic acid and benzylamine, the amidation, reduction, and removal of BH3 using BF3–OEt2 were carried out sequentially, resulting in an 73% isolated yield of 2h.
 | ||
| Scheme 6 Conversion of borane–amine to free amine. aYield of 2h when amidation, reduction and removal of BH3 are carried out sequentially in a one-pot reaction. | ||
The tandem conversion of the carboxylic acid to the borane–amine followed by the process for the decomplexation of BH3 using BF3–OEt2 was additionally applied to the synthesis of racemic cinacalcet. The chiral (R)-cinacalcet is used as a calcimimetic agent. In our procedure 3-(3-(trifluoromethyl)phenyl)propanoic acid and (±)1-(1-naphthyl)ethylamine were subjected to the tandem amidation/reduction conditions to provide the corresponding borane–amine 4a–BH3. Curiously, the NMR characterization of this adduct showed the presence of 2 components. This is thought to be caused by a sort of isomerism arising from the adjacent chiral center and borane-coordinated amine, as the two components were separable by column chromatography. The decomplexation protocol using BF3–OEt2 was applied to this two component mixture whereupon the peaks recoalesced to a single species which was identified as racemic cinacalcet (4a). The product 4a was isolated in 94% yield, which is an improvement over similar protocols.25 The decomplexation of 4a–BH3 to 4a did require a somewhat longer reaction time (12 h) and the use a stronger acid (6 M) in the workup procedure compared with the conversion of 2h–BH3 to 2h (Scheme 7).
 | ||
| Scheme 7 Synthesis of racemic cinacalcet (4a). | ||
Conclusions
In conclusion, we have developed open-air processes for the reduction of amides and a one-pot, reductive amination of carboxylic acids to the corresponding amines or borane–amines using borane–ammonia with titanium tetrafluoride as the activator in refluxing dichloroethane. This new synthesis tolerates amides derived from aromatic and aliphatic acids and amines as well as some functional group tolerability towards nitro, halogens, and electron donating groups. This process can also be viewed as a new process for the synthesis of borane–amines from carboxylic acids, though the amines can be readily freed from their borane complexes using boron trifluoride diethyl etherate, a process which was applied to the synthesis of racemic cinacalcet.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Purdue University Herbert C. Brown Center for Borane Research is gratefully acknowledged.Notes and references
- O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS PubMed.
- Q. Yang, Q. F. Wang and Z. K. Yu, Chem. Soc. Rev., 2015, 44, 2305–2329 RSC.
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed.
- A. Trowbridge, S. M. Walton and M. J. Gaunt, Chem. Rev., 2020, 120, 2613–2692 CrossRef CAS PubMed.
- M. Y. Kuai, Z. H. Jia, L. J. Chen, S. Gao and W. W. Fang, Eur. J. Org Chem., 2024, 27, e202300933 CrossRef CAS.
- V. K. Pandey, C. S. Tiwari and A. Rit, Org. Lett., 2021, 23, 1681–1686 CrossRef CAS PubMed.
- R. Kumar, R. K. Meher, J. Sharma, A. Sau and T. K. Panda, Org. Lett., 2023, 25, 7923–7927 CrossRef CAS PubMed.
- P. Q. Ye, Y. L. Shao, X. Z. Ye, F. J. Zhang, R. H. Li, J. N. Sun, B. H. Xu and J. X. Chen, Org. Lett., 2020, 22, 1306–1310 CrossRef CAS PubMed.
- V. Vinayagam, S. K. Sadhukhan, S. K. Karre, R. Srinath, R. K. Maroju, P. R. Karra, H. Bathula, S. Kundrapu and S. R. Surukonti, Org. Lett., 2023, 25, 4610–4614 CrossRef CAS PubMed.
- K. Matos and E. R. Burkhardt, in Pharmaceutical Process Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 127–143 Search PubMed.
- E. W. Baxter and A. B. Reitz, in Organic Reactions, 2002, pp. 1–714 Search PubMed.
- A. F. Abdel-Magid and S. J. Mehrman, Org. Process Res. Dev., 2006, 10, 971–1031 CrossRef CAS.
- E. Podyacheva, O. I. Afanasyev, A. A. Tsygankov, M. Makarova and D. Chusov, Synthesis, 2019, 51, 2667–2677 CrossRef CAS.
- N. U. D. Reshi, V. B. Saptal, M. Beller and J. K. Bera, ACS Catal., 2021, 11, 13809–13837 CrossRef CAS.
- T. Irrgang and R. Kempe, Chem. Rev., 2020, 120, 9583–9674 CrossRef CAS PubMed.
- S. Bhattacharyya, A. Chatterjee and J. S. Williamson, Synlett, 1995, 10, 1079–1080 CrossRef.
- E. L. Stoll, T. Tongue, K. G. Andrews, D. Valette, D. J. Hirst and R. M. Denton, Chem. Sci., 2020, 11, 9494–9500 RSC.
- K. G. Andrews, D. M. Summers, L. J. Donnelly and R. M. Denton, Chem. Commun., 2016, 52, 1855–1858 RSC.
