
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium complexes with triazenide ligands bearing an N-heterocyclic moiety, and their catalytic properties in the reduction of nitroarenes†
Christian A. Romero-Soto‡
a,
Ana L. Iglesias‡ b,
Amor F. Velázquez-Hama,
Juan P. Camarena-Díaza,
Erick Correa-Ayalaa,
Jessica L. Gomez-Lopeza,
Daniel Cháveza,
Adrián Ochoa-Terána,
Gerardo Aguirrea,
Arnold L. Rheingoldc,
Douglas B. Grotjahn§
d,
Miguel Parra-Hakea and
Valentín Miranda-Soto*a
b,
Amor F. Velázquez-Hama,
Juan P. Camarena-Díaza,
Erick Correa-Ayalaa,
Jessica L. Gomez-Lopeza,
Daniel Cháveza,
Adrián Ochoa-Terána,
Gerardo Aguirrea,
Arnold L. Rheingoldc,
Douglas B. Grotjahn§
d,
Miguel Parra-Hakea and
Valentín Miranda-Soto*a
aTecnológico Nacional de México/Instituto Tecnológico de Tijuana/Centro de Graduados e Investigación en Química, Blvd. Alberto Limón Padilla S/N, 22454 Tijuana, BC, Mexico. E-mail: vmiranda@tectijuana.mx
bFacultad de Ciencias de la Ingeniería y Tecnología, Universidad Autónoma de Baja California, Mexico
cDepartment of Chemistry and Biochemistry, University of California at San Diego, CA 92093, USA
dDepartment of Chemistry and Biochemistry, San Diego State University, CA 92182, USA
First published on 31st July 2024
Abstract
A series of ruthenium complexes of formulae [RuCl(triazenide)(p-cymene)] have been synthesized using as ligand a triazenide monofunctionalized with an N-heterocyclic moiety. Nuclear magnetic resonance, high resolution mass spectrometry and X-ray diffraction were used to characterize the triazenide ligands and their complexes. In addition, these ruthenium complexes catalyzed the reduction of nitrobenzene to aniline in the presence of sodium borohydride and ethanol as solvent at room temperature. Notably, complex 5 was especially active in the reduction of nitroarenes substituted at the aromatic ring with electron-withdrawing or electron-donating fuctional groups affording the desired arylamines in good to excellent yields (80–100%). The role of the N-heterocyclic moiety on catalysis was explored.
Introduction
Aryl amines are an important motif found in the structure of many significant molecules used as dyes, pharmaceuticals, and agrochemicals.1 The most straightforward methodology to obtain aryl amines is a two-step pathway that includes nitration of arenes and subsequent reduction of nitroarenes.2 The use of iron, ferrous salts, or iron catalysts in aqueous acid is a chemical process, referred as the Béchamp reduction, which has been used in order to achieve the reduction of nitroarenes to the corresponding aryl amines.3 However, the main drawback of this methodology is the production of a lot of metallic waste along with the elevated temperature required for an efficient reduction.4a,g In contrast, catalytic hydrogenation is a green methodology for reduction of nitroarenes that has been extensively reviewed in the last decade.4 These methodologies are predominantly based on heterogeneous catalysis, and have been classified based on the source of hydrogen utilized during the reduction and the mechanism involved in the process. In comparison, homogeneous catalytic systems have been less studied, probably due to some disadvantages associated with their industrial application. Nevertheless, there are several examples of homogeneous catalysts based on iron,5 cobalt,6 nickel,7 molybdenum,8 manganese,9 palladium,10 rhodium,11 and ruthenium complexes.12 Among these, ruthenium complexes stand out for being able to reduce nitroarenes using as hydrogen source sodium borohydride, hydrogen gas or alcohols. In the sodium borohydride methodology, the ligands more frequently found in the ruthenium complexes are imines, amide–phosphines, aryloxazolines, and quinolates. These are five or six-membered chelating ligands with N–O or N–C coordinating atoms. However, there are no examples for the reduction of nitroarenes performed by ruthenium complexes containing four-membered chelating ligands with N–N atom donors, bearing a pedant-heterocyclic base. Exploring the catalytic properties of iridium, rhodium and ruthenium–triazenide complexes13 in the transfer hydrogenation of unsaturated molecules, we envision that either complex A or B, with the triazenide bearing an N-heterocyclic moiety, could be useful in the reduction of nitroarenes. Therefore, in this work, we report the structural characterization, catalytic behaviour, and reactivity of four ruthenium complexes with triazenide ligands bearing an N-heterocyclic moiety (Chart 1).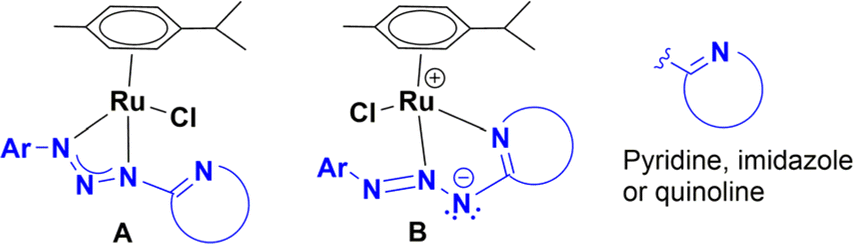 | ||
| Chart 1 Four (A) and five-membered (B) chelating triazenide ligands in ruthenium complexes. | ||
Results and discussion
Precursors of triazenide ligands functionalized with heterocycles like pyridine 1 and quinoline 2 were synthesized by the traditional procedure of N-coupling of diazonium salts (Scheme 1).14,15 Triazenides 1 and 2 were fully characterized by spectroscopic techniques. On the other hand, ligands 3 and 4 functionalized with imidazole moieties have been previously reported by our group13b and were synthesized by the reaction of 2-lithioimidazoles with 4-azidotoluene, followed by hydrolysis.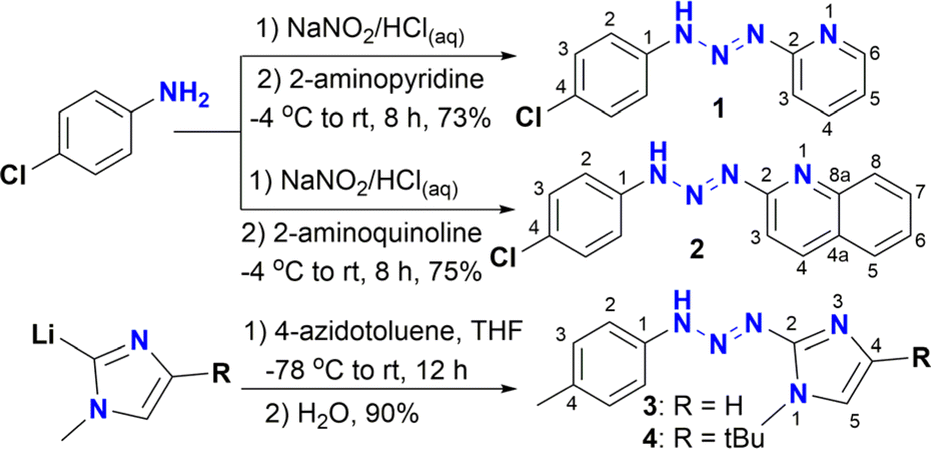 | ||
| Scheme 1 Synthesis of triazenes 1–4. | ||
The 1H NMR spectra of triazenes 1 and 2 show the characteristic NH signal of the diazoamine group as a broad singlet at 11.51 and 13.12 ppm, respectively. The expected A2B2 system for the aromatic rings is observed for 2 as two doublets at 7.52 and 7.63 ppm (J = 8.6 Hz), whilst for 1, the signal for proton H4 of the pyridine ring overlaps with one of the A2B2 signals and has the appearance of a triplet centered at 7.54 ppm. The resonances for the aromatic protons of pyridine and quinolone rings are observed in the region of 6.97–8.93 ppm and 7.45 to 8.34 ppm, respectively.
The 13C {1H} NMR spectra of compounds 1 and 2 show the typical signals for aromatic carbons from 109.0 to 154.7 ppm and 110.5 to 153.9 ppm, respectively. Interestingly, for triazene 1, the aromatic carbon C1 is absent from both 13C {1H} NMR and APT spectra, even if other NMR solvents are used in its analysis. Finally, running a 13C proton-coupled NMR experiment shows the signal as a singlet at 148.2 ppm, which is under the signal for carbon C6 of the pyridine ring in the regular 13C experiment (Fig. S5 and S6†).
