
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel environmental applications of green tea: sensing and remediation of Ag+ in aqueous system†
Ankita Doia,
Mainak Ganguly *b and
Priyanka Sharmab
*b and
Priyanka Sharmab
aDepartment of Biosciences, Manipal University Jaipur, Jaipur, 303007, Rajasthan, India
bDepartment of Chemistry, Manipal University Jaipur, Jaipur, 303007, Rajasthan, India. E-mail: mainak.ganguly@jaipur.manipal.edu
First published on 1st October 2024
Abstract
The strong fluorescence of green tea was quenched with Fe3+ because of ligand-to-metal charge transfer and subsequent formation of magnetite (Fe3O4) nanoparticles (heavy metal effect). Ag+ restored the lost fluorescence by confining iron particles (capped with Cl−) with the formation of AgCl. Thus, toxic Ag was sensed in the aqueous system with a linear detection range of 10−4 M to 10−7 M and a detection limit of 4.1 × 10−9 M. The sensing protocol was applied for natural samples to detect Ag+. Gallic acid was found to be the pivotal component in the tea extract used to design a sensing platform. The company of the green tea were also varied and obtained comparable results.
Introduction
Silver ions have a significant role in the metabolism of copper. Ag+ plays an important role in the glycolysis process for pyruvate and lactate formation. Silver ions have been known for their antibacterial activities since ancient times; hence the silver vessels are often used to hoard liquids or water to prevent it from any microbial infection. Ions of silver with micromolar concentrations (1 to 10 μM) are adequate to destroy bacteria in water.1,2 Salts of Ag such as AgNO3 are used in medicine, pharmacology, the electric industry, etc. Earlier silver nitrate and iodide were used in black and white photography.3 Ag+ and Ag nanoparticles (colloidal silver) are well-used in nanotechnology. It was reported that 10 ppm (10 mg L−1) of the quantity of silver has 90% of the Ag+ ions and only 10% Ag NPs.4 Some studies showed that silver ions have better antibacterial activity than AgNPs. It was reported that AgNPs produce Ag+ ions.1However, pollution due to industries is a matter of great concern all over the world. Pollution due to heavy metals is one of them. Not only industrial waste but also electronic waste, which kept on piling, emerged heavy metals from semiconductors and batteries. Among these metals, silver is one of them polluting the soil5 and water sources due to leaching. Silver ions and nanoparticles are found to be quite toxic. Depending on the doses and size, Ag has been proven to be toxic to bacteria,6 yeast, algae,7 crustaceans,8 and humans. They can cause alterations in the enzymes of the liver, levels of neurotransmitters, loss in weight, lethargy, or even death.9 Thus, Ag+ sensing was an active field of research. Mehta et al. and Sharma et al. summarized the sensing of Ag+ employing various existing methods.10,11
Green tea (GTE) is a well-known beverage in our daily life. It is also used in cosmetics,12 food,13 traditional medicines14 etc. However, in the synthesis of the nanoparticles, its use is growing day by day, because of the reducing and stabilizing properties of the polyphenols of GTE. Gottimukkala and Hao et al. reported the synthesis of iron nanoparticles using GTE, which is black and has a zero oxidation state of iron.15 Plachtová et al. synthesized iron and iron-oxide nanoparticles for the elimination of malachite green dye from water along with ecotoxicology.16
Iron-based nanoparticles are widely used for environmental remediation, e.g., toxic dye removals, removal of heavy metals, etc.10 due to electrostatic attraction, hydrogen bonding, etc. However, in recent times food sources are also used in fluorescence to detect pollutants such as heavy metals, etc. Reversibly, different nanoparticles are used for the detection of components of food sources.17 He et al. used green tea carbon dots for Fe3+ detection.18 Similarly, Patra et al. used green tea carbon dots for chromium(VI) detection.19
Conversely, ionic metals such as Cu2+ are used for fluorescence ionic probe formation and detection of herbicides such as glyphosate in green tea.20 Sequentially, components of green tea such as tannic acid used for the synthesis of Fe3O4-based ethanol sensing.21
However, iron nanoparticles for sensing applications have not been available in the literature. In the present work, Ag+ sensing selectively and sensitively employing iron hydrosol, passivated with green tea (GTEFe) fluorometrically is demonstrated for the first time. No report is so far available for the fluorometric use of green tea and iron hydrosol with environmental applications. The sensing of Ag+ was also compared with different groups (Table S1, ESI†).
