DOI:
10.1039/D4RA05117C
(Paper)
RSC Adv., 2024,
14, 29919-29933
New synthetic chitosan Schiff bases bearing pyranoquinolinone or benzonaphthyridine and their silver nanoparticles derivatives with potential activity as antioxidant and molecular docking study for EGFR inhibitors†
Received
15th July 2024
, Accepted 9th September 2024
First published on 20th September 2024
Abstract
In this study, two new carboxaldehydes 3, and 4 were synthesized by Vilsmeier–Haack formylation of 6-butyl-benzo[h][1,6]naphthyridine-2,5-dione 2 and 6-butyl-pyrano[3,2-c]quinolinone 1, respectively. Structures of newly synthesized compounds were achieved by IR, 1H NMR, 13C NMR, mass techniques, and elemental analyses. The two synthesized carboxaldehydes 3 and 4 were used as precursors for the synthesis of two new chitosan-based Schiff bases, CS1 and CS2. The new chitosan Schiff bases were grafted on silver nanoparticles, providing CS1/Ag and CS2/Ag structures. However, CS1 and CS2 and their silver nanoparticles were characterized by FT-IR, XRD, SEM-EDX, XRF, TEM, TGA, and DSC. The target compounds CS1, CS2, CS1/Ag, and CS2/Ag were assessed as radical scavengers against 1,1-diphenyl-2-picrylhydrazyl radicals (DPPH%). The results showed that CS1 and CS2 had a better ability to scavenge DPPH radical than its unmodified chitosan. CS1/Ag and CS2/Ag, combining the unique properties of silver and Schiff bases, displayed excellent antioxidant activity (IC50, 59.13, and 32.54 μg mL−1, respectively). In addition, the previous compounds were tested in vitro for inhibition of epidermal growth factor receptor (EGFR) tyrosine kinase using the EGFR kinase assay kit (Cat. #40321). In particular, compound CS1/Ag displayed potent inhibitory activity towards EGFR with IC50 20.45 μg mL−1 compared to reference drug sorafenib (IC50 = 0.76 μg mL−1). The bioactivity of new chitosan Schiff bases was studied by molecular docking to see how they bind with the EGFR receptor. The results implied that CS1 has a higher binding energy than CS2 and CS regarding EGFR kinase, which agreed with the results obtained from the experimental EGFR inhibition assay.
1 Introduction
Chitosan, as a second biopolymer in nature, has a linear structure consisting of [β-(1,4)-2-amino-doxy-D-glucopyranose], which is derived from the N-deacetylation of chitin.1,2 Chitosan is an important polymer with a wide range of functionalities because it is biodegradable, non-toxic, and biocompatible.3–5 The abundant supply of chitosan, the presence of reactive hydroxyl and amino sets, and the variety of modifications successfully made chitosan a viable candidate in a diversity of applications, such as biomaterials for tissue engineering, drug delivery applications,6 and agriculture.7 Besides, chitosan derivatives have exposed antioxidant,8 anticancer activities with less toxicity on no tumor cells,9,10 and epidermal growth factor receptor (EGFR) inhibitory activity.11
Among the different chemical modifications of chitosan (CS) is the reaction of aldehydes and ketones with their amino groups, established as the Schiff base modification.12–14 Numerous derivatives of CS Schiff bases have various biomedical activities, including an antitumor and antioxidant.15,16 It was found that some chitosan Schiff bases exhibited inhibitory activities towards (acetylcholinesterase (AchE), butyrylcholinesterase (BchE), glutamine synthetase (GSI), and sphingomyelinase (SMASE)).17,18
Besides, the azo aromatics have revealed great biological and therapeutic activities. Therefore, naphthyridines were estimated as HIV-1 integrase inhibitors, fibroblast growth factor receptor (FGFR) tyrosine kinase, human cytomegalovirus (HCMV), and enzyme acetylcholinesterase.19–27 Furthermore, benzo[h][1,6]naphthyridine demonstrates high affinity. 5-Hydroxytryptamine receptor 4 (5-HT4) ligands and acts as protein kinase CK2 inhibitors for curing cancer, mammalian target of rapamycin (mTOR), and 3-phosphoinositide-dependent protein kinase-1 inhibitors (PDK1).28 As well, it shows anticancer activity against several cell lines, including human alveolar basal epithelial cells (A-549), colon adenocarcinoma (HCT-15), breast cancer cell line (T-47D), and human liver cancer cell line (HepG-2).28,29
In the current medical era, pyrano[3,2-c]quinolinones emerged as a promising privileged moiety in biological and therapeutic activities. Pyranoquinolinones are known for their great cytotoxic ability toward several cell lines (HepG-2, A-549, MCF-7).30–32 Interestingly, pyranoquinolinone derivatives possess high antioxidant activity.33,34 It is an important motif found in plenty of alkaloids having pharmaceutical applications.35 For example, huajiaosimuline and zanthosimuline have inhibitor activity towards multidrug-resistant KB-VI tumor cells.36 Also, pyranoquinolinone derivatives have emerged as notable topoisomerase IIB inhibitors.37,38
We therefore aimed to enhance the overall biological potency of chitosan via its condensation reactions with novel carboxaldehydes, 6-butyl-benzo[h][1,6]naphthyridine-3-carbaldehyde 3, and 6-butyl-pyrano[3,2-c]quinoline-3-carbaldehyde 4. In addition, we have incorporated silver nanoparticles into the synthesized Schiff bases, hoping that the formed compounds will be eco-friendly favorable pharmacological candidates and discover new anticancer and antioxidant drugs. Recently, well-known silver nanoparticles have gained much concern in the nanomedicine field. Also, silver nanoparticles display efficient inhibitory activities of cancer cells, inhibition of angiogenesis, and antioxidants.39,40 Modern studies exhibit the ability of silver nanoparticles (Ag NPs) to induce cytotoxicity in tumor cells by various methods such as DNA damage, oxidative stress, apoptosis, or cell cycle arrest.41 Remarkably, chitosan Schiff base loaded with Ag NPs exposes antimicrobial, and anticancer activity.42–44 Hence, we hypothesized that incorporating silver nanoparticles into the newly synthesized Schiff bases might significantly increase their biological activities.
The new synthesized chitosan Schiff base derivatives were characterized using different analytical methodologies, including Fourier transform infrared (FTIR), X-ray diffraction (XRD), scanning electron microscope (SEM), energy-dispersive X-ray spectroscopy (EDX), X-ray fluorescence (XRF), transmission electron microscopy (TEM), thermogravimetric analyzer (TGA) and differential scanning calorimetric analyses (DSC). As a result of the previous studies, which showed the activity of chitosan derivatives, pyranoquinolines, and benzonaphthyridines as antioxidants and enzyme inhibitors for cancer therapy, we chose to investigate antioxidant efficiencies on DPPH of the synthesized chitosan Schiff bases. As well, these compounds were assayed for their potential inhibitory activity towards the epidermal growth factor receptor (EGFR). The molecular docking study of the new compounds was carried out.
2 Materials and methods
2.1 . Materials
Chitosan (CS) low molecular weight (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–190000 Da, 75% deacetylated) (Iceland), DMF, and phosphorus oxychloride were purchased from Sigma-Aldrich. Silver nitrate and ammonium acetate were provided by Merck. Sodium citrate, sodium carbonate, glacial acetic acid, methanol, and ethanol were purchased by a local company (Pio Chem., Cairo, Egypt).
000–190000 Da, 75% deacetylated) (Iceland), DMF, and phosphorus oxychloride were purchased from Sigma-Aldrich. Silver nitrate and ammonium acetate were provided by Merck. Sodium citrate, sodium carbonate, glacial acetic acid, methanol, and ethanol were purchased by a local company (Pio Chem., Cairo, Egypt).
2.2. Synthetic procedure
2.2.1. 6-Butyl-pyrano[3,2-c]quinolinone (1). Compound 1 was synthesized correspondingly to the reported literature method.45
2.2.2. 6-Butyl-4-hydroxybenzo[h][1,6]naphthyridine-2,5(1H,6H)-dione (2). A mixture of compound 1 (2.85 g, 10 mmol) with ammonium acetate (6.16 g, 80 mmol) was stirred and heated for 8 h at 200 °C under free-solvent conditions.29 The completion of the reaction was achieved by TLC. After that, the pale brown residue so obtained was filtered, followed by three 10 mL methanol washes, dried, and crystallized from glacial acetic acid to give compound 2 as brown colour; yield 2.16 g (76%); m.p. 274–276 °C. IR (KBr, cm−1): 3397, 3296 (OH, NH), 3089 (CHaromatic), 2961, 2936, 2873 (CHaliphatic), 1704 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Oα-pyridone), 1683 (COγ-pyridone), and 1639 (COquinolinone), 1613 (CCaromatic). 1H NMR (500 MHz, DMSO-d6) δH: 0.89 (t, J = 7.15 Hz, 3H, CH3butyl), 1.34–1.38 (m, 2H, CH2butyl), 1.53–1.55 (m, 2H, CH2butyl), 4.17 (t, J = 7.15 Hz, 2H, NCH2butyl), 5.06 (s, 1H, C3–H), 7.32 (t, J = 7.15 Hz, 1H, Ar–H), 7.58 (d, J = 8.5 Hz, 1H, Ar–H), 7.72 (t, J = 7.6 Hz, 1H, Ar–H), 7.81 (s, 1H, NH), 7.99 (d, J = 8.0 Hz, 1H, Ar–H), 8.61 (s, 1H, OH). 13C NMR (125 MHz, DMSO-d6) δC: 14.2 (s, C4′), 20.1 (s, C3′), 29.7 (s, C2′), 40.3 (C1′), 82.0, 100.1, 115.8, 115.9, 123.5, 123.9, 124.3, 134.2, 138.8, 158.7, 158.8, 160.1. Mass spectrum, m/z (Ir %): 285.11 [M+ + 1; 33], 284.18 [M+; 100], 256.05 [M+–CO; 44], 214.10 (33), 199.66 (95), 145.09 (62), 131.62 (35). Analysis calculated for C16H16N2O3 (284.1): C, 67.59; H, 5.67; N, 9.85%. Found: C, 67.55; H, 5.71; N, 9.83%.
Oα-pyridone), 1683 (COγ-pyridone), and 1639 (COquinolinone), 1613 (CCaromatic). 1H NMR (500 MHz, DMSO-d6) δH: 0.89 (t, J = 7.15 Hz, 3H, CH3butyl), 1.34–1.38 (m, 2H, CH2butyl), 1.53–1.55 (m, 2H, CH2butyl), 4.17 (t, J = 7.15 Hz, 2H, NCH2butyl), 5.06 (s, 1H, C3–H), 7.32 (t, J = 7.15 Hz, 1H, Ar–H), 7.58 (d, J = 8.5 Hz, 1H, Ar–H), 7.72 (t, J = 7.6 Hz, 1H, Ar–H), 7.81 (s, 1H, NH), 7.99 (d, J = 8.0 Hz, 1H, Ar–H), 8.61 (s, 1H, OH). 13C NMR (125 MHz, DMSO-d6) δC: 14.2 (s, C4′), 20.1 (s, C3′), 29.7 (s, C2′), 40.3 (C1′), 82.0, 100.1, 115.8, 115.9, 123.5, 123.9, 124.3, 134.2, 138.8, 158.7, 158.8, 160.1. Mass spectrum, m/z (Ir %): 285.11 [M+ + 1; 33], 284.18 [M+; 100], 256.05 [M+–CO; 44], 214.10 (33), 199.66 (95), 145.09 (62), 131.62 (35). Analysis calculated for C16H16N2O3 (284.1): C, 67.59; H, 5.67; N, 9.85%. Found: C, 67.55; H, 5.71; N, 9.83%.
