
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective synthesis of cyclic alcohols from cycloalkanes using nickel(II) complexes of tetradentate amidate ligands†
Anjana Rajeeva,
Sethuraman Muthuramalingamb and
Muniyandi Sankaralingam *a
*a
aBioinspired & Biomimetic Inorganic Chemistry Lab, Department of Chemistry, National Institute of Technology Calicut, Kozhikode, Kerala 673601, India. E-mail: msankaralingam@nitc.ac.in; sankarjan06@gmail.com
bInstitut de Química Computacional i Catalisi; (IQCC), Departament de Química, Universitat de Girona, Girona E-17003, Catalonia, Spain
First published on 24th September 2024
Abstract
Selective functionalisation of hydrocarbons using transition metal complexes has evoked significant research interest in industrial chemistry. However, selective oxidation of unactivated aliphatic C–H bonds is challenging because of the high bond dissociation energies. Herein, we report the synthesis, characterisation and catalytic activity of nickel(II) complexes ([Ni(L1–L3)(OH2)2](ClO4)2 (1–3)) of monoamidate tetradentate ligands [L1: 2-(bis(pyridin-2-ylmethyl)amino)-N-phenylacetamide, L2: 2-(bis(2-pyridin-2-ylmethyl)amino)-N-(naphthalen-1-yl)acetamide, L3: N-benzyl-2-(bis(pyridin-2-ylmethyl)amino)acetamide] in selective oxidation of cycloalkanes using m-CPBA as the oxidant. In cyclohexane oxidation, catalysts showed activity (TON) in the order 1 (654) > 2 (589) > 3 (359) with a high A/(K + L) ratio up to 23.6. Using catalyst 1, the substrate scope of the reaction was broadened by including other cycloalkanes such as cyclopentane, cycloheptane, cyclooctane, adamantane and methylcyclohexane. Further, the Fenton-type reaction in the catalytic cycle was discarded based on the relatively high 3°/2° ratio of 8.6 in adamantane oxidation. Although the formation of chlorinated products during the reactions confirmed the contribution of the 3-chlorobenzoyloxy radical mechanism, the high alcohol selectivity obtained for the reactions indicated the participation of nickel-based oxidants in the oxidation process.
1. Introduction
Modern catalytic approaches introduced a phenomenal change in synthetic and industrial chemistry by offering proficient and sustainable methods for synthesising organic molecules of paramount importance.1,2 In this context, harnessing transition metal catalysts in functionalisation reactions has sparked increasing research interest.3–5 The advent of transition metal-based catalysis opened up new possibilities for organic transformations that were once thought to be unattainable. For example, the oxidation of a hydrocarbon such as cyclohexane produces cyclohexanol and cyclohexanone, which are high-value-added compounds with versatile applications.6 However, this oxidation is a strenuous task due to the high C–H bond dissociation energy of cyclohexane and requires multi-step procedures as well as rigorous reaction conditions.6,7 Conversely, various catalysts, especially, 3d-metal molecular catalysts are known to mediate cyclohexane oxidation efficiently under mild reaction conditions in the presence of a suitable oxygen source.4,8–21 The catalytic potential of nickel complexes in cyclohexane oxidation was first demonstrated using complex [Ni(TPA)(OAc)(H2O)](BPh4) (I) (TPA = tris(2-pyridylmethyl) amine) and meta-chloroperbenzoic acid (m-CPBA).11 Later, several notable attempts using nickel complexes with diverse ligands were reported, among which nickel(II) complexes of tetradentate ligands (I–VI) (Scheme 1) showed promising activity in terms of turnover number (TON) and alcohol selectivity (A/(K + L); A = cyclohexanol, K = cyclohexanone, L = caprolactone).4,11–17 Also, pyridine is present as the donor moiety in most of these complexes and its replacement by phenolic or oxazoline groups has resulted in improved activity (catalysts II and VI).12,16 Nevertheless, systematic tuning of ligands by introducing distinct donor moieties and evaluating the catalytic properties of corresponding complexes has been less examined in cyclohexane oxidation mediated by nickel(II) complexes. Moreover, ligands that can stabilise or facilitate the formation of reactive nickel oxygen species need to be synthesised for developing more selective and potential catalysts.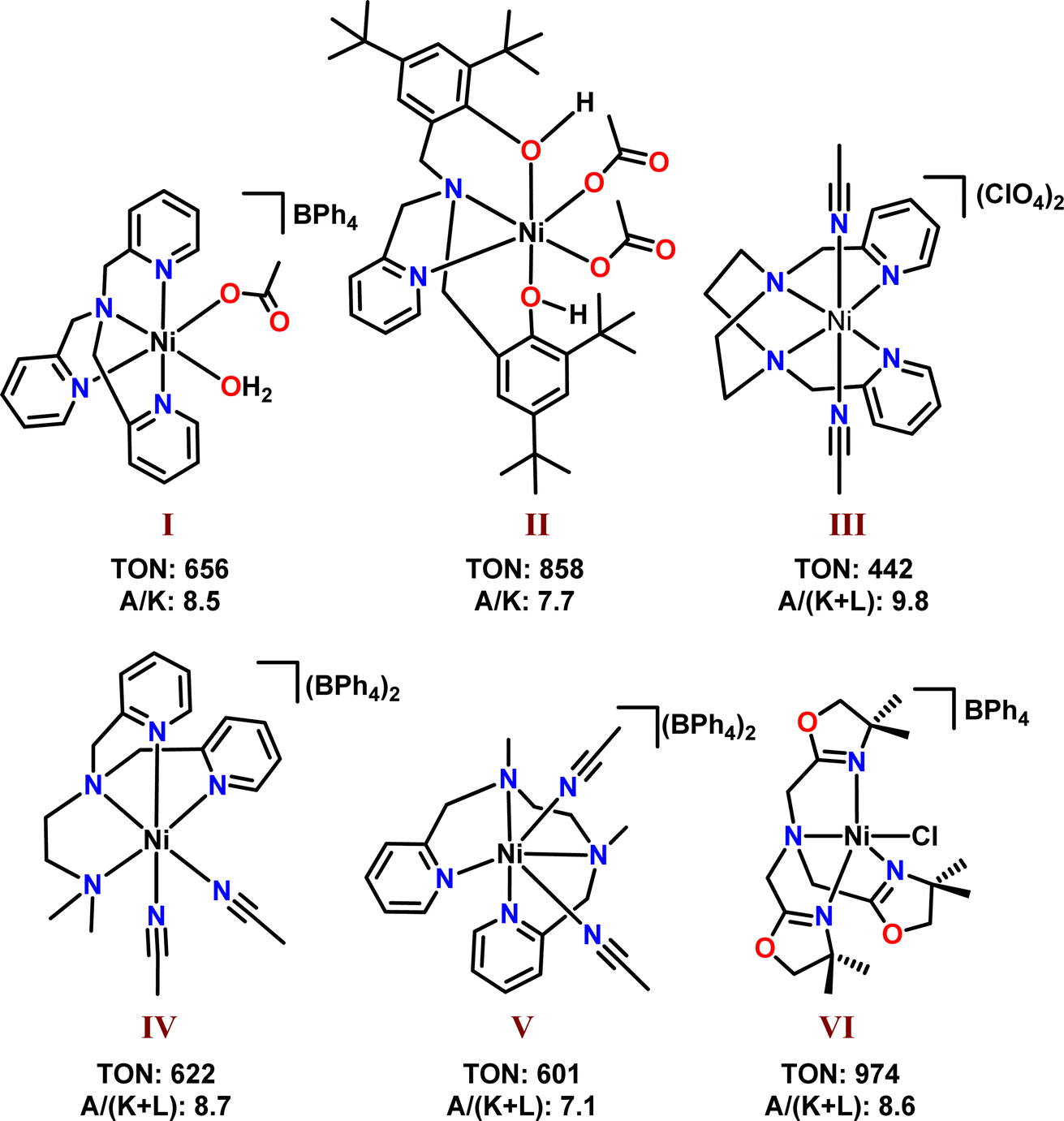 | ||
| Scheme 1 Notable reported nickel(II) complexes of tetradentate ligands as the catalysts for cyclohexane oxidation. | ||
Intriguingly, amidate ligands are an exemplary choice for developing competent catalysts due to their ability to stabilise higher oxidation states of nickel, which is a key factor in governing the catalytic potential of many complexes.22–27 For instance, McDonald et al. and Company et al. have extensively worked on generating, characterising and exploring the reactivity of different high-valent nickel species supported by amidate ligands.22–27 Notably, we recently reported that nickel(II) complexes with aminoquinoline-based monoamidate pincer ligands exhibit superior catalytic activity in the oxidation of cycloalkanes compared to other nickel catalysts containing tridentate ligands using m-CPBA as the oxidant.28 The latest studies on the mechanism of cyclohexane oxidation by nickel(II) complexes confirm the involvement of the 3-chlorobenzoyloxy radical pathway and a partial contribution of nickel-based oxidant in the catalytic cycle.29–31
Also, it is noted that the steric factors of the catalysts play a pivotal role in the activity and therefore, it is essential to delve deeper into the impact of ligand choices in the catalytic performances of nickel(II) complexes. For this reason, we sought to develop nickel(II) complexes (1–3) containing monoamidate tetradentate ligands, [2-(bis(pyridin-2-ylmethyl)amino)-N-phenylacetamide (L1; [Ni(L1)(H2O)2](ClO4)2 (1)), 2-(bis(2-pyridin-2-ylmethyl)amino)-N-(naphthalen-1-yl)acetamide (L2; [Ni(L2)(H2O)2](ClO4)2 (2)), and N-benzyl-2-(bis(pyridin-2-ylmethyl)amino)acetamide (L3; [Ni(L3)(H2O)2](ClO4)2 (3))] and probed their catalytic potential in cycloalkane oxidation using m-CPBA as the oxidant. All the complexes catalyzed the oxidation of cyclohexane and produced cyclohexanol with excellent selectivity. High 3°/2° obtained in the oxidation substrates such as adamantane and methylcyclohexane ruled out the possibility of the hydroxyl radical mechanism in the oxidation reaction. In addition, the formation of chlorinated products during the oxidation of other cycloalkanes (cyclopentane, cycloheptane and cyclooctane) revealed the involvement of the free radical mechanism. However, higher selectivity achieved for the cyclic alcohols implies that the contribution of a metal-based oxidant cannot be discarded in the catalytic cycle.
