
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, anticancer and in silico assessment of 8-caffeinyl chalcone hybrid conjugates†
Mohammad Navid Soltani Rad *ab,
Somayeh Behrouz*ab,
Maedeh Charbaghia,
Marzieh Behrouza,
Elham Zarenezhadc and
Ali Ghanbariasadd
*ab,
Somayeh Behrouz*ab,
Maedeh Charbaghia,
Marzieh Behrouza,
Elham Zarenezhadc and
Ali Ghanbariasadd
aDepartment of Chemistry, Shiraz University of Technology, Shiraz, 71555-313, Iran. E-mail: soltani@sutech.ac.ir; behrouz@sutech.ac.ir; Fax: +98-71-3735-4520; Tel: +98-71-3735-4500
bMedicinal Chemistry Research Laboratory, Novel Technology for Health Research Center, Shiraz University of Technology, Shiraz, 71555-313, Iran
cNon-communicable Diseases Research Center, Fasa University of Medical Sciences, Fasa, Iran
dDepartment of Medical Biotechnology, School of Medicine, Fasa University of Medical Sciences, Fasa, Iran
First published on 22nd August 2024
Abstract
In this paper, we report the design, synthesis, and characterization of novel 8-caffeinyl chalcone hybrid conjugates, which were studied for their anticancer properties, toxicity, and in silico behavior. The synthesized compounds consist of 8-caffeinyl and chalcone structures with diverse substituents. The synthesis involved three main stages: bromination of caffeine to produce 8-BC, synthesis of chalcones, and subsequent coupling of these chalcones with 8-BC. The anticancer activity of the resulting compounds was evaluated in vitro against breast cancer MCF-7 and melanoma A-375 cell lines, revealing certain compounds to have significant efficacy compared to the reference drug methotrexate. Toxicity assessments using a healthy cell line indicated that most compounds displayed some level of toxicity, with only a few exceptions. Molecular docking studies indicated robust binding affinities of selected compounds to B-RAF kinase and hDHFR enzymes. In silico analyses of pharmacokinetic and physicochemical properties demonstrated that the majority of the compounds adhered to Lipinski's rule of five. Furthermore, density functional theory (DFT) studies were performed to gain deeper insights into the properties of the intermediates used throughout the research.
1. Introduction
Cancer, the second leading cause of death globally following cardiovascular disease, is a complex condition characterized by the uncontrolled proliferation of abnormal cells in the body.1 In 2017 alone, it resulted in over 9.6 million deaths worldwide, with projections indicating that global cancer-related mortality could reach 22 million by 2030.2 Despite significant advancements in understanding cancer development and treatment in recent years, the disease remains challenging to manage due to factors such as tumor heterogeneity, genetic mutations, and its ability to affect various cells and organs.3 Cancer is a dynamic ailment that evolves over time, accumulating new mutations, making clinical management a persistent challenge in the 21st century. Treatment options include radiation therapy, surgery, chemotherapy, and proton therapy, with the choice of therapy tailored to the cancer type and stage.4 While surgery is typically the primary treatment, advanced stages often require a combination of radiotherapy and chemotherapy. However, these treatments can cause significant damage to surrounding healthy tissues, leading to long-term complications such as neurotoxicity, cardiotoxicity, and infertility. Furthermore, chemotherapeutic agents have been associated with various drawbacks such as lack of selectivity, low efficacy, high prices, inconsistency, and