2.2.3. 6-Butyl-4-hydroxy-2,5-dioxo-1,2,5,6-tetrahydrobenzo[h][1,6]naphthyridine-3-carbaldehyde (3). To cold dry DMF (25 mL) in an ice bath, POCl3 (7 mL, 77 mmol) was added dropwise with stirring at 0 °C for 30 min.; then, the mixture was stirred at room temperature for 30 min. A solution of compound 2 (2.84 g, 10 mmol) in dimethyl formamide (25 mL) was added dropwise with continuous stirring. After completion of addition, the mixture was agitated at room temperature for 30 minutes and then heated to 60 °C for 6 hours. The resulting solution was poured into ice water (300 mL) and neutralized with Na2CO3. The produced residue was filtered off, dried, and recrystallized from glacial acetic acid to give a pale-yellow powder. Yield 2.34 g (75%); m.p. 198–202 °C. IR (KBr, cm−1): 3340, 3215 (OH, NH), 2967, 2900, 2868 (CHaliphatic), 1739 (COald.), 1724 (COα-pyridone), 1670 (COquinolinone), and 1621 (CCaromatic). 1H NMR (500 MHz, DMSO-d6) δH: 0.96 (t, J = 8.0 Hz, 3H, CH3butyl), 1.40–1.45 (m, 2H, CH2butyl), 1.59–1.64 (m, 2H, CH2butyl), 4.32 (t, J = 8.0 Hz, 2H, NCH2butyl), 7.49 (t, J = 8.0 Hz, 1H, Ar–H), 7.76 (d, J = 8.0 Hz, 1H, Ar–H), 7.91 (t, J = 8.0 Hz, 1H, Ar–H), 8.16 (d, J = 8.0 Hz, 1H, Ar–H), 9.93 (s, 1H, HCO), 10.35 (s, 2H, OH + NH, exchangeable in D2O). 13C NMR (101 MHz, DMSO-d6) δC: 14.1 (s, C4′), 20.0 (s, C3′), 29.6 (s, C2′), 42.1 (C1′), 94.2, 98.8, 112.9, 116.1, 123.9, 124.9, 135.6, 139.6, 159.6, 160.8, 160.9, 161.5, 189.9 (COald.). Mass spectrum, m/z (Ir %): 312.29 [M+; 11.83], 283.83 [M+–CO; 100], 226.82 (42), 199.40 (77), 184.96 (27), 145.10 (36). Analysis calculated for C17H16N2O4 (312.33): C, 65.38; H, 5.16; N, 8.97%. Found: C, 65.28; H, 5.26; N, 8.90%.
2.2.4. 6-Butyl-4-chloro-2,5-dioxo-5,6-dihydro-2H-pyrano[3,2-c]quinoline-3-carbaldehyde (4). Phosphorus oxychloride (7 mL, 77 mmol) was added dropwise to a stirred cold DMF (30 mL) in an ice bath; then, the mixture was stirred at room temperature for 30 min. A solution of compound 1 (2.85 g, 10 mmol), in DMF (20 mL) was added dropwise with continuous stirring. After the end of addition, the reaction mixture was stirred at room temperature for 30 min and heated at 60 °C for 6 hours. The resulting mixture was poured into ice-cooled water (300 mL) and neutralized with Na2CO3. The precipitate was filtered off, dried, and recrystallized from isopropyl alcohol to give yellow powder. Yield 2.4 g (72.5%); m.p. 148–150 °C. IR (KBr, cm−1): 2959, 2931, 2872 (CHaliphatic), 1776 (COα-pyrone), 1730 (COald.), 1659 (COquinolinone), 1613 (CCaromatic). 1H NMR (500 MHz, DMSO-d6) δH: 0.93 (t, J = 10.0 Hz, 3H, CH3butyl), 1.38–1.45 (m, 2H, CH2butyl), 1.59–1.64 (m, 2H, CH2butyl), 4.30 (t, J = 10.0 Hz, 2H, NCH2butyl), 7.52 (t, J = 10.0 Hz, 1H, C9–H), 7.83–7.93 (m, 2H, C7–H + C8–H), 8.10 (d, J = 10.0 Hz, 1H, C10–H), 9.99 (s, 1H, HCO). 13C NMR (125 MHz, DMSO-d6) δC: 13.6 (s, C4′), 19.4 (s, C3′), 29.1 (s, C2′), 41.9 (C1′), 100.5, 112.7, 116.5, 124.4, 124.7, 128.6, 135.9, 138.6, 158.2, 160.9, 162.7, 173.6, 184.7 (CHO). Mass spectrum, m/z (Ir %): 333.02 [M+ + 2; 14], 331.06 [M+; 45], 303.32 [M+–(HCO + H); 90], 218.38 (40), 131.52 (68). Analysis calculated for C17H14ClNO4 (331.75): C, 61.55; H, 4.25; Cl, 10.69; N, 4.22%. Found: C, 61.47; H, 4.35; Cl, 10.60; N, 4.31%.
2.2.5. Synthesis of chitosan Schiff base (CS1). 1 g of CS was dissolved in about 20 mL of 10% acetic acid and 30 mL of ethanol. The mixture was stirred for 6 hours at room temperature to obtain a homogenous solution. Then, a predetermined amount of compound 3 (2.5 wt% with respect to Cs) dissolved in 10 mL ethanol/acetic acid mixture (50/50, v/v) was added gradually to the previous solution of CS. The mixture was poured into a circular bottom flask and refluxed at 60 °C with continuous stirring for 15 h, A yellow precipitate was attained after cooling, washed with methanol, and dried. The obtained Cs Schiff base is coded here as CS1, from the reaction of CS with 6-butyl-4-hydroxy-2,5-dioxo-1,2,5,6-tetrahydrobenzo[h][1,6]naphthyridine-3-carbaldehyde (3)
2.2.6. Synthesis of chitosan Schiff base (CS2). The method adopted for the synthesis of CS2 is as mentioned for the synthesis of CS1. A brown gel of CS2 is produced after cooling. The final product obtained is washed with methanol. The obtained Cs Schiff base is coded here as CS2, from the reaction of CS with 6-butyl-4-chloro-2,5-dioxo-5,6-dihydro-2H-pyrano[3,2-c]quinoline-3-carbaldehyde (4).
2.2.7. Synthesis of CS Schiff bases/Ag NPs (CS1/Ag and CS2/Ag). Firstly, Ag NPs were prepared by adding 1 mL of AgNO3 (0.25 mol L−1) and 1 mL of sodium citrate (0.2 mol L−1) in 100 mL distilled water and then mixed with chitosan Schiff base under stirring (at 70 °C for 8 h). The colour of the mixture turns brown, indicating the reduction of Ag+ ions into metallic Ag. After 8 h the mixture was cooled. Finally, CS Schiff base/Ag-NPs collected by centrifugation and then dried at 100 °C overnight.
2.3. Characterization techniques
Melting points were assessed by a digital Stuart-SMP3 apparatus. Fourier transform infrared spectra of synthesized compounds were reported on the FT-IR Nicolet IS10 spectrophotometer (cm−1) at the faculty of education, chemistry department, Ain Shams University, using KBr disks, at ranges from 500 to 4000 cm−1. 1H NMR (500 MHz) and 13C NMR (125 MHz) spectra were measured on the Bruker spectrometer (δ) at the National Research Centre using DMSO-d6 as a solvent and tetramethyl silane as an internal standard. A Bruker Axs D8 Advance X-ray diffractometer with CuKα irradiation (λ = 0.145060 nm) was used for XRD data. Whereas the average crystal size (D in nm) of CS1 was calculated using the Debye-Scherrer equation, D = Kλ/βcosθ, where the constant K = 0.91, λ = 0.15406 nm, β (in radians) is full width half maximum, and θ (deg) is peak position (radius). Scanning electron microscope (SEM) using an SEM model, FESEM, QUANTA FEG 250, FEI, Netherlands was used to the surface morphology of samples. A JEOL (JEM-210, Japan) transmission electron microscope (TEM) was used to confirm the formation of Chitosan Schiff bases. Thermo Scientific Fisher Niton XL2 XRF analyzer was used for X-ray fluorescence spectrometer (XRF) data. TGA measurements were performed on a Shimadzu-50 thermal analyzer from room temperature up to 800 °C at a heating rate of 10 °C min−1. Differential scanning calorimetric analyses (DSC) were accomplished with a DSC evo 131-stram, France, in a nitrogen atmosphere with a heating flow of 10 °C min−1 from −170 to 500 °C. Elemental microanalyses were measured on a PerkinElmer CHN-2400 analyzer at the Chemical War Department, Ministry of Defense, Cairo, Egypt.
2.4. Biological activities
2.4.1. Antioxidant. A methanol solution of 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical (0.004% w/v) and the tested compound was prepared and stored at 10 °C in the dark. Then, A 40-μL aliquot of the prepared compound with various concentrations (0.5 to 1000 μg mL−1) was added to 3 mL of DPPH solution. After incubation for 16 minutes at room temperature, the absorbance (A) of the reaction mixture was computed at 515 nm using a UV-visible spectrophotometer (Milton Roy, Spectronic 1201). Measurements were also made of the absorbance of the DPPH radical without the control and the reference control (ascorbic acid). All the determinations were performed three times, and the average was computed. The antioxidant activity of the DPPH radical was estimated in accordance with the formula:| | |
Antioxidant activity % = [{(Ac − At)/Ac} × 100]
| (1) |
where Ac = absorbance of the control at t = 0 min and At = absorbance of the sample + DPPH at t = 16 min.46,47
2.4.2. EGFR inhibition assay. The epidermal growth factor receptor (EGFR) kinase inhibition activities of the target compounds were evaluated by the EGFR Kinase Assay Kit (Catalog # 40321_BPS Bioscience, San Diego, CA). The EGFR Kinase Assay Kit is packaged in a practical 96-well format with enough cleansed recombinant EGFR enzyme, EGFR substrate, ATP, and Kinase Buffer 1 for 100 enzyme tests. The target compounds were formulated into solutions of different concentrations (0.25, 0.5, 1, 2, 3.9, 7.8, 15.6, 31.25, 62.5, 125, 250, and 500 μg mL−1), and three independent experiments were performed for each group.48 Sorafenib was also tested at the same concentration as the positive control (reference standard). The IC50 values were calculated using GraphPad Prism 5.0 software.Protocol: All samples and controls should be tested in duplicate. ATP, PTK substrate Poly (Glu:Tyr 4:1) (10 mg mL−1), and 5× kinase buffer 1 were liquified. The master mixture was prepared (25 μl per well); consequently, 25 mL was added to every well. 5 mL of inhibitor solution was added to each well identified as a “test inhibitor”. Conversely, 5 mL of the same inhibitor-free solution (inhibitor buffer) was added to the “Reference Control” and “Blank”. 3 mL of 1× kinase buffer was developed by mixing 600 mL of 5× kinase buffer 1 with 2400 mL of water. About 20 mL of 1× kinase buffer 1 was added to the wells designated as “Blank”. EGFR enzyme was thawed on ice. Upon the first thaw, a brief tube containing the enzyme was spun to recover the full content of the tube. The amount of EGFR required for the assay was calculated, and the enzyme was diluted to 1 ng μl−1 with 1× kinase buffer 1. The reaction was initiated by adding 20 mL of diluted EGFR enzyme to the wells designated “positive control” and “test inhibitor control.” After incubation for 40 minutes at 30 °C, the Kinase-Glo Max reagent was defrosted. Following the 40 minute reaction, each well got 50 mL of Kinase-Glo Max reagent. Plates were coated with aluminum foil and incubated at room temperature for 15 min. Finally, luminescence was measured using the microplate reader.49
2.5. Molecular docking
The docking simulation was carried out using AutoDockVina.50,51 The crystal structure of the EGFR-T790M/V948R protein was obtained from the protein data bank using the PDB code 7ZYQ. All ligands and receptors were investigated for docking with rigid protein geometry by Auto Dock Tools version 1.5.6.52–54 The docking cavity was defined according to the interactions of the protein with the co-crystalized ligand, which is also used as a reference ligand. The grid box with dimensions of 14 × 24 × 14 points at X = 42.296, Y = −0.11672, and Z = −1.851 with 1.0 Å spacing was placed to make the entire binding cavity involved. The co-crystalized ligand was redocked to the receptor to validate the docking parameters. The 2D and 3D images were produced by Discovery Studio and Chimera.55
3 Results and discussion
3.1. Chemistry
Here, we discuss an affordable and effectively operated lactone-to-lactam evolution to generate 6-butyl-4-hydroxybenzo[h][1,6]naphthyridine-2,5-dione 2.The reaction takes place between n-butyl pyranoquinolinone 1 and ammonium acetate at 200 °C in good yield (76%) as shown in Scheme 1.29 The structure of compound 2 was approved via spectral data. The IR spectrum of compound 2 displayed two adsorption bands at 3397 and 3296 cm−1 attributed to OH and NH groups, respectively. Also, the presence of a stretching band for cyclic lactam carbonyl at 1704 cm−1,29 which is lower than the cyclic lactone carbonyl group in the starting compound 1. 1H NMR declared the appearance of a new singlet signal at 7.81 ppm due to NH. 13C NMR spectrum revealed sixteen signals, which is agreeable with the number of carbon atoms in its molecular formula. The mass spectrum of compound 2 exhibited a molecular ion peak [M+] as the base peak at m/z 284.18 (100%).
