
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparative study of metal–organic frameworks synthesized via imide condensation and coordination assembly†
Qiao Liu‡
 ,
Jordon S. Hilliard,
Zhongzheng Cai§
and
Casey R. Wade*
,
Jordon S. Hilliard,
Zhongzheng Cai§
and
Casey R. Wade*
Department of Chemistry and Biochemistry, The Ohio State University, 100 West 18th Ave, Columbus, OH 43210, USA. E-mail: wade.521@osu.edu
First published on 30th August 2024
Abstract
A series of metal–organic frameworks (1-XDI) have been synthesized by imide condensation reactions between an amine-functionalized pentanuclear zinc cluster, Zn4Cl5(bt-NH2)6, (bt-NH2 = 5-aminobenzotriazolate), and organic dianhydrides (pyromellitic dianhydride (PMDA), naphthalenetetracarboxylic dianhydride (NDA), 3,3′,4,4′-biphenyltetracarboxylic dianhydride (BPDA) and 4,4′-(hexafluoroisopropylidene)diphthalic anhydride (HFIPA)). The properties of the 1-XDI MOFs have been compared with analogues (2-XDI) prepared using traditional coordination assembly. The resulting materials have been characterized by ATR-IR spectroscopy, acid-digested 1H NMR spectroscopy, elemental analysis, and gas adsorption measurements. N2 adsorption isotherm data reveal modest porosities and BET surface areas (30–552 m2 g−1). All of the new 1-XDI and 2-XDI MOFs show selective adsorption of C2H2 over CO2 while 2-PMDI and 2-BPDI exhibit high selectivity toward C3H6/C3H8 separation. This study establishes imide condensation of preformed metal–organic clusters with organic linkers as a viable route for MOF design.
Introduction
The advent of metal–organic frameworks (MOFs) and covalent organic frameworks (COFs) has stimulated intense interest in the design and study of new porous materials for applications in gas separation,1–3 heterogeneous catalysis,4,5 and energy storage.6,7 Porous materials are typically classified based on their structure, composition, and method of self-assembly. For example, MOFs are a subset of coordination polymers (CPs) characterized by 2D or 3D extended structures containing coordination compounds (i.e. metal–ligand bonds) and the potential for guest-accessible porosity. Reversible coordinative bond formation during MOF self-assembly often results in highly crystalline products, but crystallinity is not requisite based on the IUPAC definition.8 The intrinsic disorder in amorphous MOFs limits precise structural characterization, making them less appealing for fundamental studies. Nevertheless, amorphous MOFs often exhibit gas accessible porosity and relatively high surface areas and have gained increasing attention for a range of applications.9–12 COFs rely on condensation reactions between organic building units to form covalent linkages such as boroxines, imines, triazines, or a variety of other functional groups13,14 Crystallinity is considered to be a distinguishing feature of COFs among other types of porous organic materials.15,16 Recently, hybrid materials such as metal-covalent organic frameworks (MCOFs) have emerged.17 MCOFs begin to blur the lines between the MOF and COF distinctions by utilizing covalent bond formation between organic linker groups and preformed metal coordination compounds for self-assembly (Table S1†).18–27 MOFs assembled from pentanuclear Zn benzotriazolate clusters, also known as Kuratowski-type clusters, have garnered significant attention for applications in CO2 capture,28–32 gas separation,33–35 catalysis,36–41 and small-molecule binding and activation.42–47 Although Kuratowski-type clusters are readily synthesized as discrete molecular species, they have not proven to be broadly accessible in MOF self-assembly reactions. As a result, only a relatively small number of MOFs containing Kuratowski-type clusters have been reported.48–55 This synthetic difficulty has motivated us to explore alternative synthetic routes for the assembly of porous materials containing Kuratowski-type clusters.Herein, we describe the synthesis and gas adsorption properties of a series of Zn benzotriazolate MOFs assembled using either imide condensation (1-XDI) or traditional coordination assembly (2-XDI, Scheme 1). The imide route relies on condensation of commercially available organic dianhydrides with an amine-functionalized Kuratowski-type cluster (1-NH2, Scheme 1). Although this method of self-assembly resembles MCOFs, the resulting materials are aptly categorized as MOFs given their composition and guest-accessible porosity. Moreover, three of the four new 1-XDI MOFs are found to be amorphous, but exhibit modest Brunauer–Emmett–Teller (BET) surface areas and adsorb CO2 and light hydrocarbon gases at room temperature. A traditional MOF solvothermal synthesis, or coordination assembly route, employing bis-benzotriazolate linkers (H2bbt-XDI) and Zn salts has been used to screen the synthesis of an analogous series of MOFs (2-XDI). This route also affords amorphous or poorly crystalline products that exhibit gas accessible porosity. Comparison of the 1-XDI and 2-XDI MOFs provides insight into the influence of the different synthetic methods on the composition and gas adsorption properties of the resulting porous materials.
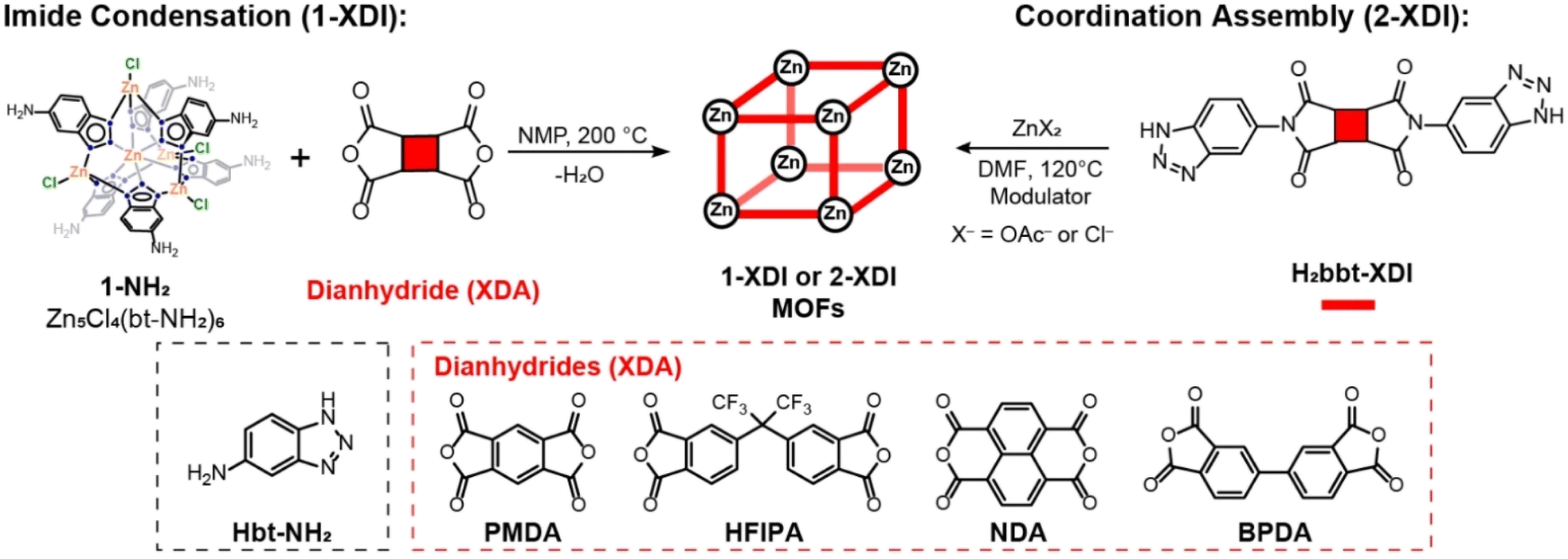 | ||
| Scheme 1 Synthetic routes to 1-XDI and 2-XDI MOFs. | ||
Results and discussion
Synthesis and characterization of 1-XDI MOFs
The amine-functionalized Kuratowski cluster 1-NH2 [Zn5Cl4(bt-NH2)6] was prepared from reaction of 5-aminobenzotriazole (Hbt-NH2) with ZnCl2 according to a modified literature procedure (Scheme 1).56 1-NH2 was obtained as an amorphous, pale yellow solid and characterized by electrospray ionization mass spectrometry (ESI-MS, positive ion mode) and 1H NMR spectroscopy. The former shows a series of ion signals in the m/z = 1230–1480 range that correspond to acetone and H2O adducts of the 1-NH2 cluster (Fig. S1 and Table S3†). The 1H NMR spectrum measured in acetone-d6 exhibits two sets of resonances associated with the bt-NH2 ligands in a ∼4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Fig. S2†). Both sets of signals are shifted downfield with respect to the free ligand, consistent with ligand complexation in the Zn5 cluster. The appearance of only two distinct ligand species is surprising given that the Zn5 cluster can form several different isomers that differ in the relative orientations of the amine groups (Fig. S3†). However, the large number of adducts observed in the ESI-MS data suggests that rapid exchange of solvent ligands may influence the number of signals observed in the NMR spectrum. Notably, there is only one report of crystallographically characterized Zn5 clusters containing monosubstituted benzotriazolate ligands.57 These clusters exhibit C1 symmetry with respect to the ligand orientations. Moreover, they were formed in situ during MOF assembly reactions and not characterized by NMR spectroscopy. Solubility tests reveal that 1-NH2 is insoluble in water, slightly soluble in methanol and ethanol, and very soluble in polar aprotic solvents such as acetone, N-methylpyrrolidone (NMP), dimethyl sulfoxide (DMSO), and N,N-dimethylformamide (DMF). Attempts to obtain single crystals of 1-NH2 for structural characterization were unsuccessful.