Ruthenium(II) triazenide complexes
Reaction of ligand precursors 1 or 2 with one half equivalent of [RuCl2(p-cymene)]2, in acetonitrile at room temperature in the presence of triethylamine, resulted in the formation of new half sandwich Ru(II) complexes 5 and 6 (Scheme 2). These compounds were isolated as air and moisture stable microcrystalline orange solids. On the other hand, complexes 7 and 8 were synthesized using a different methodology according to Scheme 2. Treatment of a solution of 3 or 4 in dry THF with K[N(SiMe3)2] led to the formation of potassium triazenide, which can be directly converted into complexes 7–8 by a transmetallation reaction with [RuCl2(p-cymene)]2. All complexes were characterized by 1H NMR and 13C {1H} NMR spectroscopy, HRMS, and single crystal X-ray diffraction analysis. The assignments are supported by 1H–1H gCOSY, 1H–13C gHSQC and 1H–13C gHMBC correlation experiments. The 1H and 13C {1H} NMR of complexes 5 and 6 show a very similar spectra, as can be expected from their proposed structures, and suggest a chiral environment around the ruthenium centre since the p-cymene aromatic protons appear as four distinct doublets in the region of 5.48 and 6.07 ppm (J = 5.6 Hz) for 5 (Fig. S34†).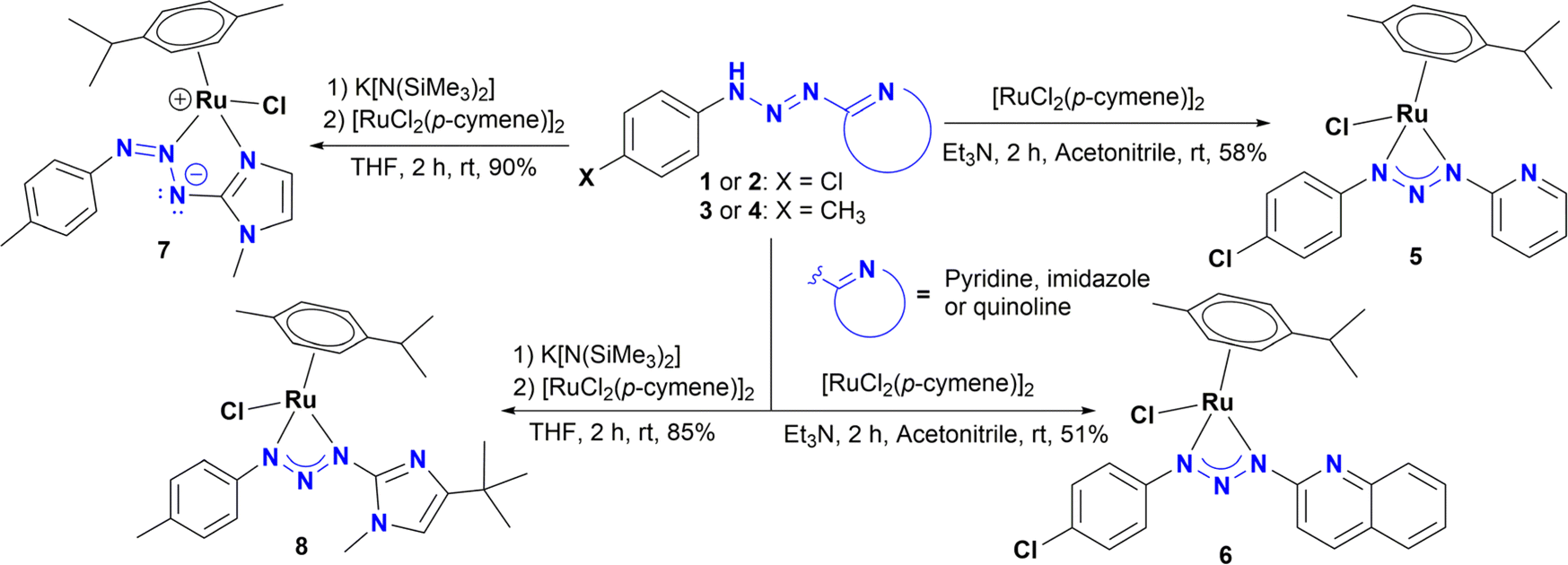 | ||
| Scheme 2 Synthesis of complexes 5–8. | ||
These resonances are slightly shifted downfield in complex 6 due to the presence of the quinoline ring and appear in the region of 5.56 and 6.17 ppm when it is analysed in CD2Cl2 (Fig. S52†). For 6 in DMSO-d6, the latter signals are slightly overlapped with an additional downfield shift of 5.83 to 6.23 ppm (Fig. S60†). Likewise, the isopropyl methyl protons of the p-cymene are not equivalent and appear as two doublets at 1.25 and 1.26 ppm (J = 6.4 and 6.9 Hz) for both 5 and 6. This behaviour has been observed in related asymmetric Ru(II)(p-cymene) complexes.12d,f–h,13d Interestingly, analysing the microcrystalline solid in solution by 1H NMR showed 6 as the major species along another minor species (∼10%) with a similar set of NMR resonances, which may be a coordination isomer of 6 (Fig. S53†).
In general, for compounds 5 and 6, all 1H NMR signals are slightly shifted up field compared to the free ligand 1 and 2 (Fig. S45 and S61,† respectively). However, an interesting feature in complexes 5 and 6 is the appearance of the aromatic A2B2 system, which exhibits a second-order effect and is observed as a multiplet centered at 7.11 for 5, and 7.37 ppm for 6 (with Δν/J = 1.9 and 1.6 Hz), respectively. The 13C {1H} NMR analysis of 5 and 6 corroborated the asymmetric nature of the complexes. The spectrum shows six signals for the aromatic p-cymene carbons, which are seen between 79.8 and 103.1 ppm for 5 and 80.3 and 103.7 ppm for 6, as well as the two diastereotopic methyl carbons of the iPr group, which appear between 22.7 and 23.0 ppm for both complexes (Fig. S35, S36 and S54†). In addition, the carbon resonances (C2) of the heterocyclic moieties in complexes 5 and 6 are shifted downfield (∼4 ppm) in comparison with the C2 in triazenes 1 and 2. In contrast, there is a slight up-fied shift (≥2.10 ppm) of the aryl carbon resonances (C1) for 5 and 6, compared to the free triazenes 1 and 2.
The 1H NMR and 13C NMR spectra of 8 are similar to those of complexes 5 and 6, suggesting the same coordination mode. Particularly, the signals for the p-cymene protons also appear as four doublets in the region of 5.50 and 6.04 ppm (J = 5.9 Hz), as well as two doublets (J = 7.0 Hz) for the methyl groups of the iPr at 1.28 and 1.30 ppm (Fig. S75†). However, the splitting pattern for the aromatic hydrogens is different. For 8 the A2B2 system is clearly observed as two doublets at 7.10 and 7.22 ppm (J = 8.3 Hz), in agreement with related Ir(III) and Rh(III) triazenide complexes.13a,b In addition, three singlets are observed at 3.52, 2.29 and 1.29 ppm, which are assigned to N–CH3, tBu and H3C–Ph, respectively. In contrast, the signals for the p-cymene ring in 7 are quite different from all other complexes, which suggests a different coordination mode. Thus, the A2B2 system of the p-cymene collapses into a pseudotriplet centered at 5.66 ppm and a slightly broad multiplet at 5.38 ppm, whereas the methyl groups of the iPr give only one doublet at 1.19 ppm (J = 6.9 Hz) (Fig. S69†). Moreover, as observed in complex 8, the signals for the A2B2 system of the aromatic ring for 7 also appear as two doublets at 7.75 and 7.12 ppm (J = 8.3 Hz). Lastly, the proton resonance for the imidazole moiety appears as two doublets at 6.72 and 7.14 ppm (J = 1.8 Hz).
In the 13C {1H} NMR spectrum for 8, the p-cymene CH carbons appear between 80.0 and 81.7 ppm. However, the same resonances for complex 7 are shifted downfield to 83.1 and 87.6 ppm compared to all other complexes. These differences in chemical shifts might be a consequence of the coordination of the imidazole ring. Another fact that supports this assumption is the resonance of C5, which are shifted upfield at 117.6 ppm compared to 120.9 ppm in the free triazene 3 (151.0 ppm), as well as the significant downfield shift of C2 (161.2 ppm) compared to the free ligand 3 (151.0 ppm). In contrast, there is no evidence for nitrogen–imidazole coordination in complex 8, whose 13C NMR data shows no significant changes in chemical shift of the C5 resonance (114.4 ppm) or in C2 (145.9 ppm) in comparison with the same signals in the free triazene 4 (114.1) and 146.8 ppm, respectively (Fig. S27 and S76†).13b
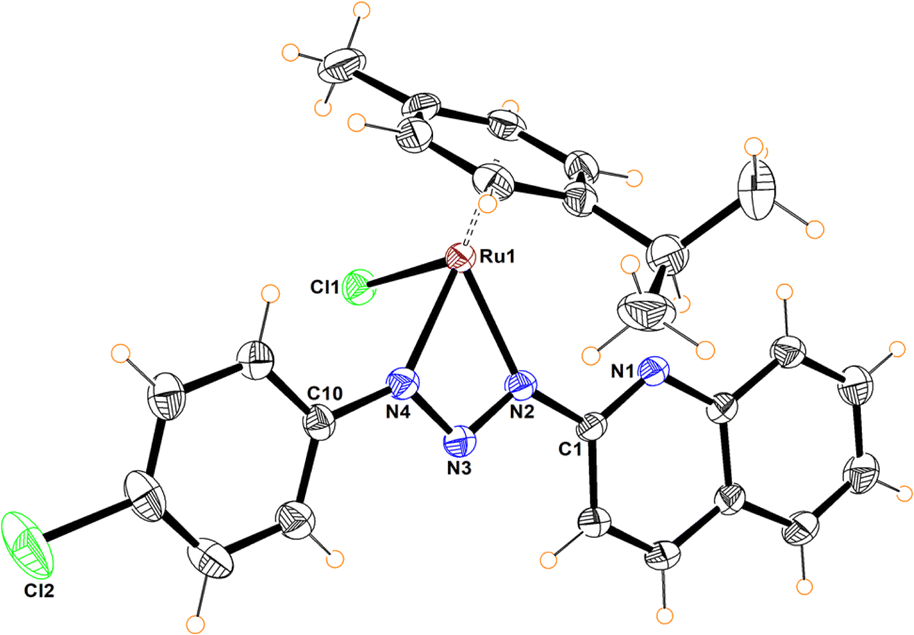