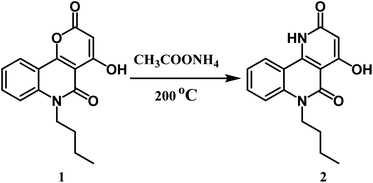 |
| | Scheme 1 Synthesis of 6-butyl-benzo[h][1,6]naphthyridine-2,5-dione 2. | |
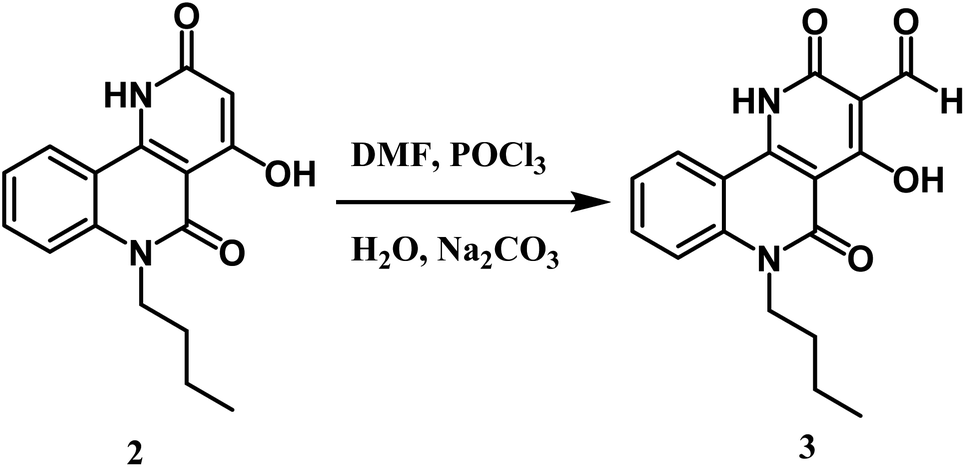
Formylation of compound 2 using the Vilsmeier–Haack reaction led to benzo[h][1,6]naphthyridine-3-carbaldehyde 3 (Scheme 2). The IR spectrum revealed three characteristic absorption bands assigned to three carbonyl groups at 1739 (HCO), 1724 (COα-pyridone), and 1670 cm−1 (COquinolone). While the 1H NMR spectrum of 3 demonstrated the absence of the singlet proton of C-3 at 5.06 ppm and displayed a distinguishable singlet signal ascribed to the aldehydic proton at δ 9.93 ppm, it also showed the existence of a more downfield chemical shift of OH at 10.35 ppm than starting compound 2, which happens because of the electron-withdrawing effect of the CHO group. 13C NMR confirmed the presence of formyl carbon signal at δ 189.9 ppm. The mass spectrum of compound 3 presented a molecular ion peak at m/z 312 (11%) (M+) and the base peak at m/z 284 (100%) assigned to [M+–CO].
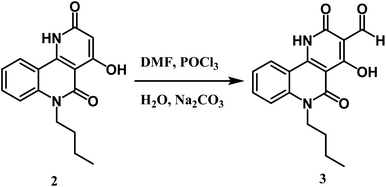 |
| | Scheme 2 Formylation reaction of 6-butyl-benzo[h][1,6]naphthyridine-2,5-dione 2. | |
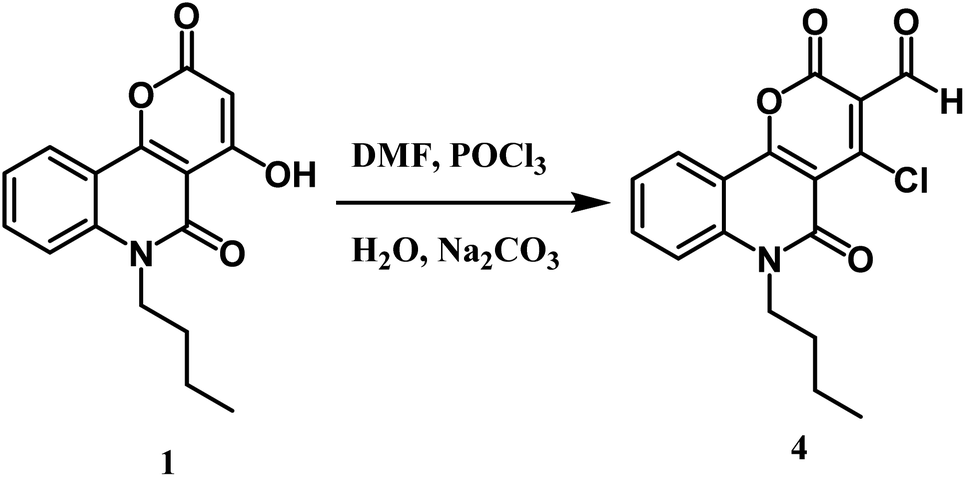
Furthermore, the novel 6-butyl-pyrano[3,2-c]quinoline-3-carbaldehyde 4 was efficiently synthesized from Vilsmeier–Haack formylation of 6-butyl-pyrano[3,2-c]quinoline-2,5-dione 1 (Scheme 3). The IR spectrum of the product proved the absence of the singlet proton at C-3 and the appearance of a new stretching vibration band at 1730 cm−1 due to the carbonyl group of the aldehydic group. 1H NMR spectrum exhibited the appearance of a singlet CHO signal at 9.99 ppm, as well as verifying the absence of the OH group. Our product gave a negative result with iron III chloride solution and a positive result with the Beilstein test. The previous observations indicate the presence of a chlorine atom instead of phenolic O–H. 13C NMR spectrum of synthesized compound 4 revealed a new sp2 hybridized carbon atom at 184.7 ppm attributed to the aldehyde group. Besides, the mass spectrum of compound 4 presented the molecular ion peak [M+] at m/z = 331 (45%) and [M+ + 2] at m/z = 333 (14%) as assumed for compounds containing one chlorine.
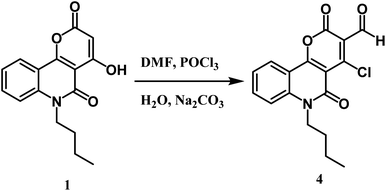 |
| | Scheme 3 Synthesis of the novel 6-butyl-pyrano[3,2-c]quinoline-3-carbaldehyde 4. | |
3.2. Synthesis of the novel chitosan Schiff bases CS1 and CS2
Scheme 4 illustrates the reaction of chitosan with the two carboxaldehydes 3, and 4 via the Schiff base condensation reaction. The chitosan amino group and aldehydic group of 3 or 4 reacted by nucleophilic attack, forming the azomethine group.
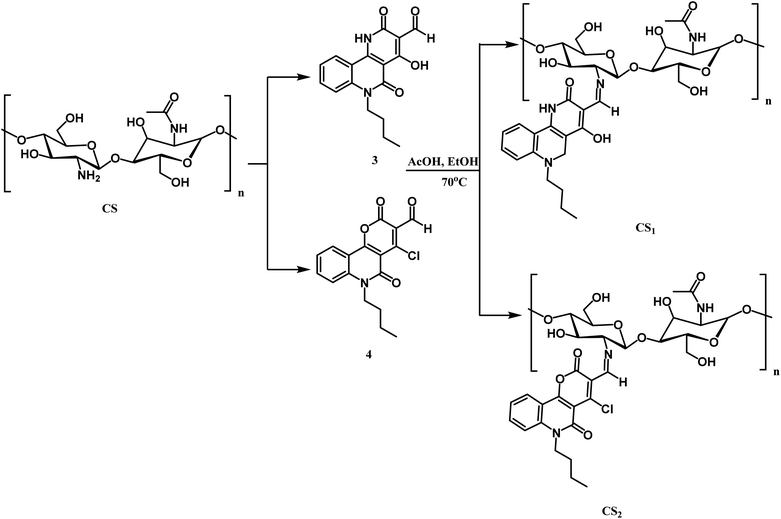 |
| | Scheme 4 Synthetic pathway for chitosan Schiff bases CS1 and CS2 formation. | |
3.3. Characterization of the prepared chitosan Schiff bases and their silver nanocomposites
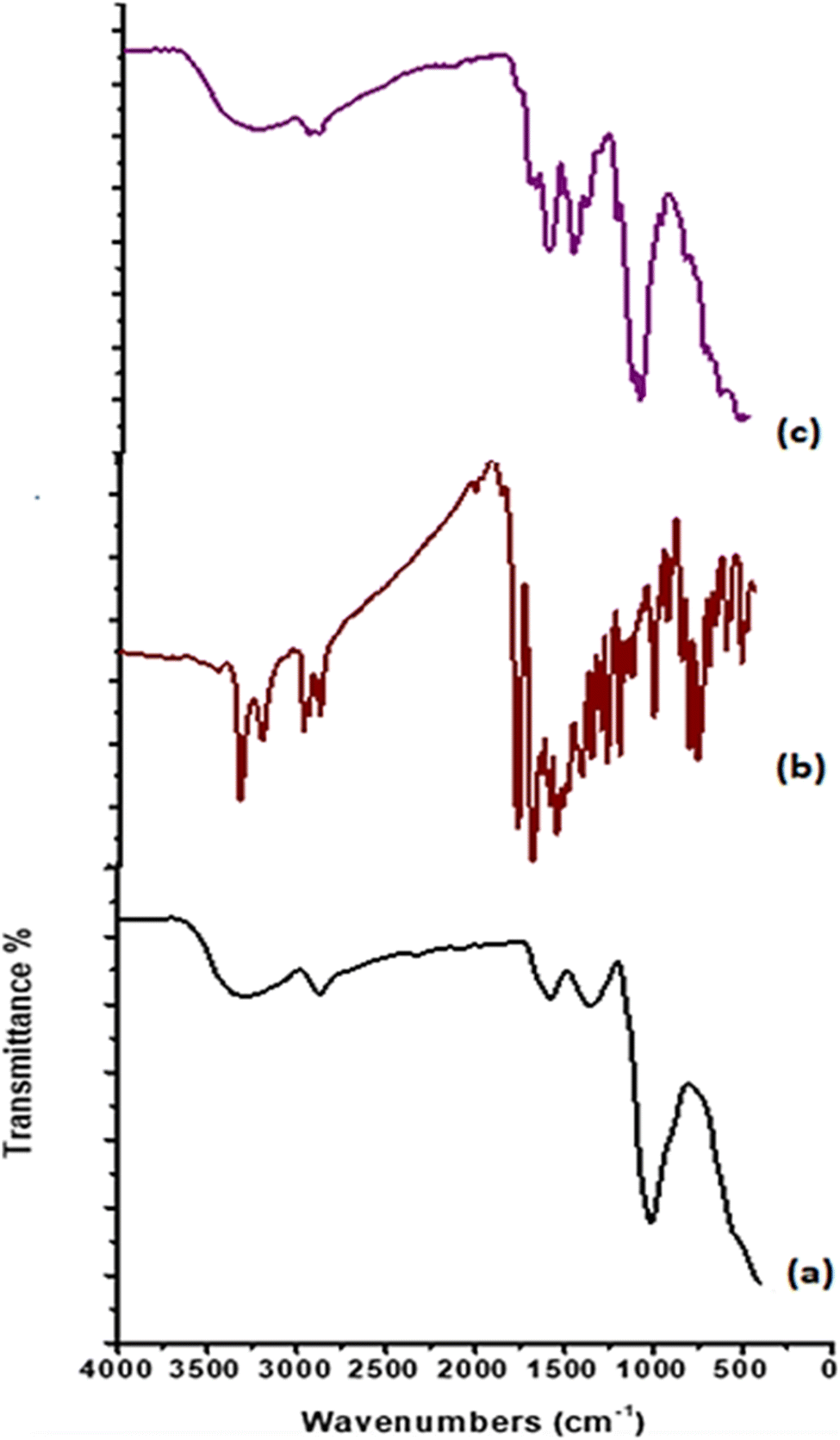
3.3.1. FT-IR analysis. The FTIR spectra for CS, CS1, and CS2 are compared as shown in Fig. 1a–c. As detected from the FT-IR chart for pure CS, it was observed that stretching vibration bands at 3355 and 3285 cm−1 correspond to OH and NH2 groups, in addition to stretching vibration band at 2870 cm−1 for –CH2. Furthermore, peaks at 1645 and 1584 cm−1 referred to stretching bands of CO amide and NH amide bending. The band at 1418 cm−1 indicated the presence of NH2 bending. Otherwise, the peak at 1321 cm−1 is attributed to the stretching band C–N. An asymmetric stretching absorbance band at 1149 cm−1 is assigned to the C–O–C bridge. While bands at 1057, 1023, and 989 cm−1 are indicated to be C–O–H and C–O groups. After the formation of chitosan-based Schiff bases, new absorption bands appeared at 1662 cm−1 for CS1 and 1649 cm−1 for CS2, assigned to the imine bond –CHN.56 Also, stretching vibration band was observed for CHaromatic at 1614 cm−1 for CS1 and 1618 cm−1 for CS2. Moreover, absorption bands at 3446 cm−1 (NH, OH), 2961 cm−1 (CHaliphatic), 1776 cm−1 (COα-pyridone), 1745 (COquinolinone), 1558 (NHbending), and 1048 cm−1 (C–O str) are attributed to CS1. Vibration peaks at 3206 cm−1 (NH), 2925, 2873 cm−1 (CHaliphatic), 1700 cm−1 (COα-pyrone), 1649 cm−1 (COquinolinone), 1552 (NHbending), and 1063 cm−1 (C–O str) corresponding to CS2.15
