
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenzimidazole–dioxoisoindoline conjugates as dual VEGFR-2 and FGFR-1 inhibitors: design, synthesis, biological investigation, molecular docking studies and ADME predictions†
Heba T. Abdel-Mohsen *a and
Amira M. Nageebb
*a and
Amira M. Nageebb
aChemistry of Natural and Microbial Products Department, Pharmaceutical and Drug Industries Research Institute, National Research Centre, Dokki, P.O. 12622, Cairo, Egypt. E-mail: hebabdelmohsen@gmail.com; ht.abdel-mohsen@nrc.sci.eg
bHigh Throughput Molecular and Genetic Technology Lab, Center of Excellence for Advanced Sciences, Biochemistry Department, Biotechnology Research Institute, National Research Centre, Dokki, P.O. 12622, Cairo, Egypt
First published on 12th September 2024
Abstract
In this investigation, a new series of benzimidazole–dioxo(benzo)isoindoline hybrids 8a–o were rationally designed and synthesized. All the synthesized hits 8a–o were investigated for their VEGFR-2 inhibitory activity at 10 μM. The conjugates 8l and 8m demonstrated potent inhibitory activity of 87.61 and 80.69%, respectively. Further evaluation of 8l and 8m on FGFR-1 at 10 μM demonstrated % inhibition = 84.20 and 76.83%, respectively. Investigation of the growth inhibitory activity of the synthesized hits on NCI cancer cell lines showed that the benzimidazole–dioxobenzoisoindoline hybrid 8m exhibits the highest antiproliferative activity. It displayed growth inhibitory activity reaching 89.75%. Examination of the effect of 8m on the cell cycle of MCF7 cell line revealed its ability to induce arrest of the cell cycle at the G2/M phase and its potential to induce the apoptosis of the same cell line. Additionally, the benzimidazole–dioxobenzoisoindoline hybrid 8m had potent anti-migratory properties as evidenced by the delay in the wound closure in reference to untreated control cells. Molecular docking of 8m in the binding pockets of the VEGFR-2 and FGFR-1 proved its ability to occupy the binding pockets of both targets in type II inhibitor binding mode and to perform the essential interactions with the crucial amino acids in the binding pockets of both targets. Moreover, ADME prediction studies demonstrated the drug like properties of the synthesized benzimidazole–dioxoisoindolines 8a–o.
1. Introduction
Each year, more and more people are diagnosed with cancer,1 which presents a problem for world health.1,2 Genetic changes and/or overexpression of certain protein kinases (PKs) are linked to many cancer forms.3,4 As a result, PKs have therefore received a lot of interest recently as potential biological targets for the discovery of new tailored medications for the prevention and control of cancer progression.3–8Numerous studies have reported a link between the pathophysiology of cancer and the development of new blood vessels (angiogenesis)6,9 which is necessary for the growth and dissemination of cancer by supplying it with nutrients and removing waste. Thus, a popular strategy for treating cancer is to target the protein kinases that start and maintain the angiogenic process.10 A tyrosine kinase system called vascular endothelial growth factor (VEGF) and its receptors (VEGFRs) play a critical role in angiogenesis in both healthy and pathological circumstances.10–12 In contrast to healthy tissues, VEGFR-2 is overexpressed in a number of cancer types.13,14 Additionally, the FGFR family is a group of four isoforms, FGFR-1 to FGFR-4, that are expressed on the cell membrane and take part in a number of critical physiological and pathological processes, including angiogenesis, cell migration and survival.15 Different forms of solid tumors have been shown to overexpress FGFRs.16 Therefore, it is thought that inhibiting FGFRs using small molecules is an appealing strategy for the development of innovative anticancer agent.17–20
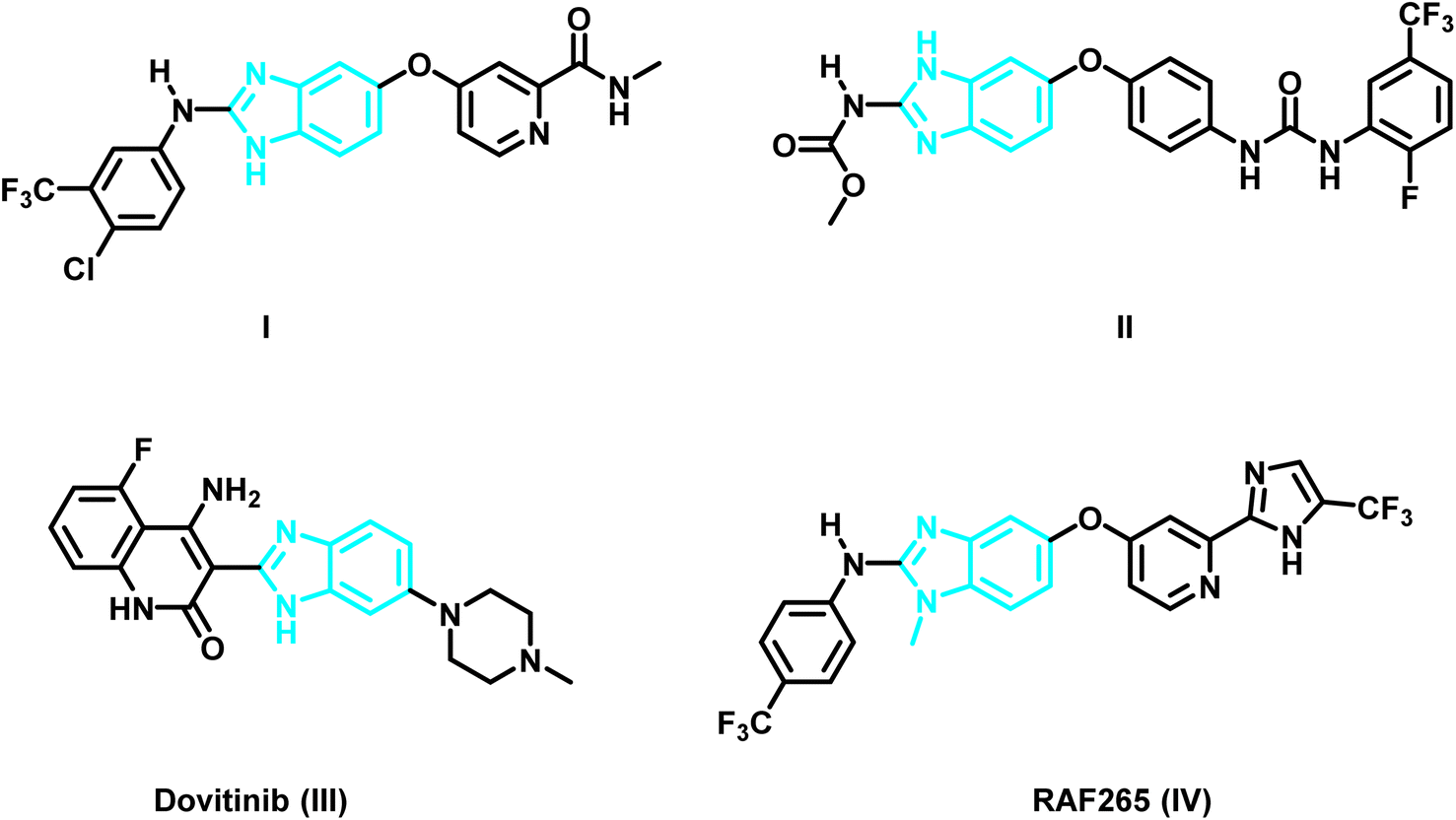
As time went on, researchers became interested in benzimidazoles as a scaffolding core that could be customized for the creation of new chemotherapeutic agents.19–24 For example, the protein data bank shows the crystal structure of the benzimidazole derivative I (PDB ID: 2QU5) (Fig. 1 and 2) in the active pocket of VEGFR-2. It demonstrated a Ki of 8.7 nM against VEGFR-2.25 Masaichi Hasegawa et al. revealed a novel series of benzimidazole–ureas as inhibitors of VEGFR-2. The derivative II showed IC50 = 3.5 nM and its crystal structure in the active site of VEGFR-2 is depicted in the PDB under the ID: 2OH4.26 Dovitinib (III), a multi-kinase inhibitor that inhibits VEGFR1-3, FGFR1-3, and PDGFR, has also been used to treat a number of tumor types, including renal cell carcinoma, gastrointestinal stromal tumors, colorectal cancer, and ovarian cancer.27,28 The X-ray crystal structure of Dovintib with the FGFR-1 active site was reported by Bunney, T. et al. (PDB ID: 5AM6).29 Additionally, melanoma and colorectal cancer were found to be sensitive to the dual BRAF/VEGFR-2 inhibitor RAF265 (IV) (PDB ID: 5CT7)30 (Fig. 1).
 | ||
| Fig. 1 Benzimidazole-based multi-kinase inhibitors. | ||
 | ||
| Fig. 2 Suggested strategy for the design of benzimidazole–dioxo(benzo)isoindoline conjugates VI. | ||
Recently, our research group have identified the essential pharmacophoric features of type II VEGFR-2, FGFR-1 and BRAF inhibitors.19 Taking the crystal structure of the benzimidazole derivative I in the binding site of VEGFR-2 (PDB ID: 2QU5) as an example,25 the derivative binds to the inactive form of VEGFR-2 in type II inhibitors binding mode. The pyridine moiety occupies the hinge region where it is involved in hydrogen bonding, through the nitrogen of the pyridine and the carboxamide NH, with Cys919, while fused benzene ring of the benzimidazole system acts as a spacer enabling the 2-amino-imidazole moiety to be involved through one of the two N–H groups and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group in hydrogen bonding with Glu885 and Asp1046, respectively, while the phenyl group at the two position is settled in hydrophobic back pocket forming hydrophobic interactions with the amino acids lining this pocket (Fig. 2).
N group in hydrogen bonding with Glu885 and Asp1046, respectively, while the phenyl group at the two position is settled in hydrophobic back pocket forming hydrophobic interactions with the amino acids lining this pocket (Fig. 2).