other issues. Hence, efforts are ongoing to develop more effective and less toxic drugs to address the limitations associated with current cancer treatments.5Chalcones, specifically 1,3-diphenylprop-2-en-1-one, are a type of open-chain flavonoid featuring an α,β-unsaturated carbonyl group. They serve as fundamental structures in various natural compounds and play a vital role as precursors for the biosynthesis of flavonoids and isoflavonoids from both natural and synthetic sources within the flavonoid family.6 These compounds come in trans and cis isomeric forms, with the trans isomer being more thermodynamically stable. They are predominantly found in plant species like Angelica, Glycyrrhiza, Humulus, and Scutellaria, which have a history of use in traditional folk remedies.7 Many derivatives of chalcones have been created and approved for medicinal use. Researchers have been intrigued by chalcones due to their simple chemistry, ease of synthesis, and the ability to produce a wide range of derivatives by replacing multiple hydrogen atoms.8 Because of their abundance in plants and ease of production, these compounds have attracted significant attention for potential therapeutic applications. Numerous chalcone derivatives have been synthesized in laboratories, showcasing diverse biological activities such as anti-diabetic, anti-neoplastic, antihypertensive, anti-retroviral, anti-inflammatory, and many others (Fig. 1).9 In recent years, several comprehensive reviews have emphasized the biological importance of chalcone derivatives, particularly those containing N-heterocyclic compounds known for their exceptional biological properties.10
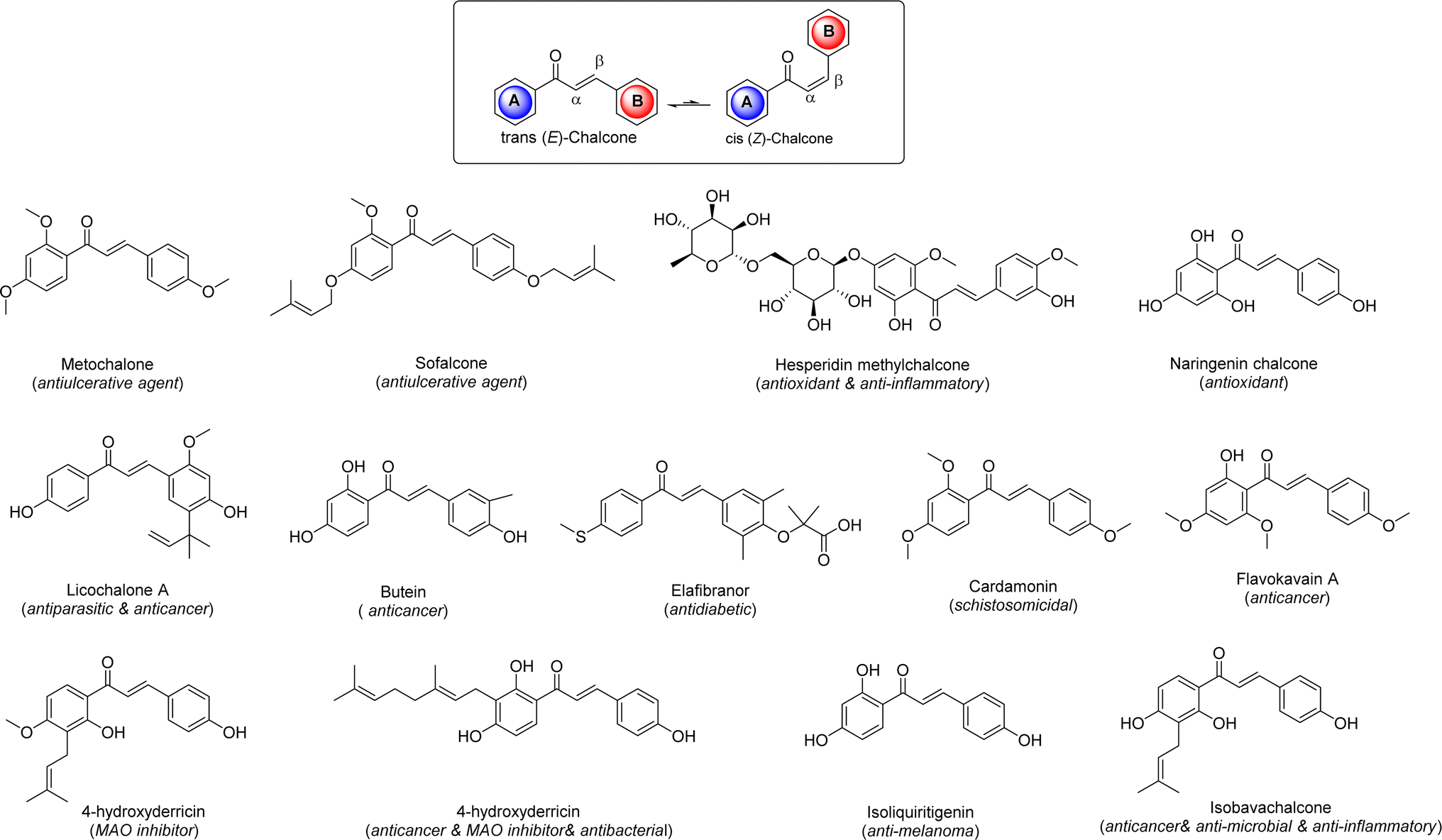 | ||
| Fig. 1 The structures of chalcone and chalcone scaffold-based drugs. | ||
Xanthine alkaloids are a group of compounds with notable significance due to their wide-ranging biological activities and pharmacological effects.11 These alkaloids, including caffeine, theophylline, paraxanthine, and theobromine (Fig. 2), are found in various plants such as coffee, tea, and cacao. They act as central nervous system stimulants, diuretics, and bronchodilators, making them commonly used in medicine and as psychoactive substances. Xanthine alkaloids also play a crucial role in plant defense mechanisms and have antioxidant properties. Their impact on human health and their diverse physiological effects makes xanthine alkaloids a subject of significant scientific interest and research.12
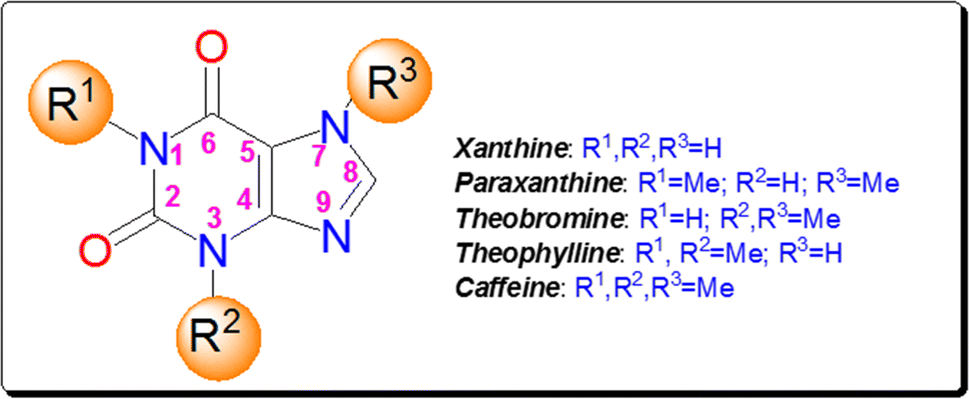 | ||
| Fig. 2 The structures of xanthine and its natural methylated derivatives. | ||
Caffeine stands out as the most well-known and widely used natural xanthine alkaloid in human society, with established biological properties.13 Unlike other N-methyl xanthines, caffeine can be uniquely modified at its C8 site by a select few electrophiles or active species.14 Notably, functionalizing the C8 position of caffeine leads to significant changes in its biological characteristics, offering a range of medicinal properties as illustrated in Fig. 3. A recent study has classified C8-modified caffeine derivatives into C8–C, C8–O, C8–S, and C8–N based on the type of atom linked to C8.15 Among these derivatives, C8–O analogues exhibit remarkable biological activities, including anticancer, antimicrobial, analgesic, MAO and adenosine inhibitors.16–18 Contrary to C8–C, C8–S, and C8–N modified derivatives, the synthesis of C8–O analogues has been rare. Fig. 4 showcases the structures and biological activities of some C8–O derivatives 1–3.