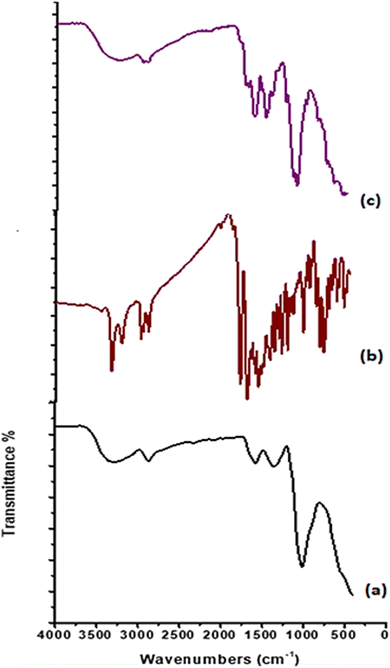 |
| | Fig. 1 FT-IR profiles of (a) CS, (b) CS1, and (c) CS2. | |
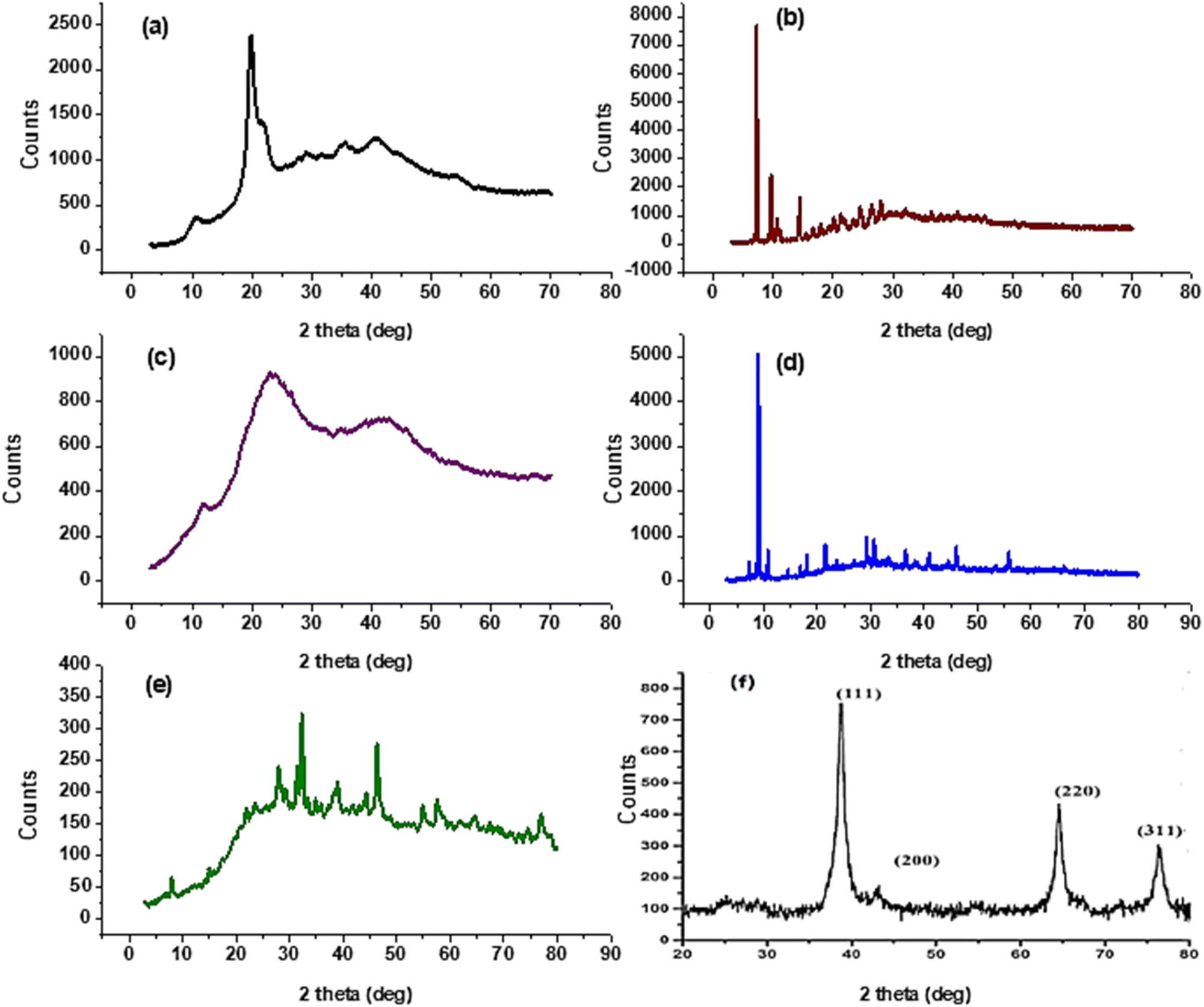
3.3.2. X-ray diffraction studies. XRD patterns of CS, CS1, and CS2 are displayed in Fig. 2a–c. The CS sample shows two characteristic peaks with 2θ (deg) = 10.6 and 19.7° with interplane distances 8.3, 4.4 A°, which signify the semicrystalline nature of CS due to intermolecular and intramolecular hydrogen bonds.57,58 After forming CS1, the peaks at 2θ (deg) = 10.6 and 19.7° disappeared instead; the appearance of new peaks showed at 7.2, 9.7, 10.7, 14.4, 15.4, 16.7, 18, 20.1, 21.4, 23.4, 24.6, 26.4, 31.9, 36.4, 37.9, 40.8, 44, and 45.1°. This shows that CS1 is of a higher semicrystalline nature with a crystal size equal to 54.7 nm. But conjugation of CS with compound 4 to give CS2, the peaks at 2θ (deg) = 10.6 and 19.7° become border and less intense. This result exhibited that CS2 has an amorphous nature. This can be due to the deformation of strong hydrogen bonds of chitosan morphology with substituted pyranoquinoline carbaldehyde 4.59 The XRD pattern of Ag NPs is displayed in Fig. 2d–e. The diffractogram of CS1/Ag declares peaks at 2θ (deg) = 38.2, 44.5, 55.8, and 64.1°, which are indexed as (111), (200), (142), and (220) planes of face-centred cubic Ag NPs that verify the silver reference file of JCPDS 89-3722 (Fig. 2d).60 Whereas CS2/Ag presents diffraction peaks at 2θ (deg) = 38.8, 44.3, 54.9, and 63.9° which were assigned to (111), (200), (142), and (220) planes (Fig. 2e). Such data approved the dispersion of Ag NPs into CS Schiff bases lattice.61–63
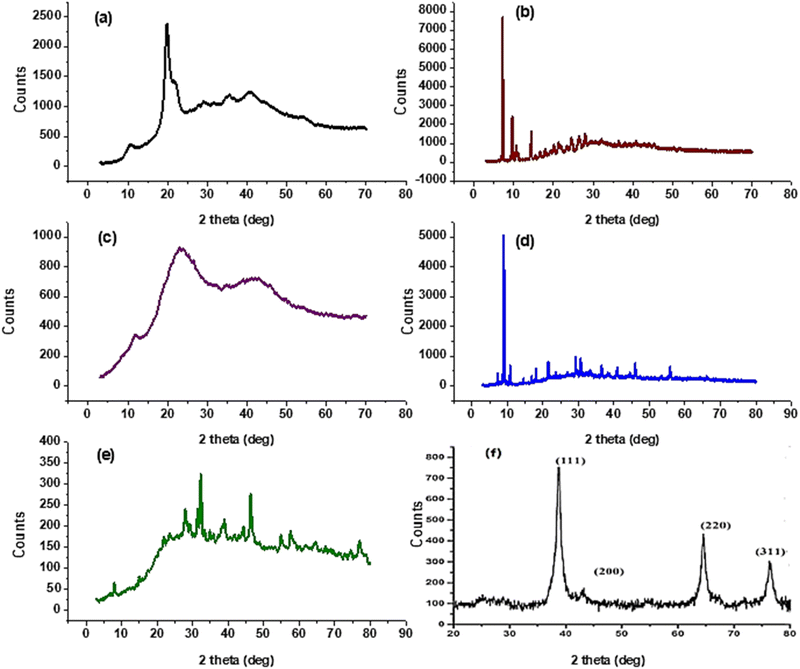 |
| | Fig. 2 X-ray diffraction patterns of (a) CS, (b) CS1, (c) CS2, (d) CS1/Ag, (e) CS2/Ag, and (f) Ag NPs. | |
3.3.3. SEM analysis. The surface morphology of CS, CS1, and CS2 was illustrated in Fig. 3. As shown in Fig. 3a, the CS surface is relatively aggregates of irregular shapes.64 But CS1 exhibited a rough surface of rod structure (Fig. 3b), and CS2 displayed nondimensional particles spread on a large surface layer (Fig. 3c). This offers new functions and bonding.65 According to Fig. 3b and c, the modification of chitosan morphology verified the configuration of chitosan-based Schiff bases. Actually, with increasing roughness of the chitosan Schiff base surface, the biological activities improve. On the other hand, Fig. 3d and e illustrate the presence of light particles dispersed homogeneously in CS1/Ag and CS2/Ag, respectively, pointing to the incorporation of Ag NPs in the CS Schiff bases matrix. For CS1/Ag, a rough surface appears with the spreading of irregular light particles of Ag NPs through the whole matrix, whereas less cavities with some roughness and round particles of Ag NPs are observed for CS2/Ag.62–66
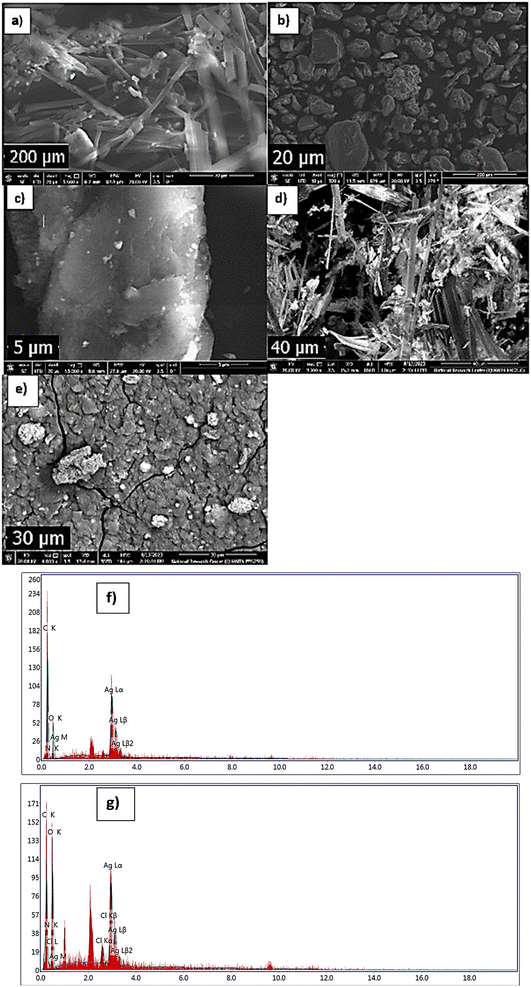 |
| | Fig. 3 SEM morphologies of (a) CS, (b) CS1, (c) CS2, (d) CS1/Ag, and (e) CS2/Ag, and EDX images of (f) CS1/Ag, and (g) CS2/Ag. | |
3.3.4. Energy dispersive spectroscopy EDX and X-ray fluorescence spectroscopy XRF. Energy dispersive spectroscopy EDX analysis was executed to examine the elemental composition of CS1/Ag and CS2/Ag (Fig. 3). Fig. 3f for CS1/Ag displayed the presence of C, N, and O elements and characteristic peaks of Ag NPs at ∼3 Kev. Also, EDX analysis of CS2/Ag shown in Fig. 3g demonstrated the presence of C, N, O, and Cl elements and strong peaks at ∼3 Kev for Ag NPs.67,68 According to XRF analysis, CS1/Ag and CS2/Ag contained an excessive proportion of Ag NPs (92.6 and 94.3%, respectively).