In reference to the identified essential fragments on one hand19 and the binding mode of the benzimidazole derivative I in the VEGFR-2's active site on the other hand,25 we have reported a new series of 2,5-diaryl benzimidazoles of potent activity on diverse kinases. Compound V is a representative example from the series and it showed IC50 values of 0.93, 3.74, 0.25 μM, against VEGFR-2, FGFR-1 and BRAF, respectively. Additionally, potential activity on NCI cancer cell lines was detected with GI50 up to 0.66 μM.19
As a continuation of our previous work, the present approach aims at replacement of the benzylidene moiety of compound V with dioxo(benzo)isoindoline moiety for the design of a new class of benzimidazole–dioxo(benzo)isoindoline conjugates of the general structure VI (Fig. 2) as type II VEGFR-2 and FGFR-1 inhibitors. The design approach considers that the benzimidazole nucleus's NH group will remain free such that the benzimidazole would be settled in target kinases' gate area and are expected to be stabilized by the engagement of the NH and CN groups through hydrogen bonding with Glu885 and Asp1046 of VEGFR-2's, and Glu531 and Asp641 of FGFR-1's binding pockets, respectively. Additionally, the 5-position would be functionalized by the inclusion of a dioxo(benzo)isoindoline moieties to fit in the hinge region of the target kinases where the carbonyl group would participate in hydrogen bonding with the key amino acids Cys919 (VEGFR-2) and Ala564 (FGFR-1) in the hinge region. In the meantime, the two position of the benzimidazole scaffold would be decorated with hydrophobic aryl moieties to be directed toward the hydrophobic back pocket of the target kinases (Fig. 2).
After the proposed compounds are synthesized, their inhibitory activity on VEGFR-2 will be evaluated. Next, the inhibitory potency of the most potent candidate on FGFR-1 would be evaluated. In parallel, each of the synthesized candidates would be evaluated for its ability to inhibit the growth of various cancer cell lines. The most effective derivative would next be examined for its effects on the cell cycle and apoptosis of a selected cancer cell line. Additionally, a wound healing assay would be employed to further demonstrate the antiangiogenic ability of the most potent candidate. Moreover, molecular docking of the most effective candidate on the target kinases would be carried out to prove the design strategy.31 Ultimately, the submission of the synthesized derivatives to the SwissADME webtool would forecast their physicochemical features.32
2. Results and discussion
2.1. Chemistry
The initial 2-substituted-benzimidzole-5-carboxylic acids 4a–e were prepared by reacting the benzoic acid derivative 3 with different aryl aldehyde sodium bisulfite adducts 2a–e. Then, through a reaction with ethanol in the presence of sulfuric acid, the benzimidazole derivatives 4a–e were transformed into the ester congeners 5a–e. The reaction of 5a–e with hydrazine hydrate produced the hydrazide derivatives 6a–e. Finally, the hydrazides 6a–e and acid anhydrides 7a–c were allowed to react in acetic acid to produce the target hybrids 8a–o (Scheme 1). | ||
| Scheme 1 Pathway for the synthesis of the benzimidazole–dioxo(benzo)isoindolines 8a–o. | ||
2.2. Biology
2.2.1.1 VEGFR-2 inhibitory activity. The capacity to inhibit VEGFR-2 at a concentration of 10 μM was tested for the designed and synthesized 2-(substituted)-N-(1,3-dioxoisoindolin-2-yl)-1H-benzo[d]imidazole-5-carboxamide 8a–o, sorafenib and the findings are shown in Table 1.
|
|||
|---|---|---|---|
| Compound ID | R | R1 | % Inhibition |
| 8a | 4-Cl | H | 5.23 ± 0.37 |
| 8b | 3-OMe | H | 37.94 ± 2.92 |
| 8c | 4-OMe | H | 71.11 ± 5.63 |
| 8d | 2,5-(OMe)2 | H | 43.48 ± 3.38 |
| 8e | 3,4,5-(OMe)3 | H | 48.90 ± 4.14 |
| 8f | 4-Cl | COOH | 53.54 ± 5.04 |
| 8g | 3-OMe | COOH | 60.37 ± 5.57 |
| 8h | 4-OMe | COOH | 34.54 ± 2.36 |
| 8i | 2,5-(OMe)2 | COOH | 24.87 ± 2.19 |
| 8j | 3,4,5-(OMe)3 | COOH | 38.56 ± 2.25 |
| 8k | 4-Cl | — | 21.16 ± 1.96 |
| 8l | 3-OMe | — | 87.61 ± 5.68 |
| 8m | 4-OMe | — | 80.69 ± 6.43 |
| 8n | 2,5-(OMe)2 | — | 47.19 ± 3.56 |
| 8o | 3,4,5-(OMe)3 | — | 9.87 ± 0.85 |
| Sorafenib | 99.63 ± 7.89 | ||

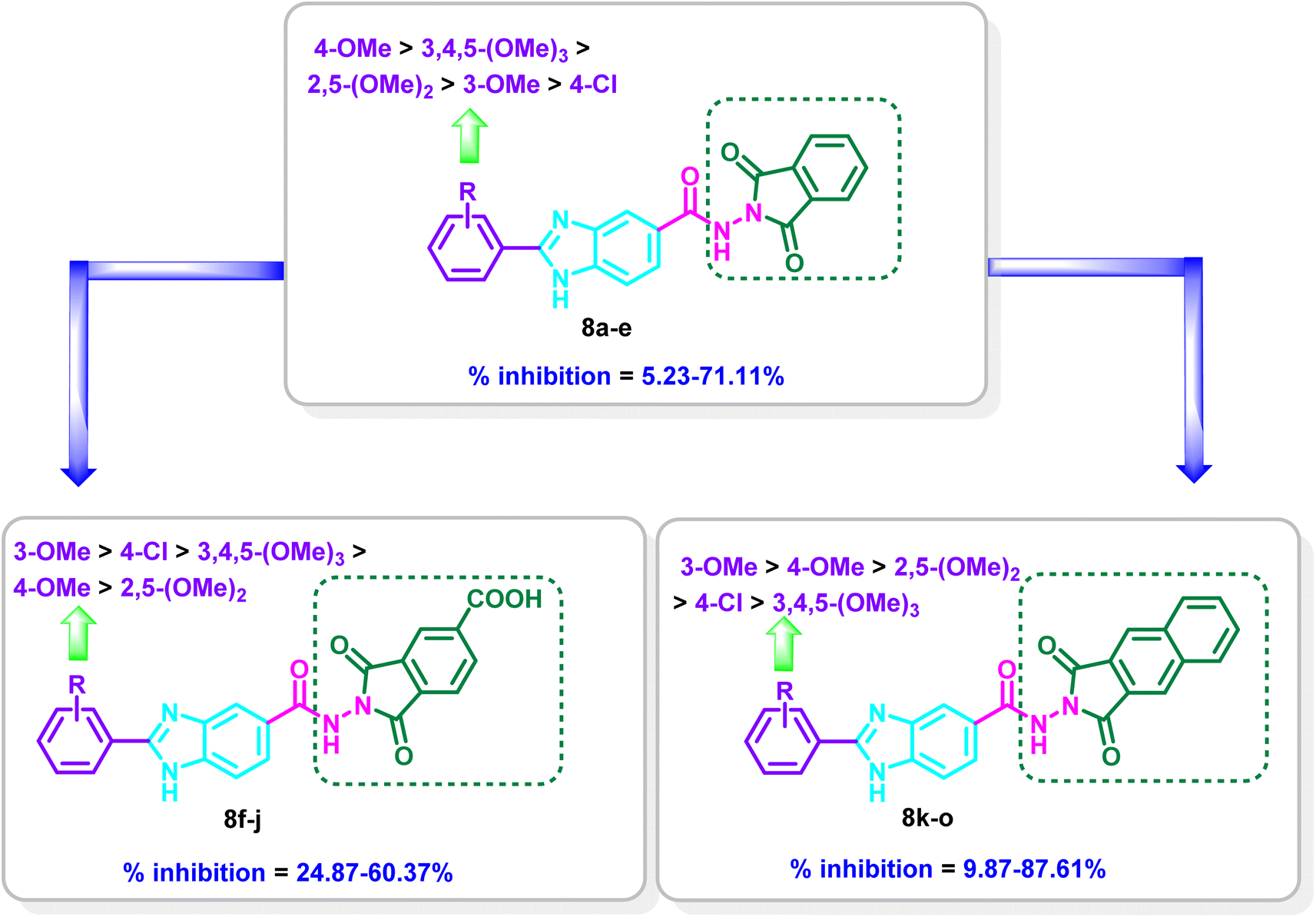
The nature of the substituents on the 2-aryl moiety of the benzimidazole core as well as on the dioxoisoindole moiety has a considerable impact on the degree of VEGFR-2 inhibition. In series 8a–e, the 4-methoxyphenyl-benzimidazole derivative 8c displayed the most potent inhibitory activity with % inhibition = 71.11% (Table 1 and Fig. 3).
 | ||
| Fig. 3 Structure–activity relationship of the benzimidazoles–dioxo(benzo)isoindoline conjugates 8a–o on the inhibition percentage of VEGFR-2. | ||
Moderate inhibitory activity on VEGFR-2 was observed when the 4-methoxyphenyl group was replaced with 3-methoxyphenyl, 2,5-dimethoxyphenyl or 3,4,5-trimethoxyphenyl moiety in 8b, 8d and 8e, respectively (% inhibition = 37.94–48.90%). On the other hand, the inhibitory action was drastically reduced when the 4-methoxyphenyl group in 8c was replaced with the 4-chlorophenyl group in 8a (71.11% inhibition in 8c versus 5.23% inhibition in 8a, respectively) (Table 1 and Fig. 3).
Introduction of a carboxylic group at the 5-position of the dioxoisoindoline moiety to yield series 8f–j resulted in increasing the potency for 8f (% inhibition = 53.54%) and 8g (% inhibition = 60.36%) in reference to 8a (% inhibition = 5.23%) and 8b (% inhibition = 37.94%), while a decrease in the potency was observed for 8h–j (% inhibition = 24.87–38.56%) in comparison to 8c–e (% inhibition = 43.48–71.11%). In series 8f–j, compound 8g incorporating 3-methoxyphenyl moiety displayed the most potent inhibitory activity (% inhibition = 60.37%). Meanwhile, the 4-chlorophenyl derivative 8f, the 4-methoxyphenyl derivative 8h, 2,5-dimethoxyphenyl 8i and 3,4,5-trimethoxyphenyl derivative 8j showed reduction in potency (% inhibition = 24.87–53.54%) (Table 1 and Fig. 3).