:1 ratio (Fig. S2†). Both sets of signals are shifted downfield with respect to the free ligand, consistent with ligand complexation in the Zn5 cluster. The appearance of only two distinct ligand species is surprising given that the Zn5 cluster can form several different isomers that differ in the relative orientations of the amine groups (Fig. S3†). However, the large number of adducts observed in the ESI-MS data suggests that rapid exchange of solvent ligands may influence the number of signals observed in the NMR spectrum. Notably, there is only one report of crystallographically characterized Zn5 clusters containing monosubstituted benzotriazolate ligands.57 These clusters exhibit C1 symmetry with respect to the ligand orientations. Moreover, they were formed in situ during MOF assembly reactions and not characterized by NMR spectroscopy. Solubility tests reveal that 1-NH2 is insoluble in water, slightly soluble in methanol and ethanol, and very soluble in polar aprotic solvents such as acetone, N-methylpyrrolidone (NMP), dimethyl sulfoxide (DMSO), and N,N-dimethylformamide (DMF). Attempts to obtain single crystals of 1-NH2 for structural characterization were unsuccessful.
Solvothermal reaction of 1-NH2 with dianhydrides PMDA, NDA, HFIPA, or BPDA in anhydrous NMP solvent at 200 °C results in imide condensation and formation of the corresponding 1-XDI MOFs (Scheme 1). Solubility tests reveal that the solid products are insoluble in common organic solvents such as NMP, DMSO, DMF, N,N-dimethylacetamide (DMA), methanol, and ethanol, suggesting hyper-crosslinked structures. The powder X-ray diffraction (PXRD) patterns of 1-HFIPI, 1-NDI and 1-BPDI are indicative of amorphous, aggregated structures, which is unsurprising given the variable connectivity and unsymmetric nature of the 1-NH2 cluster (Fig. 1a). However, the PXRD pattern of 1-PMDI exhibits several broad reflections in the 2θ = 5–25° range, revealing the presence of a poorly crystalline phase. Importantly, these reflections do not match the crystalline phase of the H2bbt-PMDI linker (Fig. S21†). The synthesis of 1-PMDI is reproducible, and the PXRD patterns show the same crystalline phase over multiple batches (Fig. S5†). Unfortunately, the poor crystallinity and limited number of reflections in the PXRD pattern have hampered attempts to index the data and gain any further insight into the structure of the crystalline phase (see Part 4 in ESI† for more details).
 | ||
| Fig. 1 (a) Overlay of PXRD patterns of 1-XDI MOFs. (b) Overlay of ATR-IR spectra of 1-XDI MOFs. Characteristic imide IR bands are highlighted in blue. (c) N2 adsorption isotherms for 1-XDI MOFs measured at 77 K. (d) Overlay of PXRD patterns of 2-XDI MOFs. (e) Overlay of ATR-IR spectra of 2-XDI MOFs. Characteristic imide IR bands are highlighted in blue. (f) N2 adsorption isotherms for 2-XDI MOFs measured at 77 K. | ||
The 1-XDI MOFs proved amenable to acid digestion using methods developed for other Kuratowski cluster-based MOFs.28,30 The digestion process cleaves the Zn-benzotriazolate bonds and solubilizes the ligand or linker fragments, which can then be identified and quantified by solution-state NMR analysis. The 1H NMR spectra of the as-synthesized 1-XDI products each exhibits a set of major aromatic resonances (Fig. 2) that match those observed for the authentic bis-imide ligands (Fig. S16–S19†). Remarkably, diimide condensation with the 1-NH2 clusters appears to be nearly quantitative (>90%) and only small amounts of unreacted Hbt-NH2 ligand are observed in the 1H NMR spectra. The spectra also show some minor impurities that are attributed to incomplete imide condensation giving rise to amide/carboxylic acid species. In support of the NMR characterization, the ATR-IR spectra of 1-HFIPI, 1-PMDI and 1-BPDI show the expected phthalic imide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O symmetric and asymmetric stretching bands at 1731 and 1780 cm−1 (Fig. 1b). 1-NDI also displays characteristic CO symmetric and asymmetric stretching bands at lower energy (1717 cm−1 and 1675 cm−1). Based on the 1H NMR and ATR-IR data, the 1-XDI MOFs exhibit a very high degree of imide connectivity between clusters. This result is somewhat unexpected given the amorphous nature of the samples. Nevertheless, the imide connections between neighboring 1-NH2 clusters are likely facilitated by reversible amide formation at the high reaction temperature as well as the conformational flexibility of the amide intermediate. Dissociation of the benzotriazolate ligands from the 1-NH2 cluster could also facilitate diimide linker condensation and MOF assembly. However, the benzotriazolate ligands are considerably inert owing to their μ3-bridging mode, and to the best of our knowledge, there is no precedent for ligand exchange in Kuratowski cluster-based MOFs or molecular clusters. Further mechanistic studies will be necessary to better understand the assembly process, but the preceding data show that imide condensation provides a viable route to porous polymer networks with Kuratowski clusters.