3.3.5. Transmission electron microscopy (TEM). Fig. 4a and b illustrate TEM images for CS Schiff bases loaded with Ag NPs. The images exhibited spherical black spots, representing that Ag NPs dispersed homogeneously into the polymeric lattice of CS Schiff bases. Silver nanoparticles are observed with relatively different particle sizes of 4–19 nm for CS1/Ag and an average size of 5–17 nm for CS2/Ag. Moreover, some aggregation was noticed inside the polymer structure of silver nanoparticles in both samples.
 |
| | Fig. 4 TEM of (a) CS1/Ag, and (b) CS2/Ag. | |
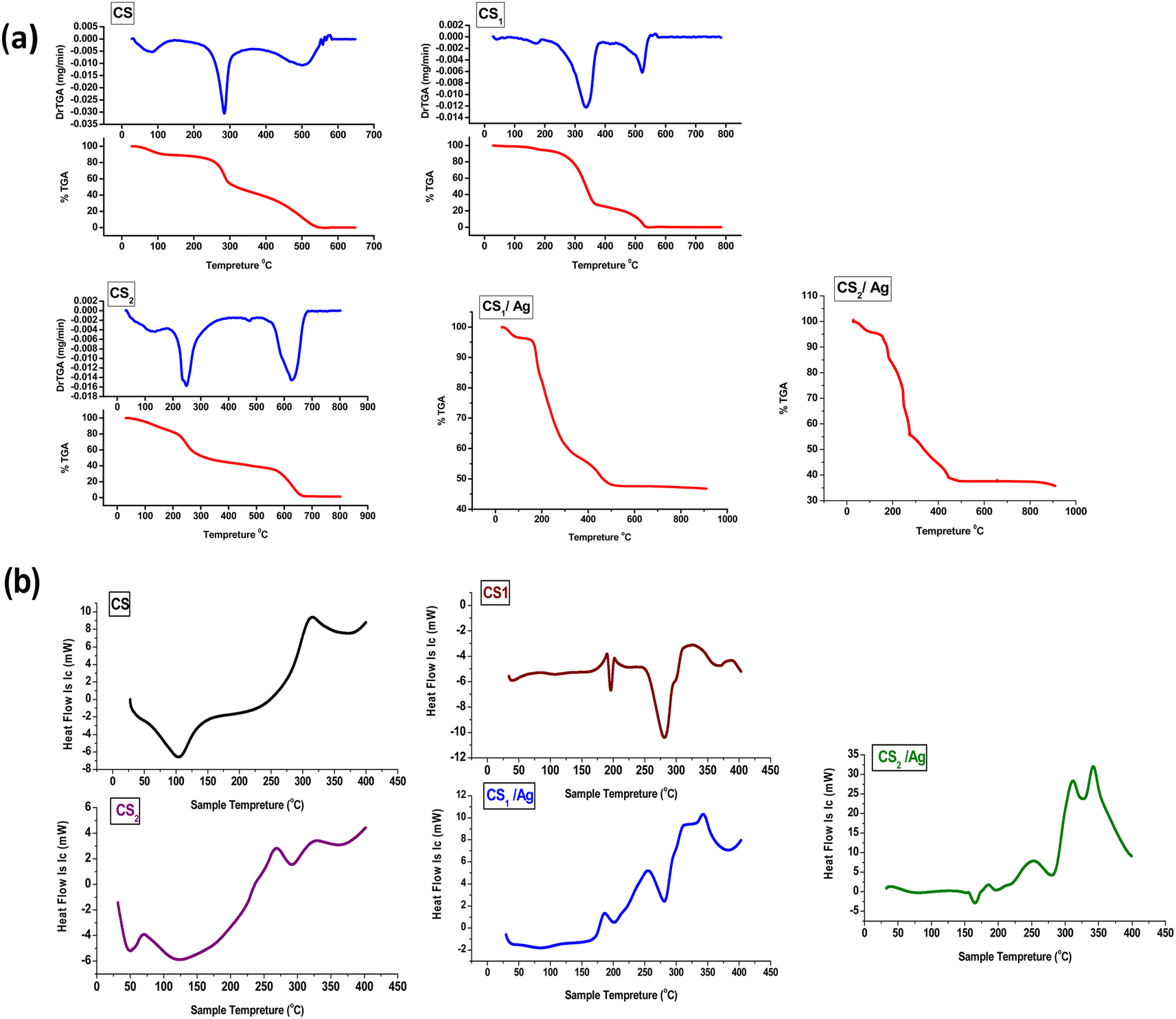
3.3.6. TGA analysis. TGA is used to study the thermal behavior of chitosan and display how chemical reactions affect it, as shown in Fig. 5a. Respectively, chitosan showed weight loss in three steps in the temperature range of 28.26–584 °C. The first step, which was because of the loss of free H2O molecules, occurred in the range of 28.26 to 125.84 °C with a weight loss of 10%.69 The second step of weight reduction is the oxidative decomposition of the chitosan backbone's pyranose ring.70 The degradation occurred between 125 and 321 °C, losing 40% of its weight. The third step, referring to the degeneration of saccharide rings, occurred from 321 to 584 °C with a weight loss of ∼48.8%.71 Fig. 5a depicts the decomposition of chitosan Schiff bases, also shown in three stages. The weight loss of the first stage was 5, 13% at the temperature range 29–187 °C, 31–168 °C for CS1 & CS2, respectively. CS1 absorbs water higher than CS; this may be due to the reduction of chitosan crystallinity by benzo[h][1,6]naphthyridine-3-carbaldehyde moiety to the polymer chain. However, CS2 displayed lower water-holding capacity than CS itself. The second stage occurs in the range 187–373, and 168–483 °C, with weight loss of 67% and 46% for CS1 and CS2, respectively. This main degradation stage is pointed to dehydration, cleavage of imine linkage, loss of heterocyclic rings attached to the CS backbone, and depolymerization of CS. The last stage's residual decomposition reactions occur around 373–561 °C and 483–734 °C, with losing 27 and 38% of the weight of CS1 and CS2, respectively. This decomposition may be assigned to the decomposition of a crosslinking structure formed during getting rid of remaining unreacted groups of the CS chain. CS1/Ag and CS2/Ag undergo heat degradation through three stages, which produce weight loss in the first stage of 5% and 5.95% at a range of 28.26–152.8 °C and 28.26–149 °C, respectively. In the second stage, at 1–371.56 °C and 149–276 °C, the weight loss is 38.9% and 40% for CS1/Ag and CS2/Ag, respectively. In the last stage, elevating the temperature range of 371.56–665.5 °C and 276–900 °C for CS1/Ag and CS2/Ag, respectively, a residual weight of about 10% and 20% was observed. CS1/Ag and CS2/Ag showed the highest thermal stability. This may be correlated to the spreading of Ag NPs, which slows down the rate of thermal degradation of the polymeric matrix.72,73
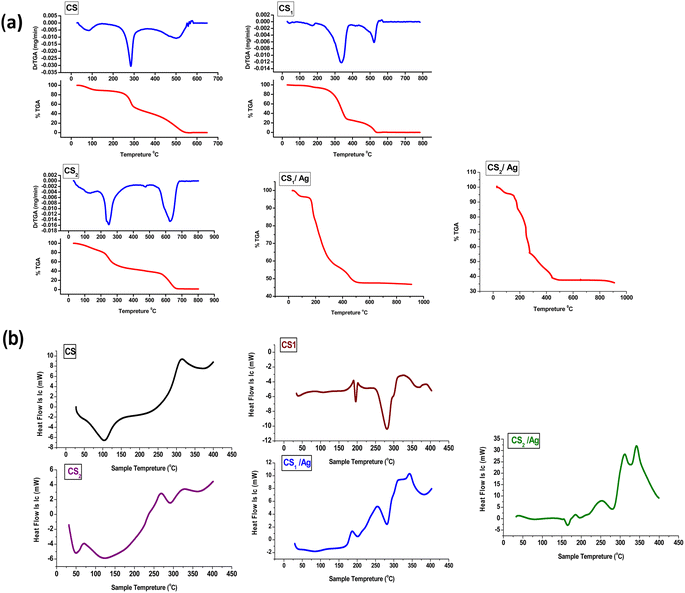 |
| | Fig. 5 (a) TGA and (b) DSC curves of CS, CS1, CS2, CS1/Ag and CS2/Ag. | |
3.3.7. DSC analysis. As shown in Fig. 5b, the CS curve displays an endothermic peak at 104 °C due to the loss of bound water. Another broad exothermic peak at 307 °C refers to the thermal cleavage of polymers.74 The DSC curve of CS1 presents sharp endothermic peaks at 195 and 281 °C and an exothermic peak at 323 °C. The sharpness of endothermic peaks indicates that the polymer Schiff base possesses crystalline and pure properties.75 The second chitosan Schiff base CS2 curve exhibits an endothermic peak at 122 and 292 °C while displaying an exothermic peak at 328 °C. On the other hand, the chitosan Schiff base-loaded Ag NPs (CS1/Ag) curve reveals broad endothermic peaks at 85 and 282 °C, as well as a sharp endothermic peak at 282 °C and an exothermic peak at 342 °C due to compound decomposition. The DSC curve for CS2/Ag exhibits a sharp endothermic peak at 166, 283, and 328 °C, and the curve demonstrates sharp exothermic peaks at 340 °C. The results point to the chitosan Schiff bases CS1 and CS2 having higher thermostability than CS itself due to the exothermic peaks of CS1 and CS2 display shifting to a higher temperature in DSC curves.76 Also, chitosan Schiff-loaded silver nanoparticles CS1/Ag and CS2/Ag have a higher degree of thermostability than CS1 and CS2 because nanoparticles possess cross-links, which decrease flexibility and segmental motions of the polymer chain.77,78
3.4. Biological activities
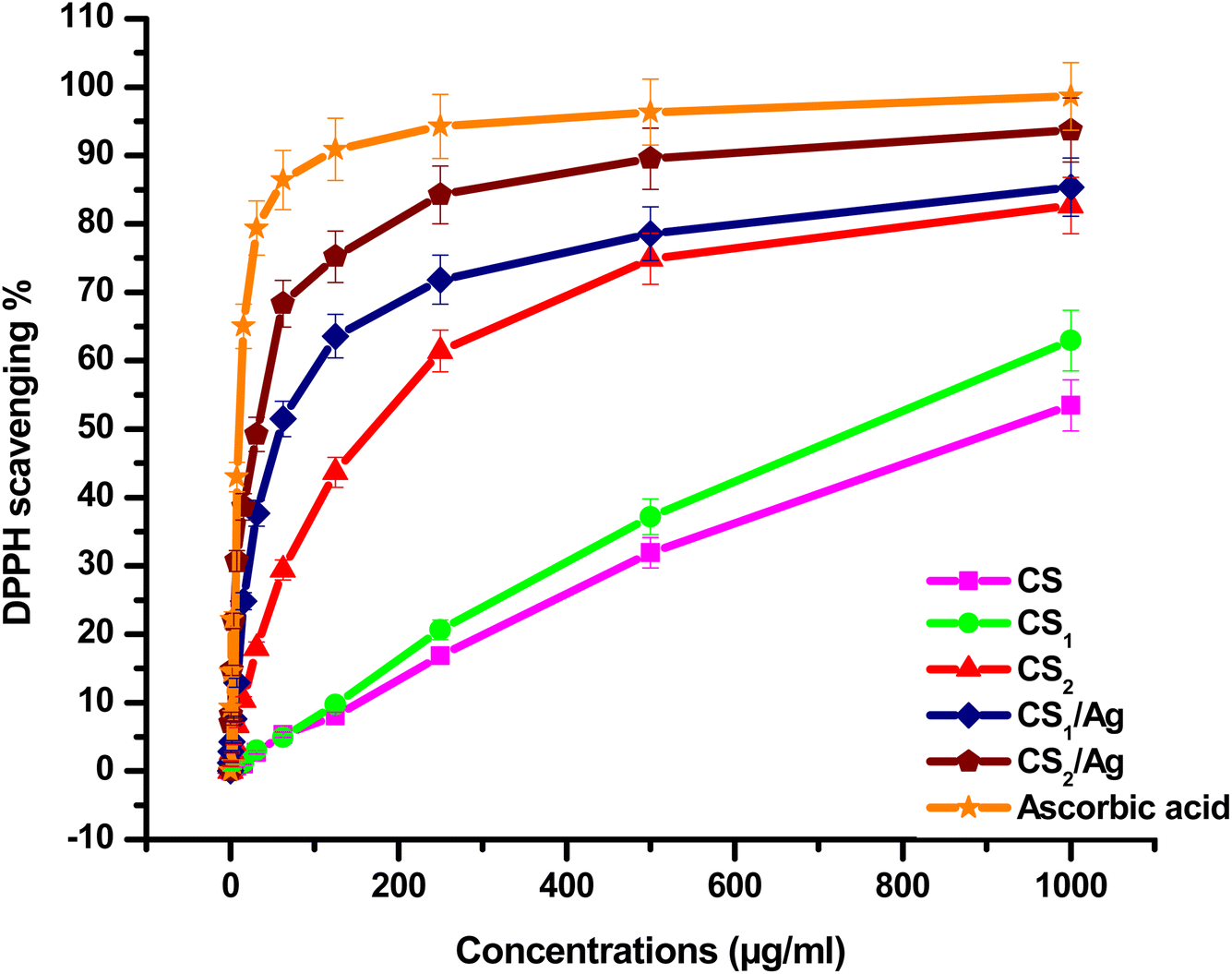