Replacement of 1,3-dioxoisoindolin-2-yl in series 8a–e with 1,3-dioxo-1,3-dihydro-2H-benzo[f]isoindol-2-yl in series 8k–o resulted mainly in increasing the % inhibition. The derivative 8o is the only exception where a decrease in the potency was found relative to 8e (% inhibition = 9.87% in 8o versus 48.90% in 8e). Compound 8l displayed the most potent activity on VEGFR-2 with % inhibition = 87.61%. Shifting of the 3-methoxy group to the 4-position to give 8m revealed a slight decrease in the inhibitory activity (inhibition % = 80.69%), while the 2,5-dimethoxyphenyl derivative 8n displayed moderate activity of 47.19% and the 3,4,5-trimethoxyphenyl derivative 8o revealed weak % inhibition of 9.87% (Table 1 and Fig. 3).
2.2.1.2 Determination of inhibitory activity of 8l and 8m on FGFR-1. Compounds 8l, 8m and sorafenib where further analysed for their inhibitory activity on FGFR-1 at 10 μM and the % inhibition where depicted in Table 2.
|
|||
|---|---|---|---|
| Compound ID | R | % Inhibition of VEGFR-2 | % Inhibition of FGFR-1 |
| 8l | 3-OMe | 87.61 ± 5.18 | 84.20 ± 7.43 |
| 8m | 4-OMe | 80.69 ± 6.41 | 76.83 ± 5.06 |
| Sorafenib | — | 99.63 ± 7.89 | 92.37 ± 3.84 |

The benzimidazole–dioxobenzoisoindolines 8l and 8m demonstrated promising inhibitory activity on FGFR-1 with % inhibition = 84.20 and 76.83%, respectively. It is obvious that compound 8l demonstrated slight better inhibitory activity on VEGFR-2 and FGFR-1 over 8m.
2.2.2.1 Antiproliferative activity. The capacity of each target benzo[d]imidazole-1,3-dioxoisoindoline hybrids 8a–o to suppress the growth of NCI 60 cancer cell lines at 10 μM concentration was tested at NCI, and the findings are shown in Table 3.
| Cell line | GI% | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 8a | 8b | 8c | 8d | 8e | 8f | 8g | 8h | 8i | 8j | 8k | 8l | 8m | 8n | 8o | |
| a Showed no GI%.b Not detected. | |||||||||||||||
| Leukaemia | |||||||||||||||
| CCRF-CEM | 8.31 | 4.56 | —a | 5.49 | — | — | — | 16.52 | 4.00 | — | 34.48 | 21.28 | 45.83 | 23.52 | 5.97 |
| HL-60(TB) | — | 11.16 | — | 4.67 | 14.40 | — | 6.30 | 14.05 | 0.13 | 11.13 | 7.15 | 17.62 | 54.16 | — | 7.12 |
| K-562 | 11.40 | 9.03 | 3.99 | 5.26 | 3.22 | — | 3.90 | 3.13 | — | — | 7.72 | 10.61 | 55.96 | 5.51 | 4.09 |
| MOLT-4 | 0.17 | 4.97 | 7.00 | — | 6.94 | — | — | 0.82 | 5.04 | 4.70 | 27.46 | 27.04 | 75.51 | 5.04 | 10.84 |
| RPMI-8226 | 6.77 | 5.32 | 1.28 | 6.77 | — | 2.62 | 1.15 | 9.54 | 8.29 | — | 45.06 | 41.00 | 32.35 | 7.78 | 13.64 |
| SR | 18.06 | 22.26 | ndb | 8.63 | 26.01 | nd | 11.56 | 16.30 | 6.50 | 23.39 | nd | nd | 61.77 | nd | nd |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||||||||||
| Non-small cell lung cancer | |||||||||||||||
| A549/ATCC | 5.59 | 1.51 | — | 9.11 | 5.20 | — | — | 3.13 | 5.73 | 11.81 | 21.23 | — | 40.46 | — | 3.02 |
| EKVX | 13.33 | 4.13 | — | 10.43 | — | 4.11 | 1.71 | 0.72 | 6.38 | 6.40 | 0.60 | 0.21 | 13.77 | — | 2.63 |
| HOP-62 | 6.21 | 4.22 | — | 0.18 | 0.17 | — | — | — | 1.63 | 4.66 | — | — | 25.04 | 7.00 | 1.40 |
| HOP-92 | — | — | — | 2.01 | — | — | — | 17.03 | 1.51 | 0.40 | — | — | — | 3.75 | — |
| NCI-H226 | 10.20 | — | — | 3.78 | — | — | — | 16.02 | 4.32 | — | 7.71 | 1.96 | 21.07 | — | — |
| NCI-H23 | 10.05 | 0.67 | 1.28 | 3.23 | — | 1.71 | — | 0.49 | — | — | 1.64 | 4.46 | 20.56 | 1.15 | 2.27 |
| NCI-H322M | 0.39 | 2.08 | — | 3.29 | 5.92 | — | — | — | — | 1.10 | — | 3.95 | nd | — | — |
| NCI-H460 | 8.80 | — | — | — | — | — | — | — | — | — | 12.76 | 2.16 | 52.04 | — | — |
| NCI-H522 | 12.03 | 7.22 | 5.00 | 11.56 | 3.79 | 0.83 | 6.58 | 7.54 | 9.35 | 5.68 | 5.96 | 6.63 | 25.52 | 7.41 | — |
|
|||||||||||||||
| Colon cancer | |||||||||||||||
| COLO 205 | — | — | — | — | — | — | — | — | — | — | — | — | 11.29 | 1.62 | — |
| HCC-2998 | — | — | — | — | — | — | — | — | — | — | — | 1.26 | — | — | — |
| HCT-116 | 12.07 | — | — | — | — | — | — | 0.17 | — | — | 5.85 | — | 30.91 | 3.56 | — |
| HCT-15 | 9.99 | 5.11 | 1.92 | 1.06 | 5.00 | — | — | — | 3.67 | 0.04 | 7.15 | 10.36 | 44.24 | — | 1.86 |
| HT29 | 5.42 | — | — | 7.25 | — | — | — | 0.49 | 0.57 | 3.24 | 4.90 | 0.12 | 39.62 | 1.29 | — |
| KM12 | — | — | — | — | — | — | — | — | — | — | — | 7.80 | 16.31 | 3.35 | — |
| SW-620 | 5.76 | — | — | 4.35 | nd | — | — | — | 3.04 | nd | — | — | 19.15 | — | — |
|
|||||||||||||||
| CNS cancer | |||||||||||||||
| SF-268 | 1.96 | — | — | 1.79 | — | — | — | — | 0.28 | — | — | 1.55 | 24.65 | — | — |
| SF-295 | 16.71 | 8.43 | — | 4.43 | — | 1.00 | — | 0.34 | 7.93 | 8.17 | 19.28 | 21.59 | 37.80 | 0.56 | — |
| SF-539 | 12.81 | 5.54 | — | 1.70 | — | — | — | 1.83 | 0.27 | 2.75 | 34.04 | 31.92 | 55.89 | 13.55 | 6.46 |
| SNB-19 | nd | 9.50 | — | nd | 1.58 | — | 0.20 | nd | nd | 4.55 | 18.95 | 14.83 | 25.73 | 3.45 | — |
| SNB-75 | 23.91 | 0.40 | 1.87 | 12.75 | 15.04 | 0.54 | — | 11.30 | 15.67 | 16.96 | 24.10 | 9.07 | nd | 11.24 | 1.41 |
| U251 | — | — | — | 0.78 | 5.21 | — | — | — | 5.29 | 3.11 | 27.61 | 23.51 | 23.39 | 5.12 | 4.94 |
|
|||||||||||||||
| Melanoma | |||||||||||||||
| LOX IMVIL | 13.08 | 6.16 | 2.80 | 6.09 | nd | 1.01 | — | 8.81 | — | nd | 19.88 | 2.64 | 51.38 | 1.03 | 6.44 |
| MALME-3M | 3.60 | 4.11 | — | 6.47 | — | 0.52 | — | 6.43 | 1.19 | 4.28 | 9.94 | 15.60 | nd | 2.51 | — |
| M14 | 6.96 | — | — | 1.40 | — | — | — | 5.31 | — | — | — | 6.20 | 39.09 | 1.20 | — |
| MDA-MB-435 | 5.47 | — | — | 1.21 | — | — | — | — | — | — | 8.04 | 1.69 | 89.75 | 9.40 | 2.65 |
| SK-MEL-2 | — | — | — | 4.74 | — | — | — | — | 0.22 | — | — | 0.66 | 21.17 | — | — |
| SK-MEL-28 | 3.13 | 3.73 | 6.94 | — | — | — | — | — | — | — | 8.53 | 6.26 | 19.54 | — | — |
| SK-MEL-5 | 12.63 | 4.51 | 4.22 | 1.58 | — | 2.10 | — | 6.39 | 5.73 | 0.36 | 23.53 | 14.42 | 58.49 | 7.56 | 3.64 |
| UACC-257 | 10.51 | — | 6.70 | 4.44 | 11.51 | 0.88 | — | 10.88 | — | 9.84 | 6.74 | 1.17 | 13.91 | 2.77 | — |
| UACC-62 | 24.94 | 16.32 | nd | 11.89 | 4.96 | nd | 4.25 | 6.82 | 5.82 | 3.64 | nd | 12.21 | 40.93 | nd | nd |
|
|||||||||||||||
| Ovarian cancer | |||||||||||||||
| IGROV1 | — | 2.59 | — | — | — | — | — | — | — | — | — | — | nd | — | — |
| OVCAR-3 | — | — | — | — | — | — | — | — | — | — | 2.26 | — | 61.37 | 1.40 | — |
| OVCAR-4 | 3.78 | — | — | — | — | — | 5.64 | — | — | 0.93 | 0.90 | 4.52 | 18.02 | — | — |