Results and discussion
Green tea extract (GTE)
Green tea has polyphenols, which incorporate phenolic acids, flavonoids, flavanols, flavandiols, and flavonoids. These compounds might tally up to 30% of total dry weight. Out of total green tea polyphenols (GTPs), the majority of them are flavonols, normally regarded as catechins. In addition, there are phenolic acids such as gallic acids and distinguishing amino acids like theanine.Most goods made from green tea are extracts, either liquid or powder, with varying percentages of polyphenols (45–90%) and caffeine (0.4–10%). Epigallocatechin, epicatechin, EGCG, and epicatechin-3-gallate are the four primary types of catechins found in green tea.22
Various components are present in GTE. The colour of the GTE is greenish-yellow. The GTE extract was stored at 4 °C in the refrigerator for further usage. The GTE showed good fluorescence λex = 274 and λmax = 400 nm. The UV-Vis λex = 274 nm [Fig. 1].
 | ||
| Fig. 1 (a) Fluorescence spectra of GTE; (b) UV-Vis absorbance of GTE. | ||
GTEFe hydrosol
Fe3O4 nanoparticles (black hydrosol, GTEFe) were synthesized from green tea extract, made via submerging 250 mg green tea leaves (Symphony) for 10 h at room temperature (25 °C) and filtered through a Whatman filter paper. The procedure was green and energetically favourable. The green tea extract (GTE) was greenish-yellow in colour. No heat or external energy was supplied to obtain the extract. Moreover, our synthetic protocol was simple, one pot, and cost-effective. Synthesized Fe3O4 nanoparticles were non-magnetic, evident from using a powerful magnet (Scheme 1). | ||
| Scheme 1 Schematic representation for the formation of GTEFe. | ||
Sensing of Ag+
After the addition of the Fe3+, the fluorescence was strongly quenched with the formation of GTEFe at λem 400 nm. Such quenched fluorescence was restored after the addition of Ag+ in GTEFe to form AgGTEFe (λem 460 nm). Ag+-induced fluorescence enhancement was quite selective and sensitive. Other metal ions in lieu of Ag+ (Hg2+, Ba2+, Cu2+, Ni2+, Na+, Ca2+, Al3+, Fe3+, K+, Cr3+ and Zn2+) were used. However, no enhancement was observed for other metal ions, unlike Ag+ (Fig. 2 and S1, ESI†). We also performed chloroauric acid in lieu of silver nitrate. However, fluorescence enhancement was observed for silver only. The blue shift for GTE was 67 nm for GTEFe and the red shift of 116 nm for AgGTEFe. Increased stroke shift was associated with increased selectivity of the analytes. Tunable stokes shifts warrant novel fluorescent probes for accuracy and precision in sensing for the upcoming generation applications.23 From the absorption spectra, it was observed that the λmax of GTE at 274 nm was blue shifted (λmax 270 nm) in GTEFe and further blue shifted (λmax 264 nm) in AgGTEFe with hypsochromic shift (Fig. S2, ESI†). Not only enhancement of fluorescence was observed by Ag+ in GTEFe, but also the highest absorbance at λmax 264 nm was noticed with Ag+ (in comparison to the addition of other metal ions) (Fig. S3, ESI†). The quantum yield for the GTE hydrosol was 1.68%, while the quantum yield for AgGTEFe was 2.02%. | ||
| Fig. 2 (a) Fluorescence spectra of GTE, GTEFe, AgGTEFe; (b) bar diagram regarding [I] of GTEFe in the presence of different metal ions; [Ag+] = 10−3 M. | ||
This proposed sensing platform was also compared with other reported platforms (Table S1, ESI†).