2. Experimental sections
2.1. Materials
All reagents used to synthesise ligands and complexes were analytical grade and used as such except for m-CPBA. Aminomethyl pyridine (99%), diethyl ether (99%), m-CPBA (77%), hydrogen peroxide (30%) (H2O2), carbon tetrabromide (CBr4) and acetonitrile (CH3CN) HPLC grade were purchased from Merck. Aniline (99%), pyridine-2-carboxaldehyde (99%), sodium borohydride (98%), and nickel(II) perchlorate hexahydrate were procured from ThermoFisher Scientific. Chloroacetyl chloride (98%) and tert-butyl hydroperoxide (70%) (t-BuOOH) were purchased from Spectrochem. Triethylamine and naphthylamine were purchased from TCI Chemicals. Benzylamine was purchased from SRL India. Methanol, dichloromethane (DCM), chloroform, and acetone were purchased from Qualigens. Methanol was distilled over magnesium turnings (10 equiv.) and iodine (1 equiv.). Acetone and dichloromethane were distilled using calcium chloride and calcium hydride, respectively. The m-CPBA was purified by dissolving 30 g in 200 mL of diethyl ether and washing with phosphate buffer (pH 7.4, 150 mL). The organic portions were collected by extraction and dried to obtain m-CPBA with >90% purity.Caution note! As dried m-CPBA is shock-sensitive, suitable precautions should be taken when it is handled.
2.2. Physical measurements
1H-NMR/13C-NMR spectra of ligands were collected on Jeol 500 MHz spectrometer. UV-visible spectra of complexes were recorded in Agilent 8454 diode array spectrometer in acetonitrile at room temperature. The attenuated total reflection infrared (ATR-IR) spectra of samples were obtained using Jasco 4700 ATR-FTIR spectrometer at room temperature. Electrospray ionisation mass spectrometry (ESI-MS) data of the complexes were measured by Bruker Daltonics-esquire 6000. After catalysis, product analysis including identification and quantification was done using an Agilent 8860 GC series gas chromatograph equipped with an FID detector, HP-5 GC column (30 m × 0.32 mm × 0.25 μm). Post-reaction mixture of cyclohexane oxidation in the presence of CBr4 was analysed using an Agilent 7820A gas chromatograph equipped with an HP-5 capillary column 30 m × 0.32 mm × 0.25 μm and a flame ionisation detector. High-resolution mass spectrometry (HRMS) data of the m-CBA adduct was recorded using Waters Synapt XS HRMS instrument.2.3. Single crystal X-RD measurements
The X-ray intensity data were collected on a D8 QUEST ECO three-circle diffractometer system equipped with a Ceramic X-ray tube (Mo Kα, λ = 0.71073 Å) and a doubly curved silicon crystal Bruker Triumph monochromator. Single crystals were mounted using a fiber loop and optically centred. Multi-scan was used for the absorption correction. The structures were solved by direct methods with the program SHELXL-2019/1 (Sheldrick, 2019) and refined by full-matrix least-squares, based on F2 method using the program SHELXL-2017/1 (Sheldrick, 2017).2.4. Generation of steric maps
To calculate the steric map SambVca 2.1A web application was used which can be found at https://www.aocdweb.com/OMtools/sambvca2.1/index.html.32 In the web user interface, the required complex was oriented in a Cartesian frame with a chosen point at the origin (Ni), a second point along the z-axis (coordinating nitrogens from the pyridine rings in the same axis) and a third point in the xz plane (one of the pyridine nitrogens in the z-direction). After alignment of the complex, nickel and acetonitrile or water molecules, that must be ignored from the steric map calculations were deleted, and the first coordination sphere around the metal was analysed.2.5. Synthesis of ligands and complexes
Synthesis of all the ligands L1–L3 was done in three steps by following/modifying the procedures in the literature and step 1 is the same for all the ligands.33–37 Complexes were prepared by treating the required ligand with nickel(II) perchlorate hexahydrate in methanol at room temperature.2.5.1.1. Synthesis of 2-(bis(pyridin-2-ylmethyl)amino)-N-phenylacetamide (L1). Step 1: synthesis of bis(2-pyridylmethyl)amine: synthesis was done by following the procedure available in the literature.33 Aminomethyl pyridine (0.514 mL, 5 mmol) was added to a methanolic solution of pyridine-2-carboxaldehyde (0.475 mL, 5 mmol) and stirred for 12 h. Afterwards, sodium borohydride (0.226 g, 6 mmol) was added to the reaction mixture under ice-cold conditions and stirred for another 12 h at room temperature. The crude reaction mixture was concentrated under reduced pressure and extracted with DCM (100 mL × 3). Organic fractions were collected, dried over sodium sulfate and concentrated to obtain a yellowish-brown oil. Yield: 0.95 g, 95%.
Step 2: synthesis of 2-chloro-N-phenylacetamide: synthesis was done by adapting the existing procedure in the literature.34 Aniline (0.24 mL, 2.65 mmol) was dissolved in dry DCM under nitrogen atmosphere and stirred at ice-cold condition. To this, triethylamine (TEA, 0.154 mL, 1.1 mmol) in dry DCM was initially added and then chloroacetylchloride (0.35 mL, 4.42 mmol) in dry DCM was added dropwise. The resulting reaction mixture was allowed to stir for 1 h. After that, the crude reaction mixture was concentrated under reduced pressure and extracted with DCM (50 mL × 3). Organic fractions were collected and dried using sodium sulfate and concentrated to obtain the product as a white solid. Yield: 0.46 g, 92%.
Step 3: synthesis of 2-(bis(pyridin-2-ylmethyl)amino)-N-phenylacetamide (L1): synthesis was done by following the existing procedure in the literature.34 A mixture of 2-chloro-N-phenylacetamide (0.25 g, 1.48 mmol), bis(2-pyridylmethyl)amine (0.29 g, 1.48 mmol), N,N-diisopropylethylamine (0.287 g, 2.22 mmol) and potassium iodide (245 mg) was degassed for 30 min. Then, the reaction mixture was refluxed and stirred for 12 h under nitrogen atmosphere. The solvent from the reaction solution was removed using a rotary evaporator to obtain a brown oil, which was purified using silica column chromatography using DCM-methanol (v/v = 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as eluent. Yield: 0.43 g, 90%. ATR-IR, cm−1 (Fig. 2 (top)): 3329 cm−1 (N–H), 1671 cm−1 (C
:1) as eluent. Yield: 0.43 g, 90%. ATR-IR, cm−1 (Fig. 2 (top)): 3329 cm−1 (N–H), 1671 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). 1H NMR (500 MHz, CDCl3) (Fig. S1†): (δH ppm) 10.86 (s, 1H), 8.60 (d, 2H), 7.76 (d, 2H), 7.62 (t, 2H), 7.34–7.29 (m, 4H), 7.16 (dd, 2H), 7.07 (t, 1H), 3.95 (s, 4H), 3.47 (s, 2H).
O). 1H NMR (500 MHz, CDCl3) (Fig. S1†): (δH ppm) 10.86 (s, 1H), 8.60 (d, 2H), 7.76 (d, 2H), 7.62 (t, 2H), 7.34–7.29 (m, 4H), 7.16 (dd, 2H), 7.07 (t, 1H), 3.95 (s, 4H), 3.47 (s, 2H).