3.4.1. Antioxidant activity. Oxidation is an important activity for providing energy essential to biological processes in microorganisms. However, some oxidants and free radicals become very toxic, which causes tissue and cell damage. Antioxidants are known as compounds that inhibit or delay cellular damage and free radical reactions. The antioxidant activity of the substance is measured by its ability to transfer hydrogen atoms or single electrons to 2,2-diphenyl-1-picrylhydrazyl (DPPH) to convert the methanolic solution of DPPH from blue or violet color to yellow hydrazine derivative.79 Fig. 6 demonstrated the scavenging activity of chitosan and its Schiff bases CS1, CS2, CS1/Ag, and CS2/Ag, comparing with a positive control (ascorbic acid) for DPPH radical. The achieved results hinted out that all the target compounds showed moderate to good DPPH scavenging ability compared to the ascorbic acid. By increasing concentration from 7.8 to 1000 μg mL−1, the radical scavenging activity increases CS (0.67 to 53.47%), CS1 (0.86 to 62.94%), CS2 (6.73 to 82.68%), CS1/Ag (12.93 to 85.58%), and CS2/Ag (30.73 to 93.71%). Moreover, IC5O for CS, CS1, CS2, CS1/Ag, and CS2/Ag was found to be 919.49, 749.13, 169.46, 59.13, and 32.54 μg mL−1, respectively. It is observed that CS2/Ag showed relatively high antioxidant activity. Furthermore, CS1/Ag and CS2/Ag displayed scavenging capacity higher than CS1 and CS2. This referred to the presence of Ag NPs, which increase the antioxidant ability of the CS Schiff bases.44,80–82 On the other hand, CS1 and CS2 are more active than CS itself. This may be due to the phenolic group of benzo[h][1,6]naphthyridine and pyranoquinolinone moieties, which are condensed with chitosan to afford imine groups.15,83,84
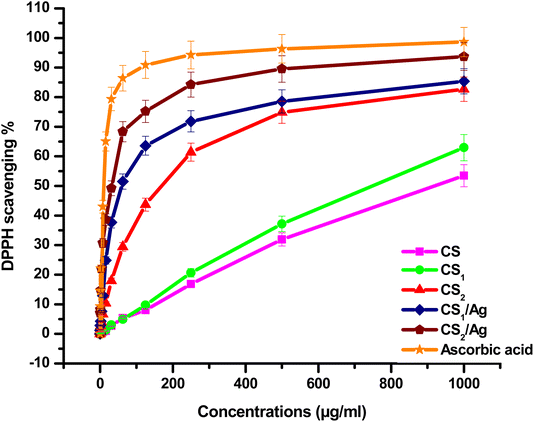 |
| | Fig. 6 DPPH-radical scavenging ability of (a) CS, (b) CS1, (c) CS2, (d) CS1/Ag, and (e) CS2/Ag at different concentrations (0–1000 μg mL−1). | |
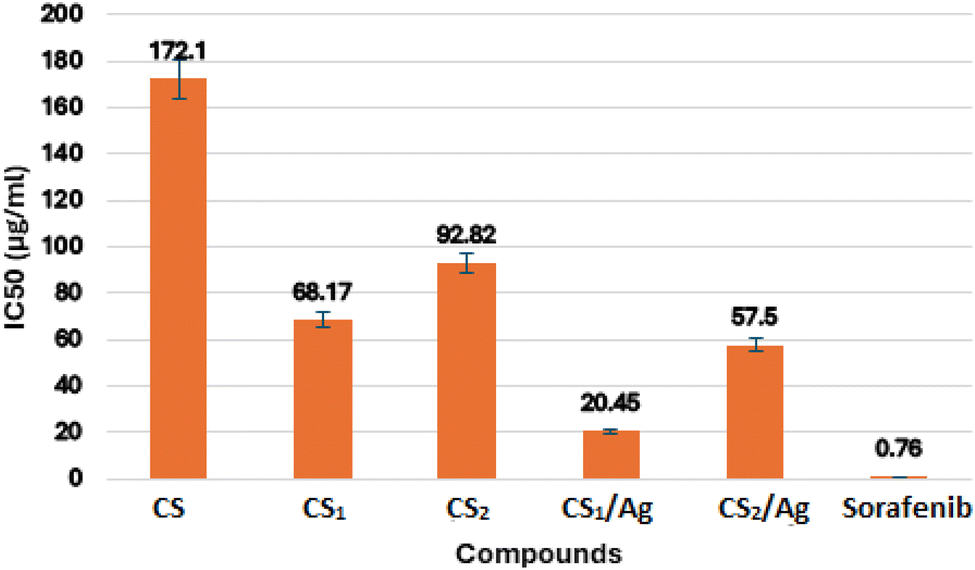
3.4.2. In vitro EGFR kinase assay. In this study, the potential inhibiting activity of the recently prepared compounds towards EGFR kinase was assessed using the reference drug sorafenib. The results were informed as 50% inhibition concentration values (IC50) that were disclosed in Fig. 7. The IC50 values of the investigated compounds ranged from 20 to 172 μg mL−1. CS1/Ag was the most effective EGFR inhibitor in this study compared to positive control sorafenib (IC50 = 0.76 μg mL−1), while compound CS2/Ag and CS1 revealed moderate inhibitory activity. Moreover, both compounds CS and CS2 displayed less inhibition potency. From the results above, the presence of an azomethine group in chitosan Schiff bases CS1 and CS2 enhances inhibitory activity, which is higher than chitosan alone.85–88 Also, the dispersion of Ag NPs into CS Schiff bases resulted in an improvement of the EGFR inhibitory activity, which may be because of the electronic effect of the metal center.15,16,44,84
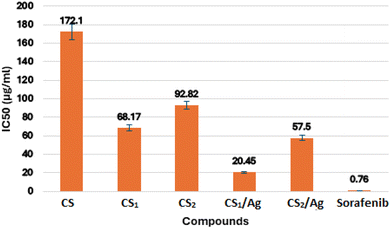 |
| | Fig. 7 Virtual IC50 values of the target compounds (a) CS, (b) CS1, (c) CS2, (d) CS1/Ag, (e) CS2/Ag and (f) sorafenib against EGFR kinase. | |
4 Molecular docking study
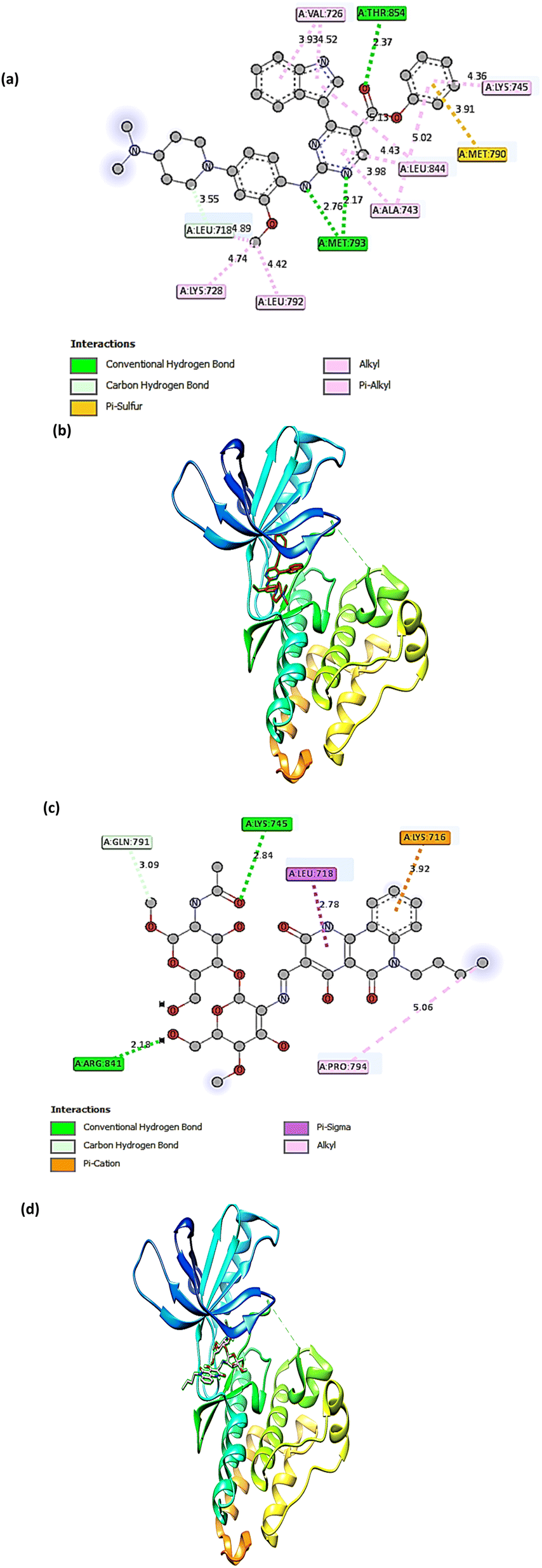
The molecular docking technique has emerged as a crucial strategy offering the most promising route for drug discovery.89 It is employed to predict the degree of binding affinity between a ligand molecule and a therapeutic target. Compared to traditional technologies, this computational tool is more affordable, efficient, and time-saving. This study was applied for CS, CS1, and CS2 to explore their binding mode towards the epidermal growth factor receptor kinase. The docking results revealed that our studied compounds occupy the same binding cavity as that occupied by the known potent EGFR inhibitor (V58,90 currently in the pre-clinical phase), as shown in Table 1. Through examination of the binding interactions of V58 to the active site of the EGFR, it showed strong bond interactions through two hydrogen bonds with key amino acids MET 793 and THR 854. In addition, it formed several Pi-interactions, including Pi-sulphur (MET 790) and six Pi–alkyl bonds (VAL 726, LYS 728, ALA 743, LYS 745, and LEU 844) (Fig. 8a and b). Compound CS1 displayed a better binding interaction with the enzyme active site among all compounds compared to the reference compound, as evidenced by the highest binding energy of −8 kcal mol−1 (Table 1). Compound CS1 disclosed several modes of interactions, including hydrogen bonds with LYS 745, ARG 841 through CO, and the OH groups of the chitosan ring, respectively. Furthermore, some Pi-interactions were observed, including Pi-cation with LYS 716 through the benzo ring of the benzonaphthyridine compound, Pi-sigma with LEU 718 through the naphthyridine moiety, and Pi-alkyl with the butyl group of the naphthyridine ring through the amino acid PRO 794 as presented in Fig. 8c and d. The unique interaction of CS1, which comprises both hydrogen bonding and Pi interactions, indicates enhanced binding selectivity, which may describe its stronger inhibitory effect than CS and CS2 (Fig. 7). Although CS2 had a lower docking score than CS1, its performance is better than CS with the EGFR enzyme. Also, the proposed binding mode of CS2 demonstrated an affinity value of −6.5 kcal mol−1 with one hydrogen bond and 2Pi–alkyl bonds with CYS 797. Besides, it formed alkyl bonds through LEU 844 and van der Waals interactions with key residues ASP 800, GLY 796, GLU 804, and HIS 805 (Fig. 9a and b). Finally, the binding energy of CS (−6.1 kcal mol−1) (Fig. 9c and d) is slightly lower than that of CS1 and CS2. From the above results, the new synthesized Schiff base CS1 showed higher affinity towards EGFR kinase than CS2 and CS, which correlated well with the results obtained from experimental enzyme inhibition.