| OVCAR-5 | — | — | — | 0.02 | — | — | — | — | — | 3.24 | 6.42 | — | — | 4.29 | — |
| OVCAR-8 | 1.22 | — | — | 0.27 | — | — | — | 0.54 | 1.76 | 1.10 | 10.05 | 9.38 | 20.59 | — | — |
| NCI/ADR-RES | 3.78 | — | — | — | — | — | — | 3.20 | — | — | — | 6.74 | 15.77 | — | — |
| SK-OV-3 | 13.01 | 5.76 | — | — | 5.23 | — | — | 17.39 | — | 6.07 | — | — | 19.85 | — | — |
|
|||||||||||||||
| Renal cancer | |||||||||||||||
| 786-0 | 2.21 | 15.91 | — | — | — | — | — | — | — | — | 33.50 | 23.26 | 28.18 | 0.15 | 4.55 |
| A498 | — | 23.81 | — | — | 5.65 | — | — | — | — | 9.02 | — | — | — | — | — |
| ACHN | 3.57 | 0.66 | — | 0.08 | — | — | — | — | — | — | 5.56 | — | 14.44 | 0.56 | — |
| CAKI-1 | 14.61 | 7.21 | 1.61 | 11.95 | 12.53 | — | — | 8.60 | 16.73 | 10.33 | 2.24 | — | 46.89 | 4.01 | — |
| RXF 393 | 2.39 | 35.48 | 3.30 | — | — | — | — | 11.61 | — | — | 56.78 | 20.04 | 60.70 | — | 8.94 |
| SN12C | nd | 4.88 | — | nd | — | — | — | nd | nd | 11.01 | 3.15 | 3.47 | 2.96 | 0.98 | — |
| TK-10 | — | — | — | — | — | — | — | — | — | 4.82 | — | — | — | — | — |
| UO-31 | 12.64 | 17.10 | — | 6.47 | 11.83 | 0.65 | 7.75 | 6.90 | 8.39 | 14.38 | — | 4.31 | nd | 11.97 | — |
|
|||||||||||||||
| Prostate cancer | |||||||||||||||
| PC-3 | 6.14 | — | 5.34 | 3.02 | — | 4.74 | — | 6.70 | — | — | 7.42 | 1.90 | 32.87 | 7.50 | 1.89 |
| DU-145 | — | — | — | — | — | — | — | — | — | — | 6.11 | 6.23 | 3.98 | — | — |
|
|||||||||||||||
| Breast cancer | |||||||||||||||
| MCF7 | 29.97 | 12.29 | 10.13 | 13.97 | 3.73 | 18.37 | 6.70 | 10.92 | 13.11 | 4.45 | 17.44 | 13.20 | 72.06 | 11.26 | 20.18 |
| MDA-MB-231/ATCC | 16.75 | 2.75 | — | 7.21 | nd | — | — | 6.65 | 9.78 | nd | — | — | — | — | — |
| HS 578T | 11.91 | 8.98 | — | — | nd | — | 0.37 | — | 1.76 | nd | 38.46 | 6.10 | 42.19 | 12.33 | 3.82 |
| BT-549 | 10.35 | — | — | — | — | — | — | — | — | — | 41.45 | 15.93 | 39.33 | 1.30 | — |
| T-47D | 12.15 | 15.55 | nd | — | — | nd | 1.54 | 2.11 | 5.26 | 8.11 | nd | 17.60 | 28.76 | nd | nd |
| MDA-MB-468 | 1.73 | — | — | — | 5.40 | — | — | 4.23 | — | 2.65 | — | — | 69.10 | — | — |
| Mean GI% | 6.25 | 1.04 | — | 0.81 | — | — | — | 1.91 | — | 1.78 | 8.39 | 6.72 | 31.21 | 1.42 | — |
The synthesized hybrids 8a–o demonstrated mild to strong inhibitory effects on a variety of cancer types. According to Table 3, compound 8m exhibits mainly a moderate to strong GI% against the examined cell lines. It has GI% greater than 50% against the leukemia cell lines HL-60(TB), K-562, MOL-4, and SR. NCI-H460 cell line from non-small cell lung cancer and SF-539 from CNS cancer are additionally sensitive to 8m. Furthermore, 8m showed moderate to strong activity on the melanoma cell lines LOX IMVIL, MDA-MB-435, and SK-MEL-5 with GI% = 51.38, 89.75 and 58.49%, respectively. Furthermore, it had strong action (GI% > 60%) against OVCAR-3 and RXF 393 from ovarian and renal carcinoma, respectively. On MCF7 and MDA-MB-468 breast cancer cell lines, 8m also showed strong inhibitory action with GI% = 72.06 and 69.10%, respectively (Table 3). Based on the results presented in Table 3, we can conclude that cell lines derived from leukemia, melanoma and breast cancer are the most sensitive to 8m.
2.2.2.2 Evaluation of the cytotoxic activity of 8m on MCF-7 cell line. Encouraged by the potent activity of 8m on diverse NCI cancer cell lines, 8m was further assayed for its growth inhibitory activity on MCF-7 cell line using SRB assay33 and the IC50 was depicted in Table 4. It was found that 8m showed potent IC50 of 5.03 μM in reference to sorafenib which showed IC50 = 3.24 μM.
2.2.2.3 Cell cycle analysis. The MCF-7 cell line's cell cycle progression was tested using the representative benzimidazole–dioxobenzoisoindoline derivative 8m at 5.0 μM concentration. Fig. 4 and Table 5 shows the findings of breast cancer MCF-7 cells before and after treatment with 8m. After treatment, the proportion of MCF-7 cells that had accumulated in the G1 phase dropped from 57.89 to 51.04%. Contrarily, the total proportion of cells in the G2/M phase rose from 18.23% to 25.26%. These findings show that 8m causes the division of MCF-7 cell line to stop at the G2/M phase. Additionally, due to cell death, there was a slight elevation in the percentage of cells that accumulated in the sub-G1 phase, going from 1.63% in the control cells to 2.08% in the 8m treated cells.
 | ||
| Fig. 4 Compound 8m's effect on MCF-7 cells' cell cycle stages. | ||
| Comp | % G0/G1 | % S | % G2/M | % Sub-G1 |
|---|---|---|---|---|
| Control | 57.89 | 23.88 | 18.23 | 1.63 |
| 8m | 51.04 | 23.70 | 25.26 | 2.08 |
2.2.2.4 Apoptosis assay. The potential of 8m to stimulate the apoptosis of MCF-7 cell line at 5 and 10 μM (Fig. 5) was investigated. According to the findings, at 5 μM, 8m showed a slight elevation of the percentage of cells in the early and late apoptosis stages (0.28 and 0.99%, respectively) as well as in the necrosis stage (2.00%) in reference to untreated cells. At 10 μM, 8m potentiate the early and late apoptosis as the percentage of cells rose from 0.09% and 0.42% in control cells to 5.24% and 31.23%, respectively, following treatment with 8m (Fig. 5).
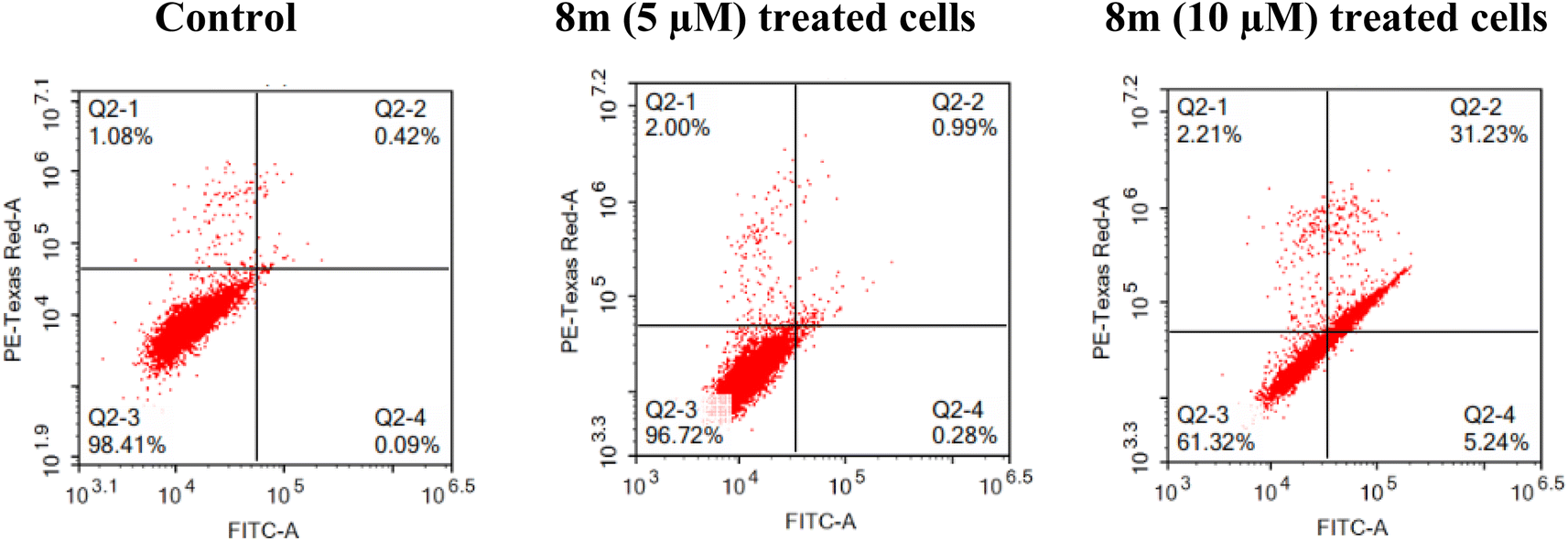 | ||
| Fig. 5 Effect of 8m on the percentage of annexin V-FITC-positive staining in MCF-7 cell line, the four quadrants are: Q2-1 (necrotic), Q2-2 (late apoptotic), Q2-3 (viable); Q2-4 (early apoptotic). | ||
2.2.2.5 Wound healing assay. Compound 8m's impact on the migration of MCF-7 cells at 5 μM (IC50 concentration) was tested by the employment of wound healing assay. In this experiment, untreated cells and 8m-treated cells were both examined regularly after making horizontal scratches into the confluent monolayer. Fig. 6 was used to display the results. A delay in the scratch closer following treatment with 8m was observed as evidenced by a distance of 0.71, 0.57 and 0.46 mm in 8m treated cells in comparison to 0.56, 0.39 and 0.22 mm in control cells after 24 h, 48 h and 72 h, respectively. This result highlights how treatment with 8m altered the migration of MCF-7 cells in comparison to control cells which supports the anti-migratory properties of 8m.