The molecular structures of compounds 5–7 were determined by X-ray diffraction analysis (Fig. 1–3). Complexes 5 and 6 crystallized in the orthorhombic crystalline system, while 7 in the monoclinic system with different space groups (P212121 for 5 and 6; P21/n for 7). These half sandwich Ru(II) complexes adopt a pseudo-octahedral “piano-stool” geometry with the p-cymene acting as the seat, a chloro ligand and the N,N donor atoms completing the coordination sphere. This three-legged structure is commonly observed in other related half-sandwich ruthenium N,N chelating ligand complexes.13c,d
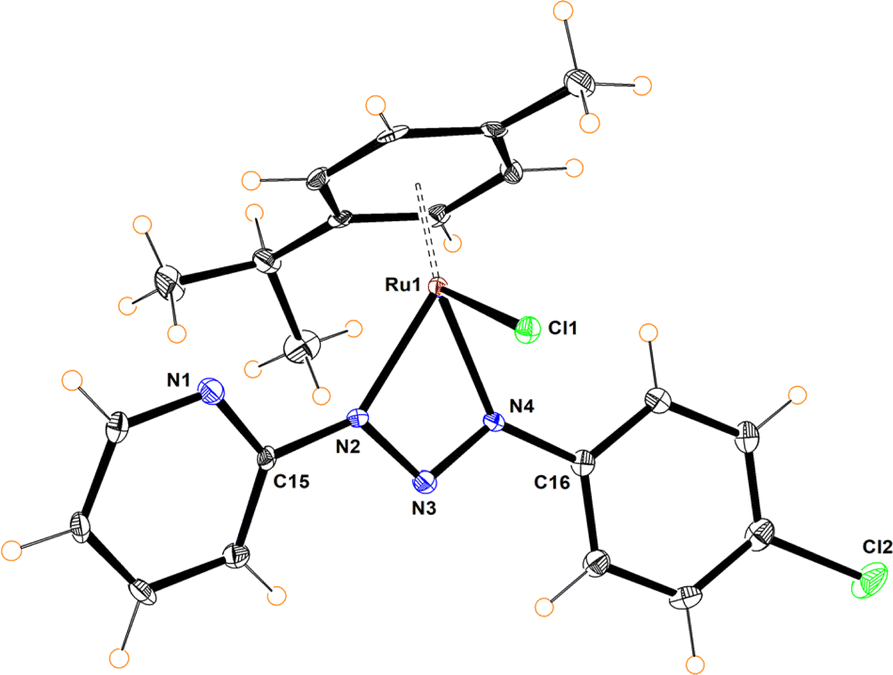 | ||
| Fig. 1 Molecular structure of 5. Selected bond distances (Å): N(2)–N(3), 1.325(4); N(3)–N(4), 1.320(4); Ru(1)–N(2), 2.067(3); Ru(1)–N(4), 2.081(4); Ru(1)–Cl(1), 2.3985(10). Selected bond angles (°): N(2)–Ru(1)–N(4), 59.16(11); N(2)–Ru(1)–Cl(1), 86.26(10); N(4)–Ru(1)–Cl(1), 83.09(1). N(2)–N(3)–N(4), 101.5(3). | ||
 | ||
| Fig. 2 Molecular structure of 6. Selected bond distances (Å): N(2)–N(3), 1.316(5); N(3)–N(4), 1.301(5); Ru(1)–N(2), 2.081(4); Ru(1)–N(4), 2.101(4); Ru(1)–Cl(1), 2.398(1). Selected bond angles (°): N(2)–Ru(1)–N(4), 58.7(1); N(2)–Ru(1)–Cl(1), 88.1(1); N(4)–Ru(1)–Cl(1), 83.2(1). N(2)–N(3)–N(4),103.1(4). | ||
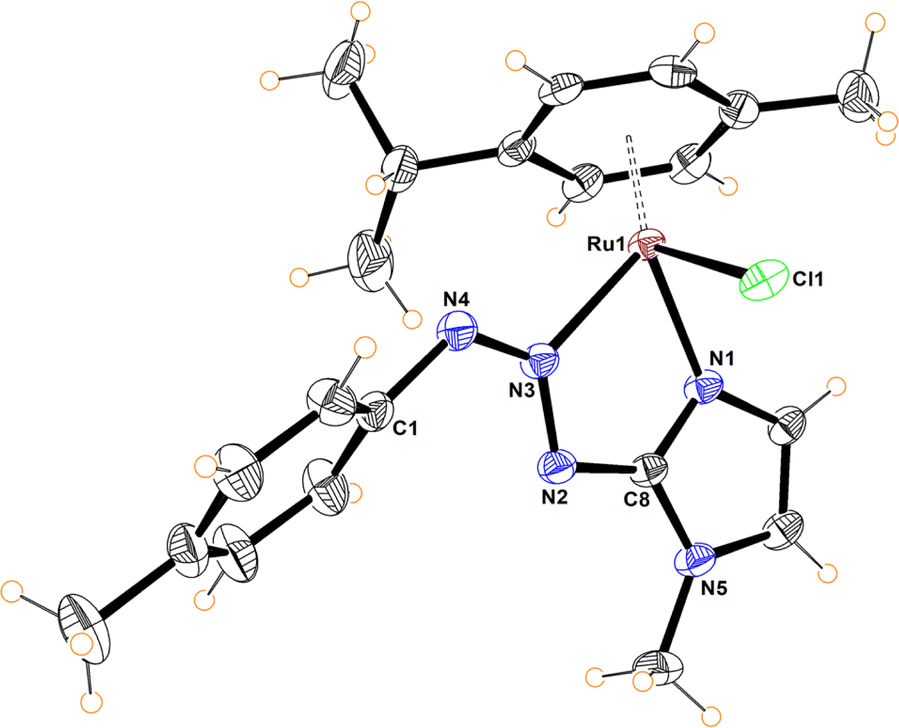 | ||
| Fig. 3 Molecular structure of 7. Selected bond distances (Å): N(3)–N(4), 1.284(2); N(2)–N(3), 1.348(2); N(3)–Ru(1), 2.103(2); N(1)–Ru(1), 2.053(2); Cl(1)–Ru(1), 2.4195(6). Selected bond angles (°): N(3)–Ru(1)–N(1) 75.39(7)°; N(3)–Ru(1)–Cl(1), 85.64(5); N(1)–Ru(1)–Cl(1), 84.17(5), N(2)–N(3)–N(4), 121.1(2). | ||
For complexes 5 and 6, the metal coordinates through the terminal nitrogen atom forming a four-membered chelate with the bidentate triazenide ligand. The octahedral distortion in these compounds is caused primarily by the small-bite angle of the coordinating ligand, which is in the range of 58.47 and 59.16°, in agreement with other reported systems, while the N–Ru–Cl angle is close to the expected 90°. The similar N(2)–N(3) and N(3)–N(4) bond distances in the triazenide (∼1.31 to ∼1.32 Å) indicates π-delocalization over the metal-triazenide system for 5 and 6.16 In contrast, in complex 7 the ligand coordinates through N(1) of the imidazole and N(3) of the triazenide system, forming a five-membered chelate ring with a bite angle of 75.39(7)° (Fig. 3).12b,13b,17 Furthermore, the N(3)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(4) double bond distance of 1.284(2) Å is comparable to analogous Rh(III) and Ir(III) Cp* half sandwich triazenide complexes, where the ligand is coordinated through the imidazole ring.13b
N(4) double bond distance of 1.284(2) Å is comparable to analogous Rh(III) and Ir(III) Cp* half sandwich triazenide complexes, where the ligand is coordinated through the imidazole ring.13b
The Ru–N bond lengths of complexes 5–7 are in the range of 2.067(3) Å to 2.103(2) Å, while the Ru1–Cl1 bond distances are between 2.398(10) to 2.4195(6) Å, and are consistent with Ru–N and Ru–Cl1 bond length values reported for other half-sandwich ruthenium complexes with N,N ligands.13c,18
Lastly, the most abundant peak in the HRMS (ESI-TOF) mass spectra of these complexes is generally associated with [M + H]+, which is seen at 503.0327 (5), 553.0467 (6), 486.0967 (7), and 542.1615 (8) amu (Fig. S43, S67, S74, and S80,† respectively). In addition, for compounds 5 and 6 the spectra show the formation of the adduct [M + Na]+ at 525.0157 and 575.0291 amu. This adduct is also present in the mass spectrum at 255.0408 and 305.5092 amu for ligands 1 and 2, respectively, and may be a consequence of using sodium formate as calibrant.
Catalysis
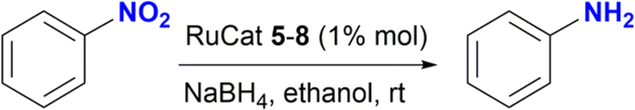
The catalytic performance of ruthenium(II) complexes 5–8 was evaluated in the reduction of nitroarenes in the presence of NaBH4, with nitrobenzene as the model substrate. As shown in Table 1, all complexes were very active in the reduction of nitrobenzene to aniline with excellent yields in the range of 90–99% in 30 min (Table 1, entries 2–5). No reaction was observed in the control experiment performed without the ruthenium catalyst (Table 1, entries 1 and 6). The use of [RuCl2(p-cymene)]2 as catalyst afforded the expected product in 13% yield (Table 1, entry 7). Moreover, the homogeneity of the catalytic reaction was tested using the mercury drop experiment in the reduction of nitrobenzene with catalyst 5, which gave a >99% of aniline as the product with no loss of efficiency (Table 1, entry 12). On the other hand, decreasing the catalyst loading to 0.5% mol gave 90% yield of aniline with the formation of 10% of azobenzene (Table 1, entry 13 and Fig. S114–S116†).
|
|||
|---|---|---|---|
| Entry | Catalyst | Time (min) | Yield (%) |
| a Reaction conditions: nitrobenzene (0.3 mmol), NaBH4 (1.2 mmol), ethanol (2 mL), room temperature. Yields were determined by GC-MS.b Mercury drop test.c Catalyst charge of 0.5, 10% of azobenzene was also detected. | |||
| 1 | — | 30 | 1 |
| 2 | 5 | 30 | >99 |
| 3 | 6 | 30 | 97 |
| 4 | 7 | 30 | 95 |
| 5 | 8 | 30 | 90 |
| 6 | — | 60 | 1 |
| 7 | [RuCl2(p-cymene)]2 | 60 | 13 |
| 8 | 5 | 60 | 100 |
| 9 | 6 | 60 | 100 |
| 10 | 7 | 60 | 100 |
| 11 | 8 | 60 | 100 |
| 12 | 5 | 30 | >99b |
| 13 | 5 | 30 | 90c |
As shown in Table 1, no significant differences in the catalytic activity of complexes 5–8 can be observed, especially at 60 min (Table 1, entries 8–11). Therefore, another model substrate (4-aminonitrobenzene) was evaluated (Table S6†). Furthermore, to better assess the role of the heterocyclic moiety on catalysis, complexes 10 (synthesized from triazene 9) and 1113d were introduced as controls for catalysts 5–8, in which the heterocyclic moiety was replaced in the structure of complexes by a phenyl or p-tolyl fragment (Chart 2).