O symmetric and asymmetric stretching bands at 1731 and 1780 cm−1 (Fig. 1b). 1-NDI also displays characteristic CO symmetric and asymmetric stretching bands at lower energy (1717 cm−1 and 1675 cm−1). Based on the 1H NMR and ATR-IR data, the 1-XDI MOFs exhibit a very high degree of imide connectivity between clusters. This result is somewhat unexpected given the amorphous nature of the samples. Nevertheless, the imide connections between neighboring 1-NH2 clusters are likely facilitated by reversible amide formation at the high reaction temperature as well as the conformational flexibility of the amide intermediate. Dissociation of the benzotriazolate ligands from the 1-NH2 cluster could also facilitate diimide linker condensation and MOF assembly. However, the benzotriazolate ligands are considerably inert owing to their μ3-bridging mode, and to the best of our knowledge, there is no precedent for ligand exchange in Kuratowski cluster-based MOFs or molecular clusters. Further mechanistic studies will be necessary to better understand the assembly process, but the preceding data show that imide condensation provides a viable route to porous polymer networks with Kuratowski clusters.
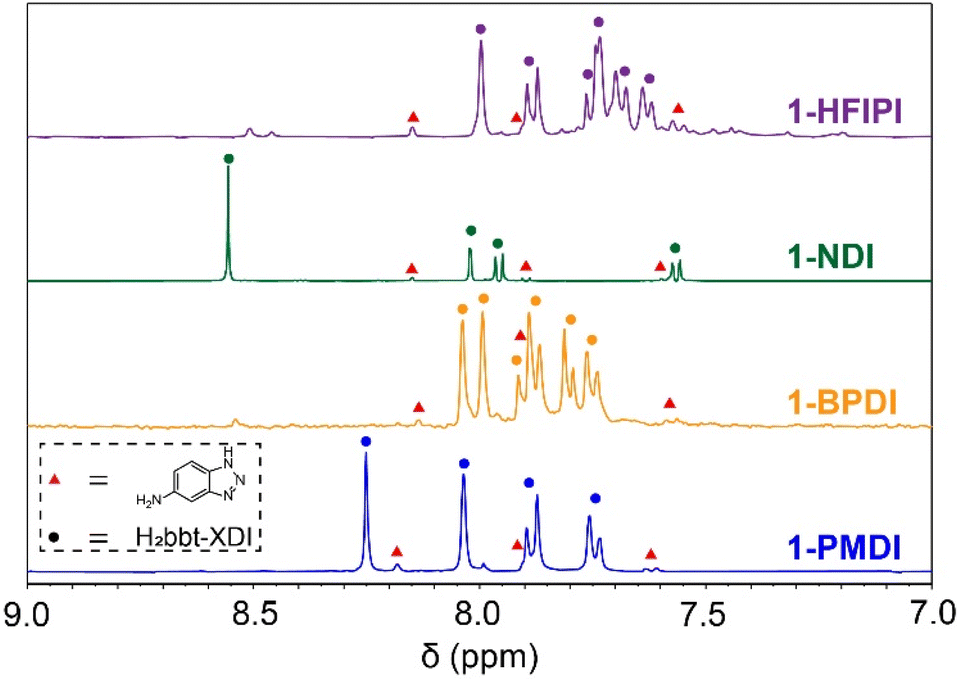 | ||
| Fig. 2 Acid-digested 1H NMR spectra of 1-XDI MOFs. Peak assignments for H2bbt-XDI and Hbt-NH2 are labelled with coloured circles and red triangles, respectively. | ||
Resonances associated with the presence of NMP guest molecules residing within the pores are also observed in the acid-digested 1H NMR spectra of the as-synthesized 1-XDI MOFs. The amount of NMP was determined by integration of the 1H NMR spectra and closely matches the predicted solvent content from thermogravimetric analysis (TGA) (Fig. S6–S9†). The TGA data also show good thermal stability with the onset of thermal degradation ≥400 °C for all of the MOFs. The MOFs were solvent exchanged with methanol and subsequently desolvated by heating at 100 °C under high vacuum for 24 h. Acid-digested 1H NMR analysis reveals some hydrolysis of the imide linkers, particularly for 1-BPDI and 1-HFIPI, as a result of the solvent exchange and thermal activation (Fig. S10–S13†). Combustion elemental analysis (C, H, N) and inductively coupled plasma atomic emission spectroscopy (ICP-OES, Zn) were carried out on the activated 1-XDI MOFs (Tables S4–S7†). The results fit reasonably well to the empirical formulas expected for 1-NH2 clusters connected by diimide linkers and show only modest deviations (<2.5 wt%) from the predicted compositions. N2 adsorption isotherm measurements reveal BET surface areas of 30 m2 g−1, 447 m2 g−1, 408 m2 g−1 and 483 m2 g−1 for activated 1-PMDI, 1-HFIPI, 1-NDI and 1-BPDI, respectively (Fig. 1c). Some batch-to-batch variability was observed in the surface areas, but they remained reasonably consistent (Fig. S14†). Moreover, the surface areas of the 1-XDI MOFs are comparable to, or even higher than, many reported porous organic polyimides and MCOFs (Tables S1 and S2†).19–21,24 DFT pore size distributions calculated from the N2 adsorption data show the presence of small micropores around 5–6 Å and a smaller volume fraction of micropores with ∼12 Å diameter (Fig. S15†). The relatively small micropores and the similarity in pore size distribution despite different linker lengths support the formation of highly disordered, crosslinked networks in the 1-XDI MOFs.
Synthesis and characterization of 2-XDI MOFs
The predominantly amorphous nature of the 1-XDI MOFs prompted us to investigate a coordinative assembly synthesis approach in hopes of obtaining crystalline analogues. Bis-benzotriazolate linkers (H2bbt-XDI) were synthesized by condensation reactions between Hbt-NH2 and the corresponding dianhydrides (Scheme 1). The new bis-imide compounds were characterized by CHN elemental analysis and 1H NMR and ATR-IR spectroscopy (Fig. S16–S20†). The linkers were subsequently screened in solvothermal reactions with ZnCl2 or Zn(OAc)2 using DMF solvent and acid or base modulators (Table 1). Solid products were obtained for the solvothermal reactions of Zn(OAc)2 with H2bbt-PMDI and H2bbt-BPDI. PXRD analysis of the corresponding materials, 2-PMDI and 2-BPDI, shows the presence of poorly crystalline phases that do not match those of the diimide linkers (Fig. 1d and S21, S22†). Unfortunately, we have thus far been unable to elucidate the structures of these products owing to their poor crystallinity and small crystallite size (see Part 4 in ESI†). Solvothermal reactions with H2bbt-HFIPI and H2bbt-NDI provided only clear solutions or amorphous solids under the screening conditions (Fig. S23 and S24†). The 2-XDI syntheses, including those affording amorphous products, were scaled-up to generate 2-PMDI, 2-BPDI, 2-NDI and 2-HFIPI.| Condition number (n) | Metal salt | Modulator |
|---|---|---|
| 1 | Zn(OAc)2 | Acetic acid |
| 2 | Zn(OAc)2 | None |
| 3 | Zn(OAc)2 | Triethylamine |
| 4 | ZnCl2 | Acetic acid |
| 5 | ZnCl2 | None |
| 6 | ZnCl2 | Triethylamine |
TGA analysis of the 2-XDI products after desolvation under high vacuum at 100 °C for 24 h reveals that they are generally less stable than the 1-XDI analogues with the onset of thermal degradation increasing in the order 2-NDI (310 °C) < 2-BPDI (350 °C) < 2-PMDI (390 °C) < 2-HFIPI (400 °C) (Fig. S25–S28†). Small mass losses of 2–6 wt% are observed below 100 °C and attributed to a small amount of water adsorbed upon exposure to ambient conditions. N2 adsorption isotherms (77 K) measured after desolvation show modest porosity with calculated BET surface areas of 171, 295, 141, and 552 m2 g−1 for 2-PMDI, 2-BPDI, 2-NDI and 2-HFIPI, respectively (Fig. 1f). The DFT pore size distributions are similar to those observed for the 1-XDI MOFs and indicate the presence two types of micropores that are 5–6 Å and ∼12 Å in diameter (Fig. S29†). Notably, 2-PMDI exhibits increased porosity compared to 1-PDMI, consistent with structural differences reflected in the PXRD patterns.