Sensitivity
Not only selectivity but also sensitivity is a vital factor for sensing applications. A monotonous increase of fluorescence in the range of 10−7 M to 10−3 M of [Ag+] was found. When [Ag+] > 10−3 M, a decrease in fluorescence was observed due to the heavy metal effect.24–26 At 10−2 M of [Ag+], the different signatures of the fluorescence spectrum were observed with quenched fluorescence intensity. A linear detection range, 10−4 M to 10−7 M by plotting fluorescence intensity vs. [Ag+] with a limit of detection (LOD) of 4.1 × 10−9 M was observed. I0 and I were fluorescence intensity before and after Ag+ treatment on GTEFe, respectively (Fig. 3). The absorbance at λmax 264 nm was increased gradually with increased [Ag+] in AgGTEFe (Fig. S5, ESI†). Silver nitrate is the only commonly available salt that is soluble in water. We added sodium salts with different counter anions in AgGTEFe and different extent of fluorescence was observed as in Fig. S6, ESI.† | ||
| Fig. 3 (a) Fluorescence spectra of GTE at different [Ag+]; (b) plot of [I] vs. [Ag+] and linear detection range of Ag+ detection (inset). | ||
Effect of pH
pH has a significant role in the emissive behaviour of GTEFe in the presence of Ag+. GTEFe (as mentioned in the Experimental section) had a pH of 4. Then titration of Ag+ was performed (Fig. 2). GTEFe at pH 2, 6, 8, 10, and 12 was made. pH was adjusted with dilute HCl and NaOH solution. No buffer was introduced with GTEFe. pH 4 had maximum enhancement with Ag+. The order of fluorescence intensities is as follows, pH 4 > pH 2 > pH 6 = pH 8 > pH 10 > pH 12. Higher pH produced AgOH,27 hindering the binding with Fe to display low fluorescence (Fig. 4). The fluorescence of AgGTEFe was maximum at pH 4. At higher pH, fluorescence decreased due to the formation of AgOH. As mentioned in the mechanism section, the fluorescence enhancement was related to the formation of AgCl. Thus, AgCl formation was hindered at higher pH. At strongly acidic pH also fluorescence enhancement was low the due to protonation of polyphenols in GTE.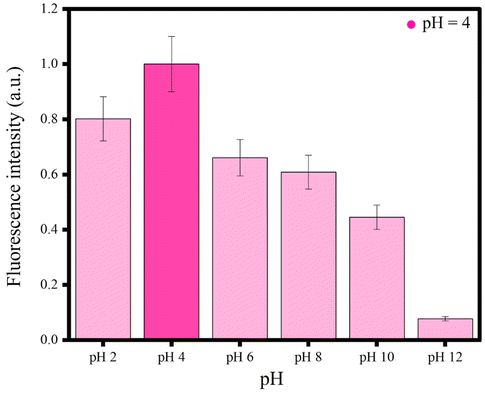 | ||
| Fig. 4 AgGTEFe at different pHs. | ||
Characterisation
The XRD spectra lacked characteristic diffraction peaks which indicated that the GTEFe nanoparticles were amorphous by nature.28,29 The broad peak at 2θ in between 20° and 30° corresponded to covered organic materials from the reaction, which were responsible for stabilizing the synthesized Fe particles (Fig. 5).28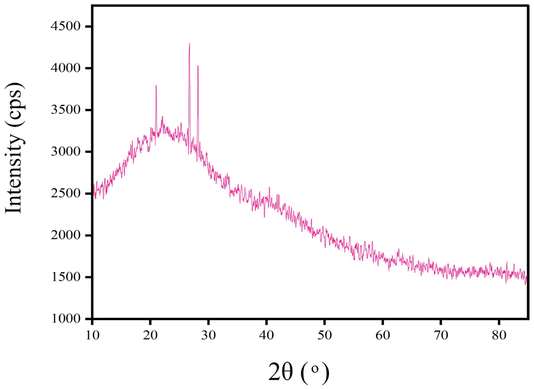 | ||
| Fig. 5 XRD pattern of GTEFe. | ||
The FTIR spectrum of the GTEFe nanoparticles exposed the estimated bands of v(Fe–OH) and v(Fe–O). The band at 624 cm−1 corresponds to the vibration of the Fe–O bond whereas 1621 and 3196 cm−1 bands represent v(Fe–OH). The v(Fe–O) band represents the presence of magnetite (Fig. 6).30
 | ||
| Fig. 6 FTIR spectrum of iron oxide (Fe3O4) nanoparticles. | ||
Particles were spheroids with a diameter of ∼70 nm, observed from FESEM images (Fig. 7).
 | ||
| Fig. 7 FESEM of GTEFe at (a) low resolution and (b) high resolution. | ||
TEM image indicated approximately 50 nm spherical particles (Fig. 8).
 | ||
| Fig. 8 TEM of GTEFe at (a) high resolution and (b) low resolution. | ||
The oxidation states of silver and iron in AgGTEFe were investigated. In this regard, XPS (Fig. 9) and XRD (S5, ESI†) were analysed. From XPS analysis, it was observed that in AgGTEFe, Ag was at +1 oxidation state. Binding energies of 367.3 eV and 373.7 eV corresponded to Ag(I)3d5/2 and Ag(I)3d3/2, respectively.31,32 The XPS peak of iron is comparatively broad due to the presence of Fe3+ and Fe2+. Binding energy 710.1 eV was due to Fe(II)2p3/2 while binding energy 711.4 eV was due to Fe(III)2p3/2.33,34 However, the peak of Fe(III)2p1/2 was not prominent for AgGTEFe in the presence of ionic silver. XRD patterns also had parity with XPS data. The 2θ values 38°, 44°, 64° and 77° corroborated (111), (200), (210), (311) of Ag2O, respectively.35–37 Magnetite is an inverse spinel mixed metal oxide consisting of Fe2+, Fe3+, and O2.38 The broad XRD 2θ peak around 20° to 30° corresponded amorphous nature of iron and 2θ 29.4° indicated (220) planes of magnetite (Fig. S5, ESI†).39,40 Thus, XRD and XPS analyses confirmed the presence of Fe3O4 and Ag2O. The particles in AgGTEFe were magnetic, attracted by a strong magnet (unlike GTEFe). So, the FESEM of the sample could not be obtained FESEM due to its magnetic nature. It is more likely that zero-valent iron nanoparticles in the hydrosol of AgGTEFe were converted to iron oxide during the sample preparation (washing and drying) of XRD and XPS.