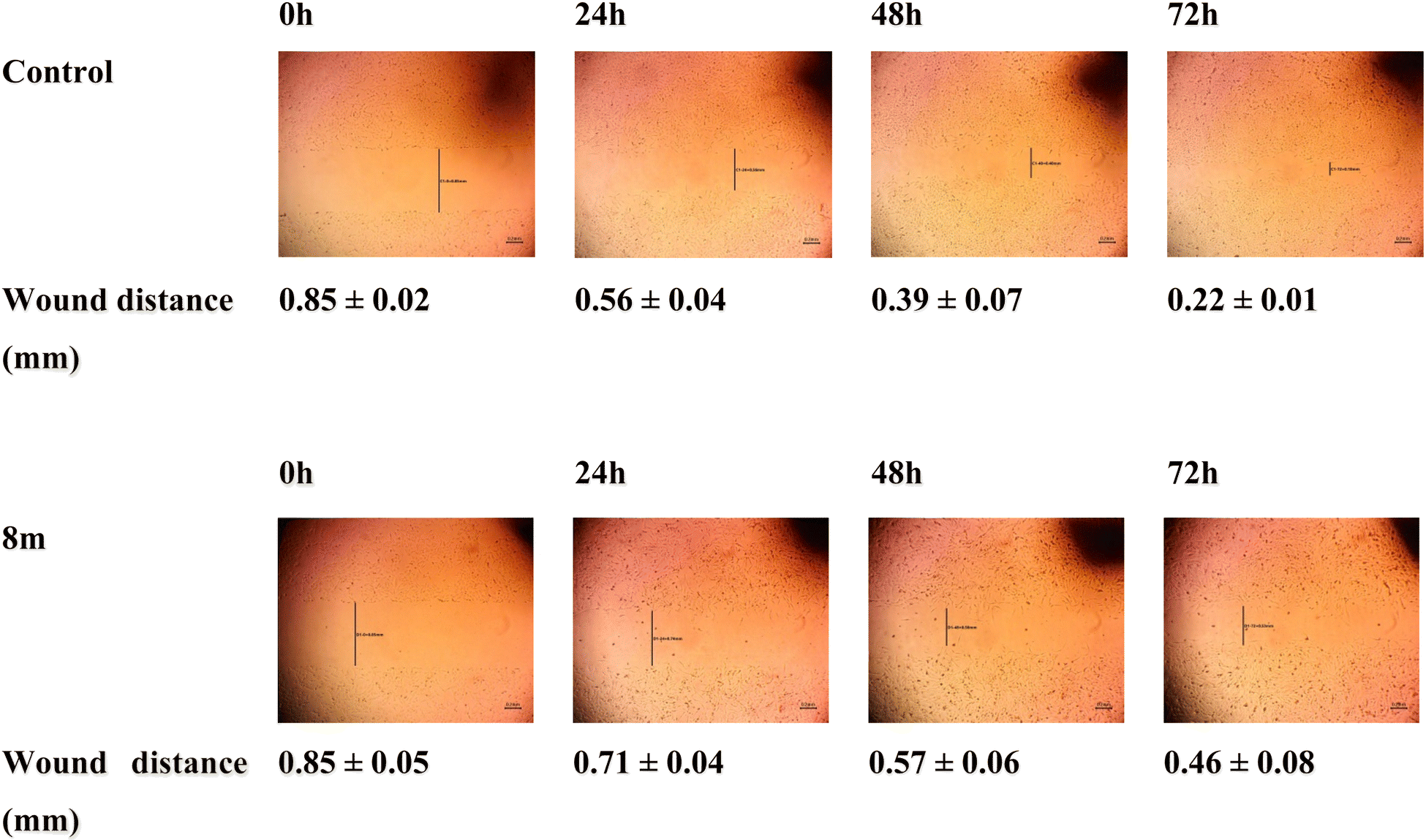 | ||
| Fig. 6 Daily measurement of the wound distances of MCF-7 cell line before and after treatment with 8m. | ||
2.3. Molecular docking simulation
Employing Autodock Vina,31 the benzimidazole–dioxo(benzo)isoindoline conjugates 8c and 8m were docked in the binding sites' of VEGFR-2 (PDB ID: 4ASD)34 and FGFR-1 (PDB ID: 4V01).35 In order to visualise the findings of the docking experiments, a free BIOVIA Discovery Studio Visualizer was utilised (https://discover.3ds.com/discovery-studio-visualizer-download). The co-crystallized ligand was self-docked to verify the validation of the docking methods.36The validation experiment displayed that the docked poses are superimposed on the co-crystalized ligands and have the ability to reproduce all the binding interactions with the key amino acids with docking energy scores of −12.20 and −13.40 kcal mol−1 in VEGFR-2 and FGFR-1, respectively (For further details see ESI†). Docking of 8c and 8m demonstrated binding energy scores of −10.10 and −10.70 kcal mol−1, respectively on VEGFR-2 binding energy scores of −9.2 and −9.51 kcal mol−1, respectively on FGFR-1.
As can be seen in Fig. 7 and 8, the dioxobenzoisoindoline moiety is positioned in the ATP binding site where one CO group establish hydrogen bonding with the essential amino acids Cys919 and Ala564 in the VEGFR-2's and FGFR-1's binding sites, respectively. Additionally, the dioxobenzoisoindoline moiety is involved in hydrophobic interactions with Leu840, Val848, Ala866, Phe918, Cys919 and Leu1035 in VEGFR-2 and Leu484, Val492, Ala512 and Leu630 in FGFR-1.
 | ||
| Fig. 7 3D presentation of 8m in the binding site of VEGFR-2. | ||
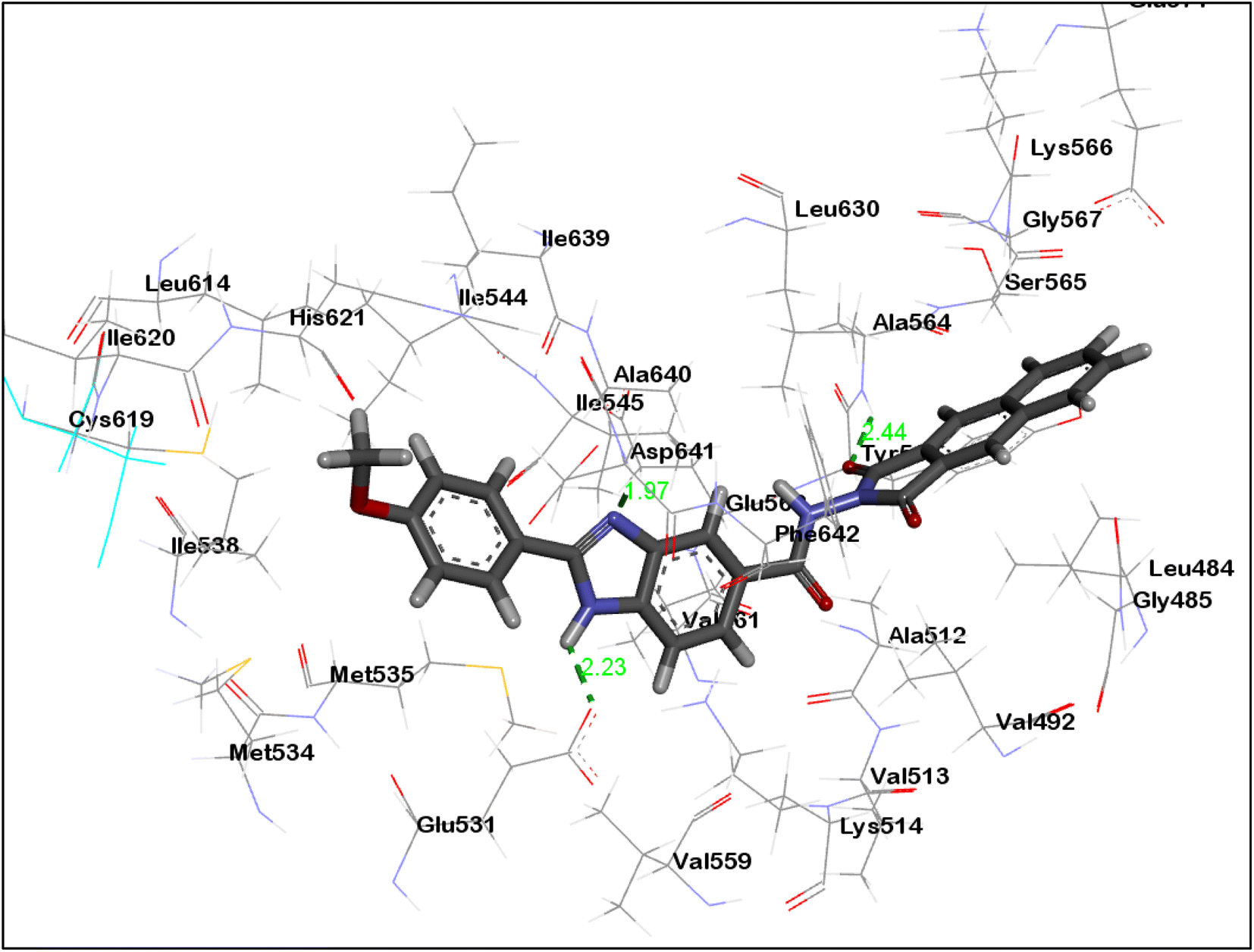 | ||
| Fig. 8 3D presentation of 8m in the binding site of FGFR-1. | ||
The benzimidazole moiety is positioned at the interface between the gate area and hydrophobic pocket forming through the NH group of the benzimidazole moiety hydrogen bonding with Glu885 and Glu531 residues in VEGFR-2 and FGFR-1, respectively. Simultaneously, the CN group forms hydrogen bonding with Asp1046 and Asp641 residues of VEGFR-2 and FGFR-1, respectively. Moreover, the benzimidazole core creates hydrophobic interactions with the amino acids Lys868, Leu889, Val899, Val916 and Cys1045 in the gate area of VEGFR-2 as well as with the amino acids Lys514, Met535, Ile545, Val561, Leu614 and Ala640 in FGFR-1.
The 4-methoxyphenyl group at the 2-position of the benzimidazole scaffold is settled in the hydrophobic pocket creating hydrophobic interactions with the amino acids Ile888, Leu889 and Ile892 in VEGFR-2 and Cys619 and His621 in FEGR-1 (Fig. 7 and 8).