2.5.1.2. Synthesis of 2-(bis(2-pyridin-2-ylmethyl)amino)-N-(naphthalen-1-yl)acetamide (L2). Step 2: synthesis of 2-chloro-N-(naphthalen-1-yl)acetamide: synthesis was done by adapting the procedure in the literature.35,36 Chloroacetylchloride (0.352 mL, 4.42 mmol) in dry DCM was slowly added to a mixture of 1-naphthylamine (0.53 g, 3.7 mmol) and triethylamine (0.62 mL, 4.42 mmol) in dry DCM at ice-cold condition under nitrogen atmosphere. Then, the reaction mixture was stirred for 20 h at room temperature. The solvent was evaporated under reduced pressure and the residue was extracted with DCM (50 mL × 3). The organic fractions were collected and concentrated to get the product as white crystalline material. Yield: 0.6 g, 74%.
Step 3: synthesis of 2-(bis(2-pyridin-2-ylmethyl)amino)-N-(naphthalen-1-yl)acetamide (L2): synthesis was done by following the procedure in the literature.36 A mixture of 2-chloro-N-(naphthalen-4-yl)acetamide (0.548 g, 2.50 mmol), bis(2-pyridylmethyl)amine (0.49 g, 2.45 mmol), N,N-diisopropylethylamine (0.325 g, 2.52 mmol) and potassium iodide (13 mg) was degassed for 30 min. Then, the reaction mixture was refluxed and stirred for 12 h under nitrogen atmosphere. The solvent from the reaction solution was removed using a rotary evaporator to obtain a brown oil, which was purified using silica column chromatography using chloroform-methanol (v/v = 30:1) as eluent. Yield: 0.66 g, 70%. ATR-IR, cm−1 (Fig. S2†(top)): 3275 cm−1 (N–H), 1682 cm−1 (CO). 1H NMR (500 MHz, CDCl3) (Fig. S3†): (δH ppm) 11.09 (s, 1H), 8.49 (d, 2H), 8.41 (d, 1H), 8.11 (d,1H), 7.85 (d, 1H), 7.65–7.60 (m, 3H), 7.56–7.49 (m, 2H), 7.45 (t, 1H), 7.38 (d, 2H), 7.14 (dd, 2H), 4.07 (s, 4H), 3.60 (s, 2H).
2.5.1.3. Synthesis of N-benzyl-2-(bis(pyridin-2-ylmethyl)amino)acetamide (L3). Step 2: synthesis of N-benzyl-2-chloroacetamide: synthesis was done by modifying the procedure available in the literature.34,37 Benzylamine (0.655 mL, 6 mmol) was dissolved in dry DCM at ice-cold condition under nitrogen atmoshphere. To this, triethylamine (0.35 mL, 2.5 mmol) in dry DCM was initially added followed by the drop-wise addition of chloroacetylchloride (0.796 mL, 10 mmol) in dry DCM. The reaction mixture was stirred in an ice bath for 1 h and an additional 2 h at room temperature. The solvent from the reaction solution was reduced under pressure and the residue was extracted with chloroform (100 mL × 3). The organic fractions were collected, dried over sodium sulfate, and concentrated to get the product as white crystalline material. Yield: 0.8 g, 72%.
Step 3: synthesis of N-benzyl-2-(bis(pyridin-2-ylmethyl)amino)acetamide (L3): synthesis was done by following the procedure used for synthesising L1 and L2.34,36 A mixture of N-benzyl-2-(bis(pyridin-2-ylmethyl)amino)acetamide (0.5 g, 2.72 mmol), bis(2-pyridylmethyl)amine (0.54 g, 2.72 mmol), N,N-diisopropylethylamine (0.53 g, 4.08 mmol) and potassium iodide (226 mg) was degassed for 30 min. Then, the reaction mixture was refluxed and stirred for 12 h under nitrogen atmosphere. The solvent from the reaction solution was removed using a rotary evaporator to obtain a brown oil, which was purified using silica column chromatography using DCM–methanol (v/v = 48:2) as eluent. Yield: 0.48 g, 51%. ATR-IR, cm−1 (Fig. S4† (top)): 3365 cm−1 (N–H), 1656 cm−1 (CO). 1H NMR (500 MHz, CDCl3) (Fig. S5†): (δH ppm) 9.15 (s, 1H), 8.26 (d, 2H), 7.41 (t, 2H), 7.18 (d, 4H), 7.14–7.11 (m, 1H), 7.07 (d, 2H), 6.97 (dd, 2H), 4.35 (s, 2H), 3.73 (s, 4H), 3.28 (s, 2H). 13C NMR (125 MHz, CDCl3) (Fig. S6†): (δC ppm) 171.32, 157.95, 149.29, 138.47, 136.50, 128.45, 127.92, 127.10, 123.23, 122.39, 60.53, 58.10, 43.26.
[Ni(L1)(H2O)2](ClO4)2 (1): yield = 75%. ATR-IR, cm−1 (Fig. 2 (bottom)): 3343 cm−1 (N–H), 1649 cm−1 (CO), 1055 cm−1, 963 cm−1, 617 cm−1 (ClO4−). ESI-MS m/z = 489.1 {[Ni(L1)]2+ + ClO4−}+ (calcd. = 489.05) (Fig. S7†).
[Ni(L2)(H2O)2](ClO4)2 (2): yield = 84%. ATR-IR, cm−1 (Fig. S2† (bottom)): 3245 cm−1 (N–H), 1643 cm−1 (CO), 1055 cm−1, 972 cm−1, 626 cm−1 (ClO4−). ESI-MS m/z = 539.1 {[Ni(L2)]2+ + ClO4−}+ (calcd. = 539.06) (Fig. S8†). Single crystals of [Ni(L2)(CH3CN)2](ClO4)2 (2a) suitable for X-ray analysis were obtained in the diffusion method, where an acetonitrile solution of 2 in a vial was kept in another vial filled with diethyl ether for 1–2 days.
[Ni(L3)(H2O)2](ClO4)2 (3): yield = 60%. ATR-IR, cm−1 (Fig. S4† (bottom)): 3369 cm−1 (N–H), 1641 cm−1 (CO), 1055 cm−1, 963 cm−1, 621 cm−1 (ClO4−). ESI-MS m/z = 503.1 {[Ni(L3)]2+ + ClO4−}+ (calcd. = 503.06) (Fig. S9†). Single crystals of [Ni(L3)(H2O)(CH3CN)](ClO4)2 (3a) suitable for X-ray analysis were grown in the diffusion method, where a methanol–acetonitrile (v/v = 1:1) solution of 3 in a vial was kept in another vial filled with diethyl ether for 1–2 days.