- M.-C. Fu, R. Shang, W.-M. Cheng and Y. Fu, Angew. Chem., Int. Ed., 2015, 54, 9042–9046 CrossRef CAS PubMed.
- L. Ouyang, R. Miao, Z. H. Yang and R. S. Luo, J. Catal., 2023, 418, 283–289 CrossRef CAS.
- M. Minakawa, M. Okubo and M. Kawatsura, Tetrahedron Lett., 2016, 57, 4187–4190 CrossRef CAS.
- K. G. Andrews, R. Faizova and R. M. Denton, Nat. Commun., 2017, 8, 15913 CrossRef PubMed.
- L. Zhu, L. S. Wang, B. J. Li, W. Li and B. Q. Fu, Catal. Sci. Technol., 2016, 6, 6172–6176 RSC.
- T. V. Q. Nguyen, W. J. Yoo and S. Kobayashi, Adv. Synth. Catal., 2016, 358, 452–458 CrossRef CAS.
- I. Sorribes, K. Junge and M. Beller, J. Am. Chem. Soc., 2014, 136, 14314–14319 CrossRef CAS PubMed.
- R. Coeck, J. Meeprasert, G. N. Li, T. Altantzis, S. Bals, E. A. Pidko and D. E. De Vos, ACS Catal., 2021, 11, 7672–7684 CrossRef CAS.
- B. Emayavaramban, P. Chakraborty and B. Sundararaju, ChemSusChem, 2019, 12, 3089–3093 CrossRef CAS PubMed.
- W. P. Liu, B. Sahoo, A. Spannenberg, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2018, 57, 11673–11677 CrossRef CAS PubMed.
- T. Toyao, S. Siddiki, Y. Morita, T. Kamachi, A. S. Touchy, W. Onodera, K. Kon, S. Furukawa, H. Ariga, K. Asakura, K. Yoshizawa and K. Shimizu, Chem.–Eur. J., 2017, 23, 14848–14859 CrossRef CAS PubMed.
- Y. P. Shi, P. C. J. Kamer and D. J. Cole-Hamilton, Green Chem., 2017, 19, 5460–5466 RSC.
- C. Qiao, X. F. Liu, X. Liu and L. N. He, Org. Lett., 2017, 19, 1490–1493 CrossRef CAS PubMed.
- I. Sorribes, J. R. Cabrero-Antonino, C. Vicent, K. Junge and M. Beller, J. Am. Chem. Soc., 2015, 137, 13580–13587 CrossRef CAS PubMed.
- A. A. Nuñez, G. R. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2007, 3154–3156 RSC.
- G. W. Gribble, P. D. Lord, J. Skotnicki, S. E. Dietz, J. T. Eaton and J. L. Johnson, J. Am. Chem. Soc., 1974, 96, 7812–7814 CrossRef CAS.
- P. Marchini, G. Liso, A. Reho, F. Liberatore and F. M. Moracci, J. Org. Chem., 1975, 40, 3453–3456 CrossRef CAS.
- G. Trapani, A. Reho and A. Latrofa, Synthesis, 1983, 1983, 1013–1014 CrossRef.
- Q. W. Zhou, W. Meng, J. Yang and H. F. Du, Angew. Chem., Int. Ed., 2018, 57, 12111–12115 CrossRef CAS PubMed.
- W. Y. Liao, Y. F. Chen, Y. X. Liu, H. F. Duan, J. L. Petersen and X. D. Shi, Chem. Commun., 2009, 6436–6438 RSC.
- S. Sato, T. Sakamoto, E. Miyazawa and Y. Kikugawa, Tetrahedron, 2004, 60, 7899–7906 CrossRef CAS.
- P. V. Ramachandran, A. A. Alawaed and H. J. Hamann, Org. Lett., 2023, 25, 6902–6906 CrossRef CAS PubMed.
- P. V. Ramachandran, A. A. Alawaed and H. J. Hamann, Org. Lett., 2023, 25, 4650–4655 CrossRef CAS PubMed.
- P. V. Ramachandran, A. A. Alawaed and A. Singh, Molecules, 2023, 28, 4575 CrossRef CAS PubMed.
- A. A. Alawaed and P. V. Ramachandran, Org. Biomol. Chem., 2024, 22, 1915–1919 RSC.
- Y. Zang, Q. Sui, Q. Xu, M. Ma, G. Li and F. Zhu, Tetrahedron Lett., 2023, 124, 154598 CrossRef CAS.
- Y. Pan, Z. Luo, J. Han, X. Xu, C. Chen, H. Zhao, L. Xu, Q. Fan and J. Xiao, Adv. Synth. Catal., 2019, 361, 2301–2308 CrossRef CAS.
- P. V. Ramachandran, H. J. Hamann and R. Lin, Dalton Trans., 2021, 50, 16770–16774 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Optimization details, experimental procedures, crystallization details, product characterization, and 1H, 11B, 13C, and 19F NMR spectra of products. CCDC 2373783. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra05888g |
| This journal is © The Royal Society of Chemistry 2024 |