2.4. ADME properties prediction
In order to analyze the drug-likeness of the designed and synthesized benzimidazole-dioxo(benzo)isoindoline conjugates 8a–o, they were submitted to free SwissADME webtool and certain findings were chosen to be displayed in Table 6.32 The information received showed that the majority of the hybrids presented meet Lipinski's rule of 5.37 The conjugates 8i, 8j and 8o are the only exceptions. The conjugates 8a–f and 8k–o are anticipated to be well absorbed from GIT.38 At the meantime, none of the candidates 8a–o are expected to cross the blood–brain barrier, demonstrating that the central nervous system won't be affected. The synthesized candidates show reasonable bioavailability scores and synthetic viability.39 In addition, the bioavailability radar chart of the synthesized candidate (see ESI,† and Fig. 9) proved that 8a–o exhibit ideal size, lipophilicity, polarity, flexibility and solubility for oral bioavailability. The only characteristic that deviates slightly from its ideal value is the degree of saturation.| Compound ID | MW | #Rotatable bonds | #H-bond acceptors | #H-bond donors | MR | TPSA | iLOGP | GI absorption | BBB permeant | Pgp substrate | Lipinski #violations | Bioavailability score | Synthetic accessibility |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 8a | 416.82 | 4 | 4 | 2 | 114.53 | 95.16 | 2.16 | High | No | No | 0 | 0.55 | 2.62 |
| 8b | 412.4 | 5 | 5 | 2 | 116.01 | 104.39 | 2.27 | High | No | No | 0 | 0.55 | 2.72 |
| 8c | 412.4 | 5 | 5 | 2 | 116.01 | 104.39 | 2.14 | High | No | No | 0 | 0.55 | 2.7 |
| 8d | 442.42 | 6 | 6 | 2 | 122.5 | 113.62 | 2.75 | High | No | Yes | 0 | 0.55 | 2.92 |
| 8e | 472.45 | 7 | 7 | 2 | 128.99 | 122.85 | 2.62 | High | No | Yes | 0 | 0.55 | 3.1 |
| 8f | 460.83 | 5 | 6 | 3 | 121.49 | 132.46 | 1.46 | High | No | No | 0 | 0.55 | 2.82 |
| 8g | 456.41 | 6 | 7 | 3 | 122.97 | 141.69 | 1.75 | Low | No | No | 0 | 0.55 | 2.94 |
| 8h | 456.41 | 6 | 7 | 3 | 122.97 | 141.69 | 1.65 | Low | No | No | 0 | 0.55 | 2.91 |
| 8i | 486.43 | 7 | 8 | 3 | 129.46 | 150.92 | 1.7 | Low | No | No | 1 | 0.55 | 3.13 |
| 8j | 516.46 | 8 | 9 | 3 | 135.95 | 160.15 | 2.1 | Low | No | Yes | 2 | 0.17 | 3.31 |
| 8k | 466.88 | 4 | 4 | 2 | 132.03 | 95.16 | 2.6 | High | No | No | 0 | 0.55 | 2.85 |
| 8l | 462.46 | 5 | 5 | 2 | 133.52 | 104.39 | 2.62 | High | No | No | 0 | 0.55 | 2.95 |
| 8m | 462.46 | 5 | 5 | 2 | 133.52 | 104.39 | 2.46 | High | No | No | 0 | 0.55 | 2.93 |
| 8n | 492.48 | 6 | 6 | 2 | 140.01 | 113.62 | 3.02 | High | No | Yes | 0 | 0.55 | 3.15 |
| 8o | 522.51 | 7 | 7 | 2 | 146.5 | 122.85 | 3.01 | High | No | Yes | 1 | 0.55 | 3.34 |
 | ||
| Fig. 9 Bioavailability radar chart from SwissADME free web tool of 8l and 8m. | ||
3. Conclusion
In this study, a new series of benzimidazole–dioxo(benzo)isoindoline hybrids 8a–o were rationally constructed and synthesized. We examined the VEGFR-2 inhibitory activity of each synthesized hit at a dose of 10 μM. Potent inhibitory activity was observed by the conjugates 8l and 8m with % inhibition = 87.61% and 80.69%, respectively. Additionally, 8l and 8m revealed potent FGFR-1 inhibition % of 84.20% and 76.83%. Examining the target hits' growth inhibitory effectiveness on NCI cancer cell lines revealed that the most effective hybrid is 8m. It demonstrated growth inhibition % up to 89.75%. Further studies proved that 8m caused G2/M phase cell cycle arrest and potentiated the apoptosis of MCF7 cell line. Furthermore, 8m demonstrated strong antimigratory properties, as evidenced by the delayed wound closure compared to untreated cells. Furthermore, the ability of benzimidazole–dioxobenzoisoindoline hybrid 8m to occupy the binding sites of VEGFR-2 and FGFR-1 and perform the necessary interactions with the crucial amino acids in those binding pockets was demonstrated by molecular docking simulations. Additionally, ADME prediction studies showed that the target benzimidazole–dioxoisoindoline hybrids 8a–o may had desired drug-like properties.4. Experimental
4.1. Chemistry
4.1.3.1 2-(4-Chlorophenyl)-N-(1,3-dioxoisoindolin-2-yl)-1H-benzo[d]imidazole-5-carboxamide (8a). Yield = 51%; mp > 300 °C; 1H NMR (400 MHz; DMSO-d6) δH 7.68 (d, 3J = 8.4 Hz, 2H), 7.88 (d like, 3J = 8.4 Hz, 1H) 7.97–8.05 (m, 4H), 8.19 (br., 1H), 8.24 (d, 3J = 8.4 Hz, 2H), 8.37 (br., 1H), 11.30 (s, 1H), 13.38 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 112.18, 119.51, 124.34, 128.87, 128.99, 129.68, 129.98, 135.67, 135.91, 166.06, 166.23 ppm; anal. calcd for C22H13ClN4O3: C, 63.39; H, 3.14; N, 13.44; found: C, 63.56; H, 3.27; N, 13.76.
4.1.3.2 N-(1,3-dioxoisoindolin-2-yl)-2-(3-methoxyphenyl)-1H-benzo[d]imidazole-5-carboxamide (8b). Yield = 58%; mp > 300 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.88 (s, 3H), 7.12 (dd, 3J = 8.0 Hz, 4J = 2.0 Hz, 1H), 7.51 (t, 3J = 8.0 Hz, 1H), 7.75–7.83 (m, 3H), 7.88 (d, 3J = 8.0 Hz, 1H), 7.97–8.00 (m, 2H), 8.02–8.05 (m, 2H), 8.24–8.32 (m, 1H), 11.31 (s, 1H), 13.30 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.37, 111.79, 116.49, 119.14, 122.47, 123.89, 124.60, 129.56, 130.28, 130.86, 135.44, 153.68, 159.71, 165.63, 165.80 ppm; anal. calcd for C23H16N4O4: C, 66.99; H, 3.91; N, 13.59; found: C, 66.71; H, 4.23; N, 13.32.
4.1.3.3 N-(1,3-dioxoisoindolin-2-yl)-2-(4-methoxyphenyl)-1H-benzo[d]imidazole-5-carboxamide (8c). Yield = 51%; mp > 300 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.86 (s, 3H), 7.15 (d, 3J = 8.8 Hz, 2H), 7.71 (d, 3J = 8.8 Hz, 1H), 7.85 (dd, 3J = 8.4 Hz, 4J = 1.6 Hz, 1H), 7.97–8.00 (m, 2H), 8.01–8.05 (m, 2H), 8.17 (d, 3J = 8.8 Hz, 2H), 8.24 (br., 1H), 11.28 (s, 1H), 13.13 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.43, 114.55, 121.89, 122.00, 123.89, 124.30, 128.46, 128.53, 129.58, 130.79, 132.90, 135.44, 153.90, 161.18, 165.66, 165.83, 168.72 ppm; anal. calcd for C23H16N4O4: C, 66.99; H, 3.91; N, 13.59; found: C, 66.70; H, 3.78; N, 13.82; HRMS (+) ESI m/z calculated for C23H17N4O4 [M + H]+: 413.1250, found: 413.1236.
4.1.3.4 2-(2,5-Dimethoxyphenyl)-N-(1,3-dioxoisoindolin-2-yl)-1H-benzo[d]imidazole-5-carboxamide (8d). Yield = 49%; mp > 300 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.82 (s, 3H), 4.00 (s, 3H), 7.11 (dd, 3J = 8.8 Hz, 4J = 3.2 Hz, 1H), 7.22 (d, 3J = 9.2 Hz, 1H), 7.79 (d, 3J = 8.4 Hz, 1H), 7.87 (dd, 3J = 8.4 Hz, 4J = 1.2 Hz, 1H), 7.90 (d, 4J = 3.2 Hz, 1H), 7.97–7.99 (m, 2H), 8.02–8.04 (m, 2H), 8.33 (s, 1H), 11.28 (s, 1H), 12.74 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.60, 56.24, 113.58, 113.87, 117.78, 117.92, 121.84, 123.88, 124.50, 128.53, 129.58, 130.78, 132.94, 135.43, 151.28, 151.37, 153.25, 165.63, 166.00, 168.71 ppm; anal. calcd for C24H18N4O5: C, 65.15; H, 4.10; N, 12.66; found: C, 65.38; H, 3.85; N, 12.89.
4.1.3.5 N-(1,3-dioxoisoindolin-2-yl)-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-5-carboxamide (8e). Yield = 52%; mp > 300 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.76 (s, 3H), 3.93 (s, 6H), 7.57 (s, 2H), 7.76 (d, 3J = 8.4 Hz, 1H), 7.88 (dd, 3J = 8.4 Hz, 4J = 1.6 Hz, 1H), 7.97–8.00 (m, 2H), 8.02–8.05 (m, 2H), 8.26 (br., 1H), 11.32 (s, 1H), 12.06 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 56.18, 60.27, 104.33, 115.32, 122.20, 123.94, 124.58, 124.78, 129.61, 135.50, 139.53, 153.37, 153.84, 165.71, 165.86 ppm; anal. calcd for C25H20N4O6: C, 63.56; H, 4.27; N, 11.86; found: C, 63.78; H, 4.50; N, 11.53.
4.1.3.6 2-(2-(4-Chlorophenyl)-1H-benzo[d]imidazole-5-carboxamido)-1,3-dioxoisoindoline-5-carboxylic acid (8f). Yield = 55%; mp = 291–293 °C; 1H NMR (400 MHz; DMSO-d6) δH 7.68 (d, 3J = 8.4 Hz, 2H), 7.76 (d like, 3J = 6.4 Hz, 1H), 7.88 (dd, 3J = 8.4 Hz, 4J = 0.8 Hz, 1H), 8.16 (d, 3J = 7.6 Hz, 1H), 8.24 (d, 3J = 8.8 Hz, 2H), 8.29 (br., 1H), 8.37 (s, 1H), 8.48 (dd, 3J = 8.0 Hz, 4J = 1.2 Hz, 1H), 11.41 (s, 1H), 13.41 ppm (br., 2H); 13C NMR (100 MHz; DMSO-d6) δC 123.89, 124.39, 124.60, 128.44, 128.54, 129.21, 129.95, 132.72, 135.19, 136.14, 137.13, 164.82, 164.86, 165.64, 165.71 ppm; anal. calcd for C23H13ClN4O5: C, 59.95; H, 2.84; N, 12.16; found: C, 59.71; H, 3.12; N, 12.00.