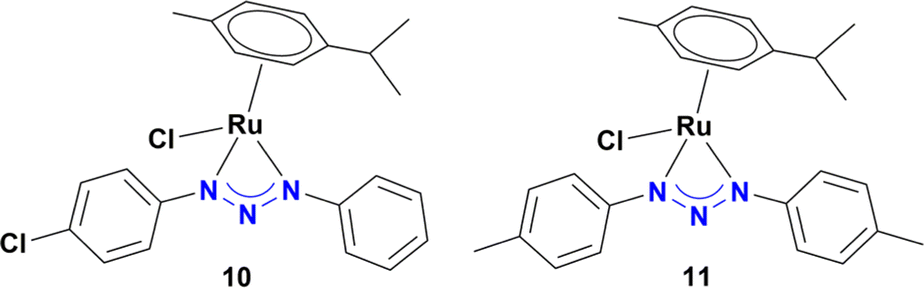 | ||
| Chart 2 Ru(II) complexes used as controls. | ||
We expanded the scope of the catalytic reduction to a series of nitroarenes using 5 as the catalyst (1% mol), at room temperature in the presence of four equivalents of NaBH4. As shown in Table 2, catalyst 5 is found to be very active in the reduction of a diverse group of nitroarenes. Electron-withdrawing and electron-donating substituents on the aromatic ring provided the expected products in good to excellent yields (Table 2, 78–100%).
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yield (%) |
| a Reaction conditions: nitroarene (0.3 mmol), catalyst 5 (0.003 mmol), NaBH4 (1.2 mmol), ethanol (2 mL) room temperature. Yields were determined by GC-MS.b Isolated yield, 1H NMR of products can be found in ESI.c 95% reduction to 4-aminobenzyl alcohol was completed after 120 min. | |||
| 1 | 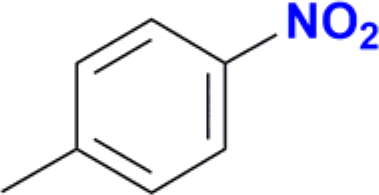 |
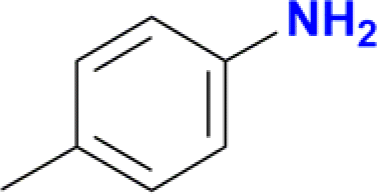 |
100 |
| 2 | 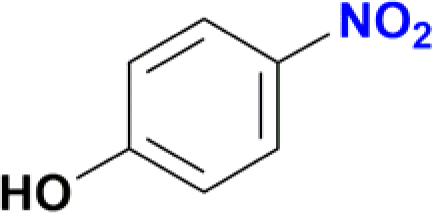 |
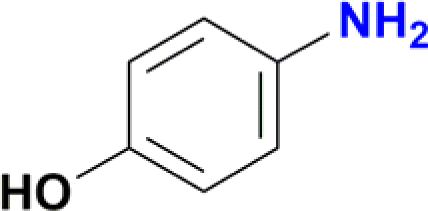 |
78 |
| 3 | 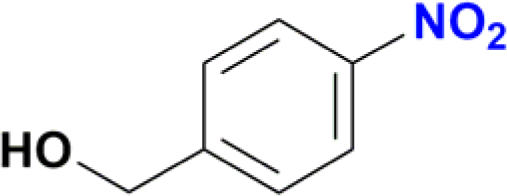 |
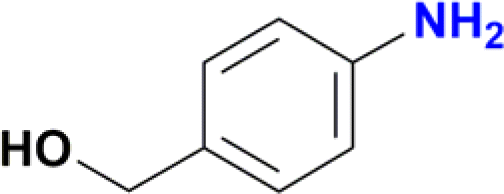 |
100 |
| 4 | 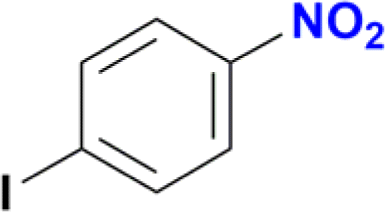 |
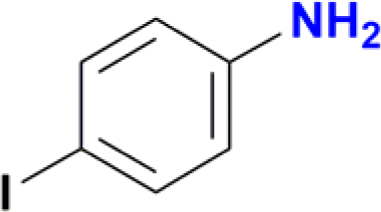 |
100 |
| 5 | 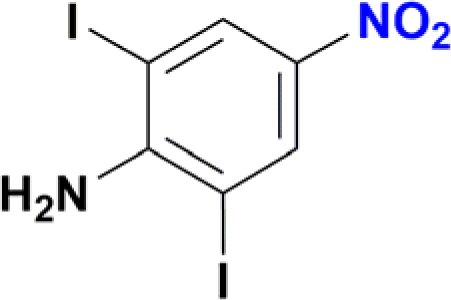 |
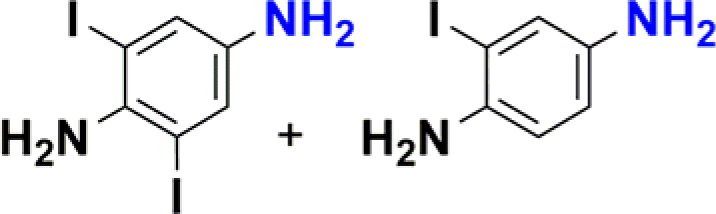 |
78/22 |
| 6 | 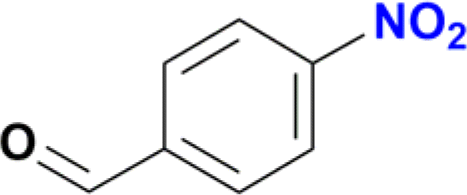 |
 |
100 |
| 7 | 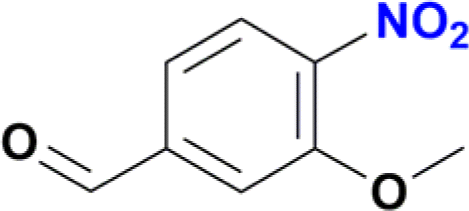 |
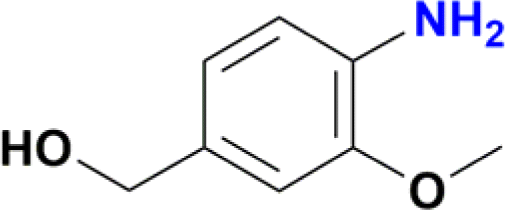 |
100 |
| 8 | 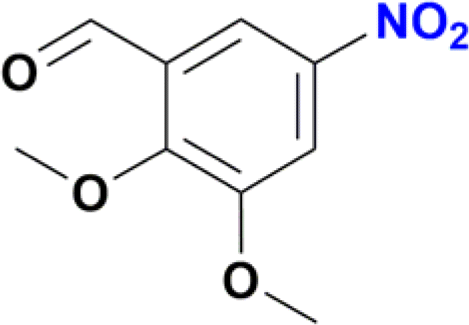 |
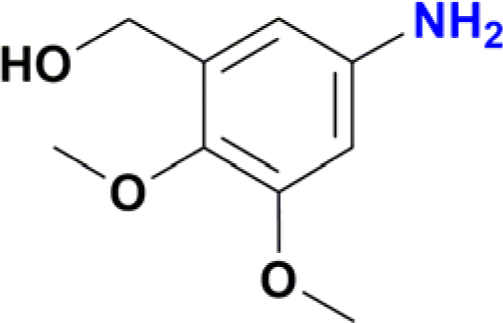 |
100 |
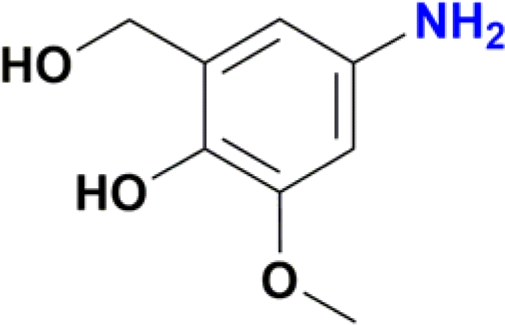
| 9 | 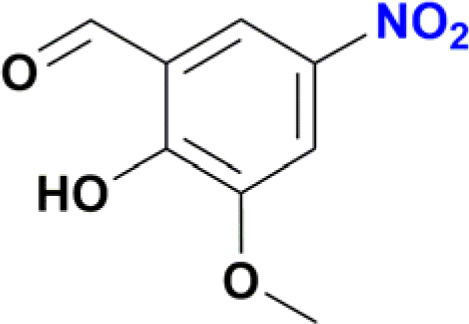 |
 |
79b |
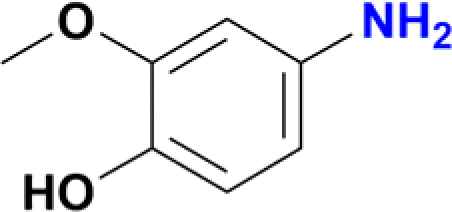
| 10 | 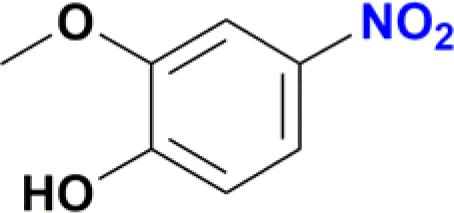 |
 |
80b |
| 11 | 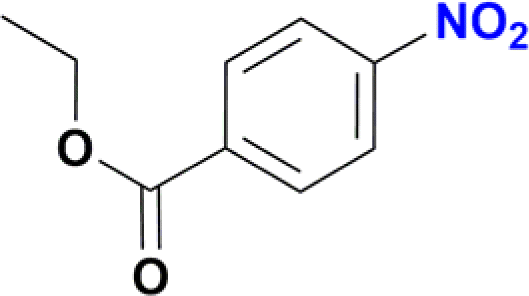 |
 |
43/57c |
| 12 | 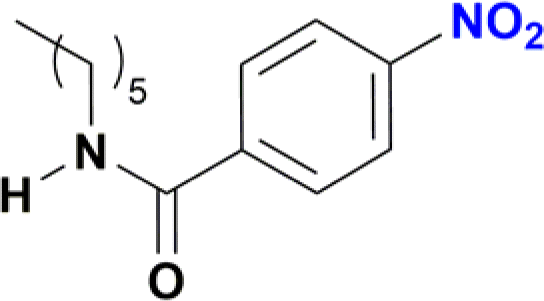 |
 |
97 |
| 13 | 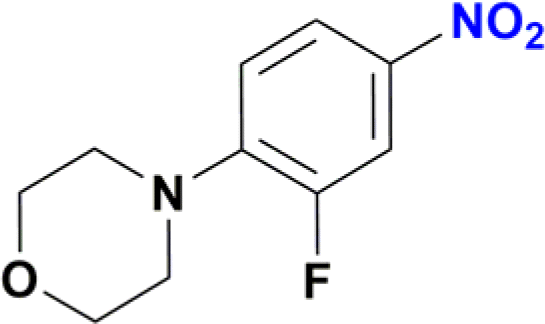 |
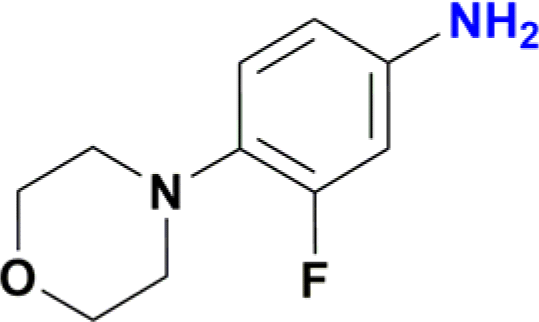 |
97 |
| 14 | 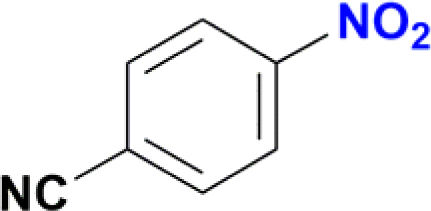 |
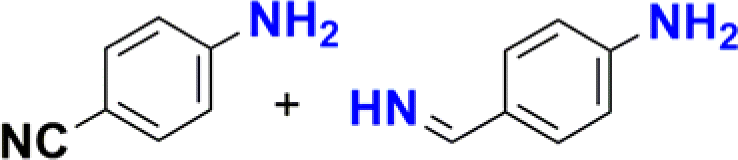 |
8/30 |
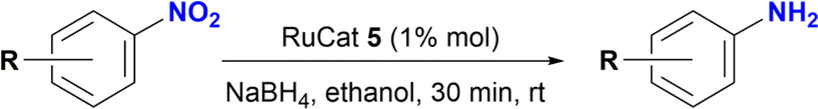
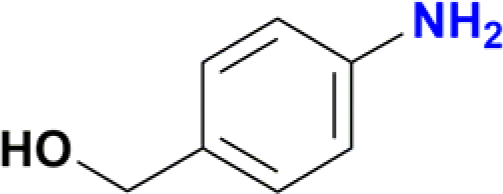
The reduction of the NO2 group in the presence of other functionality groups such as –OH, –COOEt and –CONHR was also explored. In the case of the aldehyde group, the carbonyl was reduced along with the NO2 group, as observed in other Ru(II) p-cymene catalytic systems (Table 2, entries 3–6).12f–k
The degree of substitution pattern on the aromatic ring had no effect on catalytic efficiency. For example, compound 5 was able to reduce the nitro group in the presence of aldehyde and methoxy groups, with the aldehyde also being reduced to the alcohol with 100% yield (Table 2, entries 6–8). In this case, reduction of the aldehyde is attributed to the NaBH4 itself, which was used in excess. Noteworthy, the presence of a hydroxyl group in para-position to the –NO2, led to the same conversion of the expected amine (∼80% ca.), regardless of the presence of other functional groups or degree of substitution (entries 2 and 9–10, Table 2). In contrast, the reduction of the (4-nitrophenyl)methanol proceeded smoothly with a 100% yield (Table 2, entry 3).12f,19