A combination of acid-digested 1H NMR analysis, ATR-IR spectroscopy, CHN elemental analysis, and ICP-OES have been used to gain insight into the composition of the 2-XDI MOFs. The acid-digested 1H NMR spectra for 2-PMDI, 2-BPDI, and 2-NDI confirm the presence of intact [bbt-XDI]2− linkers (Fig. S30–S32†). However, 2-HFIPI shows a complex mixture of amide species resulting from hydrolysis of the imide linkers during the solvothermal reaction (Fig. S33†). This conclusion is supported by the ATR-IR spectra of the 2-XDI MOFs (Fig. 1e). 2-PMDI and 2-BPDI exhibit intense features at 1731 and 1780 cm−1 that correspond to phthalimide CO symmetric and asymmetric stretching bands. The six-membered imide ring in 2-NDI displays characteristic CO symmetric and asymmetric stretching bands at lower energy (1717 cm−1 and 1675 cm−1). Bands corresponding to a C–N–C stretching mode and out of plane bending of the imide ring are also observed at 1365 and 738 cm−1, respectively. On the other hand, the ATR-IR spectrum for 2-HFIPI shows an intense CO stretch at 1674 cm−1 that can be attributed to amide species arising from hydrolysis of the H2bbt-HFIPI linker.
Since the 2-XDI MOFs were prepared from Zn2+ salts via coordination assembly and the structures could not be determined using X-ray or electron diffraction, the identity of the metal building units remains ambiguous. The elemental analysis data show significant deviations from the C, H, N, and Zn mass percent compositions expected for the presence of Zn5 clusters derived from 1-NH2 (Tables S8–S11†). Moreover, the deviations for the 2-XDI MOFs are considerably larger than those observed for the 1-XDI series, and there is no clear trend indicating the 2-XDI materials have a consistent metal:linker stoichiometry among the series. Based on literature precedent, other metal building units such as Zn9 clusters or infinite chains may also form during solvothermal assembly.32,48–54,58,59 This leaves open the possibility that the materials are assembled from a less commonly observed SBU.
Gas adsorption properties of 1-XDI and 2-XDI MOFs
CO2, C2H2, C2H4, C2H6, C3H6, and C3H8 gas adsorption isotherms (300 K) were measured for the activated 1-XDI and 2-XDI MOFs to investigate their potential for selective CO2/C2H2, C2H4/C2H6, and C3H6/C3H8 adsorptive separation processes. From these data, the corresponding ideal adsorbed solution theory (IAST) selectivity values have been calculated for binary, equimolar gas mixtures at 1 bar and 300 K (Tables S13 and S14†). Consistent with their relatively low surface areas, the 1-XDI and 2-XDI materials all exhibit gas adsorption capacities of less than 2 mmol g−1 at 1 bar (Tables S15 and S16†). With the exception of 1-PMDI, the gas uptake profiles and capacities of the 1-XDI MOFs are nearly identical across all tested gases (Fig. 3 and S38†). This behavior is consistent with their similar pore size distributions and comparable BET surface areas (Fig. 1c and S15†). 1-PMDI exhibits negligible adsorption of all of the gases studied, which is in accordance with its non-porous structure. The remaining 1-XDI MOFs exhibit favorable adsorption of C2H2 over CO2 with modest IAST selectivities in the 2.3–2.9 range but show little to no selectivity for C2H4/C2H6 or C3H6/C3H8 separation (Fig. 3).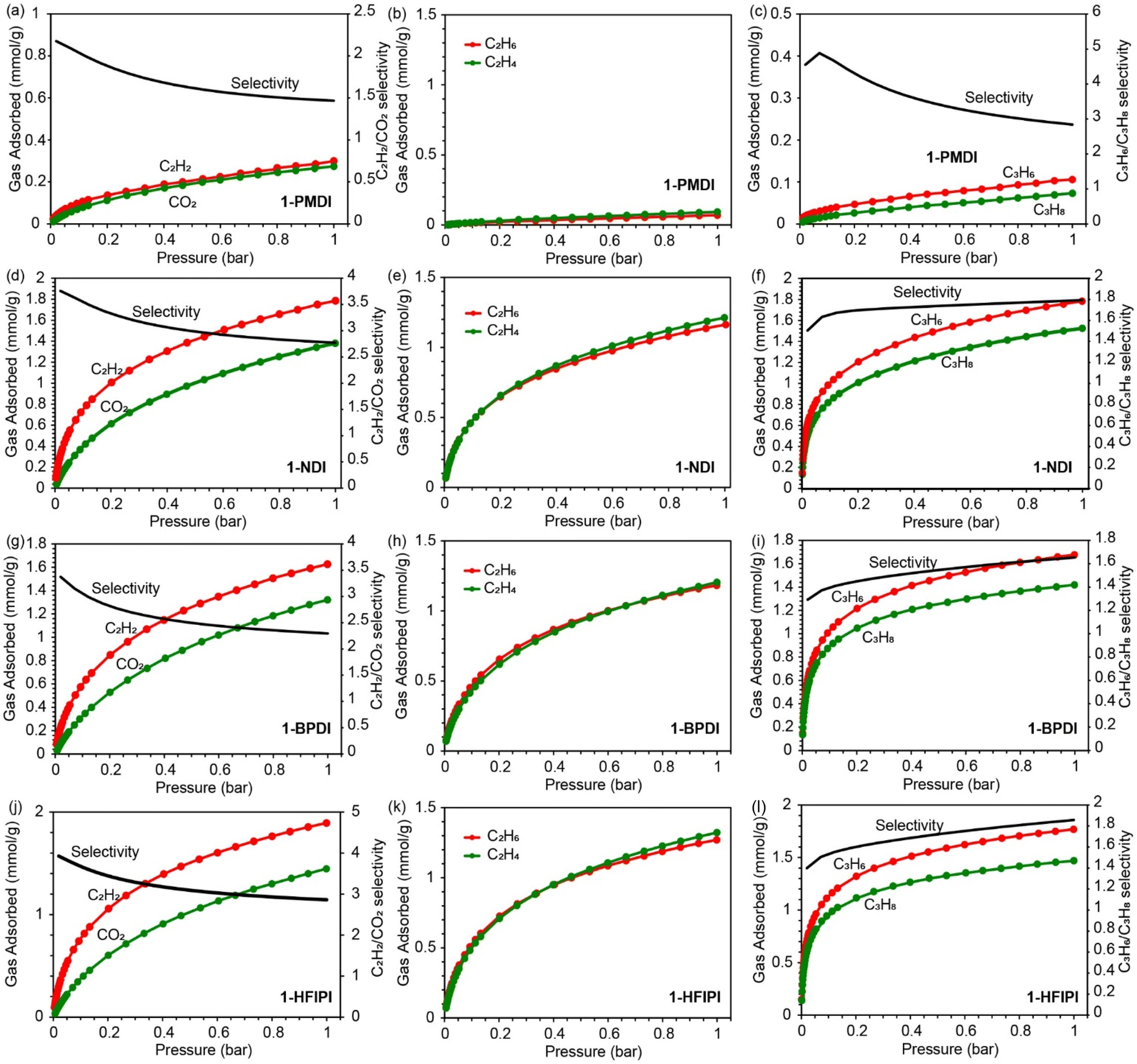 | ||
| Fig. 3 (a)–(l) C2H2 and CO2, C2H4 and C2H6, C3H6 and C3H8 adsorption isotherms (left y-axis) and predicted IAST selectivity at 300 K (right y-axis) for 1-XDI MOFs. | ||
The 2-XDI materials exhibit more variability in their gas adsorption profiles with similar or slightly lower capacities than the 1-XDI analogues (Fig. 4 and S39†). They also show modest IAST selectivity values (2.3–3.4) for adsorption of C2H2 over CO2. Despite its low surface area (171 m2 g−1), crystalline 2-PMDI has the steepest C2H2 uptake profile among the series. For the larger C2 and C3 gases, the order of gas uptake at 1 bar follows the trend suggested by the BET surface areas: 2-HFIPI > 2-BPDI > 2-PMDI > 2-NDI. No significant selectivity is observed for C2H4/C2H6 adsorption, but all of the 2-XDI materials show some selectivity for adsorption of C3H6 over C3H8. In particular, 2-PMDI and 2-BPDI show high calculated IAST selectivity values of 10 and 6, respectively, up to 1 bar total pressure (Fig. 4). This selectivity is comparable to other porous materials reported for C3H6/C3H8 separation, including AGTU-3a (7.0),60 MAF-23-O (8.8),61 Ni-NP (10.5),62 and Ni-MOF-74 (10).63 However, these selectivities are much lower than those of state-of-the-art materials such as Co-gallate (333) and UTSA-400 (>107) (Table S17†).64,65
 | ||
| Fig. 4 (a)–(l) C2H2 and CO2, C2H4 and C2H6, C3H6 and C3H8 adsorption isotherms (left y-axis) and predicted IAST selectivity at 300 K (right y-axis) for 2-XDI MOFs. | ||
Despite their amorphous or poorly crystalline structures, the 1-XDI and 2-XDI materials both show selective C2H2 adsorption with similar C2H2/CO2 capacities and IAST selectivity values in the 2.3–3.4 range. While neither are on par with the top-performing materials for C2H2/CO2 separation, they are comparable to CAU-10H (2.5),66 [Ni(tzba)0.5(F)(bpy)] (2.2),67 [Ca(dtztp)0.5] (1.7),68 and NUM-14a (3.3) (Table S18†).69 The most striking difference in adsorption properties between the 1-XDI and 2-XDI series is the relatively high C3H6/C3H8 selectivity of 2-PMDI and 2-BPDI. Although the origin of this difference remains unclear, it is notable that these are the only members of the two series that are both crystalline and porous. In this regard, crystalline frameworks resulting from the traditional MOF solvothermal synthesis route appear to offer some advantages toward adsorbent design for gas separation.