 | ||
| Fig. 9 XPS of AgGTEFe (a) wide angle, (b) Fe and (c) Ag. | ||
DLS of freshly prepared GTEFe after 30 min of sonication was performed. The 100% particles of GTEFe were with particle size 410.9 nm ± 147.8 nm. Similarly, DLS of freshly prepared AgGTEFe after 30 min of sonication. The 86.5% particles of AgGTEFe had a particle size of 242.1 ± 72.08 nm and 13.5% particles of AgGTEFe had a particle size of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 774 ± 2461 nm. Both Ostwald ripening and digestive ripening were driving forces for the final size (Fig. S7, ESI†).41,42
774 ± 2461 nm. Both Ostwald ripening and digestive ripening were driving forces for the final size (Fig. S7, ESI†).41,42
The stability of GTEFe hydrosol was not great. A zeta potential of 4.8 mV indicated that the synthesized nanoparticles were passivated by the negatively charged capping agent of tea extract (oxidized form of polyphenol). On the contrary, AgGTEFe formed a precipitate with zeta potential 0 mV. Keeping this idea in mind, GTEFe as a nanotrap was designed to remove Ag+ from water. Two pieces of cotton of equal mass (0.5 g) were taken. One piece of cotton wool was dipped in 10 mL GTEFe and dried in air. GTEFe-impregnated cotton wool was put inside the funnel. The 10−3 M Ag+ solution was passed through unmodified and modified cotton wool separately. Unmodified cotton wool showed no significant change in the concentration of Ag+. However, modified cotton wool trapped Ag+ and the filtrate had a concentration 40 times lower (2.5 × 10−5 M) (Fig. 10).
 | ||
| Fig. 10 Removal of [Ag+] via cotton wool with and without the impregnation of GTEFe. | ||
The polyphenol present in the GTE could form a complex with Fe3+ resulting in fluorescence quenching in GTEFe due to charge transfer from ligand to metal.43 After that, Fe3+ was reduced to Fe0 (and subsequently converted to Fe3O4 due to aerial oxidation) and fluorescence quenching was observed due to the heavy metal effect.25,26,44
Mechanism
The inherent emissive property of GTE was lost with the addition of Fe3+. As Fe3+ itself has absorbance, the addition of Fe3+ might prevent the emission of the fluorescence.45,46 The added Fe3+ was converted to Fe0 and iron oxide47,48 With fluorescence quenching due to the heavy metal effect.24–26When Fe3+ was added to GTE, the fluorescence was remarkably quenched due to the absorption of energy by the ionic iron. Liu et al. illustrated the fluorescence quenching of the fluorophore via heavy metal ions with the absorption of energy of electronic transition (from conduction band to valence band).49
However, GTE contains polyphenols with reducing and stabilizing properties.50 GTE reduced Fe and formed GTE-passivated Fe3O4 nanoparticles. As a corollary, polyphenols in GTE were converted to their quinone form. From UV-Vis spectroscopy, it was observed that the absorption peaks of GTE and GTEFe were overlapped, indicating the chance of non-radiating energy transfer from GTE to Fe3O4 nanoparticles and quenching was observed.
The RP (radiating plasmon) model states that the metal structures's optical properties can be computed via Mie theory, electrodynamics, and/or Maxwell's equations. The metal colloids's extinction might result from either scattering or absorption, depending on the size, form of the particle, and Mie theory. The model predicted that nanoparticles often extinguish fluorescence as absorption predominates over scattering. Consideringly, it was implied that the “lossy surface waves” that cause the quenching of fluorescence generated electron oscillations that were unable to emit to the far field due to the impossibility of wavevector matching.51,52 In our present study, the emission from GTE in the proximity of Fe3O4 nanoparticles (in situ generated) became trapped plasmons, producing “lossy surface waves” and quenching of fluorescence was observed.