2.6. Catalytic oxidation of cycloalkanes
:MeCN mixture (v/v = 3:1) at 60 °C under nitrogen atmosphere for 2 h. Initially, the nickel catalyst (1–3) (0.15 mM) was taken in a reaction tube in a DCM:MeCN mixture (3:1 v/v) and cyclohexane (2.8 M) was added to it. Further, the oxidant m-CPBA (0.35 M) was added using a syringe under nitrogen atmosphere and the reaction mixture was stirred for 2 h at 60 °C. Then, the reaction mixture was cooled down to room temperature and internal standard (bromobenzene) was added to the mixture. It was then passed through a silica plug and eluted with diethyl ether. The products were identified using an Agilent 8860 GC series gas chromatograph equipped with an FID detector, HP-5 GC column (30 m × 0.32 mm × 0.25 μm). Product quantification was done using calibration curves of authentic samples obtained by following the temperature program with a split ratio of 25:1, inlet temperature = 130 °C, initial temperature = 40 °C, heating rate = 0.1 °C min−1 to 42 °C, 8 °C min−1 to 190 °C (hold 1 min). FID temperature = 280 °C.28:MeCN mixture (v/v = 3:1) at 60 °C under nitrogen atmosphere for 2 h. Further, the oxidant m-CPBA (0.35 M) was added dropwise using a syringe. Then, the reaction mixture was cooled down to room temperature and internal standard (bromobenzene) was added to it. It was then passed through a silica plug and eluted with diethyl ether. The products were identified using an Agilent 8860 GC series gas chromatograph equipped with an FID detector, HP-5 GC column (30 m × 0.32 mm × 0.25 μm) and quantified using calibration curves of authentic samples obtained by following the temperature program with a split ratio of 25:1, inlet temperature = 130 °C, initial temperature = 40 °C, heating rate = 0.1 °C min−1 to 42 °C, 8 °C min−1 to 190 °C (hold 1 min). FID temperature = 280 °C.28:MeCN mixture (v/v = 3:1) at 60 °C under nitrogen atmosphere for 2 h. Firstly, the nickel catalyst (0.15 mM) was taken in a reaction tube containing the solvent followed by the addition of adamantane (0.27 M). Further, the oxidant m-CPBA (0.18 M) was added dropwise using a syringe under nitrogen atmosphere and the reaction mixture was stirred for 2 h at 60 °C. Then, the reaction mixture was cooled down to room temperature and internal standard (bromobenzene) was added to it. It was then passed through a silica plug and eluted with diethyl ether. The products were identified using an Agilent 8860 GC series gas chromatograph equipped with an FID detector, HP-5 GC column (30 m × 0.32 mm × 0.25 μm) and quantified using calibration curves of authentic samples obtained by following the temperature program with a split ratio of 25:1: inlet temperature = 130 °C, initial temperature = 60 °C, heating rate = 10 °C min−1 to 130 °C, then to 135 °C with a heating rate = 0.5 °C min−1 and finally to 220 °C at a heating rate = 20 °C min−1, FID temperature = 280 °C.28:MeCN mixture (3:1 v/v) and cyclohexane (2.8 M) was added to it. Then, m-CPBA (0.35 M) was added using a syringe under nitrogen atmosphere followed by the addition of CBr4 (0.35 M). The reaction mixture was stirred for 2 h at 60 °C. After that, the reaction mixture was cooled to room temperature and passed through a silica plug and eluted with diethyl ether. The mixture was analysed using an Agilent 7820A gas chromatograph equipped with an HP-5 capillary column 30 m × 0.32 mm × 0.25 μm and a flame ionisation detector.3. Results and discussion
3.1. Synthesis of ligands and complexes
The ligands L1–L3 (Scheme 2) were synthesised by adapting the procedures available in the literature and involved the dehydrohalogenation reaction between bis(2-pyridylmethyl)amine and the required acetamide precursor (Fig. S1–S6†).33–37 | ||
| Scheme 2 Tetradentate ligands employed in this study. | ||
The corresponding nickel(II) complexes (1–3) were prepared by reacting the ligands with nickel(II) perchlorate hexahydrate in distilled methanol. All the complexes were isolated as blue powder with good yields. Synthesised ligands and complexes were characterised with an array of advanced analytical techniques such as 1H NMR, ATR-IR and UV-vis spectroscopies, ESI-MS and single crystal XRD. Based on these analyses, complexes were formulated as [Ni(L1)(H2O)2](ClO4)2 (1), [Ni(L2)(H2O)2](ClO4)2 (2), [Ni(L3)(H2O)2](ClO4)2 (3).
3.2. Characterisation of complexes
 | ||
| Fig. 1 UV-vis absorption spectra of complexes 1–3 (10 × 10−3 M) recorded in acetonitrile at room temperature. | ||
 | ||
| Fig. 2 ATR-IR spectra of ligand L1 (top) and complex 1 (bottom). | ||
In addition, a shoulder peak around 762–799 nm was also observed due to the spin-forbidden transition 3A2g(F) → 1E1g(D). The band corresponding to 3A2g(F) → 3T1g(P) (ν3) transition was not observed experimentally probably due to the overlapping with the ligand-based high-energy transitions. However, using λ(nm) values of ν1 and ν2 transitions, ν3 λ(nm) has been calculated for all the complexes (1, 343 nm; 2, 345 nm; 3, 326 nm, Table 1).
O stretching frequency in all the ligands (1656–1682 cm−1) and respective complexes (1641–1649 cm−1) implies that the CO group of the ligands remains intact during complex formation. Moreover, all the complexes showed a broad intense peak at 1055 cm−1, a sharp peak around 617–621 cm−1 and a weaker absorption peak around 963–972 cm−1, which can be ascribed to the presence of uncoordinated ClO4− ions.33,38,39 | ||
| Fig. 3 ORTEP diagrams of the cationic part of complexes (a) 2a and (b) 3a with labelling schemes. Displacement ellipsoids are shown at 30% probability level. Hydrogen atoms (except from the amide group and water molecule) are omitted for clarity. | ||
| 2a | 3a | ||
|---|---|---|---|
| Bond lengths [Å] | |||
| Ni1–N5 | 2.028(3) | Ni1–N4 | 2.042(2) |
| Ni1–N4 | 2.089(4) | Ni1–N3 | 2.079(2) |
| Ni1–N3 | 2.075(4) | Ni1–N2 | 2.090(2) |
| Ni1–N2 | 2.090(3) | Ni1–N1 | 2.081(2) |
| Ni1–N1 | 2.057(4) | Ni1–O1 | 2.0649(19) |
| Ni1–O1 | 2.065(3) | Ni1–O(w) | 2.072(2) |
|
|||
| Bond angles [°] | |||
| N5–Ni1–N4 | 90.76(12) | N4–Ni1–N3 | 100.01(10) |
| N5–Ni1–N3 | 97.49(14) | N4–Ni1–N2 | 177.00(10) |
| N5–Ni1–N2 | 174.26(13) | N4–Ni1–N1 | 99.06(9) |
| N5–Ni1–N1 | 100.70(14) | N3–Ni1–N2 | 79.77(9) |
| N4–Ni1–N2 | 94.39(12) | N3–Ni1–N1 | 160.80(9) |
| N3–Ni1–N4 | 90.03(13) | N1–Ni1–N2 | 81.30(9) |
| N3–Ni1–N2 | 80.02(12) | N4–Ni1–O1 | 93.17(8) |
| N3–Ni1–N1 | 161.81(12) | N3–Ni1–O1 | 94.44(8) |
| N1–Ni1–N4 | 89.81(13) | N2–Ni1–O1 | 83.88(9) |
| N1–Ni1–N2 | 81.86(12) | N1–Ni1–O1 | 86.69(8) |
| N5–Ni1–O1 | 92.10(11) | N4–Ni1–O(w) | 88.51(9) |
| N4–Ni1–O1 | 176.28(13) | N3–Ni1–O(w) | 85.77(9) |
| N3–Ni1–O1 | 91.95(14) | N2–Ni1–O(w) | 94.45(10) |
| N2–Ni1–O1 | 82.86(11) | N1–Ni1–O(w) | 92.53(9) |
| N1–Ni1–O1 | 87.31(14) | O1–Ni1–O(w) | 178.24(9) |
| 2a | 3a | |
|---|---|---|
| a R1 = [Σ(‖Fo| − |Fc‖)/Σ|Fo|]; wR2 = {[Σ(w(Fo2 − Fc2)2)/Σ(wFo4)]1/2}. | ||
| Empirical formula | C28H28Cl2N6NiO9 | C23H27Cl2N5NiO10 |
| Formula weight (g mol−1) | 722.17 | 663.10 |
| Temperature (K) | 100(2) | 100(2) |
| Wavelength/(Å) | 0.71073 | 0.71073 |
| Crystal size (mm) | 0.140 × 0.140 × 0.200 | 0.100 × 0.130 × 0.240 |
| Crystal habit | Violet prism | Blue prism |
| Crystal system | Triclinic | Monoclinic |
| Space group | P -1 | P1 21/c1 |
| a (Å) | 10.8951(6) | 17.786(3) |
| b (Å) | 12.6583(7) | 7.4291(13) |
| c (Å) | 12.8328(7) | 21.345(4) |
| α (°) | 87.684(2) | 90 |
| β (°) | 77.714(2) | 101.411(5) |
| γ (°) | 72.944(2) | 90 |
| V (Å3) | 1652.77(16) | 2764.6(8) |
| Z | 2 | 4 |
| ρcald (g cm−3) | 1.451 | 1.593 |
| ε (mm−1) | 0.808 | 0.959 |
| F(000) | 744 | 1368 |
| θ (min, max) (°) | 2.31, 27.55 | 2.98, 27.57 |
| Reflections collected | 52485 |
97866 |
| Independent reflections, R (int) | 7553, 0.0184 | 6383, 0.0822 |
| Max. and min. Transmission | 0.8950 and 0.8550 | 0.9100 and 0.8020 |
| Data/restraints/parameters | 7553/13/555 | 6383/12/403 |
| Goodness-of-fit on F2 | 1.043 | 1.034 |
| R1, wR2 (I > 2σ(I))a | 0.0706, 0.2081 | 0.0429, 0.0942 |
| R1, wR2 (all data) | 0.0761, 0.2147 | 0.0633, 0.1045 |
It is noteworthy that, unlike several amidate ligands, the amide nitrogen in L2 and L3 did not undergo deprotonation (even in the presence of TEA) during complex formation to coordinate with the nickel(II) centre, instead, the carbonyl oxygen participates in the coordination in both 2a and 3a.28,40 A similar coordination behaviour was observed in the crystal structure of a zinc complex of L2.36 Nickel(II) centre in both complexes resides in a distorted octahedral environment coordinated with three nitrogen atoms and one carbonyl oxygen atom of the respective ligand along with either two acetonitrile molecules (2a) or one acetonitrile molecule and a water molecule (3a). The acetonitrile molecules are coordinated during the crystallisation process. Bond angles of 2a and 3a fall in the range 79.77(9)–100.70(14) and 160.80(9)–178.24(9), which deviate significantly from the ideal octahedral bond angles of 90° and 180°.