1-Iodo-4-nitrobenzene was readily reduced to 4-iodoaniline in 30 minutes with quantitative yield (Table 2, entry 4).12e,20a,b On the other hand, the reaction towards the 2,6-diiodo-4-nitroaniline was not chemoselective, since a mixture of the dehydrohalogenated aniline and the desired product was obtained (Table 2, entry 5 and Fig. S146†). Longer reaction time increased the conversion to the mono iodo-diaminobenzene (63%), while decreasing the yield of the desired 2,6-diiodobenzene-1,4-diamine (35%). For analogous derivatives, a similar behaviour was observed because the hydrogenolysis of the carbon–halogen bond is enhanced by amino substitution in the aromatic ring.20c
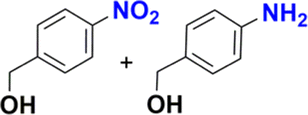
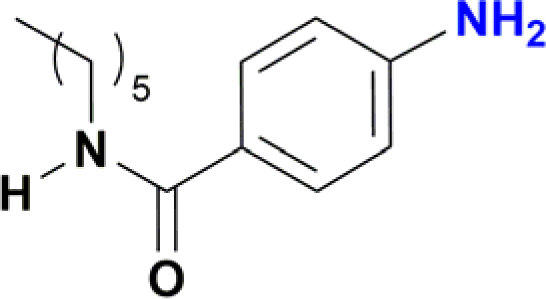
The reduction of nitroarenes in the presence of ester, amide and ciano groups was also performed. For example, ethyl-4-nitrobenzoate¶ afforded the product 4-amino-benzyl-alcohol with 57% yield (Table 2, entry 11), the remaining 43% corresponds to the reduction of only the ester group, with the –NO2 remaining unaffected (Fig. S157–S159†). In this instance both functional groups are reduced, contrary to the behaviour reported in a previous work.12d Furthermore, in another related Ru(II) system12b only the –NO2 group is reduced, in the 4-nitrobenzoyl chloride substrate. On the other hand, the reduction of N-hexyl-4-nitrobenzamide|| is chemoselective towards the nitro group, yielding the expected aniline with 97% conversion (Table 2, entry 12 and Fig. S160–S162†). For multiple bond substituted substrates, like 4-nitrobenzonitrile, the reduction did not proceed as expected with only 8% conversion to the corresponding aromatic amine (Table 2, entry 14). Furthermore, the partial hydrogenation of the nitrile bond to imine ensued (30%), with no concomitant reduction of the nitro group, and starting substrate (62%, Fig. S166–S169†).
Unlike the behaviour seen for the ester group (vide supra), longer reaction times resulted in additional products observed by GC-MS, including the formation of the amide derivative, but as previously observed without the reduction of the nitro group (Fig. S170†). Finally, the nitro group of 4-(2-fluoro-4-nitrophenyl)morpholine, an important pharmaceutical intermediate for the synthesis of the antibiotic Linezolid, was reduced with 97% yield (Table 2, entry 13 and Fig. S163†).
Mechanism insights studies
To determine a plausible mechanism for the reduction of nitroarenes, and ascertain the active species, several experiments were performed. Previous work showed that ruthenium metal hydrides behave as active species in the reduction of nitroarenes under homogeneous catalytic conditions.12a–c,f Therefore, the formation of hydrides was explored in alcohols with and without NaBH4, as well as in the presence of KOH. For instance, we found that an orange solution of 5 in methanol changed its color, slowly over time (∼48 h), to give a red-dark solution, which was analysed by 1H NMR. Interestingly, no traces of hydrides were detected; instead, a new organometallic species was found (12), which was formulated as the coordination isomer of 5 (Scheme 3). Furthermore, complex 6 dissolved in methanol behaves as 5, giving the coordination isomer 13 (Scheme 3).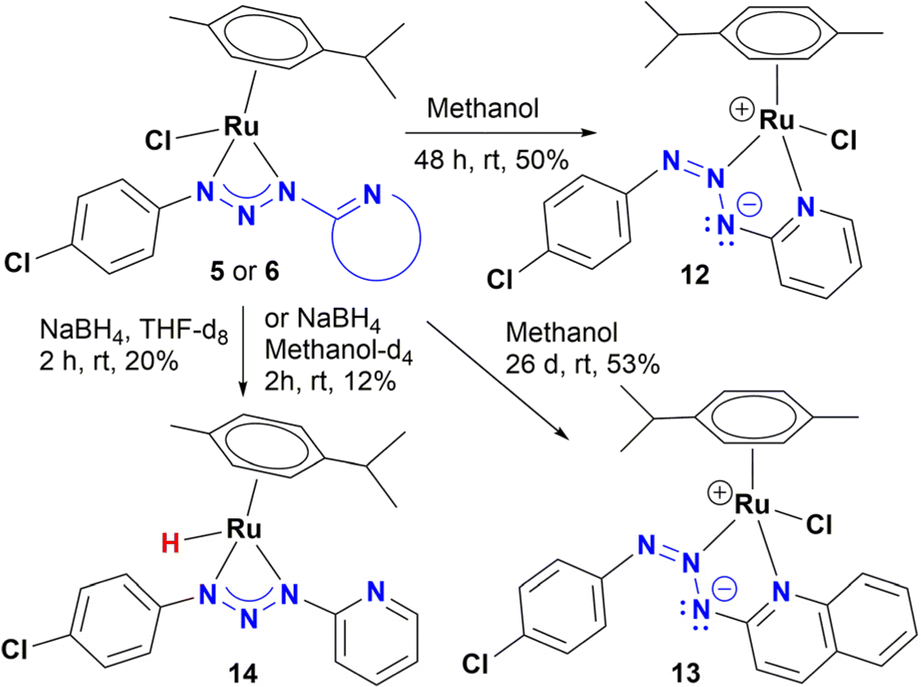 | ||
| Scheme 3 Exploration of ruthenium hydride formation. | ||
Compounds 12 and 13 were fully characterized by NMR, MS and X-ray diffraction analysis. The crystalline structure of both complexes shows the coordination of the heterocyclic moiety to give a five-membered chelate where the central nitrogen of the triazenide system coordinates to the ruthenium metal ion (Fig. 4 and S98†).