4.1.3.7 2-(2-(3-Methoxyphenyl)-1H-benzo[d]imidazole-5-carboxamido)-1,3-dioxoisoindoline-5-carboxylic acid (8g). Yield = 63%; mp > 300 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.88 (s, 3H), 7.12 (dd, 3J = 8.0 Hz, 4J = 2.0 Hz, 1H), 7.50 (t, 3J = 8.0 Hz, 1H), 7.77–7.82 (m, 3H), 7.89 (d, 3J = 8.4 Hz, 1H), 8.15 (d, 3J = 7.6 Hz, 1H), 8.30 (br., 1H), 8.38 (s, 1H), 8.48 (dd, 3J = 8.0 Hz, 4J = 1.2 Hz, 1H), 11.41 (s, 1H), 13.33 ppm (br., 2H); 13C NMR (100 MHz; DMSO-d6) δC 55.39, 111.80, 116.51, 119.16, 122.21, 123.92, 124.41, 124.51, 129.98, 130.29, 130.85, 132.73, 136.17, 137.22, 153.64, 159.72, 164.87, 164.92, 165.70, 165.81 ppm; anal. calcd for C24H16N4O6: C, 63.16; H, 3.53; N, 12.28; found: C, 63.35; H, 3.39; N, 12.44.
4.1.3.8 2-(2-(4-Methoxyphenyl)-1H-benzo[d]imidazole-5-carboxamido)-1,3-dioxoisoindoline-5-carboxylic acid (8h). Yield = 69%; mp > 300 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.86 (s, 3H), 7.15 (d, 3J = 8.8 Hz, 2H), 7.71 (d, 3J = 8.4 Hz, 1H), 7.85 (dd, 3J = 8.4 Hz, 4J = 1.2 Hz, 1H), 8.15–8.18 (m, 3H), 8.24 (br., 1H), 8.37 (s, 1H), 8.48 (dd, 3J = 8.0 Hz, 4J = 1.2 Hz, 1H), 11.37 (s, 1H), 13.07 ppm (br., 2H); 13C NMR (100 MHz; DMSO-d6) δC 55.44, 114.55, 121.98, 123.93, 124.17, 124.41, 128.52, 129.99, 132.75, 136.18, 137.18, 153.97, 161.16, 164.89, 164.94, 165.70, 165.88 ppm; anal. calcd for C24H16N4O6: C, 63.16; H, 3.53; N, 12.28; found: C, 63.46; H, 3.81; N, 11.96.
4.1.3.9 2-(2-(2,5-Dimethoxyphenyl)-1H-benzo[d]imidazole-5-carboxamido)-1,3-dioxoisoindoline-5-carboxylic acid (8i). Yield = 65%; mp > 300 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.82 (s, 3H), 4.00 (s, 3H), 7.11 (dd, 3J = 8.8 Hz, 4J = 3.2 Hz, 1H), 7.22 (d, 3J = 9.2 Hz, 1H), 7.78 (d, 3J = 8.4 Hz, 1H), 7.87 (dd, 3J = 8.4 Hz, 4J = 1.2 Hz, 1H), 7.90 (d, 4J = 3.2 Hz, 1H), 8.15 (d, 3J = 7.6 Hz, 1H), 8.33 (s, 1H), 8.37 (s, 1H), 8.48 (dd, 3J = 8.0 Hz, 4J = 1.2 Hz, 1H), 11.36 (s, 1H), 12.48 (br., 1H), 13.84 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.73, 56.35, 113.71, 113.96, 117.81, 118.11, 122.00, 124.03, 124.52, 130.08, 132.84, 136.32, 137.29, 151.50, 153.36, 164.98, 165.02, 165.85, 166.22 ppm; anal. calcd for C25H18N4O7: C, 61.73; H, 3.73; N, 11.52; found: C, 61.52; H, 3.98; N, 11.76.
4.1.3.10 1,3-Dioxo-2-(2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-5-carboxamido)-isoindoline-5-carboxylic acid (8j). Yield = 68%; mp > 300 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.76 (s, 3H), 3.93 (s, 6H), 7.57 (s, 2H), 7.75 (d, 3J = 8.4 Hz, 1H), 7.88 (dd, 3J = 8.8 Hz, 4J = 1.6 Hz, 1H), 8.16 (d, 3J = 7.5 Hz, 1H), 8.26 (br., 1H), 8.38 (s, 1H), 8.49 (dd, 3J = 7.8 Hz, 4J = 1.2 Hz, 1H), 11.38 (s, 1H), 12.70 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 56.13, 60.22, 104.28, 120.36, 122.10, 123.91, 124.37, 124.39, 124.81, 129.98, 132.73, 136.14, 137.18, 139.46, 153.31, 164.89, 164.93, 165.67, 165.80 ppm; anal. calcd for C26H20N4O8: C, 60.47; H, 3.90; N, 10.85; found: C, 60.22; H, 3.59; N, 10.98.
4.1.3.11 2-(4-Chlorophenyl)-N-(1,3-dioxo-1,3-dihydro-2H-benzo[f]isoindol-2-yl)-1H-benzo[d]imidazole-5-carboxamide (8k). Yield = 54%; mp 220-222 °C; 1H NMR (400 MHz; DMSO-d6) δH 7.68 (d, 3J = 8.4 Hz, 2H), 7.78 (d like, 3J = 6.4 Hz, 1H), 7.93 (dd, 3J = 7.6 Hz, 4J = 1.6 Hz, 1H), 7.95 (t, 3J = 7.6 Hz, 2H), 8.25 (d, 3J = 8.8 Hz, 2H), 8.34 (br., 1H), 8.57 (d, 3J = 8.4 Hz, 2H), 8.60 (d, 3J = 7.2 Hz, 2H), 11.33 (s, 1H), 13.37 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 121.74, 125.77, 127.30, 127.57, 128.54, 128.56, 129.26, 131.62, 131.69, 135.20, 135.34, 152.61, 162.01, 165.69 ppm; anal. calcd for C26H15ClN4O3: C, 66.89; H, 3.24; N, 12.00; found: C, 66.64; H, 3.38; N, 12.26.
4.1.3.12 N-(1,3-dioxo-1,3-dihydro-2H-benzo[f]isoindol-2-yl)-2-(3-methoxyphenyl)-1H-benzo[d]imidazole-5-carboxamide (8l). Yield = 55%; mp 215–217 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.89 (s, 3H), 7.12 (dd, 3J = 8.0 Hz, 4J = 2.0 Hz, 1H), 7.51 (t, 3J = 8.0 Hz, 1H), 7.77–7.83 (m, 3H), 7.92–7.96 (m, 3H), 8.33 (br.,1H), 8.55 (d, 3J = 8.0 Hz, 2H), 8.59 (d, 3J = 7.2 Hz, 2H), 11.32 (s, 1H), 13.40 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.43, 111.81, 116.52, 119.17, 121.76, 125.66, 127.32, 127.60, 130.35, 130.93, 131.65, 131.72, 135.37, 135.48, 159.77, 162.04, 165.75 ppm; anal. calcd for C27H18N4O4: C, 70.12; H, 3.92; N, 12.12; found: C, 70.42; H, 3.75; N, 12.35.
4.1.3.13 N-(1,3-dioxo-1,3-dihydro-2H-benzo[f]isoindol-2-yl)-2-(4-methoxyphenyl)-1H-benzo[d]imidazole-5-carboxamide (8m). Yield = 58%; mp 208–210 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.86 (s, 3H), 7.15 (d, 3J = 8.8 Hz, 2H), 7.73 (br., 1H), 7.90 (t, 3J = 7.6 Hz, 1H), 7.96 (t, 3J = 7.6 Hz, 2H), 8.18 (d, 3J = 8.8 Hz, 2H), 8.52–8.61 (m, 5H), 11.28 (s, 1H), 13.12 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.40, 114.49, 118.98, 121.71, 122.11, 125.19, 127.25, 127.51, 127.54, 128.42, 129.70, 131.36, 131.58, 131.62, 132.47, 135.27, 135.39, 160.71, 161.05, 161.97, 165.70 ppm; anal. calcd for C27H18N4O4: C, 70.12; H, 3.92; N, 12.12; found: C, 69.95; H, 4.20; N, 11.83; HRMS (+) ESI m/z calculated for C27H19N4O4 [M + H]+: 463.1406, found: 463.1400.
4.1.3.14 2-(2,5-Dimethoxyphenyl)-N-(1,3-dioxo-1,3-dihydro-2H-benzo[f]isoindol-2-yl)-1H-benzo[d]imidazole-5-carboxamide (8n). Yield = 50%; mp 205–207 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.83 (s, 3H), 4.01 (s, 3H), 7.11 (dd, 3J = 8.8 Hz, 4J = 2.8 Hz, 1H), 7.22 (d, 3J = 9.2 Hz, 1H) 7.76–7.83 (m, 1H), 7.90–7.92 (m, 2H), 7.95 (t, 3J = 8.0 Hz, 2H), 8.52 (d, 3J = 6.8 Hz, 1H), 8.57 (d, 3J = 8.0 Hz, 2H), 8.60 (d, 3J = 8.0 Hz, 2H), 11.27 (d, J = 15.6 Hz, 1H), 12.46 ppm (d, J = 14.4 Hz, 1H); 13C NMR (100 MHz; DMSO-d6) δC 55.57, 56.20, 113.53, 118.96, 121.71, 127.23, 127.49, 127.52, 129.68, 131.34, 131.56, 131.60, 132.45, 134.56, 135.25, 135.38, 137.69, 142.22, 145.50, 153.21, 160.70, 161.95 ppm; anal. calcd for C28H20N4O5: C, 68.29; H, 4.09; N, 11.38; found: C, 68.56; H, 4.27; N, 11.66.