3.3. Oxidation of cycloalkanes
:MeCN (v/v = 3:1) mixture at 60 °C for 2 hours. However, m-CPBA was found to be the only suitable oxidant for the reaction (Fig. S10†) and afforded cyclohexanol (A) as the major product and cyclohexanone (K) along with caprolactone (L; formed due to the Baeyer–Villiger oxidation of K) as the minor products. In addition, chlorobenzene (PhCl; formed due to the dissociation of m-CPBA or m-CPBA adduct) and chlorocyclohexane (Cy6Cl; produced by the chlorine atom abstraction of cyclohexyl radical (Cy6˙) from the solvent DCM molecule) were also identified in the reaction mixture (Fig. S10†). The TON of oxidised products reported here is the average of three determinations, and it is calculated by taking the ratio of the number of mmol of formed product to the number of mmol of catalyst used. The catalytic activity was notably affected by the catalyst loading, oxidant concentration, temperature as well as time, and thus, the reaction conditions were optimised using catalyst 1 by tuning these parameters and the results are summarised in Table 4.
| Entry | Cat. conc.b (mM) | Cy6Cl (TON) | A (TON) | K (TON) | L (TON) | Total TONf | PhCl (TON) | A/(K + L) |
|---|---|---|---|---|---|---|---|---|
| a Cy6Cl = chlorocyclohexane, A = cyclohexanol, K = cyclohexanone, L = caprolactone, PhCl = chlorobenzene.b Reaction conditions: cyclohexane (2.8 M), oxidant (0.35 M), catalyst in DCM:MeCN solvent mixture (v/v = 3:1), 60 °C, 2 h under N2.c Reaction conditions: cyclohexane (2.8 M), catalyst (0.15 mM) in DCM:MeCN solvent mixture (v/v = 3:1), 60 °C, 2 h under N2.d Reaction conditions: cyclohexane (2.8 M), oxidant (0.35 M), catalyst (0.15 mM) in DCM:MeCN solvent mixture (v/v = 3:1), 2 h under N2.e Reaction conditions: cyclohexane (2.8 M), oxidant (0.35 M), catalyst (0.15 mM) in DCM:MeCN solvent mixture (v/v = 3:1), 60 °C under N2.f TON = number of mmol of product/number of mmol of catalyst. The TON is the average of three determinations. |
||||||||
| 1 | 0.1 | 172 | 528 | 16 | 26 | 742 | 951 | 12.6 |
| 2 | 0.15 | 136 | 497 | 10 | 11 | 654 | 878 | 23.6 |
| 3 | 0.2 | 90 | 348 | 6 | 8 | 452 | 621 | 24.8 |
| 4 | 0.25 | 63 | 356 | 5 | 10 | 434 | 441 | 23.7 |
| 5 | 0.3 | 83 | 197 | 4 | 5 | 289 | 408 | 21.8 |
|
||||||||
| Oxidantc (M) | ||||||||
| 6 | 0.05 | 13 | 61 | 2 | — | 76 | 159 | 30.5 |
| 7 | 0.15 | 73 | 150 | 5 | — | 228 | 397 | 30.0 |
| 8 | 0.25 | 68 | 398 | 8 | 6 | 480 | 489 | 28.4 |
| 9 | 0.35 | 136 | 497 | 10 | 11 | 654 | 878 | 23.6 |
| 10 | 0.45 | 150 | 533 | 5 | 35 | 723 | 930 | 13.3 |
|
||||||||
| Temp.d (°C) | ||||||||
| 11 | 25 | — | — | — | — | — | 60 | — |
| 12 | 40 | 26 | 240 | 5 | 6 | 277 | 125 | 21.8 |
| 13 | 50 | 37 | 355 | 9 | 7 | 408 | 322 | 22.1 |
| 14 | 60 | 136 | 497 | 10 | 11 | 654 | 878 | 23.6 |
| 15 | 80 | 135 | 524 | 11 | 15 | 685 | 918 | 20.1 |
| 16 | 100 | 153 | 538 | 16 | 17 | 724 | 1107 | 16.3 |
|
||||||||
| Timee (h) | ||||||||
| 17 | 0.5 | 15 | 99 | 4 | — | 118 | 270 | 24.7 |
| 18 | 1.0 | 28 | 167 | 6 | 1 | 202 | 374 | 23.8 |
| 19 | 1.5 | 46 | 260 | 6 | 5 | 317 | 406 | 23.6 |
| 20 | 2.0 | 136 | 497 | 10 | 11 | 654 | 878 | 23.6 |
| 21 | 2.5 | 134 | 520 | 10 | 12 | 676 | 890 | 23.6 |
| 22 | 5 | 128 | 416 | 9 | 12 | 565 | 856 | 19.8 |
| 23 | 10 | 104 | 407 | 8 | 14 | 533 | 739 | 18.5 |
| 24 | 24 | 61 | 356 | 2 | 44 | 463 | 622 | 7.7 |
The effect of catalyst loading was studied using different catalyst concentrations (0.1–0.3 mM, Table 4, entries 1–5). Even though the achieved total TON was the highest (742) when 0.1 mM (Table 4, entry 1) of 1 was used, the obtained alcohol selectivity (A/(K + L) = 12.6) was the lowest. Upon using 0.15 mM of catalyst, a double-fold increment in the alcohol selectivity (23.6) was noted with a total TON of 654 (Table 4, entry 2). As the catalyst concentration increased, the obtained total TON decreased by maintaining the alcohol selectivity (Table 4, entries 3–5). Therefore, the optimum catalyst loading was selected as 0.15 mM. Then, the reactions were performed under different oxidant concentrations (0.05–0.45 M) and evaluated the catalytic activity (Table 4, entries 6–10). When lower oxidant concentrations were used, the total TON obtained was poor, but, excellent alcohol selectivity was observed (Table 4, entries 6 and 7). With the increment in the amount of oxidant used, an enhancement in the total TON (480–723) and a drop in the A/(K + L) ratio (28.4–13.3) were noted (Table 4, entries 8–10). Based on the results, a high total TON (654) with a good A/(K + L) ratio (23.6) was achieved using 0.35 M of m-CPBA and it was taken as the optimum oxidant concentration (Table 4, entry 9). Further, the temperature effect on the catalytic activity was monitored by performing the reaction at different temperatures (25–100 °C; Table 4, entries 11–16). The reaction at 25 °C did not afford any oxidised products, rather a small amount of chlorobenzene (PhCl) was formed. A gradual increase in the total TON (277–724) was seen when the reaction temperature was changed with in the range of 40 °C to 100 °C. However, high temperatures favoured overoxidation as well as Baeyer–Villiger oxidation and produced more amount of K and L. This, in turn, reduced alcohol selectivity at high temperatures and thus 60 °C was chosen as the suitable temperature for the reaction. In addition, the effect of reaction time was also evaluated by carrying out the reactions for various time durations (Table 4, entries 17–24). Up to 2.5 h, an improvement in the total TON was noted with good alcohol selectivity. In contrast, keeping the reaction for 5, 10 or 24 h led to decreased activity, which could be due to the evaporation of solvent or volatile oxidised products over time. The difference in the catalytic activity was only marginal when the reaction time was 2 or 2.5 h and thus the optimum time for the reaction was selected as 2 h. Under optimised conditions, a control experiment was performed without using any catalyst and the oxidised products were formed in trace amounts (Table 5, entry 1). On the other hand, using catalysts 1–3, the reaction progressed very well and afforded A, K and L along with Cy6Cl and PhCl in moderate yield (15.4–28.0%). The catalytic activity was in the order 1 > 2 > 3 in terms of total TON as well as A/(K + L) ratio (Table 5, entries 2–4).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalystb | Cy6Cl (TON) | A (TON) | K (TON) | L (TON) | Total TONc | PhCl (TON) | A/(K + L) | Yieldd (%) |
| a Cy6Cl = chlorocyclohexane, A = cyclohexanol, K = cyclohexanone, L = caprolactone, PhCl = chlorobenzene.b Reaction conditions: cyclohexane (2.8 M), oxidant (0.35 M), catalyst (0.15 mM) in DCM:MeCN solvent mixture (v/v = 3:1), 60 °C, 2 h under N2.c TON = number of mmol of product/number of mmol of catalyst. The TON is the average of three determinations.d Yield based on m-CPBA.e Values based on 0.15 mM virtual nickel catalyst. |
|||||||||
| 1 | None | 25 | 52 | 9 | — | 86e | 59 | 5.7 | 4 |
| 2 | 1 | 136 | 497 | 10 | 11 | 654 | 878 | 23.6 | 28.0 |
| 3 | 2 | 96 | 472 | 9 | 12 | 589 | 782 | 22.4 | 25.2 |
| 4 | 3 | 65 | 275 | 8 | 11 | 359 | 508 | 14.4 | 15.4 |
| 5 | 1 | 48 | 62 | 10 | 23 | 143 | 223 | 1.8 | 6.1 |