To understand further the mechanism of Ag+ adsorption with GTEFe, GTEFe was synthesized with Fe(NO3)3·9H2O in lieu of FeCl3. The restoration of fluorescence after the addition of Ag+ was also observed with GTEFe, synthesized from ferric nitrate. The fluorescence enhancement was low, and the spectral nature was broad. GTEFe (synthesized from FeCl3), being passivated with Cl−, produced AgCl, highly insoluble (1.6 × 10−10 at 25 °C) with making precipitate. Fe0 (produced in situ in the presence of GTE extract) also coagulated and settled down rendering GTE free to fluoresce. However, the unreacted metal ions present in the solution were attributed to be responsible for the red shift of the fluorescence. The increase of fluorescence of AgGTEFe [involving Fe(NO3)3·9H2O] with the addition of NaCl from outside further supported the role of Cl− on fluorescence enhancement. It is to be noted that chloride has the lowest quenching ability than bromide and iodide. That is why fluorescence restoration was not influenced due to the presence of Cl− ions.53
However, it was not the sole mechanism for fluorescence restoration, as GTEFe [synthesized from Fe(NO3)3·9H2O)] also showed somewhat fluorescence restoration. Ag+ might also bind to zero-valent iron and the drift of electron density from Fe0 to Ag+ might be the driving force of Ag+–Fe0 interaction, as Fe3+ is the most stable form of iron. As a result, the quenching effect was decreased with AgGTEFe.
GTE is used by many researchers for the synthesis of nanoparticles due to its antioxidant properties. Though green tea is a natural product containing several compounds,54 Vilchis-Nestor et al. demonstrated that gallic acid is the main compound for the antioxidant properties of green tea.55 To understand the mechanism better the experiment was repeated with an aqueous solution of pure gallic acid instead of GTE. Similar fluorometric behaviour was observed for gallic acid like GTE. In other words, the fluorescence was quenched with the addition of Fe and restored with the addition of Ag+ [Fig. S8, ESI†].
Muthusamy et al. demonstrated the formation of zero-valent metal nanoparticles from metal ion precursors by employing gallic acid. Gallic acid, as a corollary, was converted to O-quinone and capped synthesized nanoparticles.56
To be noted that GTE did not exhibit emissive behaviour with Ag+. So, restoration of fluorescence was the driving factor for fluorescence enhancement (Fig. S9, ESI†).
The process was repeated by producing tea extract obtained from the LIPTON company. The quenching for GTEFe and enhancement for AgGTEFe. So, the trends of the fluorescence behaviour were similar for GTE obtained from Symphony and LIPTON (Fig. S10, ESI†).
Effect of temperature
The fluorescence behaviour of AgGTEFe was gauged at different temperatures after ageing AgGTEFe at different temperatures for 30 min. At low temperatures (10 °C) fluorescence was increased in comparison to room temperature due to decreased Brownian motion.57 Slow increment of temperature generated a competition between increased Brownian motion and the interaction between Ag+ & GTEFe. The first factor decreased fluorescence intensity, while the second factor increased fluorescence intensity. Thus, there were no significant changes in fluorescence behaviour from 20 °C to 50 °C (concerning room temperature 30 °C). From 60 °C, fluorescence was increased and at 70 °C, the highest increment was observed. This was because of the higher Ag+ and Fe interaction, rendering GTE free. Further increase in temperature caused a huge increase in Brownian motion (Fig. 11). | ||
| Fig. 11 Plot of IT/IRT at different temperatures, (b) schematic representation of restoration of fluorescence with Ag+. | ||
Real water analysis
Natural water samples were gathered from multiple sources, such as the Yamuna River in Mathura, India, rain, tap and drinking water from Jaipur.Varying quantities of Ag+ were added to the water. The above-discussed fluorometric sensing techniques were used for Ag+ detection in natural water. The obtained data showed that the actual spiked concentrations were quite close to the estimated data.
Adsorption of Ag+ on GTEFe
For analyzing the efficacy of GTEFe in the removal of Ag+ from solution and the nature of adsorption, various adsorption isotherms were fitted with the obtained data. Langmuir adsorption isotherm indicates homogenous sites of adsorption and monolayer of adsorbate on the adsorbent following the following equation.