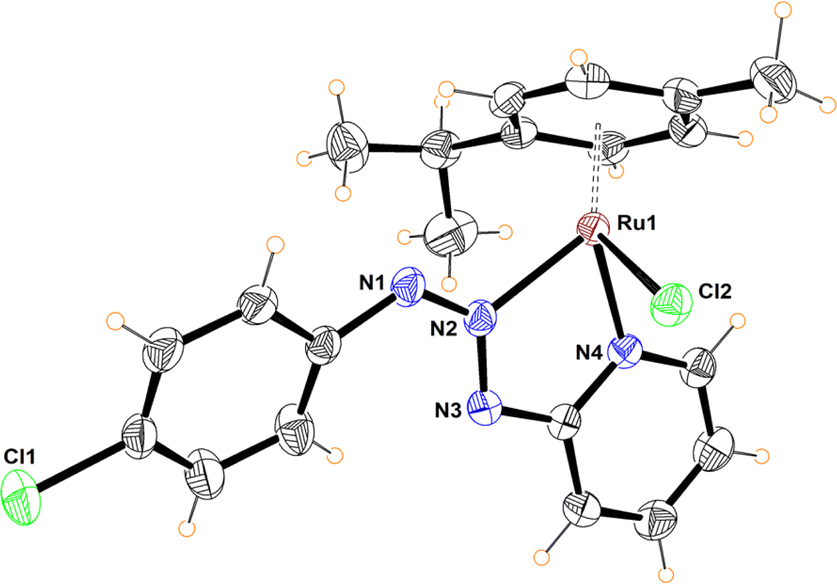 | ||
| Fig. 4 Molecular structure of complex 12. Selected bond distances (Å): N(1)–N(2), 1.285(4); N(2)–N(3), 1.337(3); Ru(1)–N(2), 2.062(3); Ru(1)–N(4), 2.060(2); Ru(1)–Cl(2), 2.4063(8). Selected bond angles (°): N(2)–Ru(1)–N(4), 75.8(1); N(2)–Ru(1)–Cl(2), 83.97(7); N(4)–Ru(1)–Cl(2), 84.56(7). N(2)–N(3)–N(4), 122.3(2). | ||
On the other hand, addition of four equivalents of NaBH4 to a solution of 5 in methanol-d4 gave a mixture of four organometallic species along unreacted complex 5 (ratio 52, 17, 15, 4, and 12%). Two of them were identified as Ru-hydrides with NMR signals at −3.04 and −5.64 ppm (Fig. S177–S178†). The NMR data of the most abundant Ru-hydride (−3.04 ppm) is consistent with the proposed structure (14) shown in Scheme 3, being the least abundant Ru-hydride a coordination isomer of 14. In contrast, if the reaction is carried out in THF-d8, only one Ru-hydride (NMR signal at −2.91 ppm, Fig. S179†) is observed together with the unreacted complex 5 (ratio of 20 to 80%, respectively). Another control experiment carried out in ethanol and KOH (10%) showed that 5 did not catalyse the reduction of nitrotoluene (Table S7†). Moreover, the equimolar reaction of 5 in either methanol, ethanol or 2-propanol did not yield 14. Based on these experiments, the mechanism shown in Scheme 4 is proposed. The catalytic cycle may include participation of the hydride 14, which has been previously synthesized and detected by NMR (Scheme 4). Thus, coordination of nitrobenzene gives species A, possibly by decoordination of one nitrogen of the triazenide ligand, which has proven to be a labile ligand in the isomerization of 5 to 12 in alcohols. Then, species A could transform into B by a hydride migration to nitrogen. This is a reaction that is probably assisted by the movement of a pair of electrons of the nitroarene as well as hydrogen donation from the solvent. Finally, elimination of water will release nitrosobenzene and allow species B to react with NaBH4 to give 14.
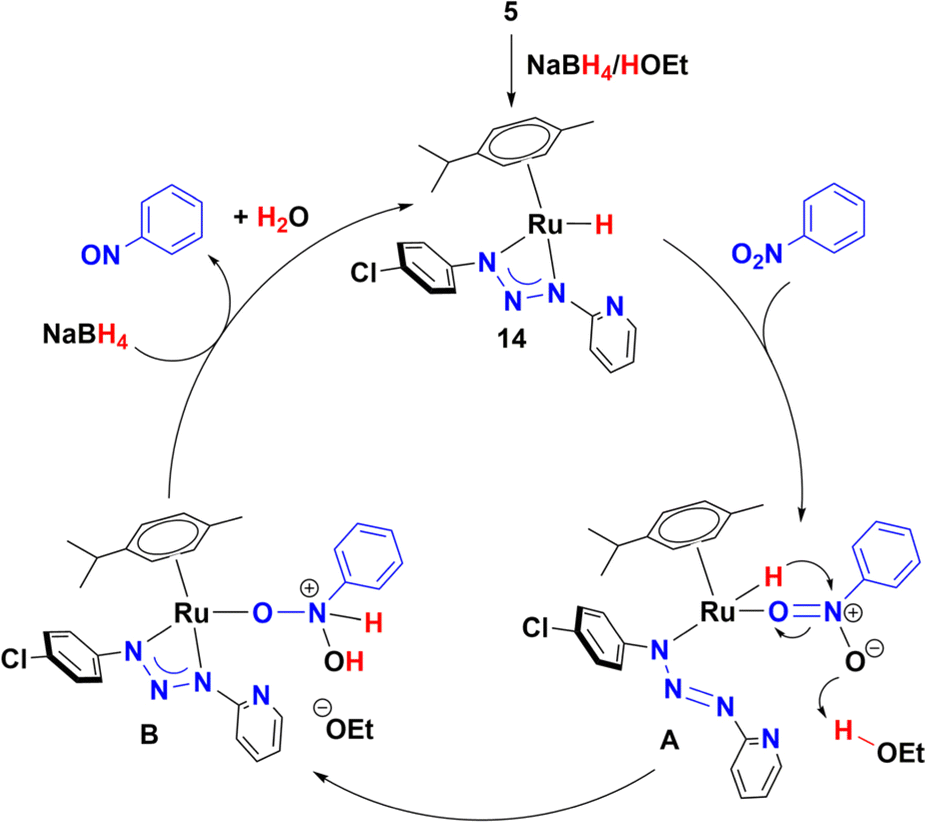 | ||
| Scheme 4 Proposed mechanism for reduction of nitrobenzene. | ||
Nitrosobenzene could be hydrogenated in a similar way to give N-phenylhydroxylamine, both compounds have been proposed as potential intermediates in a condensation mechanism proposed by Haber.4b As noted in Table 1 (entry 13), azobenzene was detected by GC-MS under catalytic conditions (Fig. S112 and S114†). Therefore, azobenzene and azoxybenzene** could be the products of the condensation of nitrosobenzene and N-phenylhydroxylamine, which is consistent with a Haber mechanism (Scheme 5).
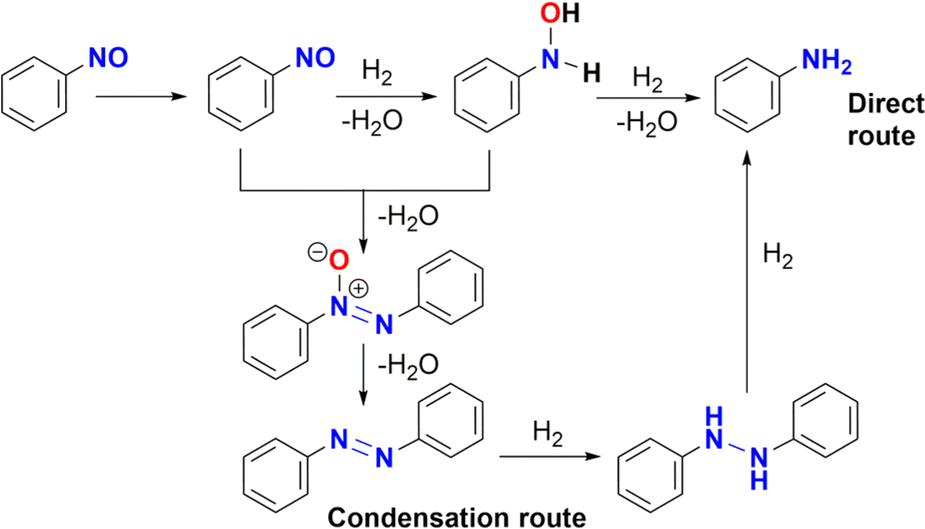 | ||
| Scheme 5 Haber mechanism for hydrogenation of nitrobenzene. | ||
Finally, the reutilization of the catalytic system was tested by addition of more substrate once the catalysis was completed in the first run (>99% yield, using 4-nitrotoluene as substrate). After the second addition of 4-nitrotoluene, no hydrogenation ensued, while the subsequent incorporation of four equivalents of NaBH4 to the reaction mixture resulted in >99% yield; one additional run afforded the same result (Chart S1†). These results mean that the catalytic system requires the subsequent addition of NaBH4 in order to be active, and can be reutilized up to three times.
In summary, the use of polar solvents like methanol or ethanol favours the isomerization of complexes 5 and 6 to their coordination isomers 12 and 13, respectively. Moreover, the addition of NaBH4 is crucial for both, catalysis and the formation of the Ru-hydride 14. Interestingly, under the same catalysis conditions the coordination isomer 12 was as effective as 5, giving the hydrogenation product of nitrobenzene with >99% yield.