Conclusions
In summary, four novel 1-XDI MOFs have been prepared by condensation of amine-functionalized Kuratowski clusters (1-NH2) with organic dianhydrides. To the best of our knowledge, this work represents the first example of incorporation of Kuratowski clusters into porous coordination polymers using a condensation assembly approach. The properties of these new materials have been compared to 2-XDI analogues synthesized by a solvothermal coordinative assembly route commonly used for MOFs. Unsurprisingly, the two different synthetic approaches do not converge to afford analogous materials. The 2-XDI MOFs differ from the 1-XDI MOFs in terms of structure, composition, and gas adsorption properties. Crystalline phases were obtained for 1-PMDI, 2-PMDI, and 2-BPDI, and PXRD data indicate that 1-PMDI and 2-PMDI adopt different framework structures. Despite extensive synthetic screening, neither route gives highly crystalline materials amenable to complete structural characterization via X-ray or electron diffraction. Based on acid-digested 1H NMR spectra, the 1-XDI and 2-XDI MOFs contain intact [bbt-XDI]2− linker species. Remarkably, the 1H NMR spectra show nearly complete conversion to diimide species in the as-synthesized 1-XDI MOFs with only small amounts of unreacted bt-NH2 groups and amide/carboxylate species.The BET surface areas of the 1-XDI MOFs are all modest and quite consistent, with the exception of 1-PMDI which is essentially nonporous. The 2-XDI MOFs show greater variability in their N2 uptake, and generally exhibit lower BET surface areas than the 1-XDI analogues. Single component gas adsorption isotherms with CO2, C2H2, C2H4, C2H6, C3H6, and C3H8 were also measured for the 1-XDI and 2-XDI MOFs. All of the materials studied showed modest IAST selectivities (2.3–3.4) for adsorption of C2H2 from C2H2/CO2 binary gas mixtures. In addition, 2-PMDI and 2-BPDI exhibit good selectivity towards C3H6/C3H8 separation, but similar behavior was not observed for the 1-PMDI and 1-BPDI MOFs.
Overall, the results of this study demonstrate that imide condensation reactions can be used to prepare porous MOFs from amine-functionalized metal clusters with a high degree of connectivity. This approach is complementary to traditional MOF solvothermal synthesis which takes place exclusively via coordinative bond formation and yields materials with different compositions and properties. Condensation assembly of MOFs opens up new possibilities for the introduction of diverse functionalities for gas separation and other applications.
Experimental section
General considerations
1H-Benzo[d][1,2,3]triazol-5-amine (Hbt-NH2) was synthesized from 4-nitro-1,2-phenylenediamine according to report procedures.70 Pyromellitic dianhydride (PMDA), naphthalenetetracarboxylic dianhydride (NDA), 3,3′,4,4′-biphenyltetracarboxylic dianhydride (BPDA), 4,4′-(hexafluoroisopropylidene)diphthalic anhydride (HFIPA) and 4-nitro-1,2-phenylenediamine were purchased from Tokyo Chemical Industry Co., Ltd. Zn(OAc)2·2H2O (Alfa Aesar), ZnCl2 (98.0% Fisher Scientific), N,N-dimethylformamide (DMF, Fisher), Triethylamine (Fisher, 99.9%), sodium nitrite, 10 wt% Pd on carbon, hydrazine and glacial acetic acid (Fisher) were used as received. N-Methyl pyrrolidone (NMP, 99%, Honeywell) was dried by distillation from calcium hydride. All other solvents and reagents were of reagent grade and used as received. Routine powder X-ray diffraction patterns for phase identification were collected using a Rigaku Miniflex 600 diffractometer with nickel-filtered Cu-Kα radiation (λ = 1.5418 Å). ESI mass spectroscopic data were collected with a Bruker Impact II QqTOF Mass Spectrometer. ATR-IR spectra were measured using a Bruker Alpha II spectrometer with a diamond ATR accessory. Solution-state NMR spectra were measured using a Bruker DPX 400 or 600 MHz spectrometer. For 1H NMR spectra, the solvent resonance was used as an internal chemical shift reference. MOF samples were digested for solution-state 1H NMR analysis by suspending 15–20 mg of solid in CF3CO2H (0.1 mL) and gently heating the suspensions until all of the solids dissolved. Dimethyl sulfoxide (DMSO-d6, 1 mL) was then added to the solutions to provide a lock signal for shimming. Solvent-suppressed 1H NMR spectra were collected using a 180° water selective excitation sculpting with default parameters and pulse shapes. Briefly, spectra were collected using selective pulses of 1 ms with the transmitter frequency set to the center of the solvent resonance. The recycle delay between scans was 2 s, 16 K points were collected, and the acquisition time was 0.7 s. Elemental microanalyses (C, H, N) and inductively coupled plasma atomic emission spectroscopy (Zn) were performed by Robertson Microlit Laboratories, Ledgewood, NJ.Gas adsorption isotherm measurements