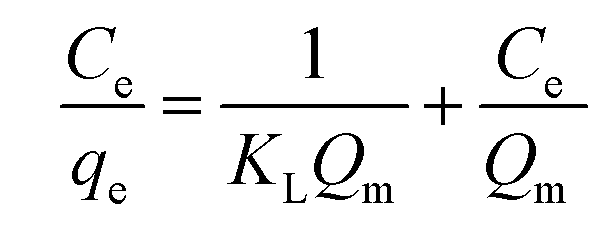 | (1) |
 | (2) |
A straight line slope, established among log(qe) and log(Ce), provides the intensity of adsorption 1/n. KF denotes Freudlich constant. Multilayered adsorption explanation is provided by Elovich adsorption isotherm.
 | (3) |
Maximum adsorption capacity (Qm) for Langmuir adsorption isotherm was 2.24 mg g−1. The coefficient of determination, R2 value came out to be 0.981. However, R2 values for Freundlich and Elovich were 0.883 and 0.948, respectively. The R2 value for Elovich came closer to the R2 value of the Langmuir plot. However, the best-fitted R2 determined that Ag forms a monolayer on the adsorbent GTEFe. Langmuir adsorption isotherm traits are better understood by dimensionless constant RL.
 | (4) |
 | ||
| Fig. 12 (a) Plot qe vs. Ce (b) Langmuir (c) Freundlich (d) Elovich isotherms for Ag+ adsorption on GTEFe. | ||
Experimental
Materials and instruments
The chemicals used in this study were of analytical calibre. In the entire study, distilled water was utilized. Glassware was washed using newly prepared aqua regia then soap-water and with plenty of distilled water. Glassware was completely dried before utilisation. Green tea was of Tea City Symphony (brand) with licence number 10013031000840. Green tea was purchased from a supermarket in Jaipur, Rajasthan. All metal salts including FeCl3 were procured from Sigma Aldrich except AgNO3. AgNO3 was purchased by Merck Specialities Private Limited respectively. Whatman paper of Whatman™ Cat no 1001 125 was used. To analyse fluorescence at room temperature Horiba FluoroMax-4 spectrometer was utilized. For FESEM, JEOL Make JSM-7610FPlus FESEM at SAIF, a high resolution (1 kV 1.0 nm, 15 kV 0.8 nm) was used. For XRD analyses, Rigaku makes an automated multipurpose X-ray Diffractometer (model: SMARTLAB) was utilized. Omicron ESCA (Electron Spectroscope for Chemical Analysis), Oxford Instrument Germany (resolution 0.60 eV) used for XPS. Model-FEI Tecnai G2 20 was used for TEM.Synthesis of GTE
Green tea (1 g) was soaked in 100 mL of distilled water for 10 h. After 10 h, the formed extract was filtered with the Whatman paper. The filtrate (extract) was stored at 4 °C and the used leaves were discarded.Synthesis of GTEFe
The 39 mL of freshly prepared green tea extract was put into a beaker and 13 mL of FeCl3 (6.1 × 10−2 M) solution was added into it (ratio 3:1). A black colour hydrosol (GTEFe) was formed.
Error analysis
All the experiments were performed thrice independently, and the error bar was calculated from the standard deviation of the obtained data.Conclusions
For the first time, green tea extract produced at room temperature was used as a sensing platform for water contaminants. The selectivity and sensitivity of the Ag+ detection for prototype applications are promising. The sensing platform was energetically favourable and cost-effective. This research will open up a new window for the scientist venturing into the field of material chemistry and environmental nanoscience. A highly used beverage has been utilized here for the first time for environmental remediation. More research is warranted to understand the effects of various components inside the tea extract for making a fluorescence platform.Data availability statement
All the data are included in ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to CSIR (Council of Scientific & Industrial Research) for financial assistance, SAIF (MUJ) and CAF (MUJ) for providing instrumental facilities.Notes and references
- A. Hamad, K. S. Khashan and A. Hadi, J. Inorg. Organomet. Polym. Mater., 2020, 30, 4811–4828 CrossRef CAS.
- S. Paul, P. Barman, N. Dey and M. Watkinson, Sens. Diagn., 2024, 3, 946–967 RSC.
- B. Tepla, K. Demnerova and H. Stiborova, J. Cult. Herit., 2020, 44, 218–228 CrossRef.
- J. R. Swathy, M. Udhaya Sankar, A. Chaudhary, S. Aigal, Anshup and T. Pradeep, Sci. Rep., 2014, 4, 7161 CrossRef CAS PubMed.
- S. I. Kolesnikov, N. I. Tsepina, L. V. Sudina, T. V. Minnikova, K. S. Kazeev and Y. V. Akimenko, Appl. Environ. Soil Sci., 2020, 2020, 9 Search PubMed.
- Y. Choi, H. A. Kim, K. W. Kim and B. T. Lee, J. Environ. Sci., 2018, 66, 50–60 CrossRef CAS PubMed.
- I. Moreno-Garrido, S. Pérez and J. Blasco, Mar. Environ. Res., 2015, 111, 60–73 CrossRef CAS PubMed.
- A. Ivask, I. Kurvet, K. Kasemets, I. Blinova, V. Aruoja, S. Suppi, H. Vija, A. Kakinen, T. Titma, M. Heinlaan, M. Visnapuu, D. Koller, V. Kisand and A. Kahru, PLoS One, 2014, 9, 14 CrossRef PubMed.