The catalytic activity was majorly influenced by the steric factors of the ligands. The m-CPBA adduct formation with the catalyst or facile approach of the substrate towards the reactive species is favoured in a less steric-hindering environment. The bis(2-pyridylmethyl)amine unit is commonly present in all the complexes and imparts the same steric hindrance. On the other hand, monocyclic aniline donor moiety in complex 1 provides less steric hindrance than dicyclic naphthyl amine moiety in complex 2 and this could be attributed to the enhanced activity of 1 over 2. Similarly, complex 3 containing monocyclic benzylamine moiety is expected to show more catalytic activity than complex 2. Nevertheless, even with the lowest steric hindrance calculated for 3 (% Vbur = 72.2) using the SambVca 2.1 web application, it was found to be the least active catalyst (Fig. 4). This is possibly due to the rapid free rotation of the benzylic CH2 group in 3, that can hamper the easy access of m-CPBA or substrate to the nickel centre and decrease the catalytic activity. Also, the present catalysts have a higher % Vbur (72.2–81.3) than our recently reported aminoquinoline-based pincer nickel(II) catalysts (% Vbur = 57.7–66.0), which correlates with the greater activity observed for the latter set of complexes.28
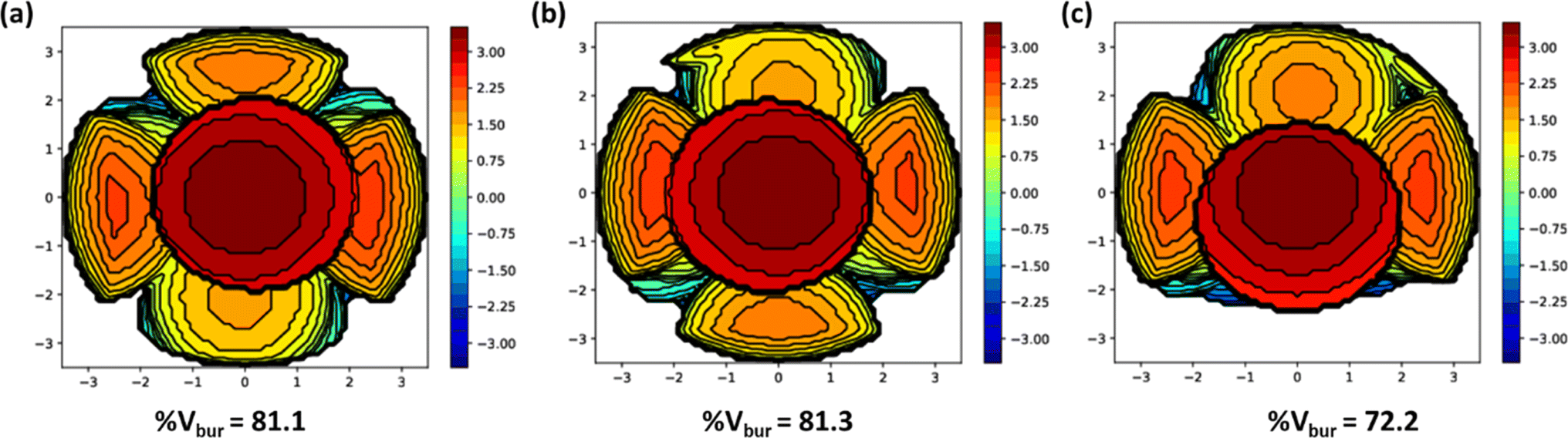 | ||
| Fig. 4 Steric maps of 1 (a), 2 (b) and 3 (c) with % Vbur generated using SambVca 2.1 web application. | ||
Moreover, compared to previously reported tetradentate nickel complexes (Scheme 1 and Table 6), the total TON and overall yield were lower when catalysts 1–3 were used.4 Despite that, remarkable improvement in the A/(K + L) ratio was observed with the present catalysts. The results suggest that the introduction of donor groups such as aniline, naphthylamine and benzylamine positively influences alcohol selectivity, but, does not enhance the overall yield.
| Catalyst | Total TON | A/(K + L) ratio | Yielda (%) | Ref. |
|---|---|---|---|---|
| a Yield based on m-CPBA concentration.b The ratio given is A/K. | ||||
| I | 656 | 8.5b | 58.7 | 11 |
| II | 858 | 7.7b | 85.8 | 12 |
| III | 442 | 9.8 | 88.3 | 13 |
| IV | 622 | 8.7 | 62.2 | 14 |
| V | 601 | 7.1 | 60.1 | 15 |
| VI | 974 | 8.6 | 97.4 | 16 |
| 1 | 654 | 23.6 | 28.0 | This work |
| 2 | 589 | 22.4 | 25.2 | This work |
| 3 | 359 | 14.4 | 15.4 | This work |
The reaction when performed under an O2 atmosphere (Table 5, entry 5) drastically reduced the product formation (TON, 143) and the A/(K + L) ratio (1.8), which could be attributed to the Russell termination of the radical chain reaction. During the oxidation process, the reactive species generate cyclohexyl radicals (Cy6˙) by abstracting a hydrogen atom from cyclohexane. Upon exposure to O2, Cy6˙ radicals react with O2 to form cyclohexyl peroxy radical species (Cy6OO˙). In the next step, radical coupling between two Cy6OO˙ species leads to the formation of Cy6OOOOCy6 tetroxide, and it eventually produces an equimolar amount of cyclohexanol and cyclohexanone. The formation of Cy6˙ radicals during the oxidation process was further confirmed by performing the reaction in the presence of CBr4 and identifying the presence of bromocyclohexane in the post-reaction mixture. This occurs due to the bromine atom abstraction from CBr4 by Cy6˙ radicals generated in the reaction (Fig. S11†).
Afterwards, the effect of the ring size in the oxidation reaction was studied using other cycloalkanes such as cyclopentane (Cy5H), cycloheptane (Cy7H) and cyclooctane (Cy8H) in the presence of catalyst 1 and the results are provided in Table 7 (Fig. S12†). The obtained total TON and overall yield were correlated to the ring size of the substrates. For example, the total TON achieved was only 385 in the case of cyclopentane, whereas in the case of cyclooctane, it reached 834. Although the bond dissociation energy of cyclopentane (95.6 kcal mol−1) is less than that of cyclohexane (99.5 kcal mol−1), the latter was more susceptible to oxidation.7 Similar observations were reported for nickel and cobalt catalysts using m-CPBA as the oxidant.28,41
| Substrateb (CynH) | CynCl (TON) | A (TON) | K (TON) | Total TONc,d | PhCl (TON) | Yielde (%) |
|---|---|---|---|---|---|---|
| a CynCl = chloro derivative of the respective substrate, A = cycloalcohol, K = cycloketone, PhCl = chlorobenzene.b Reaction conditions: substrate (2.8 M), oxidant (0.35 M), catalyst (0.15 mM) in DCM:MeCN solvent mixture (v/v = 3:1), 60 °C, 2 h under N2.c TON = number of mmol of product/number of mmol of catalyst. The TON is the average of three determinations.d TON of lactone is excluded.e Yield based on m-CPBA. |
||||||
| Cy5H | 92 | 278 | 15 | 385 | 473 | 16.5 |
| Cy6H | 136 | 497 | 10 | 654 | 878 | 28.0 |
| Cy7H | 186 | 506 | 70 | 762 | 436 | 32.6 |
| Cy8H | 228 | 532 | 74 | 834 | 380 | 35.7 |
The regioselectivity of reactive species was examined using catalyst 1 and substrates such as adamantane and methylcyclohexane with primary (1°), secondary (2°), and tertiary (3°) C–H bonds (Tables S1 and S2†). The oxidation of adamantane afforded 1-adamantanol (3°-ol), 2-adamantanol (2°-ol), 2-adamantanone (2°-one) and 1-chloroadamantane (3°-Cl) with a total TON of 337 and 3°/2° ratio of 8.6 (Table S1 and Fig. S12†). If the adamantane oxidation reaction proceeds via a Fenton-type mechanism (˙OH radical), a poor 3°/2° = 0.4–1.3 is typically observed.29 A 3°/2° ratio in the range of 4.4–6.9 was reported by Hartwig et al. stating the involvement of 3-chlorobenzoyloxy radical in the catalytic cycle.29 A much higher value in the range of 8.7–18.7 is noted in a few reports where the involvement of a nickel oxygen species was proposed.4,16 Even though the ratio determined in the present study can be used to discard the possibility of the Fenton-type mechanism, it does not ensure or exclude the presence of 3-chlorobenzoyloxy radical or the nickel-based oxidant in the catalytic cycle. Further, the oxidation of methylcyclohexane was carried out using 1 and obtained tertiary (3°-ol), secondary (o, m, p-ols and 2°-ones) and primary (1°-ol) C–H oxidised products with a preference for secondary C–H oxidation (3°-ol:2°-ols = 36:64). In contrast, the control experiment without any catalyst afforded 3°-ol majorly with poor overall yield (3°-ol:2°-ols = 82:18) (Table S2†). Similar findings have been noted in previous studies where a nickel-based oxidant was proposed.16,21
Furthermore, to identify if any nickel intermediate is formed during the catalytic reaction, the reaction of catalyst 1 and m-CPBA was examined using UV-vis spectroscopy at room temperature. However, no spectral changes were noticed in the complex spectrum (5 mM) after the addition of m-CPBA (1 equiv.). Despite that, the addition of the base, triethylamine (1 equiv.) in the reaction mixture led to a redshift in the absorption bands of complex 1 from 551 to 561 nm, 762 to 771 nm, and 880 to 934 nm (Fig. S13a†). HRMS analysis of the reaction mixture revealed the formation of an m-CBA adduct of complex 1 (m/z = 545.0895 [Ni(L1) + (m-CBA-H)]+ (calcd = 545.0890) (Fig. S13b†). In addition, the m-CBA adduct formation was further confirmed by the same spectral pattern obtained upon the reaction of 1 (5 mM) with m-CBA (1 equiv.) in the presence of triethylamine (1 equiv.) in acetonitrile at room temperature (Fig. S13a†).