Single-component gas adsorption isotherms were measured using a Micromeritics 3Flex Surface Characterization Analyzer. All measurements were performed using the highest purity gases available from Linde or Praxair (N2, NI 5.0UH-K; CO2, CD 5.0LS-K; CO2, CD 5.0LS-K; C2H2, AC 2.6AA; C2H4, EY 5.0RS-K; C2H6, ET 5.0RS-G; C3H6, PY 2.5IS-PX; C3H8, PR 2.5ISK). Prior to analysis, samples (100–200 mg) were transferred to oven-dried and tared sample tubes equipped with TranSeals (Micromeritics) and heated to 100 °C (1 °C min−1) under vacuum until the outgassing rate was less than 0.0033 mbar min−1. BET surface areas were calculated from the N2 adsorption isotherms (77 K) by fitting the data to the BET equation with the appropriate pressure range (0.0001 ≤ P/Po ≤ 0.1) determined by the consistency criteria of Rouquerol.71,72 An open-source Python-based code package PyGAPS (Python General Adsorption Processing Suite) was used for isotherm fitting and the calculation of spreading pressure to generate IAST selectivity values at different pressures.73Synthesis of H2-bbtXDI linkers
Synthesis of 1-NH2 (Zn5Cl4(bt-NH2)6)
Hbt-NH2 (1.25 g, 9.30 mmol, 6 equiv.) was dissolved in ethanol (50 mL) in a 250 mL round bottom flask. A solution of anhydrous ZnCl2 (1.06 g, 7.8 mmol, 5 equiv.) and 2,6-lutidine (0.997 g, 9.30 mmol, 6 equiv.) in ethanol (20 mL) was added dropwise with stirring. The resulting mixture was heated to reflux for 1 h resulting in precipitation of a light-yellow solid. The solid was collected by filtration, washed with hot ethanol, and dried overnight in vacuo at 100 °C to afford the product (1.23 g, 62%). 1H NMR (acetone-d6, 400 MHz) δ (ppm): 8.41 (dd, 1H, Ar–H, 3JH–H = 9 Hz), 8.18 (q, 4H, Ar–H, 3JH–H = 9 Hz), 7.66 (t, 1H, Ar–H, 3JH–H = 11 Hz), 7.39 (t, 4H, Ar–H, 3JH–H = 11 Hz), 7.17 (d, 4H, Ar–H, 3JH–H = 7 Hz), 7.09 (d, 1H, Ar–H, 3JH–H = 8 Hz), 5.39 (br, 8H, –NH2). See Fig. S1 and Table S3 in the ESI† for ESI-MS data.General procedure for 1-XDI MOF synthesis
The 1-XDI MOFs were synthesized by solvothermal reaction of 1-NH2 (1 equiv., 6 amine groups per cluster) with the corresponding dianhydride (3 equiv.) (Scheme 1). Under an N2 atmosphere, a pre-dried Schlenk flask was charged with XDA (XDA = PMDA, NDA, BPDA, HFIPA; 3 equiv., specific quantities given below) and anhydrous NMP (half of the total solvent amount given below) and fit with a vented addition funnel. The mixture was heated to 200 °C in a sand bath with stirring, and a solution of 1-NH2 (1 equiv., specific quantities given below) in NMP was added dropwise to the XDA solution over the course of 4 h via the pre-dried addition funnel. The addition funnel was quickly replaced with a glass stopper, and the reaction was heated at 200 °C for 24 hours under an N2 atmosphere. Upon cooling to room temperature, the resulting precipitate was isolated by filtration. The solid product was immersed in fresh NMP (100 mL) and heated at 120 °C for 24 hours to remove any trapped XDA. The solid was collected by filtration, washed with additional NMP (3 × 50 mL) and MeOH (3 × 50 mL). The product was dried in vacuo at 100 °C for 24 hours prior to gas adsorption isotherm measurements.1-PMDI. 800 mg 1-NH2, 413 mg PMDA, and 30 mL of NMP were used. Yield: 1.12 g. Anal. Calcd for Zn5Cl4(C22H8N8O4)3: C, 43.70; H, 1.33; N, 18.53; Zn, 18.03; found: C, 45.95; H, 1.74; N, 19.56; Zn, 19.41.
1-NDI. 400 mg 1-NH2, 254 mg NDA, and 6 mL of NMP were used. Yield: 590 mg. Anal. Calcd for Zn5Cl4(C26H10N8O4)3: C, 54.14; H, 2.05; N, 16.29; found: C, 54.30; H, 1.97; N, 16.33.
1-BPDI. 400 mg 1-NH2, 279 mg BPDA, and 16 mL of NMP were used. Yield: 650 mg. Anal. Calcd for Zn5Cl4(C28H12N8O4)3: C, 49.40; H, 1.78; N, 16.46; Zn, 16.01. Found: C, 50.51; H, 2.21; N, 16.51; Zn, 14.01.
1-HFIPI. 500 mg 1-NH2, 526 mg HFIPA, and 13 mL of NMP were used. Yield: 970 mg. Anal. Calcd for Zn5Cl4(C31H12F6N8O4)3: C, 44.82; H, 1.46; N, 13.49; Zn, 13.12. Found: C, 45.88; H, 1.66; N, 13.25; Zn, 13.43.
General procedure for 2-XDI MOF synthetic screening
H2bbt-XDI linker (10 mg, 1 equiv.) and modulator (0.2 mL) were mixed with DMF (3 mL) in a 20 mL scintillation vial, and the suspension was sonicated to ensure maximum dissolution of the linker. The corresponding Zn source (4 equiv.) was added, and the vial was sealed with a Teflon-lined cap (Qorpak, #272628). The reaction vial was then placed in a room temperature, programmable oven and heated to 120 °C for 24 hours. Upon cooling to room temperature, the resulting solids were collected by filtration and washed with DMF (3 × 10 mL) and methanol (3 × 10 mL). The products are labeled according to the screening condition number (n) in Table 1 (i.e. 2-XDI-n; XDI = PMDI, NDI, BPDI and HFIPI; n = 1, 2, 3, 4, 5, 6;).Scale-up synthesis of 2-XDI MOFs
The 2-XDI MOF syntheses were scaled up (120 mg of linker) according to the results of PXRD analysis from the screening reactions (Fig. S21–S24†). Based on these data, 2-PMDI-1, 2-NDI-5, 2-BPDI-1, and 2-HFIPI-5 were chosen for synthetic scale-up. A 100 mL screw top media jar was used as the reaction vessel.2-PMDI. Yield: 170 mg. Anal. Found: C, 43.16; H, 2.24; N, 15.66.
2-NDI. Yield: 45 mg. Anal. Found: C, 50.08; H, 2.19; N, 17.77; Zn, 13.82.
2-BPDI. Yield: 120 mg. Anal. Found: C, 48.98; H, 2.72; N, 14.95; Zn, 16.40.
2-HFIPI. Yield: 25 mg. Anal. Found: C, 45.40; H, 1.94; N, 13.70; Zn, 12.98.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Q. L.: conceptualization, investigation, formal analysis, writing; J. S. H.: investigation; Z. C.: investigation; C. R. W.: conceptualization, formal analysis, supervision, writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by an Early Career Faculty grant (80NSSC18K1504) from NASA's Space Technology Research Grants Program. We thank Chandler Greenwell, Xuetao Liu and XtalPi, Inc. for performing micro-crystal electron diffraction experiments.References
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- J. Liang, Y.-B. Huang and R. Cao, Coord. Chem. Rev., 2019, 378, 32–65 CrossRef CAS.