Conclusions
Four air-stable and easily handled Ru(II) complexes of triazenide ligands bearing different heterocyclic moieties were prepared and fully characterized by spectroscopic and spectrometric techniques. In complex 7, ligand 3 coordinates to the metal through the central nitrogen of the triazenide system and the nitrogen of the imidazole forming a five-membered chelate ring. In compounds 5, 6, and 8 a bidentate four-membered chelate ring was obtained where the ligand coordinates through the terminal nitrogen atoms of the triazenide system regardless of the heterocyclic moiety. However, a change in the coordination mode of the triazenide ligand is observed when complexes 5 and 6 were dissolved in alcohols giving the coordination isomers 12 and 13. In these complexes the triazenide ligand coordinates to the metal ion through the central nitrogen and the heterocyclic moiety forming a five-membered chelate. The molecular structures of complexes 5, 6, 7, 12, and 13 were confirmed by X-ray diffraction analysis. Complexes 5–8 were tested in the catalytic reduction of nitroarenes. All complexes were found to be highly active in the reduction of nitrobenzene to aniline. Moreover, complex 5 bearing a pyridine ring, displayed the best catalytic performance with structurally diverse substrates and a wide functional group tolerance to afford the desired aromatic amines in good to excellent yields under mild conditions. The introduction of an N-heterocycle in the structure of the ligand seems to improve the catalytic activity of the complexes, since higher yields of products can be obtained compared to the tested controls. Studies are underway to further extend the scope of substrates and to the use of greener methodologies.Experimental
Materials and methods
Unless otherwise specified, all manipulations were carried out in an argon-filled glovebox or using Schlenk techniques. Commercially available reagents were used as received from Aldrich Chemical Co. and Alfa Aesar. [RuCl2(p-cymene)]2 was prepared by published procedures;21 1-[(4-methyl)phenyl]-3-[(4-tert-butyl-2-methyl)imidazole-1-yl]triazene (3), 1-[(4-methyl)phenyl]-3-[(2-methyl)imidazole-1-yl]triazene (4), and [Ru{1,3-bis(4′-methyl phenyl)triazenide}(Cl)(p-cymene)] (11) were previously synthesized by our group.13b,d The preparation and characterization of [RuCl(ClPh-triazenide-Ph)(p-cymene)] (10) is given in the experimental and ESI.† DMSO-d6, (CD3)2CO, CDCl3 and CD2Cl2 were used as received from CIL. NMR spectra were recorded at 400 MHz with Bruker Avance III spectrometer at 30 °C unless otherwise specified. 1H and 13C NMR chemical shifts are reported in ppm referenced to residual solvent resonances (1H NMR: 7.24 for CHCl3 in CDCl3, 5.32 for CHDCl2 in CD2Cl2, 2.50 for DMSO in DMSO-d6, 2.05 for acetone-d5 in acetone-d6; 13C {1H} NMR: 77.23, 54.0, 39.51 and 29.92 for CDCl3, CD2Cl2, DMSO-d6, and acetone-d6, respectively). Coupling constants J are given in Hertz (Hz). FT-IR spectra were recorded on a PerkinElmer FT-IR 1605 spectrophotometer (ATR mode). Melting points were measured in an Electrothermal GAC 88629 apparatus. Mass spectra were obtained by direct insertion on an Agilent Technologies 5975C instrument. High Resolution Mass Spectrometry (HRMS) data were obtained in a micrOTOF-Q III MS instrument with electrospray ionization using sodium formate as calibrant. A complete set of NMR and HRMS spectra has been provided in the ESI,† as evidence of bulk purity and identity. X-ray diffraction data for crystals of compounds 5, 6, 7, 12, and 13 were collected on an Agilent Technologies Supernova AtlasS2 and a Bruker APEX II CCD diffractometers.Synthetic procedures
General procedure for the catalytic reduction of nitroarenes
A standard 20 mL vial was charged with a solution of Ru catalyst (0.003 mmol) in ethanol (2 mL), the appropriate nitroarene (0.3 mmol) and NaBH4 (0.045 g, 1.2 mmol). The reaction mixture was stirred for 30 min at room temperature. The reaction was quenched with water (2 mL), and the product was extracted with ethyl acetate (3 × 5 mL). The solvent was removed under vacuum, and the obtained residue was dissolved in methanol and analysed by CG-MS. The analytical GC-MS system used was an Agilent 7890A GC coupled to 5975C Mass detector Agilent Technologies, equipped with either a HP-5MS capillary column (30 m × 0.25 mm × 0.25 micron) or a DB-5MS UI capillary column (30 m × 0.25 mm × 0.25 micron) Agilent Technologies, Inc. An Agilent Technologies 7693 auto sampler was used to inject 1 μL of a sample solution. The ionization energy was 70 eV with a mass range of 30 to 800 m/z. The injector temperature was set at 250 °C and the detector to 230 °C. The flow rate of the carrier gas (helium) was 1.0 mL min−1 injected with a gas dilution of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50. Identification of the individual components was based on comparison with the mass spectra library (NIST08). For substrates nitrobenzene, nitroaniline, nitrophenol and nitrobenzaldehyde the initial column temperature was set at 80 °C, held for 2 min, followed by a ramp of 15 °C min−1 to 250 °C. For all other substrates, the initial column temperature was set at 80 °C, held for 2 min, followed by a ramp of 10 °C min−1 to 250 °C. Catalytic reactions of entries 9 and 10 in Table 2 were processed according to the general method but with a modification. In these cases, the crude solid obtained after removal of ethanol and water was purified by column chromatography using dichloromethane–methanol (80:20) as mobile phase. Removal of solvents under vacuum affords the expected amines. Product of entry 9 is 4-amino-2-(hydroxymethyl)-6-methoxyphenol, brown solid (40 mg, 79%). 1H NMR (400.1 MHz, DMSO-d6): δ 7.74 (d, 4JHH = 4.0 Hz, 1H, Ar), 7.29 (d, 4JHH = 4 Hz, 1H, Ar), 5.00 (br s, CH2OH, 1H), 4.34 (s, 2H, CH2OH), 3.65 (s, 3H, OCH3) ppm. Product of entry 10 is 4-amino-2-methoxyphenol, orange solid (33.3 mg, 80%). 1H NMR (400.1 MHz, DMSO-d6): δ 7.63 (d, 3JHH = 8.0 Hz, 1H, Ar), 7.28 (s, 1H, Ar), 5.97 (d, 3JHH = 8.0 Hz, 1H, Ar), 3.64 (s, 3H, OCH3) ppm.
:50. Identification of the individual components was based on comparison with the mass spectra library (NIST08). For substrates nitrobenzene, nitroaniline, nitrophenol and nitrobenzaldehyde the initial column temperature was set at 80 °C, held for 2 min, followed by a ramp of 15 °C min−1 to 250 °C. For all other substrates, the initial column temperature was set at 80 °C, held for 2 min, followed by a ramp of 10 °C min−1 to 250 °C. Catalytic reactions of entries 9 and 10 in Table 2 were processed according to the general method but with a modification. In these cases, the crude solid obtained after removal of ethanol and water was purified by column chromatography using dichloromethane–methanol (80:20) as mobile phase. Removal of solvents under vacuum affords the expected amines. Product of entry 9 is 4-amino-2-(hydroxymethyl)-6-methoxyphenol, brown solid (40 mg, 79%). 1H NMR (400.1 MHz, DMSO-d6): δ 7.74 (d, 4JHH = 4.0 Hz, 1H, Ar), 7.29 (d, 4JHH = 4 Hz, 1H, Ar), 5.00 (br s, CH2OH, 1H), 4.34 (s, 2H, CH2OH), 3.65 (s, 3H, OCH3) ppm. Product of entry 10 is 4-amino-2-methoxyphenol, orange solid (33.3 mg, 80%). 1H NMR (400.1 MHz, DMSO-d6): δ 7.63 (d, 3JHH = 8.0 Hz, 1H, Ar), 7.28 (s, 1H, Ar), 5.97 (d, 3JHH = 8.0 Hz, 1H, Ar), 3.64 (s, 3H, OCH3) ppm.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
CRS: investigation, methodology, and visualization. ALI: investigation, methodology, and writing original draft. AVH: investigation. JPCD: investigation. ECA: investigation. JLGL: validation. DC: methodology and formal analysis. AOT: validation. GA: methodology and formal analysis. ALR: methodology and formal analysis. DBG: formal analysis. MPH: formal analysis. VMS: conceptualization, funding acquisition, supervision, writing original draft, reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Tecnológico Nacional de México (TecNM, grants 6185.17P and 14438.22-P) and Consejo Nacional de Ciencia y Tecnología (CONACyT, grant 167005). CRS, AVH, JLGL, and JPCD thank CONACyT for graduate fellowship. We thank CONACyT for ITT NMR, HRMS and X-ray facilities and UNPA HRMS facilities (Grants INFR-2011-3-173395, INFR-2012-01-187686, INFR-2014-224405 and INFR-2015-252013).Notes and references
- A. S. Travis, Manufacture and uses of the anilines: A vast array of processes and products, in The Chemistry of Anilines, ed. Z. Rapport, Patai Series: The Chemistry of Functional Groups, Interscience Wiley, 2007, pp 715–782 Search PubMed.
- (a) G. Booth, Nitro Compounds, Aromatics, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Verlag GmbH & Co. KGaA, Weinheim, 2012, vol. 24, pp. 301–347 Search PubMed; (b) P. F. Vogt, Amines, Aromatic, in Ullmann's Encyclopedia of Industrial Chemistry; Wiley-VCH, Verlag GmbH & Co. KGaA, Weinheim, 2012, vol. 2, pp. 699–716 Search PubMed.
- Z. Wang, Béchamp Reduction, in Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons Inc., 2010, pp. 284–287 Search PubMed.
- (a) A. H. Romero, ChemistrySelect, 2020, 5, 13054–13075 CrossRef CAS; (b) J. Song, Z.-F. Huang, L. Pan, K. Li, X. Zhang, L. Wang and J.-J. Zou, Appl. Catal., B, 2018, 227, 386–408 CrossRef CAS; (c) D. Formenti, F. Ferretti, F. K. Scharnagl and M. Beller, Chem. Rev., 2019, 119, 2611–2680 CrossRef CAS PubMed; (d) M. Orlandi, D. Brenna, R. Harms, S. Jost and M. Benaglia, Org. Process Res. Dev., 2018, 22, 430–445 CrossRef CAS; (e) H. Göksu, H. Sert, B. Kilbas and F. Sen, Curr. Org. Chem., 2017, 21, 794–820 CrossRef; (f) H. K. Kadam and S. G. Tilve, RSC Adv., 2015, 5, 83391–83407 RSC; (g) H.-U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 CrossRef CAS.