Nevertheless, these observations do not conclusively substantiate the formation of m-CPBA or m-CBA adduct during the oxidation of cycloalkanes using catalysts 1–3. In general, our results demonstrate that catalysts 1–3 are capable of catalysing the oxidation of a series of cycloalkanes using m-CPBA as the oxidant with excellent alcohol selectivity. By considering recent advances in the mechanistic studies of this chemistry, three possible pathways are proposed for the cycloalkane oxidation catalyzed by complexes 1–3 (Scheme 3). The high A/(K + L) ratio obtained and preferential oxidation of 2° C–H bonds in methylcyclohexane direct towards the contribution of nickel-based oxidants (paths 1 and 2). Although the reports in the last decade discussed a mechanism that occurs exclusively through path 1,11,12,14,15,18–20 the latest theoretical and experimental findings validate the contribution of paths 2 and 3.13,28–31 All the pathways are expected to start with the formation of an m-CPBA adduct from the reaction of the nickel(II) complex and m-CPBA. If the oxidation proceeds via path 1, the formed m-CPBA adduct undergoes O–O homolysis to generate [(L)NiII–O˙] and this species further abstracts a hydrogen atom from the substrate to form R˙ radical. In path 2, a hydroxo species [(L)NiIII–OH] produced from the m-CPBA adduct is responsible for the generation of the substrate radical (R˙). Unlike paths 1 and 2, a non-nickel-based reactive species (3-chlorobenzoyloxy radical) is involved in path 3 for the generation of R˙ radicals. Nonetheless, in all the cases, the formed R˙ radicals either react with m-CPBA to produce cyclic alcohol or abstract a chlorine atom from the dichloromethane solvent to produce chlorocyclohexane. Even though the literature provides evidence for the aryloxy radical mechanism, efforts to trap or characterise the proposed nickel-based oxidants have not yielded success to date.
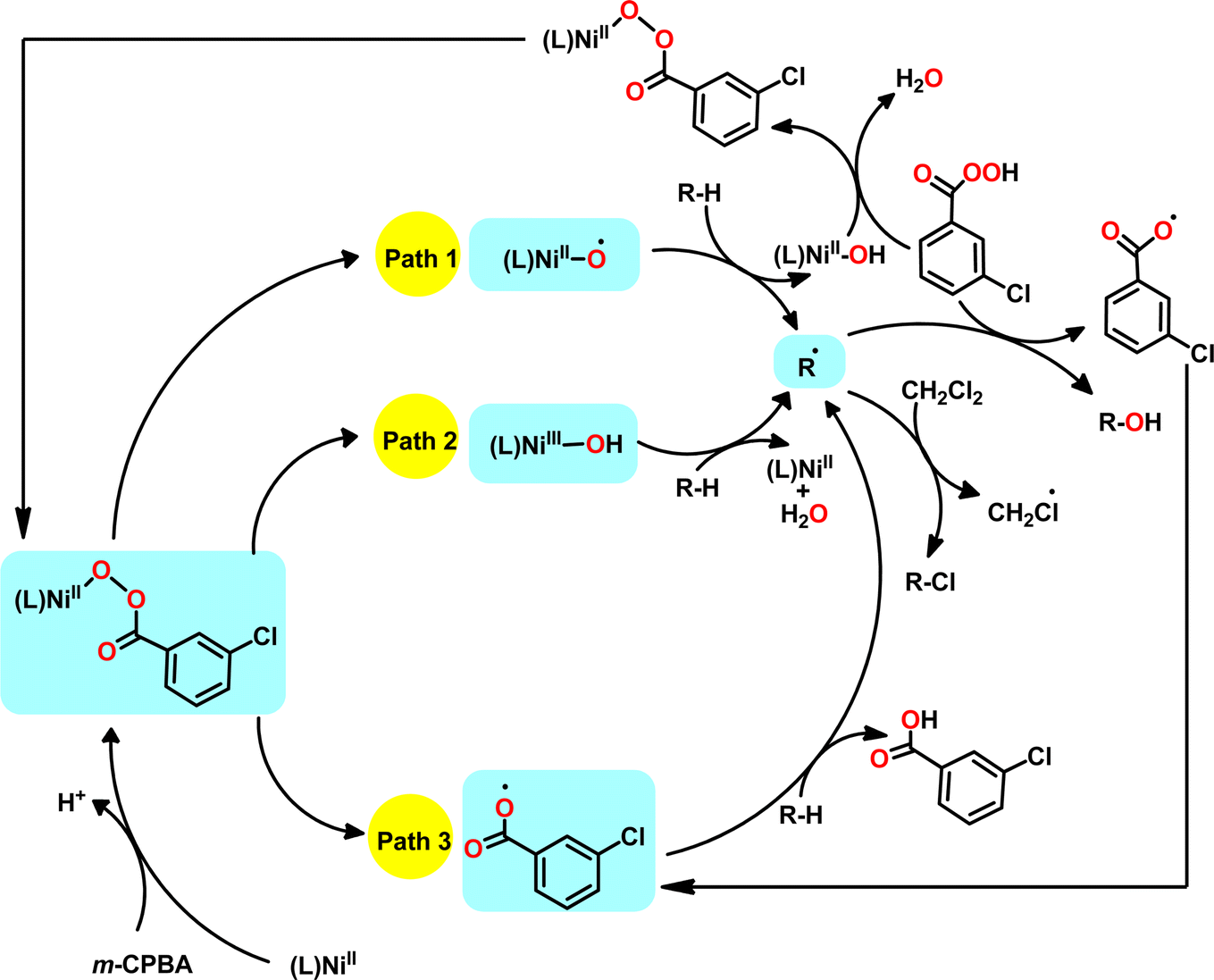 | ||
| Scheme 3 Possible mechanistic pathways of cycloalkane oxidation catalysed by complexes 1–3 using m-CPBA as the oxidant. | ||
4. Conclusion
Three monoamidate tetradentate ligands (L1–L3) and the respective nickel(II) complexes (1–3) were prepared and characterised. The catalytic potential of 1–3 was evaluated in the oxidation of cyclohexane using m-CPBA as the oxidant. Complexes showed activity in the order 1 (654) > 2 (589) > 3 (359) with a high A/(K + L) ratio up to 23.6. In addition, oxidation of other cycloalkanes such as cyclopentane, cycloheptane, cyclooctane, methylcyclohexane and adamantane was performed using catalyst 1. Strikingly, the ring size of the substrates influenced the oxidation susceptibility of the substrates due to which cyclopentane oxidation resulted in low overall product yield and cyclooctane oxidation resulted in high overall product yield. A relatively high 3°/2° ratio (8.6) determined for adamantane oxidation confirmed the non-involvement of the Fenton-type mechanism (3°/2° = 0.4–1.3) in the catalytic cycle. Similarly, the preferential oxidation of 2° C–H bonds in methylcyclohexane indicated the possible participation of nickel-based oxidant in the oxidation reaction. However, the presence of chlorinated products and a high amount of chlorobenzene in the reaction mixture support the major contribution of the 3-chlorobenzoyloxy radical mechanism.Data availability
The data that support the findings of the present study are available in the ESI† of this article. CCDC 2371833 (for 2) and 2371834 (for 3) contain the supplementary crystallographic data for this article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MS sincerely acknowledges the Department of Science and Technology for the award of the DST-Inspire Faculty Research Grant (IFA-17-CH286), the Science and Engineering Research Board (SERB) for the Core Research Grant (CRG/2023/002850) and the National Institute of Technology Calicut for the Faculty Research Grant (NIT/DEAN(R&C)/FRG/2018-19/3). A.R. acknowledges the UGC-SRF for the fellowship to pursue her PhD. We thank the CMC, NIT Calicut for NMR characterisations. S.M. acknowledges the Spain Ministry of Science (FJC2021-047874-I) for providing the fellowship to support his research work. We thank Prof. Miquel Costas, IQCC, Universitat de Girona, Spain, for helping us to record mass spectra and single crystal X-ray data of the complexes.References
- B. Gabriele, Molecules, 2022, 27, 1227 CrossRef PubMed.