4.1.3.15 N-(1,3-dioxo-1,3-dihydro-2H-benzo[f]isoindol-2-yl)-2-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole-5-carboxamide (8o). Yield = 57%; mp 252–254 °C; 1H NMR (400 MHz; DMSO-d6) δH 3.76 (s, 3H), 3.93 (s, 6H), 7.58 (s, 2H), 7.77 (d, 3J = 8.4 Hz, 1H), 7.90–7.94 (m, 3H), 8.29 (br., 1H), 8.52–8.56 (m, 4H), 11.31 (s, 1H), 13.20 ppm (br., 1H); 13C NMR (100 MHz; DMSO-d6) δC 56.12, 60.19, 104.24, 119.01, 121.70, 124.70, 125.52, 127.26, 127.54, 129.71, 131.36, 131.65, 132.45, 135.38, 139.43, 153.28, 160.70, 161.96, 165.56 ppm; anal. calcd for C29H22N4O6: C, 66.66; H, 4.24; N, 10.72; found: C, 66.39; H, 4.56; N, 11.00.
4.2. Biological activity
4.3. Molecular modeling
Molecular docking simulations were achieved utilizing Autodock Vina31 (For further information see ESI†). A free BIOVIA Discovery Studio Visualizer was utilised (https://discover.3ds.com/discovery-studio-visualizer-download) to visualize the findings.4.4. ADME prediction studies
The SMILES notations of the target benzimidazole–dioxoisoindoline conjugates 8a–o were submitted to SwissADME web tool to predict their physicochemical and drug likeness properties.32Data availability
All data are available on request.Conflicts of interest
The authors have no conflict of interest to declare.Acknowledgements
The authors are appreciative for the National Cancer Institute (NCI), located in Bethesda, Maryland, USA, for testing 8a–o for their anticancer efficacy. This paper is based upon work supported by Science, Technology & Innovation Funding Authority (STDF) under grant ID 37225. The Medical Research Ethics Committee at the National Research Centre (NRC), Egypt, has granted the current study ethical approval 315062023.References
- R. L. Siegel, A. N. Giaquinto and A. Jemal, Ca-Cancer J. Clin., 2024, 74, 12–49 CrossRef PubMed.
- H. T. Abdel-Mohsen, A. Petreni and C. T. Supuran, Arch. Pharm., 2022, 355, e2200274 CrossRef PubMed.
- R. M. Hassan, W. H. Abd-Allah, A. M. Salman, A. A. El-Azzouny and M. N. Aboul-Enein, Eur. J. Pharm. Sci., 2019, 139, 105045 CrossRef PubMed.
- C. Braicu, M. Buse, C. Busuioc, R. Drula, D. Gulei, L. Raduly, A. Rusu, A. Irimie, A. G. Atanasov, O. Slaby, C. Ionescu and I. Berindan-Neagoe, Cancers, 2019, 11, 1618 CrossRef CAS PubMed.
- M. S. Abdel-Maksoud, A. A. B. Mohamed, R. M. Hassan, M. A. Abdelgawad, G. Chilingaryan, S. Selim, M. S. Abdel-Bakky and M. M. Al-Sanea, Int. J. Mol. Sci., 2021, 22, 10491 CrossRef CAS PubMed.
- H. T. Abdel-Mohsen, M. M. Anwar, N. S. Ahmed, S. S. Abd El-Karim and S. H. Abdelwahed, Molecules, 2024, 29, 875 CrossRef CAS PubMed.
- H. T. Abdel-Mohsen, A. M. Nageeb and I. A. Y. Ghannam, Drug Dev. Res., 2024, 85, e22249 CrossRef CAS PubMed.
- R. M. Hassan, I. H. Ali, A. M. El Kerdawy, M. T. Abo-Elfadl and I. A. Y. Ghannam, Bioorg. Chem., 2024, 152, 107728 CrossRef CAS PubMed.
- K. Wada, J. Y. Lee, H. Y. Hung, Q. Shi, L. Lin, Y. Zhao, M. Goto, P. C. Yang, S. C. Kuo, H. W. Chen and K. H. Lee, Bioorg. Med. Chem., 2015, 23, 1507–1514 CrossRef CAS PubMed.
- R. H. Adams and K. Alitalo, Nat. Rev. Mol. Cell Biol., 2007, 8, 464–478 CrossRef CAS PubMed.
- C. S. Abhinand, R. Raju, S. J. Soumya, P. S. Arya and P. R. Sudhakaran, J. Cell Commun. Signaling, 2016, 10, 347–354 CrossRef PubMed.
- S. J. Modi and V. M. Kulkarni, Med. Drug Discovery, 2019, 2, 100009 CrossRef.
- D. J. Hicklin and L. M. Ellis, J. Clin. Oncol., 2005, 23, 1011–1027 CrossRef CAS PubMed.
- J. Muto, K. Shirabe, K. Sugimachi and Y. Maehara, Hepatol. Res., 2015, 45, 1–9 CrossRef PubMed.
- S. Dai, Z. Zhou, Z. Chen, G. Xu and Y. Chen, Cells, 2019, 8, 614 CrossRef CAS PubMed.
- M. A. Krook, J. W. Reeser, G. Ernst, H. Barker, M. Wilberding, G. Li, H. Z. Chen and S. Roychowdhury, Br. J. Cancer, 2021, 124, 880–892 CrossRef CAS PubMed.
- S. S. Abd El-Karim, Y. M. Syam, A. M. El Kerdawy and H. T. Abdel-Mohsen, Bioorg. Chem., 2024, 142, 106920 CrossRef CAS PubMed.
- H. T. Abdel-Mohsen, E. A. Abd El-Meguid, A. M. El Kerdawy, A. E. E. Mahmoud and M. M. Ali, Arch. Pharm., 2020, 353, e1900340 CrossRef PubMed.
- H. T. Abdel-Mohsen, M. A. Ibrahim, A. M. Nageeb and A. M. El Kerdawy, BMC Chem., 2024, 18, 42 CrossRef CAS PubMed.
- R. M. Allam, A. M. El Kerdawy, A. E. Gouda, K. A. Ahmed and H. T. Abdel-Mohsen, Bioorg. Chem., 2024, 146, 107243 CrossRef CAS PubMed.
- H. I. El Diwani, H. T. Abdel-Mohsen, I. Salama, F. A. Ragab, M. M. Ramla, S. A. Galal, M. M. Abdalla, A. Abdel-Wahab and M. A. El Demellawy, Chem. Pharm. Bull., 2014, 62, 856–866 CrossRef CAS PubMed.
- I. H. Ali, H. T. Abdel-Mohsen, M. M. Mounier, M. T. Abo-elfadl, A. M. El Kerdawy and I. A. Y. Ghannam, Bioorg. Chem., 2022, 126, 105883 CrossRef CAS PubMed.
- H. T. Abdel-Mohsen and A. El Kerdawy, Egypt. J. Chem., 2024, 67, 437–446 Search PubMed.
- H. T. Abdel-Mohsen, Y. M. Syam, M. S. Abd El-Ghany and S. S. Abd El-Karim, Arch. Pharm., 2024, e2300721, DOI:10.1002/ardp.202300721.
- M. H. Potashman, J. Bready, A. Coxon, T. M. DeMelfi Jr, L. DiPietro, N. Doerr, D. Elbaum, J. Estrada, P. Gallant, J. Germain, Y. Gu, J. C. Harmange, S. A. Kaufman, R. Kendall, J. L. Kim, G. N. Kumar, A. M. Long, S. Neervannan, V. F. Patel, A. Polverino, P. Rose, S. Plas, D. Whittington, R. Zanon and H. Zhao, J. Med. Chem., 2007, 50, 4351–4373 CrossRef CAS PubMed.
- M. Hasegawa, N. Nishigaki, Y. Washio, K. Kano, P. A. Harris, H. Sato, I. Mori, R. I. West, M. Shibahara, H. Toyoda, L. Wang, R. T. Nolte, J. M. Veal and M. Cheung, J. Med. Chem., 2007, 50, 4453–4470 CrossRef CAS PubMed.
- R. J. Motzer, C. Porta, N. J. Vogelzang, C. N. Sternberg, C. Szczylik, J. Zolnierek, C. Kollmannsberger, S. Y. Rha, G. A. Bjarnason, B. Melichar, U. De Giorgi, V. Grünwald, I. D. Davis, J. L. Lee, E. Esteban, G. Urbanowitz, C. Cai, M. Squires, M. Marker, M. M. Shi and B. Escudier, Lancet Oncol., 2014, 15, 286–296 CrossRef CAS PubMed.
- R. Porta, R. Borea, A. Coelho, S. Khan, A. Araújo, P. Reclusa, T. Franchina, N. Van Der Steen, P. Van Dam, J. Ferri, R. Sirera, A. Naing, D. Hong and C. Rolfo, Crit Rev Oncol Hematol, 2017, 113, 256–267 CrossRef PubMed.
- T. D. Bunney, S. Wan, N. Thiyagarajan, L. Sutto, S. V. Williams, P. Ashford, H. Koss, M. A. Knowles, F. L. Gervasio, P. V. Coveney and M. Katan, EBioMedicine, 2015, 2, 194–204 CrossRef PubMed.
- T. E. Williams, S. Subramanian, J. Verhagen, C. M. McBride, A. Costales, L. Sung, W. Antonios-McCrea, M. McKenna, A. K. Louie, S. Ramurthy, B. Levine, C. M. Shafer, T. Machajewski, P. A. Renhowe, B. A. Appleton, P. Amiri, J. Chou, D. Stuart, K. Aardalen and D. Poon, ACS Med. Chem. Lett., 2015, 6, 961–965 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
- M. McTigue, B. W. Murray, J. H. Chen, Y. L. Deng, J. Solowiej and R. S. Kania, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18281–18289 CrossRef CAS PubMed.
- J. A. Tucker, T. Klein, J. Breed, A. L. Breeze, R. Overman, C. Phillips and R. A. Norman, Structure, 2014, 22, 1764–1774 CrossRef CAS PubMed.
- Y. M. Syam, S. S. Abd El-Karim and H. T. Abdel-Mohsen, Arch. Pharm., 2024, e2300682, DOI:10.1002/ardp.202300682.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- W. J. Egan, K. M. Merz Jr and J. J. Baldwin, J. Med. Chem., 2000, 43, 3867–3877 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- H. T. Abdel-Mohsen, M. A. Omar, A. Petreni and C. T. Supuran, Arch. Pharm., 2022, 355, e2200180 CrossRef PubMed.
- H. T. Abdel-Mohsen, A. M. El Kerdawy, M. A. Omar, A. Petreni, R. M. Allam, H. I. El Diwani and C. T. Supuran, Eur. J. Med. Chem., 2022, 228, 114004 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05462h |
| This journal is © The Royal Society of Chemistry 2024 |