- R.-B. Lin, S. Xiang, W. Zhou and B. Chen, Chem, 2020, 6, 337–363 CAS.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- Q. Wang and D. Astruc, Chem. Rev., 2020, 120, 1438–1511 CrossRef CAS PubMed.
- H. Bin Wu and X. W. Lou, Sci. Adv., 2017, 3, eaap9252 CrossRef PubMed.
- J. Ren, Y. Huang, H. Zhu, B. Zhang, H. Zhu, S. Shen, G. Tan, F. Wu, H. He, S. Lan, X. Xia and Q. Liu, Carbon Energy, 2020, 2, 176–202 CrossRef CAS.
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. Paik Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef CAS.
- T. D. Bennett and S. Horike, Nat. Rev. Mater., 2018, 3, 431–440 CrossRef.
- Z. Yu, L. Tang, N. Ma, S. Horike and W. Chen, Coord. Chem. Rev., 2022, 469, 214646 CrossRef CAS.
- N. Ma and S. Horike, Chem. Rev., 2022, 122, 4163–4203 CrossRef CAS PubMed.
- Z. Lin, J. J. Richardson, J. Zhou and F. Caruso, Nat. Rev. Chem, 2023, 7, 273–286 CrossRef CAS PubMed.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- F. Haase and B. V Lotsch, Chem. Soc. Rev., 2020, 49, 8469–8500 RSC.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- O. M. Yaghi, M. J. Kalmutzki and C. S. Diercks, in Introduction to Reticular Chemistry, 2019, pp. 177–195 Search PubMed.
- J. Dong, X. Han, Y. Liu, H. Li and Y. Cui, Angew. Chem., Int. Ed., 2020, 59, 13722–13733 CrossRef CAS PubMed.
- H. L. Nguyen, F. Gándara, H. Furukawa, T. L. H. Doan, K. E. Cordova and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 4330–4333 CrossRef CAS PubMed.
- S. K. Elsaidi, M. H. Mohamed, J. S. Loring, B. P. McGrail and P. K. Thallapally, ACS Appl. Mater. Interfaces, 2016, 8, 28424–28427 CrossRef CAS PubMed.
- R. Jangir, A. C. Kalita, D. Kaleeswaran, S. K. Gupta and R. Murugavel, Chem.–Eur. J., 2018, 24, 6178–6190 CrossRef CAS PubMed.
- W. Rong-Jia, Z. Hou-Gan, Z. Zhi-Yin, N. Guo-Hong and L. Dan, CCS Chem., 2020, 3, 2045–2053 Search PubMed.
- H.-S. Lu, W.-K. Han, X. Yan, C.-J. Chen, T. Niu and Z.-G. Gu, Angew. Chem., Int. Ed., 2021, 60, 17881–17886 CrossRef CAS PubMed.
- X. Li, J. Wang, F. Xue, Y. Wu, H. Xu, T. Yi and Q. Li, Angew. Chem., Int. Ed., 2021, 60, 2534–2540 CrossRef CAS PubMed.
- H.-G. Zhou, R.-Q. Xia, J. Zheng, D. Yuan, G.-H. Ning and D. Li, Chem. Sci., 2021, 12, 6280–6286 RSC.
- W.-K. Han, Y. Liu, X. Yan, Y. Jiang, J. Zhang and Z.-G. Gu, Angew. Chem., Int. Ed., 2022, 61, e202208791 CrossRef CAS PubMed.
- L. Sun, M. Lu, Z. Yang, Z. Yu, X. Su, Y.-Q. Lan and L. Chen, Angew. Chem., Int. Ed., 2022, 61, e202204326 CrossRef CAS PubMed.
- R.-J. Wei, P.-Y. You, H. Duan, M. Xie, R.-Q. Xia, X. Chen, X. Zhao, G.-H. Ning, A. I. Cooper and D. Li, J. Am. Chem. Soc., 2022, 144, 17487–17495 CrossRef CAS PubMed.
- C. E. Bien, K. K. Chen, S. C. Chien, B. R. Reiner, L. C. Lin, C. R. Wade and W. S. W. Ho, J. Am. Chem. Soc., 2018, 140, 12662–12666 CrossRef CAS PubMed.
- A. M. Wright, Z. Wu, G. Zhang, J. L. Mancuso, R. J. Comito, R. W. Day, C. H. Hendon, J. T. Miller and M. Dincă, Chem, 2018, 4, 2894–2901 CAS.
- C. E. Bien, Q. Liu and C. R. Wade, Chem. Mater., 2020, 32, 489–497 CrossRef CAS.
- Z. Cai, C. E. Bien, Q. Liu and C. R. Wade, Chem. Mater., 2020, 32, 4257–4264 CrossRef CAS.
- Q. Liu, N. Hoefer, G. Berkbigler, Z. Cui, T. Liu, A. C. Co, D. W. McComb and C. R. Wade, Inorg. Chem., 2022, 61, 18710–18718 CrossRef CAS PubMed.
- J. Teufel, H. Oh, M. Hirscher, M. Wahiduzzaman, L. Zhechkov, A. Kuc, T. Heine, D. Denysenko and D. Volkmer, Adv. Mater., 2013, 25, 635–639 CrossRef CAS PubMed.
- M. H. Mohamed, Y. Yang, L. Li, S. Zhang, J. P. Ruffley, A. G. Jarvi, S. Saxena, G. Veser, J. K. Johnson and N. L. Rosi, J. Am. Chem. Soc., 2019, 141, 13003–13007 CrossRef CAS PubMed.
- Q. Liu, S. G. Cho, J. Hilliard, T.-Y. Wang, S.-C. Chien, L.-C. Lin, A. C. Co and C. R. Wade, Angew. Chem., Int. Ed., 2023, 62, e202218854 CrossRef CAS PubMed.
- E. D. Metzger, C. K. Brozek, R. J. Comito and M. Dincă, ACS Cent. Sci., 2016, 2, 148–153 CrossRef CAS PubMed.
- R. J. Comito, K. J. Fritzsching, B. J. Sundell, K. Schmidt-Rohr and M. Dincă, J. Am. Chem. Soc., 2016, 138, 10232–10237 CrossRef CAS PubMed.
- E. D. Metzger, R. J. Comito, C. H. Hendon and M. Dincă, J. Am. Chem. Soc., 2017, 139, 757–762 CrossRef CAS PubMed.
- R. J. Comito, Z. Wu, G. Zhang, J. A. Lawrence III, M. D. Korzyński, J. A. Kehl, J. T. Miller and M. Dincă, Angew. Chem., Int. Ed., 2018, 57, 8135–8139 CrossRef CAS PubMed.
- E. D. Metzger, R. J. Comito, Z. Wu, G. Zhang, R. C. Dubey, W. Xu, J. T. Miller and M. Dincă, ACS Sustain. Chem. Eng., 2019, 7, 6654–6661 CrossRef CAS.
- H. D. Park, R. J. Comito, Z. Wu, G. Zhang, N. Ricke, C. Sun, T. Van Voorhis, J. T. Miller, Y. Román-Leshkov and M. Dincă, ACS Catal., 2020, 10, 3864–3870 CrossRef CAS.
- D. Denysenko, M. Grzywa, J. Jelic, K. Reuter and D. Volkmer, Angew. Chem., Int. Ed., 2014, 53, 5832–5836 CrossRef CAS PubMed.