- V. P. Ananikov, L. L. Khemchyan, Yu. V. Ivanova, V. I. Bukhtiyarov, A. M. Sorokin, I. P. Prosvirin, S. Z. Vatsadze, A. V. Medved'ko, V. N. Nuriev, A. D. Dilman, V. V. Levin, I. V. Koptyug, K. V. Kovtunov, V. V. Zhivonitko, V. A. Likholobov, A. V. Romanenko, P. A. Simonov, V. G. Nenajdenko, O. I. Shmatova, V. M. Muzalevskiy, M. S. Nechaev, A. F. Asachenko, O. S. Morozov, P. B. Dzhevakov, S. N. Osipov, D. V. Vorobyeva, M. A. Topchiy, M. A. Zotova, S. A. Ponomarenko, O. V. Borshchev, Yu. N. Luponosov, A. A. Rempel, A. A Valeeva, A. Yu. Stakheev, O. V. Turova, I. S. Mashkovsky, S. V. Sysolyatin, V. V. Malykhin, G. A. Bukhtiyarova, A. O. Terent’ev and I. B. Krylov, Russ. Chem. Rev., 2014, 83(10), 885–985 CrossRef.
- T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2363 CrossRef PubMed.
- M. Sankaralingam, M. Balamurugan and M. Palaniandavar, Coord. Chem. Rev., 2020, 403, 213085 CrossRef.
- A. Rajeev, M. Balamurugan and M. Sankaralingam, ACS Catal., 2022, 12, 9953–9982 CrossRef.
- M. T. Musser and E. I. Du, Cyclohexanol and Cyclohexanone, Ullmann's Encycl. Ind. Chem., 2012, pp. 49–60 Search PubMed.
- Y.-R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRC Press, 1st edn, 2002 Search PubMed.
- U. Schuchardt, R. Pereira and M. Rufo, J. Mol. Catal. A: Chem., 1998, 135, 257–262 CrossRef.
- A. R. Silva, T. Mourão and J. Rocha, Catal. Today, 2013, 203, 81–86 CrossRef.
- N. M. F. Carvalho, A. Horn and O. A. C. Antunes, Appl. Catal., A, 2006, 305, 140–145 CrossRef.
- T. Nagataki, Y. Tachia and S. Itoh, Chem. Commun., 2006, 4016–4018 RSC.
- T. Nagataki and S. Itoh, Chem. Lett., 2007, 36, 748–749 CrossRef.
- T. Wada, H. Sugimoto, Y. Morimoto and S. Itoh, Polyhedron, 2022, 227, 116150 CrossRef.
- M. Balamurugan, R. Mayilmurugan, E. Suresh and M. Palaniandavar, Dalton Trans., 2011, 40, 9413–9424 RSC.
- M. Sankaralingam, P. Vadivelu and M. Palaniandavar, Dalton Trans., 2017, 46, 7181–7193 RSC.
- I. Terao, S. Horii, J. Nakazawa, M. Okamura and S. Hikichi, Dalton Trans., 2020, 49, 6108–6118 RSC.
- A. Rajeev and M. Sankaralingam, Highlights of Oxygen Atom Transfer Reactions Catalysed by Nickel Complexes, Oxygen Atom Transfer Reactions, Mechanisms of oxidation reactions, Bentham Science., 2023, pp. 62–90 Search PubMed.
- T. Nagataki, K. Ishii, Y. Tachi and S. Itoh, Dalton Trans., 2007, 1120–1128 RSC.
- M. Sankaralingam, M. Balamurugan, M. Palaniandavar, P. Vadivelu and C. H. Suresh, Chem. - Eur. J., 2014, 20, 11346–11361 CrossRef PubMed.
- M. Sankaralingam, P. Vadivelu, E. Suresh and M. Palaniandavar, Inorg. Chim. Acta, 2013, 407, 98–107 CrossRef.
- S. Hikichi, K. Hanaue, T. Fujimura, H. Okuda, J. Nakazawa, Y. Ohzu, C. Kobayashi and M. Akita, Dalton Trans., 2013, 42, 3346–3356 RSC.
- P. Pirovano, E. R. Farquhar, M. Swart and A. R. McDonald, J. Am. Chem. Soc., 2016, 138, 14362–14370 CrossRef.
- P. Pirovano, B. Twamley and A. R. McDonald, Chem. - Eur. J., 2018, 24, 5238–5245 CrossRef PubMed.
- P. Pirovano, A. R. Berry, M. Swart and A. R. McDonald, Dalton Trans., 2018, 47, 246–250 RSC.
- T. Corona, F. F. Pfaff, F. AcuÇa-ParØs, A. Draksharapu, C. J. Whiteoak, V. Martin-Diaconescu, J. Lloret-Fillol, W. R. Browne, K. Ray and A. Company, Chem. - Eur. J., 2015, 21, 15029–15038 CrossRef PubMed.
- T. Corona, A. Draksharapu, S. K. Padamati, I. Gamba, V. Martin-Diaconescu, F. Acuña-Pares, W. R. Browne and A. Company, J. Am. Chem. Soc., 2016, 138, 12987–12996 CrossRef.
- A. Juvanteny, C. Souilah, R. Quintero, C. García-Bellido, N. Pagès-Vilà, T. Corona, P. Salvador and A. Company, Inorg. Chem., 2024, 63, 14325–14334 CrossRef PubMed.
- A. Rajeev, A. T. Thomas, A. Das and M. Sankaralingam, Eur. J. Inorg. Chem., 2024, 27, e202400205 CrossRef.
- Y. Qiu and J. F. Hartwig, J. Am. Chem. Soc., 2020, 142, 19239–19248 CrossRef PubMed.
- T. Shinke, M. Itoh, T. Wada, Y. Morimoto, S. Yanagisawa, H. Sugimoto, M. Kubo and S. Itoh, Chem. - Eur. J., 2021, 27, 14730–14737 CrossRef PubMed.
- Z. Wu, D. Sun, Y. Lee, Y. Zhao, W. Nam and Y. Wang, Dalton Trans., 2023, 52, 8676–8684 RSC.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef PubMed.
- A. Das, A. Rajeev, S. Bhunia, M. Arunkumar, N. Chari and M. Sankaralingam, Inorg. Chim. Acta, 2021, 526, 120515 CrossRef.
- L. Zhang, S. Li, H. Liu, Y. Cheng, X. Wei, X. Chai and G. Yuan, Inorg. Chem., 2020, 59(23), 17464–17472 CrossRef PubMed.
- L. Ma, S. Li, H. Zheng, J. Chen, L. Lin, X. Ye, Z. Chen, Q. Xu, T. Chen, J. Yang, N. Qiu, G. Wang, A. Peng, Y. Ding, Y. Wei and L. Chen, Eur. J. Med. Chem., 2011, 46, 2003–2010 CrossRef.
- H. G. Lee, J. H. Lee, S. P. Jang, I. H. Hwang, S. Kim, Y. Kim, C. Kim and R. G. Harrison, Inorg. Chim. Acta, 2013, 394, 542–551 CrossRef.
- A. P. Singh, N. K. Kaushik, K. Verma and R. Gupta, Indian J. Chem., 2011, 50(1), 474–483 Search PubMed.
- A. E. Wickenden and R. A. Krause, Inorg. Chem., 1965, 4, 404–407 CrossRef.
- H. D. Lutz, R. A. Becker and W. Eckers, Indian J. Chem., 1983, 39, 7–14 Search PubMed.
- K. H. Bok, M. M. Lee, G. R. You, H. M. Ahn, K. Y. Ryu, S. Kim, Y. Kim and C. Kim, Chem. - Eur. J., 2017, 23, 3117–3125 CrossRef PubMed.
- Y. Morimoto, Y. Shimaoka, K. Fukui and S. Itoh, ACS Omega, 2024, 9(22), 23624–23633 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of ligands and complexes are provided in Fig. S1–S9. The catalytic oxidation of adamantane and methylcyclohexane data are provided in Tables S1 and S2. CCDC 2371833 and 2371834. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra05222f |
| This journal is © The Royal Society of Chemistry 2024 |