- (a) J. Wu and C. Darcel, J. Org. Chem., 2021, 86, 1023–1026 CrossRef CAS PubMed; (b) G. Wienhofer, M. Baseda-Kruger, C. Ziebart, F. A. Westerhaus, W. Baumann, R. Jackstell, K. Junge and M. Beller, Chem. Commun., 2013, 49, 9089–9091 RSC; (c) R. M. Deshpande, A. N. Mahajan, M. M. Diwakar, P. S. Ozarde and R. V. Chaudhari, J. Org. Chem., 2004, 69, 4835–4838 CrossRef CAS PubMed.
- (a) D. I. Ioannou, D. K. Gioftsidou, V. E. Tsina, M. G. Kallitsakis, A. G. Hatzidimitriou, M. A. Terzidis, P. A. Angaridis and I. N. Lykakis, J. Org. Chem., 2021, 86, 2895–2906 CrossRef CAS PubMed; (b) S. Kumar and R. Gupta, ChemistrySelect, 2017, 2, 8197–8206 CrossRef CAS; (c) C.-G. Chao and D. E. Bergbreiter, Catal. Commun., 2016, 77, 89–93 CrossRef CAS.
- (a) S. Dayan, N. Kayacıb, O. Dayan, N. Özdemir and K. N. Özpozan, Polyhedron, 2020, 175, 114181 CrossRef; (b) R. Lopes, M. Pereira and B. Royo, ChemCatChem, 2017, 9, 3073–3077 CrossRef CAS; (c) G. Vijaykumar and S. K. Mandal, Dalton Trans., 2016, 45, 7421–7426 RSC.
- (a) S. Liu, J. I. Amaro-Estrada, M. Baltrun, I. Douair, R. Schoch, L. Maron and S. Hohloch, Organometallics, 2021, 40, 107–118 CrossRef CAS; (b) I. Sorribes, G. Wienhöfer, C. Vicent, K. Junge, R. Llusar and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 7794–7798 CrossRef CAS PubMed.
- V. Zubar, A. Dewanji and M. Rueping, Org. Lett., 2021, 23, 2742–2747 CrossRef CAS PubMed.
- (a) N. Zengin, H. Göksu and F. Şen, Chemosphere, 2021, 282, 130887 CrossRef CAS PubMed; (b) S. T. Yang, P. Shen, B. S. Liao, Y. H. Liu, S. M. Peng and S. T. Liu, Organometallics, 2017, 36, 3110–3116 CrossRef CAS; (c) A. Krogul and G. Litwinienko, Org. Process Res. Dev., 2015, 19, 2017–2021 CrossRef CAS; (d) A. Kumar, K. Purkait, S. K. Dey, A. Sarkar and A. Mukherjee, RSC Adv., 2014, 4, 35233–35237 RSC.
- Y. Liu, W. Miao, W. Tang, D. Xue, J. Xiao, C. Wang and C. Li, Chem.–Asian J., 2021, 16, 1725 CrossRef CAS PubMed.
- (a) S. Pachisia, R. Kishan, S. Yadav and R. Gupta, Inorg. Chem., 2021, 60, 2009–2022 CrossRef CAS PubMed; (b) R. Saha, A. Mukherjee and S. Bhattacharya, Eur. J. Inorg. Chem., 2020, 4539–4548 CrossRef CAS; (c) R. Nandhini, B. S. Krishnamoorthy and G. Venkatachalam, J. Organomet. Chem., 2019, 903, 120984 CrossRef; (d) Z.-J. Yao, J.-W. Zhu, N. Lin, X.-C. Qiao and W. Deng, Dalton Trans., 2019, 48, 7158–7166 RSC; (e) S.-C. A. Lin, Y.-H. Liu, S.-M. Peng and S.-T. Liu, Mol. Catal., 2019, 466, 46–51 CrossRef CAS; (f) W.-G. Jia, S. Ling, H.-N. Zhang, E.-H. Sheng and R. Lee, Organometallics, 2018, 37, 40–47 CrossRef CAS; (g) W.-G. Jia, Z.-B. Wang, X.-T. Zhi, J.-Q. Han and Y. Sun, J. Coord. Chem., 2017, 70, 848–858 CrossRef CAS; (h) W.-G. Jia, T. Zhang, D. Xie, Q.-T. Xu, S. Ling and Q. Zhang, Dalton Trans., 2016, 45, 14230–14237 RSC; (i) W.-G. Jia, H. Zhang, T. Zhang, D. Xie, S. Ling and E.-H. Sheng, Organometallics, 2016, 35, 503–512 CrossRef CAS; (j) S. Hohloch, L. Suntrup and B. Sarkar, Organometallics, 2013, 32(24), 7376–7385 CrossRef CAS; (k) A. A. Deshmukh, A. K. Prashar, A. K. Kinage, R. Kumar and R. Meijboom, Ind. Eng. Chem. Res., 2010, 49, 12180–12184 CrossRef CAS.
- (a) L. J. Medrano-Castillo, M. A. Collazo-Flores, J. P. Camarena-Díaz, E. Correa-Ayala, D. Chávez, D. B. Grotjahn, A. L. Rheingold, V. Miranda-Soto and M. Parra-Hake, Inorg. Chim. Acta, 2020, 507, 119551 CrossRef CAS; (b) J. P. Camarena-Díaz, A. L. Iglesias, D. Chávez, G. Aguirre, D. B. Grotjahn, A. L. Rheingold, M. Parra-Hake and V. Miranda-Soto, Organometallics, 2019, 38, 844–851 CrossRef; (c) E. Correa-Ayala, C. Campos-Alvarado, D. Chávez, D. Morales-Morales, S. Hernández-Ortega, J. J. García, M. Flores-Álamo, V. Miranda-Soto and M. Parra-Hake, Inorg. Chim. Acta, 2017, 466, 510–519 CrossRef CAS; (d) E. Correa-Ayala, A. Valle-Delgado, G. Ríos-Moreno, D. Chávez, D. Morales-Morales, S. Hernández-Ortega, J. J. García, M. Flores-Álamo, V. Miranda-Soto and M. Parra-Hake, Inorg. Chim. Acta, 2016, 446, 161–168 CrossRef CAS.
- (a) A. E. Chichibábin and R. L. Persitz, J. Russ. Phys.–Chem. Soc., 1925, 57, 301–304 Search PubMed; (b) G. Vernin, C. Siv, J. Metzger and C. Párkányi, Synthesis, 1977, 10, 691–693 CrossRef.
- A. Messmer, A. Gelléri and G. Hajós, Tetrahedron, 1986, 42, 4827–4836 CrossRef CAS.
- (a) D. Lee, P. Perez, W. Jackson, T. Chin, M. Galbreath, F. R. Fronczek, R. Isovitsch and D. S. Limoto, Bioorg. Med. Chem. Lett., 2016, 26, 3243–3247 CrossRef CAS PubMed; (b) M. F. Ibárra-Vázquez, S. A. Cortes-Llamas, A. A. Peregrina-Lucano, J. G. Alvarado-Rodríguez, R. Marríquez-González, F. A. López-Dellamary, M. I. Moreno-Brambila and I. I. Rangel-Salas, Inorg. Chim. Acta, 2016, 451, 209–215 CrossRef; (c) C. GuhaRoy, R. J. Butcher and S. Bhattacharya, J. Organomet. Chem., 2008, 693, 3923–3931 CrossRef CAS; (d) S. Antoniutti, M. Bedin, J. Castro, S. García-Fontana and G. Albertin, Inorg. Chem., 2006, 45, 3816–3825 CrossRef PubMed.
- (a) Y. Hu, L. Li, A. P. Shaw, J. R. Norton, W. Sattler and Y. Rong, Organometallics, 2012, 31, 5058–5064 CrossRef CAS; (b) K. Gray, M. J. Page, J. Wagler and B. A. Messerle, Organometallics, 2012, 31, 6270–6277 CrossRef CAS.
- (a) J. Vajs, I. Steiner, A. Brozovic, A. Pevec, A. Ambriović-Ristov, M. Matković, I. Piantanida, D. Urankar, M. Osmak and J. Košmrlj, J. Inorg. Biochem., 2015, 153, 42–48 CrossRef CAS PubMed; (b) G. Albertin, S. Antoniutti, J. Castro and S. Paganelli, J. Organomet. Chem., 2010, 695, 2142–2152 CrossRef CAS; (c) M. C. Carrión, F. Sepúlveda, F. A. Jalón, B. R. Manzano and A. M. Rodríguez, Organometallics, 2009, 28, 3822–3833 CrossRef.
- P. Bhaskar, K. Chakrabarti, S. Shee, M. Maji, A. Mishra and S. Kundu, RSC Adv., 2016, 6, 100532–100545 RSC.
- (a) W. Jia, T. Du, L. Gao and J. Du, Appl. Organomet. Chem., 2016, 34, e5651 CrossRef; (b) J. Wei-Guo, C. Ming-Xia, X. Qiu-Tong, G. Li-Li and Y. Guozan, Polyhedron, 2018, 153, 69–75 CrossRef; (c) M. Pietrowski, Curr. Org. Synth., 2012, 9, 470–487 CrossRef CAS.
- M. A. Bennett, T. N. Huang, T. W. Matheson, A. K. Smith, S. Ittel and W. Nickerson, Inorg. Synth., 1982, 21, 74 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2087352 (5), 2087431 (6), 2087433 (7), 2284161 (12), 2284162 (13). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra04813j |
| ‡ These authors contributed equally to the work. |
| § Deceased. |
| ¶ Ethyl-4-nitrobenzoate (Table 2, entry 11) was generated in situ from the reaction of p-nitrobenzoyl chloride with ethanol. |
| || N-Hexyl-4-nitrobenzamide (Table 2, entry 12) was generated in situ from the reaction of p-nitrobenzoyl chloride with hexylamine. |
| ** Azoxybenzene was not detected. However, their analogues azoxytoluene and azotoluene were detected in the reduction of nitrotoluene (Fig. S171 and S174†). |
| This journal is © The Royal Society of Chemistry 2024 |