- A. M. Wright, A. J. Rieth, S. Yang, E. N. Wang and M. Dincă, Chem. Sci., 2018, 9, 3856–3859 RSC.
- R. Röβ-Ohlenroth, B. Bredenkötter and D. Volkmer, Organometallics, 2019, 38, 3444–3452 CrossRef.
- L. Li, Y. Yang, M. H. Mohamed, S. Zhang, G. Veser, N. L. Rosi and J. K. Johnson, Organometallics, 2019, 38, 3453–3459 CrossRef CAS.
- G. M. Su, H. Wang, B. R. Barnett, J. R. Long, D. Prendergast and W. S. Drisdell, Chem. Sci., 2021, 12, 2156–2164 RSC.
- B. R. Barnett, H. A. Evans, G. M. Su, H. Z. H. Jiang, R. Chakraborty, D. Banyeretse, T. J. Hartman, M. B. Martinez, B. A. Trump, J. D. Tarver, M. N. Dods, L. M. Funke, J. Börgel, J. A. Reimer, W. S. Drisdell, K. E. Hurst, T. Gennett, S. A. FitzGerald, C. M. Brown, M. Head-Gordon and J. R. Long, J. Am. Chem. Soc., 2021, 143, 14884–14894 CrossRef CAS PubMed.
- S. Biswas, M. Grzywa, H. P. Nayek, S. Dehnen, I. Senkovska, S. Kaskel and D. Volkmer, Dalton Trans., 2009, 9226, 6487–6495 RSC.
- D. Denysenko, M. Grzywa, M. Tonigold, B. Streppel, I. Krkljus, M. Hirscher, E. Mugnaioli, U. Kolb, J. Hanss and D. Volkmer, Chem.–Eur. J., 2011, 17, 1837–1848 CrossRef CAS PubMed.
- P. Schmieder, D. Denysenko, M. Grzywa, B. Baumgärtner, I. Senkovska, S. Kaskel, G. Sastre, L. van Wüllen and D. Volkmer, Dalton Trans., 2013, 42, 10786–10797 RSC.
- P. Schmieder, M. Grzywa, D. Denysenko, M. Hambach and D. Volkmer, Dalton Trans., 2015, 44, 13060–13070 RSC.
- A. J. Rieth, Y. Tulchinsky and M. Dincã, J. Am. Chem. Soc., 2016, 138, 9401–9404 CrossRef CAS PubMed.
- H. Bunzen, M. Grzywa, R. Aljohani, H.-A. A. Krug von Nidda and D. Volkmer, Eur. J. Inorg. Chem., 2019, 2019, 4471–4476 CrossRef CAS.
- K. Knippen, B. Bredenkötter, L. Kanschat, M. Kraft, T. Vermeyen, W. Herrebout, K. Sugimoto, P. Bultinck and D. Volkmer, Dalton Trans., 2020, 49, 15758–15768 RSC.
- L. Yun, C. Xin-Yi, Z. Kai, Z. Xue-Wen, Z. Dong-Dong, Z. Wei-Xiong, C. Xiao-Ming and Z. Jie-Peng, CCS Chem., 2021, 4, 1587–1596 Search PubMed.
- S. Biswas, M. Tonigold, M. Speldrich, P. Kögerler, M. Weil and D. Volkmer, Inorg. Chem., 2010, 49, 7424–7434 CrossRef CAS PubMed.
- Y.-Q. Lan, S.-L. Li, H.-L. Jiang and Q. Xu, Chem.–Eur. J., 2012, 18, 8076–8083 CrossRef CAS PubMed.
- R. Röβ-Ohlenroth, M. Kraft, H. Bunzen and D. Volkmer, Inorg. Chem., 2022, 61, 16380–16389 CrossRef PubMed.
- P. Q. Liao, H. Chen, D. D. Zhou, S. Y. Liu, C. T. He, Z. Rui, H. Ji, J. P. Zhang and X. M. Chen, Energy Environ. Sci., 2015, 8, 1011–1016 RSC.
- Z. Chang, R.-B. Lin, Y. Ye, C. Duan and B. Chen, J. Mater. Chem. A, 2019, 7, 25567–25572 RSC.
- Y. Wang, N.-Y. Huang, X.-W. Zhang, H. He, R.-K. Huang, Z.-M. Ye, Y. Li, D.-D. Zhou, P.-Q. Liao, X.-M. Chen and J.-P. Zhang, Angew. Chem., Int. Ed., 2019, 58, 7692–7696 CrossRef CAS PubMed.
- Y. Xie, Y. Shi, H. Cui, R.-B. Lin and B. Chen, Small Struct., 2022, 3, 2100125 CrossRef CAS.
- D.-L. Chen, H. Shang, W. Zhu and R. Krishna, Chem. Eng. Sci., 2014, 117, 407–415 CrossRef CAS.
- B. Liang, X. Zhang, Y. Xie, R.-B. Lin, R. Krishna, H. Cui, Z. Li, Y. Shi, H. Wu, W. Zhou and B. Chen, J. Am. Chem. Soc., 2020, 142, 17795–17801 CrossRef CAS PubMed.
- Y. Xie, Y. Shi, E. M. Cedeño Morales, A. El Karch, B. Wang, H. Arman, K. Tan and B. Chen, J. Am. Chem. Soc., 2023, 145, 2386–2394 CrossRef CAS PubMed.
- Y. Ye, S. Xian, H. Cui, K. Tan, L. Gong, B. Liang, T. Pham, H. Pandey, R. Krishna, P. C. Lan, K. A. Forrest, B. Space, T. Thonhauser, J. Li and S. Ma, J. Am. Chem. Soc., 2022, 144, 1681–1689 CrossRef CAS PubMed.
- G.-D. Wang, H.-H. Wang, W.-J. Shi, L. Hou, Y.-Y. Wang and Z. Zhu, J. Mater. Chem. A, 2021, 9, 24495–24502 RSC.
- G.-D. Wang, Y.-Z. Li, W.-F. Zhang, L. Hou, Y.-Y. Wang and Z. Zhu, ACS Appl. Mater. Interfaces, 2021, 13, 58862–58870 CrossRef CAS PubMed.
- Q. Zhang, G.-N. Han, X. Lian, S.-Q. Yang and T.-L. Hu, Molecules, 2022, 27, 5929–5939 CrossRef CAS PubMed.
- D. Graham and G. McAnally, Heterocycl. Commun., 1999, 5, 377–378 CAS.
- J. Rouquerol, P. L. Llewellyn and F. Rouquerol, Stud. Surf. Sci. Catal., 2007, 160, 49–56 CrossRef CAS.
- K. S. Walton and R. Q. Snurr, J. Am. Chem. Soc., 2007, 129, 8552–8556 CrossRef CAS PubMed.
- P. Iacomi and P. L. Llewellyn, Adsorption, 2019, 25, 1533–1542 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectroscopic (NMR, ATR-IR, ESI-MS), crystallographic (PXRD), thermogravimetric (TGA), gas adsorption, and elemental analyses. See DOI: https://doi.org/10.1039/d4ra05563b |
| ‡ Present address: Materials Physics and Applications Division, Los Alamos National Laboratory, Los Alamos, NM 87545, USA. |
| § Present address: National Engineering Laboratory of Eco-Friendly Polymeric Materials (Sichuan), College of Chemistry, Sichuan University, Chengdu 610064, China. |
| This journal is © The Royal Society of Chemistry 2024 |