Table 1 The interactions of compounds CS, CS1, CS2 and V58 with EGFR
| Compound |
Affinity (kcal mol−1) |
Amino acid |
Interaction types |
Distance (Å) |
| CS |
−6.1 |
MET 793 |
Hydrogen bond |
3.03 |
| 2.36 |
| ASP 800 |
Hydrogen bond |
2.45 |
| CYS 797 |
van der Waals |
— |
| PRO 794 |
van der Waals |
— |
| GLY 796 |
Carbon hydrogen bond |
— |
| CS1 |
−8 |
LYS 716 |
Pi–cation |
3.92 |
| LEU 718 |
Pi–sigma |
2.78 |
| LYS 745 |
Hydrogen bond |
2.84 |
| PRO 794 |
Pi–alkyl bonds |
5.06 |
| ARG 841 |
Hydrogen bond |
2.18 |
| CS2 |
−6.5 |
CYS 797 |
Hydrogen bond |
3.06 |
| CYS 797 |
2Pi–alkyl bonds |
4.42 |
| 4.67 |
| LEU 844 |
Alkyl bonds |
5.14 |
| 5.10 |
| ASP 800 |
van der Waals |
— |
| GLY 796 |
van der Waals |
— |
| GLU 804 |
van der Waals |
— |
| HIS 805 |
van der Waals |
— |
| V58 |
−9.9 |
VAL 726 |
Two Pi–alkyl bonds |
3.93 |
| 4.52 |
| LYS 728 |
Pi–alkyl bonds |
4.74 |
| ALA 743 |
Pi–alkyl bond |
3.98 |
| LYS 745 |
Pi–alkyl bond |
4.36 |
| MET 790 |
Pi–sulfur |
3,91 |
| MET 793 |
Hydrogen bond |
2.17 |
| 2.76 |
| LEU 844 |
Pi–alkyl bond |
4.43 |
| THR 854 |
Hydrogen bond |
2.37 |
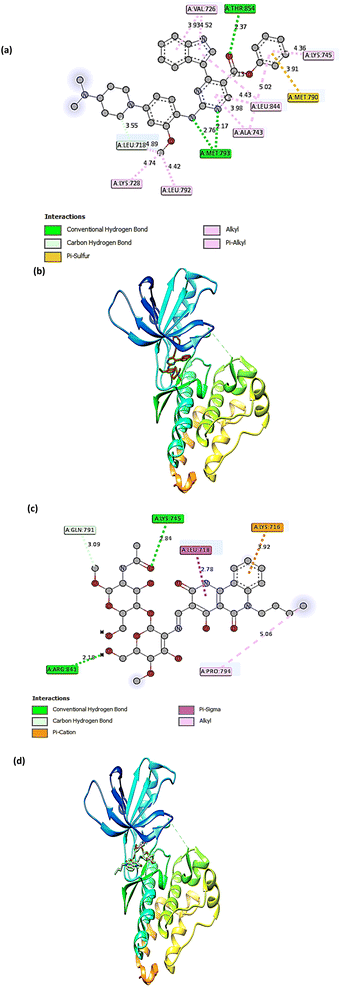 |
| | Fig. 8 The 2D and 3D interactions of the (a and b) V58, and (c and d) CS1 with active site of EGFR. | |
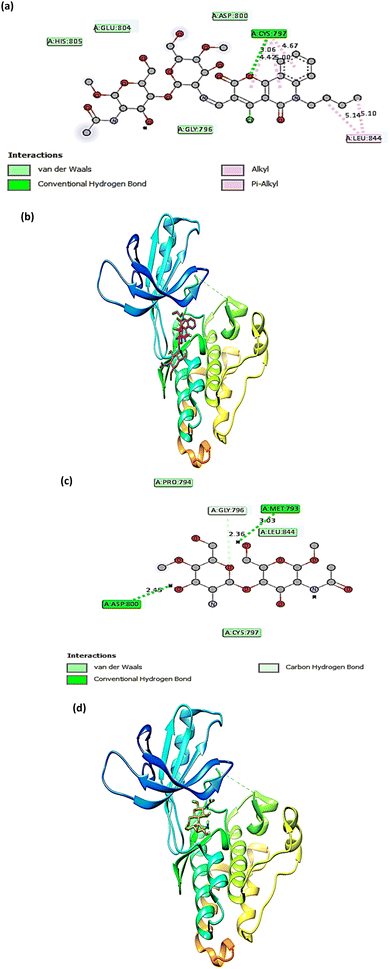 |
| | Fig. 9 The 2D and 3D interactions of the (a and b) CS2, and (c and d) CS with active site of EGFR. | |
5 Conclusions
In the present study, new chemically modified chitosan with benzonaphthyridine and pyranoquinolinone motifs was synthesized. New chitosan Schiff bases decorated with silver nanoparticles to obtain new nanocomposite. The characterization of the target compounds was approved by FT-IR, XRD, SEM, TEM, EDX, XRF, TGA and DSC. The effect of chitosan modification with benzonaphthyridine and pyranoquinolinone on its antioxidant activity was estimated using the DPPH radical scavenging assay. As a result, an enhancement in the antioxidant activity was observed after the functionalization of chitosan with benzonaphthyridine and pyranoquinolinone, which are important scaffolds in pharmaceutical chemistry. Compound CS2/Ag showed a promising antioxidant activity with IC50 = 32.54 μg mL−1. Furthermore, all the synthesized Schiff bases were screened for their inhibitory profiles against EGFR kinase, utilizing sorafenib as a reference drug. Compound CS1/Ag displayed potent inhibitory activity towards EGFR (IC50 = 20.45 μg mL−1). Accordingly, CS2/Ag and CS1/Ag compounds can be considered as a hopeful template in the field of optimization and development of new drugs as antioxidant agents and EGFR inhibitors. The experimental enzyme inhibition results were further verified by the molecular docking study, which indicated that compound CS1 has a better binding with the EGFR receptor than chitosan alone and the other Schiff base CS2.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Hany M. Hassanin: conceptualize the plan, and methodology. Shrouk M. Hassan and Mai A. Mostafa: performed the experiments, analyzed the experimental data, wrote the paper. Jehan M. Morsy and Elham S. Othman: revised the paper.
Conflicts of interest
The authors declare that they have no competing interests.
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at Ain Shams University for funding this work.
Notes and references
- H. S. Adhikari and P. N. Yadav, Int. J. Biomater., 2018, 2018(1), 2952085, DOI:10.1155/2018/2952085.
- Q. Zhao, L. Fan, Y. Liu and J. Li, A review, Food Chem., 2022, 380, 131838, DOI:10.1016/j.foodchem.2021.131838.
- M. Z. Elsabee and E. S. Abdou, Mater. Sci. Eng., C, 2013, 33, 1819–1841, DOI:10.1016/j.msec.2013.01.010.
- X. Li, K. Chen, X. Ji, X. Yuan, Z. Lei, M. W. Ullah, J. Xiao and G. Yang, Eng. Sci., 2021, 13, 106–120, DOI:10.30919/es8d1159.
- M. Sun, L. Yuan, X. Yang and L. Shao, ES Mater. Manuf., 2020, 9, 40–47, DOI:10.30919/esmm5f780.
- K. Yao, J. Li, F. Yao and Y. Yin, Chitosan-based hydrogels, Taylor & Francis Group, LLC, 2012 Search PubMed.
- H. Xie, H. Yue, W. Zhang, W. Hu, X. Zhou, P. Prinsen and R. Luque, Catal. Commun., 2018, 104, 118–122, DOI:10.1016/j.catcom.2017.09.025.
- T. M. Tamer, M. M. ElTantawy, A. Brussevich, A. Nebalueva, A. Novikov, I. V. Moskalenko, M. M. Abu-Serie, M. A. Hassan, S. Ulasevich and E. V. Skorb, Int. J. Biol. Macromol., 2023, 234, 123687, DOI:10.1016/j.ijbiomac.2023.123687.
- Z. Zhou, Y. Liu, X. Jiang, C. Zheng, W. Luo, X. Xiang, X. Qi and J. Shen, Int. J. Biol. Macromol., 2023, 224, 797–809, DOI:10.1016/j.ijbiomac.2022.10.167.
- F. Bayat, M. Pourmadadi, M. M. Eshaghi, F. Yazdian and H. Rashedi, J. Cluster Sci., 2023, 34, 2565–2577, DOI:10.1007/s10876-023-02405-y.
- S. Maya, L. G. Kumar, B. Sarmento, N. S. Rejinold, D. Menon, S. V. Nair and R. Jayakumar, Carbohydr. Polym., 2013, 93(2), 661–669, DOI:10.1016/j.carbpol.2012.12.032.
- R. Antony, S. T. D. Manickam, K. Saravanan, K. Karuppasamy and S. Balakumar, J. Mol. Struct., 2013, 1050, 53–60, DOI:10.1016/j.molstruc.2013.07.006.
- A. B. Dos Santos, J. Traverse, F. J. Cervantes and J. B. van Lier, Biotechnol. Bioeng., 2005, 89, 42–52, DOI:10.1002/bit.20308.
- R. Verma, N. R. Peters, M. D'Onofrio, G. P. Tochtrop, K. M. Sakamoto, R. Varadan, M. Zhang, P. Coffino, D. Fushman, R. J. Deshaies and R. W. King, Science, 2004, 306, 117–120, DOI:10.1126/science.1100946.
- M. A. Ali, K. A. Aswathy, G. M. Ramanujam and V. Jaisankar, Int. J. Biol. Macromol., 2023, 225, 1575–1587, DOI:10.1016/j.ijbiomac.2022.11.214.
- A. G. Hamodin, W. E. Elgammal, A. M. Eid and A. G. Ibrahim, Int. J. Biol. Macromol., 2023, 243, 125180, DOI:10.1016/j.ijbiomac.2023.125180.
- C. A. Caro, L. Lillo, F. J. Valenzuela and G. Cabello, Chem.-Biol. Interact., 2017, 263, 81–87, DOI:10.1016/j.cbi.2016.12.021.
- N. Turan, K. Buldurun, F. Türkan, A. Aras, N. Çolak, M. Murahari, E. Bursal and A. Mantarcı, Mol. Diversity, 2022, 26, 2459–2472, DOI:10.1007/s11030-021-10344-x.
- K. Ramkumar, E. Serrao, S. Odde and N. Neamati, Med. Res. Rev., 2010, 30, 890–954, DOI:10.1002/med.20194.
- B. A. Johns, J. G. Weatherhead, S. H. Allen, J. B. Thompson, E. P. Garvey, S. A. Foster, J. L. Jeffrey and W. H. Miller, Bioorg. Med. Chem. Lett., 2009, 19, 1807–1810, DOI:10.1016/j.bmcl.2009.01.089.
- J. Y. Melamed, M. S. Egbertson, S. Varga, J. P. Vacca, G. Moyer, L. Gabryelski, P. J. Felock, K. A. Stillmock, M. V. Witmer, W. Schleif, D. J. Hazuda, Y. Leonard, L. Jin, J. D. Ellis and S. D. Young, Bioorg. Med. Chem. Lett., 2008, 18, 5307–5310, DOI:10.1016/j.bmcl.2008.08.038.
- M. S. Egbertson, H. M. Moritz, J. Y. Melamed, W. Han, D. S. Perlow, M. S. Kuo, M. Embrey, J. P. Vacca, M. M. Zrada, A. R. Cortes, A. Wallace, Y. Leonard, D. J. Hazuda, M. D. Miller, P. J. Felock, K. A. Stillmock, M. V. Witmer, W. Schleif, L. J. Gabryelski, G. Moyer, J. D. Ellis, L. Jin, W. Xu, M. P. Braun, K. Kassahun, N. N. Tsou and S. D. Young, Bioorg. Med. Chem. Lett., 2007, 17, 1392–1398, DOI:10.1016/j.bmcl.2006.11.080.
- G. Falardeau, H. Lachance, A. St-Pierre, C. G. Yannopoulos, M. Drouin, J. Bédard and L. Chan, Bioorg. Med. Chem. Lett., 2005, 15, 1693–1695, DOI:10.1016/j.bmcl.2005.01.050.
- L. Chan, H. Jin, T. Stefanac, J. F. Lavallée, G. Falardeau, W. Wang, J. Bédard, S. May and L. Yuen, J. Med. Chem., 1999, 42, 3023–3025, DOI:10.1021/jm9902483.
- A. M. Thompson, C. J. Connolly, J. M. Hamby, S. Boushelle, B. G. Hartl, A. M. Amar, A. J. Kraker, D. L. Driscoll, R. W. Steinkampf, S. J. Patmore, P. W. Vincent, B. J. Roberts, W. L. Elliott, W. Klohs, W. R. Leopold, H. D. Hollis Showalter and W. A. Denny, J. Med. Chem., 2000, 43, 4200–4211, DOI:10.1021/jm000161d.
- S. Vanlaer, A. Voet, C. Gielens, M. De Maeyer and F. Compernolle, Eur. J. Org Chem., 2009, 2009, 643–654, DOI:10.1002/ejoc.200800972.
- S. Bera, K. K. Pandey, A. C. Vora and D. P. Grandgenett, J. Mol. Biol., 2011, 410, 831–846, DOI:10.1016/j.jmb.2011.01.043.
- K. Prabha, R. Satheeshkumar, V. Nasif, J. Saranya, K. Sayin, J. Natarajan, C. Chandrasekar and K. J. Rajendra Prasad, ChemistrySelect, 2022, 7, e202200288, DOI:10.1002/slct.202200288.
- M. A. Mostafa, J. Phys. Org. Chem., 2023, 36, e4429, DOI:10.1002/poc.4429.
- K. D. Upadhyay, N. M. Dodia, R. C. Khunt, R. S. Chaniara and A. K. Shah, ACS Med. Chem. Lett., 2018, 9, 283–288, DOI:10.1021/acsmedchemlett.7b00545.