- N. Hadrup and H. R. Lam, Regul. Toxicol. Pharmacol., 2014, 68, 1–7 CrossRef CAS PubMed.
- P. K. Mehta, L. N. Neupane, S. H. Park and K. H. Lee, J. Hazard. Mater., 2021, 411, 125041 CrossRef CAS PubMed.
- P. Sharma, M. Ganguly and A. Doi, Appl. Nanosci., 2024, 1–13 Search PubMed.
- Y. Q. Xu, S. Q. Chen, H. B. Yuan, P. Tang and J. F. Yin, J. Food Sci. Technol., 2012, 49, 362–367 CrossRef CAS PubMed.
- Q. V. Vuong, C. E. Stathopoulos, M. H. Nguyen, J. B. Golding and P. D. Roach, Food Rev. Int., 2011, 27, 227–247 CrossRef CAS.
- V. V Chopade, A. A. Phatak, A. B. Upaganlawar and A. A. Tankar, Pharmacogn. Rev., 2008, 2, 157–162 Search PubMed.
- D. Z. Gottimukkala and P. Harika Reddy, J. Nanomed. Biother. Discovery, 2017, 7, 1–4 Search PubMed.
- P. Plachtová, Z. Med, R. Zbo, R. S. Varma and B. Maršálek, ACS Sustain. Chem. Eng. Iron, 2018, 6, 8679–8687 CrossRef PubMed.
- A. Kushwaha, G. Singh, U. K. Gaur and M. Sharma, Mater. Adv., 2024, 5, 4378–4400 RSC.
- Y. He, S. Liu, F. Xie, Y. Zhou and X. Yang, J. Food Compos. Anal., 2024, 132, 106332 CrossRef CAS.
- P. Swagata, A. K. Golder and R. V. S. Uppaluri, Opt. Mater., 2024, 154, 115767 CrossRef.
- C. Siying, Z. Yiwan, S. Xinxiang, P. Xiutan, F. Haiyan and S. Yuanbin, Food Chem., 2004, 447, 138859 Search PubMed.
- S. Ananthi, M. Kavitha, E. R. Kumar, A. Balamurugan, Y. Al-Douri, H. K. Alzahrani, A. A. Keshk, T. M. Habeebullah, H. Abdel-Hafez Shams and N. M. El-Metwaly, Sensor. Actuator. B Chem., 2024, 352, 131071 CrossRef.
- S. M. Chacko, P. T. Thambi, R. Kuttan and I. Nishigaki, China’s Med., 2010, 5, 1–9 CrossRef PubMed.
- T. B. Ren, W. Xu, W. Zhang, X. X. Zhang, Z. Y. Wang, Z. Xiang, L. Yuan and X. B. Zhang, J. Am. Chem. Soc., 2018, 140, 7716–7722 CrossRef CAS PubMed.
- S. S. Tan, S. J. Kim and E. T. Kool, J. Am. Chem. Soc., 2011, 133, 2664–2671 CrossRef CAS PubMed.
- A. Sekar, R. Yadav and N. Basavaraj, New J. Chem., 2021, 45, 2326–2360 RSC.
- I. K. Kandela and R. M. Albrecht, Scanning, 2007, 29, 152–161 CrossRef CAS PubMed.
- I. E. Layla Badr, Chem. Phys. Lett., 2022, 800, 139681 CrossRef.
- E. C. Njagi, H. Huang, L. Stafford, H. Genuino, H. M. Galindo, J. B. Collins, G. E. Hoag and S. L. Suib, Langmuir, 2011, 27, 264–271 CrossRef CAS PubMed.
- M. A. J. Kouhbanani, N. Beheshtkhoo, S. Taghizadeh, A. M. Amani and V. Alimardani, Adv. Nat. Sci. Nanosci. Nanotechnol., 2019, 10, 015007 CrossRef CAS.
- S. Kamilah, C. Soh, A. Azzura, A. Rahman and M. Shamsuddin, Malaysian J. Anal. Sci., 2018, 22, 768–774 Search PubMed.
- H. Chen, G. Zhang, W. Zhang and W. Gao, RSC Adv., 2023, 13, 11450–11456 RSC.
- P. Postolache, V. Petrescu, D. D. Dumitrascu, C. Rimbu, N. Vrînceanu and C. R. Cipaian, Chem. Eng. Commun., 2016, 203, 649–659 CrossRef CAS.
- M. Yuan, C. Nan, Y. Yang, G. Sun, H. Li and S. Ma, ACS Omega, 2017, 2, 4269–4277 CrossRef CAS PubMed.
- G. Wang, Y. Yang, X. Xu, S. Zhang, Z. Yang, Z. Cheng, J. Xian, T. Li, Y. Pu, W. Zhou, G. Xiang and Z. Pu, Molecules, 2023, 28, 1–18 Search PubMed.
- K. Shameli, M. Bin Ahmad, A. Zamanian, P. Sangpour, P. Shabanzadeh, Y. Abdollahi and M. Zargar, Int. J. Nanomed., 2012, 7, 5603–5610 CrossRef CAS PubMed.
- K. Jyoti, M. Baunthiyal and A. Singh, J. Radiat. Res. Appl. Sci., 2016, 9, 217–227 CAS.
- S. P. Vinay, Udayabhanu, H. N. Sumedha, G. Nagaraju, S. Harishkumar and N. Chandrasekhar, Appl. Organomet. Chem., 2020, 34, e5830 CrossRef CAS.
- E. Gürsoy, G. B. Vonbun-Feldbauer and R. H. Meißner, J. Phys. Chem. Lett., 2023, 14, 6800–6807 CrossRef PubMed.
- B. Guan, D. Ding, L. Wang, J. Wu and R. Xiong, Mater. Res. Express, 2017, 4, 056103 CrossRef.
- M. E. Compeán-Jasso, F. Ruiz, J. R. Martínez and A. Herrera-Gómez, Mater. Lett., 2008, 62, 4248–4250 CrossRef.
- S. T. Gentry, S. F. Kendra and M. W. Bezpalko, J. Phys. Chem. C, 2011, 115, 12736–12741 CrossRef CAS.
- M. Ganguly, A. Pal and T. Pal, J. Phys. Chem. C, 2012, 116, 9265–9273 CrossRef CAS.
- Z. Markova, P. Novak, J. Kaslik, P. Plachtova, M. Brazdova, D. Jancula, K. M. Siskova, L. Machala, B. Marsalek, R. Zboril and R. Varma, ACS Sustain. Chem. Eng., 2014, 2, 1674–1680 CrossRef CAS.
- V. A. Lavrenko, A. I. Malyshevskaya, L. I. Kuznetsova, V. F. Litvinenko and V. N. Pavlikov, Powder Metall. Met. Ceram., 2006, 45, 476–480 CrossRef CAS.
- C. R. Lohani, J. M. Kim and K. H. Lee, Bioorganic Med. Chem. Lett., 2009, 19, 6069–6073 CrossRef CAS PubMed.
- C. R. Lohani and K. H. Lee, Sensor. Actuator. B Chem., 2010, 143, 649–654 CrossRef CAS.
- M. Ganguly, S. Dib, U. Kurien, R. B. Rangel-Alvarado, Y. Miyahara and P. A. Ariya, J. Phys. Chem. C, 2018, 122, 18690–18704 CrossRef CAS.
- M. Ganguly, S. Dib and P. A. Ariya, Sci. Rep., 2018, 8, 1–10 CAS.
- J. Liu, Q. Zhang, W. Xue, H. Zhang, Y. Bai, L. Wu, Z. Zhai and G. Jin, Nanomaterials, 2019, 9, 1294 CrossRef CAS PubMed.
- Z. Wang, Y. Huang, D. Lv, G. Jiang, F. Zhang and A. Song, Green Chem. Lett. Rev., 2019, 12, 197–207 CrossRef CAS.
- J. R. Lakowicz, Anal. Biochem., 2005, 337, 171–194 CrossRef CAS PubMed.
- S. Mamta, G. Mainak and S. Priyanka, Nanoscale Adv., 2024, 6, 4545–4566 RSC.
- R. Giri, Spectrochim. Acta, Part A, 2004, 60, 757–763 CrossRef PubMed.
- S. P. J. Namal Senanayake, J. Funct. Foods, 2013, 5, 1529–1541 CrossRef CAS.
- A. R. Vilchis-Nestor, V. Sánchez-Mendieta, M. A. Camacho-López, R. M. Gómez-Espinosa, M. A. Camacho-López and J. A. Arenas-Alatorre, Mater. Lett., 2008, 62, 3103–3105 CrossRef CAS.
- N. Muthusamy, P. Kanniah, P. Vijayakumar, U. Murugan, D. S. Raj and U. Sankaran, J. Inorg. Organomet. Polym. Mater., 2021, 31, 4693–4709 CrossRef CAS.
- M. Ganguly, A. Pal, Y. Negishi and T. Pal, Chem.–A Eur. J., 2012, 18, 15845–15855 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05545d |
| This journal is © The Royal Society of Chemistry 2024 |