- H. M. Hassanin, W. R. Abd Elmoneam and M. A. Mostafa, Med. Chem. Res., 2019, 28, 28–38, DOI:10.1007/s00044-018-2259-9.
- S. M. Hassan, J. M. Morsy, H. M. Hassanin and E. S. Othman, J. Heterocycl. Chem., 2021, 58, 305–314, DOI:10.1002/jhet.4169.
- E. S. Othman, H. Hassan and M. Abass, J. Heterocycl. Chem., 2019, 56, 3257–3266, DOI:10.1002/jhet.3721.
- S. Jadhav, R. Patil, D. Kumbhar, A. Patravale, D. Chandam and M. Deshmukha, Int. J. Pharm. Sci. Rev. Res., 2015, 35, 75–82 Search PubMed.
- H. A. Abd El-Nabi, Pharmazie, 1997, 52, 28–32 CAS.
- I. S. Chen, S. J. Wu, I. L. Tsai, T. S. Wu, J. M. Pezzuto, M. C. Lu, H. Chai, N. Suh and C. M. Teng, J. Nat. Prod., 1994, 57, 1206–1211, DOI:10.1021/np50111a003.
- H. M. Hassanin, R. A. Serya, W. R. Abd Elmoneam and M. A. Mostafa, R. Soc. Open Sci., 2018, 5, 172407, DOI:10.1098/rsos.172407.
- A. M. Saeed, I. M. Abdou, A. A. Salem, M. A. Ghattas, N. Atatreh and S. S. AlNeyadi, Open Med. Chem. J., 2020, 10, 1–14, DOI:10.4236/ojmc.2020.101001.
- S. C. Boca, M. Potara, A. M. Gabudean, A. Juhem, P. L. Baldeck and S. Astilean, Cancer Lett., 2011, 311, 131–140, DOI:10.1016/j.canlet.2011.06.022.
- A. K. Mittal, S. Kumar and U. C. Banerjee, J. Colloid Interface Sci., 2014, 431, 194–199, DOI:10.1016/j.jcis.2014.06.030.
- J. Lin, Z. Huang, H. Wu, W. Zhou, P. Jin, P. Wei, Y. Zhang, F. Zheng, J. Zhang, J. Xu, Y. Hu, Y. Wang, Y. Li, N. Gu and L. Wen, Autophagy, 2014, 10, 2006–2020, DOI:10.4161/auto.36293.
- M. M. Abdel-Aziz, M. H. A. Elella and R. R. Mohamed, Int. J. Biol. Macromol., 2020, 142, 244–253, DOI:10.1016/j.ijbiomac.2019.09.096.
- J. Venkatesan, J. Y. Lee, D. S. Kang, S. Anil, S. K. Kim, M. S. Shim and D. G. Kim, Int. J. Biol. Macromol., 2017, 98, 515–525, DOI:10.1016/j.ijbiomac.2017.01.120.
- M. A. Mostafa, M. M. Ismail, J. M. Morsy, H. M. Hassanin and M. M. Abdelrazek, Polym. Bull., 2023, 80, 4035–4059, DOI:10.1007/s00289-022-04238-7.
- T. Kappe, R. Aigner, P. Hohengassner and W. Stadlbauer, J. Prakt. Chem./Chem.-Ztg., 1994, 336, 596–601, DOI:10.1002/prac.19943360707.
- S. M. Gomha, Z. A. Muhammad, M. R. Abdel-aziz, H. M. Abdel-aziz, H. M. Gaber and M. M. Elaasser, J. Heterocycl. Chem., 2018, 55, 530–536, DOI:10.1002/jhet.3088.
- G. C. Yen and P. D. Duh, J. Agric. Food Chem., 1994, 42, 629–632, DOI:10.1021/jf00039a005.
- E. A. Abdelsalam, A. A. Abd El-Hafeez, W. M. Eldehna, M. A. El Hassab, H. M. M. Marzouk, M. M. Elaasser, N. A. Abou Taleb, K. M. Amin, H. A. Abdel-Aziz, P. Ghosh and S. F. Hammad, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 2265–2282, DOI:10.1080/14756366.2022.2104841.
- J. L. Nakamura, Expert Opin. Ther. Targets, 2007, 11(4), 463–472, DOI:10.1517/14728222.11.4.463.
- O. Trott and A. J. Olson, J. Comput. Chem., 2009, 31, 455–461, DOI:10.1002/jcc.21334.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898, DOI:10.1021/acs.jcim.1c00203.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639–1662, DOI:10.1002/(SICI)1096-987X(19981115)19:14<1639:AID-JCC10>3.0.CO;2-B.
- R. Huey, G. M. Morris, A. J. Olson and D. S. Goodsell, J. Comput. Chem., 2007, 28, 1145–1152, DOI:10.1002/jcc.20634.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791, DOI:10.1002/jcc.21256.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612, DOI:10.1002/jcc.20084.
- X. Jin, J. Wang and J. Bai, Carbohydr. Res., 2009, 344, 825–829, DOI:10.1016/j.carres.2009.01.022.
- T. Baran, E. Açıksöz and A. Menteş, Carbohydr. Polym., 2016, 142, 189–198, DOI:10.1016/j.carbpol.2016.01.057.
- X. Wang, Y. Du, L. Fan, H. Liu and Y. Hu, Polym. Bull., 2005, 55, 105–113, DOI:10.1007/s00289-005-0414-1.
- M. Timur and A. Paşa, ACS Omega, 2018, 3, 17416–17424, DOI:10.1021/acsomega.8b01872.
- J. U. Shareef, M. N. Rani, S. Anand and D. Rangappa, Mater. Today: Proc., 2017, 4(11), 11923–11932, DOI:10.1016/j.matpr.2017.09.113.
- S. Hajji, R. B. S. B. Salem, M. Hamdi, K. Jellouli, W. Ayadi, M. Nasri and S. Boufi, Process Saf. Environ. Prot., 2017, 111, 112–121, DOI:10.1016/j.psep.2017.06.018.
- R. A. Abdel-Monem, A. M. Khalil, O. M. Darwesh, A. I. Hashim and S. T. Rabi, J. Macromol. Sci., Part A: Pure Appl. Chem., 2020, 57, 145–155, DOI:10.1080/10601325.2019.1674666.
- A. H. Doctorsafaei, S. B. Burujeny, H. A. Rudbari, N. Kordestani and S. A. A. Najafabadi, Chem. Eng. J., 2020, 381, 122776, DOI:10.1016/j.cej.2019.122776.
- S. Kumar, M. Kumari, P. K. Dutta and J. Koh, Int. J. Polym. Mater., 2014, 63, 173–177, DOI:10.1080/00914037.2013.812088.
- H. Ma, A. Kong, Y. Ji, B. He, Y. Song and J. Li, J. Cleaner Prod., 2019, 214, 89–94, DOI:10.1016/j.jclepro.2018.12.217.
- S. Pal, Y. K. Tak and J. M. Song, Environ. Microbiol., 2007, 73, 1712–1720, DOI:10.1128/AEM.02218-06.
- C. Karthik, S. Suresh, S. Mirulalini and S. Kavitha, Inorg.
Nano-Met. Chem., 2020, 50, 606–612, DOI:10.1080/24701556.2020.1723025.
- A. A. Hamed, G. R. Saad, I. A. Abdelhamid, M. M. Abdel-Aziz, H. A. Taha, M. M. Abou El Dahab and M. Z. Elsabee, Polym. Bull., 2022, 79, 11259–11284, DOI:10.1007/s00289-021-03993-3.
- A. M. Omer, Z. M. Ziora, T. M. Tamer, R. E. Khalifa, M. A. Hassan and M. S. Mohy-Eldin, Molecules, 2021, 26, 449, DOI:10.3390/molecules26020449.
- P. Ferreira, J. F. J. Coelho, K. S. C. R. Dos Santos, E. I. Ferreira and M. H. Gil, J. Carbohydr. Chem., 2006, 25, 233–251, DOI:10.1080/07328300600734085.
- M. A. Hassan, T. M. Tamer, K. Valachová, A. M. Omer, M. El-Shafeey, M. S. M. Eldin and L. Šoltés, Int. J. Biol. Macromol., 2021, 166, 18–31, DOI:10.1016/j.ijbiomac.2020.11.119.
- R. A. Abdel-Monem, A. M. Khalil, O. M. Darwesh, A. I. Hashim and S. T. Rabie, J. Macromol. Sci., Part A: Pure Appl.Chem., 2020, 57, 145–155, DOI:10.1080/10601325.2019.1674666.
- A. M. Khalil, R. A. Abdel-Monem, O. M. Darwesh, A. I. Hashim, A. A. Nada and S. T. Rabie, J. Chem., 2017, 1, 1434320, DOI:10.1155/2017/1434320.
- L. Zeng, C. Qin, L. Wang and W. Li, Carbohydr. Polym., 2011, 83, 1553–1557, DOI:10.1016/j.carbpol.2010.10.007.
- E. Musellim, M. H. Tahir, M. S. Ahmad and S. Ceylan, Appl. Therm. Eng., 2018, 137, 54–61, DOI:10.1016/j.applthermaleng.2018.03.050.
- T. M. Tamer, M. A. Hassan, A. M. Omer, W. M. Baset, M. E. Hassan, M. E. El-Shafeey and M. S. M. Eldin, Process Biochem., 2016, 51, 1721–1730, DOI:10.1016/j.procbio.2016.08.002.
- J. Ma, C. Liu, R. Li and J. Wang, e-Polym., 2012, 12, 1–14, DOI:10.1515/epoly.2012.12.1.386.
- M. E. Ahmed, H. M. Mohamed, M. I. Mohamed and N. G. Kandile, Int. J. Biol. Macromol., 2020, 162, 1388–1397, DOI:10.1016/j.ijbiomac.2020.08.048.
- T. M. Tamer, K. Valachová, M. S. Mohyeldin and L. Soltes, J. Appl. Pharm. Sci., 2016, 6, 195–201, DOI:10.7324/JAPS.2016.60428.
- K. Chokshi, I. Pancha, T. Ghosh, C. Paliwal, R. Maurya, A. Ghosh and S. Mishra, RSC Adv., 2016, 6, 72269–72274, 10.1039/C6RA15322D.
- S. S. Alharthi, T. Gomathi, J. J. Joseph, J. Rakshavi, J. A. K. Florence, P. N. Sudha, P. N. Sudha, G. Rajakumar and M. Thiruvengadam, J. King Saud Univ., Sci., 2022, 34, 102177, DOI:10.1016/j.jksus.2022.102177.
- S. Hajji, S. B. Khedir, I. H. Mnif, M. Hamdi, I. Jedidi, R. Kallel, S. Boufi and M. Nasri, Subjects, 2019, 1863, 241–254, DOI:10.1016/j.bbagen.2018.10.010.
- J. Zhou, B. Wen, H. Xie, C. Zhang, Y. Bai, H. Cao, Q. Che, J. Guo and Z. Su, Food Funct., 2021, 12, 926–951, 10.1039/D0FO02768E.
- T. M. Tamer, H. Zhou, M. A. Hassan, M. M. Abu-Serie, S. Shityakov, S. M. Elbayomi, M. S. Mohy-Eldin, Y. Zhang and T. Cheang, Int. J. Biol. Macromol., 2023, 240, 124339, DOI:10.1016/j.ijbiomac.2023.124339.
- R. F. Elshaarawy, A. A. Refaee and E. A. El-Sawi, Carbohydr. Polym., 2016, 146, 376–387, DOI:10.1016/j.carbpol.2016.03.017.
- A. L. Fadli, A. Hanifah, A. Fitriani, A. Rakhmawati and W. S. B. Dwandaru, AIP Conf. Proc., 2018, 2014, 020017, DOI:10.1063/1.5054421.
- S. Govindan, E. A. K. Nivethaa, R. Saravanan, V. Narayanan and A. Stephen, Appl. Nanosci., 2012, 2, 299–303, DOI:10.1007/s13204-012-0109-5.
- I. Aranaz, A. R. Alcántara, M. C. Civera, C. Arias, B. Elorza, A. H. Caballero and N. Acosta, Polymers, 2021, 13, 3256, DOI:10.3390/polym13193256.
- Y. Gan, Y. Xu, X. Zhang, H. Hu, W. Xia, Z. Yu, T. Sun, J. Zhang, C. Wen and S. Zheng, Molecules, 2023, 28, 6962, DOI:10.3390/molecules28196962.
- T. Grabe, K. Jeyakumar, J. Niggenaber, T. Schulz, S. Koska, S. Kleinbölting, M. E. Beck, M. P. Müller and D. Rauh, ACS Med. Chem. Lett., 2023, 14, 591–598, DOI:10.1021/acsmedchemlett.2c00514.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*