 | ||
| Fig. 3 The biological profiles obtained by the C8 modification of caffeine. | ||
 | ||
| Fig. 4 The structure of some C8–O caffeine derivatives and their corresponding biological activity. | ||
A hybrid conjugate compound is an artificial combination of two or more pharmacophores that fall into a known category of compounds for producing the desired effect. Hybrid conjugates play a crucial role in various fields, including chemistry, biology, and medicine.19 These molecules combine different properties and functionalities, resulting in enhanced performance and versatility. These synthetic structures have the potential to generate novel chemotherapeutic agents that enhance efficacy and activity, improve binding affinity, extensively improve drug delivery, and reduce side effects.20 Through scientific innovation, molecular hybrids have been developed by linking different biologically active agents together with the goal of preserving the pharmacological effects of each component. In drug development, they can create novel drugs with unique properties. The ability to tailor hybrid conjugates to specific applications makes them valuable tools for researchers and developers seeking innovative solutions to complex challenges.21
Recently, we reported the synthesis of 8-caffeinyl-triazolylmethoxy hybrid conjugates 3 (Fig. 4),15 which are C8–O caffeine derivatives exhibiting significant anticancer properties. In pursuit of our ongoing research on synthesis and biological assessments of 8-caffeinyl-hybrid conjugate compounds,22–24 hereby, we present our latest work on the synthesis, evaluation of anticancer activity, and in silico analysis of novel 8-caffeinyl chalcone hybrid conjugates, the general structures of which are illustrated in Fig. 5.
 | ||
| Fig. 5 General structure of novel 8-caffeinyl chalcone hybrid conjugates. | ||
2. Results and discussion
2.1 Chemistry
For the synthesis of the title compounds, 8-BC was primarily synthesized using the standard procedure.22 Subsequently, ketone 6 and/or aldehyde 7 were prepared by reacting 4-hydroxy acetophenone and/or 4-hydroxy benzaldehyde with 8-BC using K2CO3 as a base in anhydrous DMF (Scheme 1). An attempt was made to couple ketone 6 and/or aldehyde 7 with desired aromatic aldehydes and/or aromatic methylketones using NaOH as a base in EtOH (90%) under reflux conditions (Scheme 1). Contrary to our expectations, products 4 and/or 5 were not obtained. Surprisingly, only 1,3,7-trimethyluric acid (8) was obtained as a side product. The question arises: how is it possible? It is believed that due to the electron-withdrawing effect of the carbonyl group in both structures 6 and 7, the charge density on the oxygen atom connected to the caffeinyl group is transferred to the carbonyl group through a mesomeric effect as shown in Scheme 2. This resonance effect leads to an increase in the positive charge density on C8, making it susceptible to attack by external nucleophiles such as OH− through an SNAr-type reaction, as illustrated in Scheme 2. After unsuccessful attempts to synthesize the desired compounds using the aforementioned procedure, the strategy was altered. Two sets of chalcone analogues, 9 and 10, were initially prepared using either the standard Claisen–Schmidt25 or acid-catalyzed aldol condensation methods,26 respectively (Scheme 3). In this process, 4-hydroxyacetophenone and/or 4-hydroxybenzaldehyde were condensed with aromatic aldehydes and/or aromatic methylketones containing various functional groups, utilizing NaOH in EtOH (90%) under reflux conditions. After synthesizing chalcones 9 and 10, they were combined with 8-BC using K2CO3 as a base in anhydrous DMF to produce the desired 8-caffeinyl chalcone hybrid conjugates 4a–4k and 5a–5f. These conjugates contained both electron-donating and/or electron-withdrawing groups and were obtained in good to excellent yields (Scheme 3).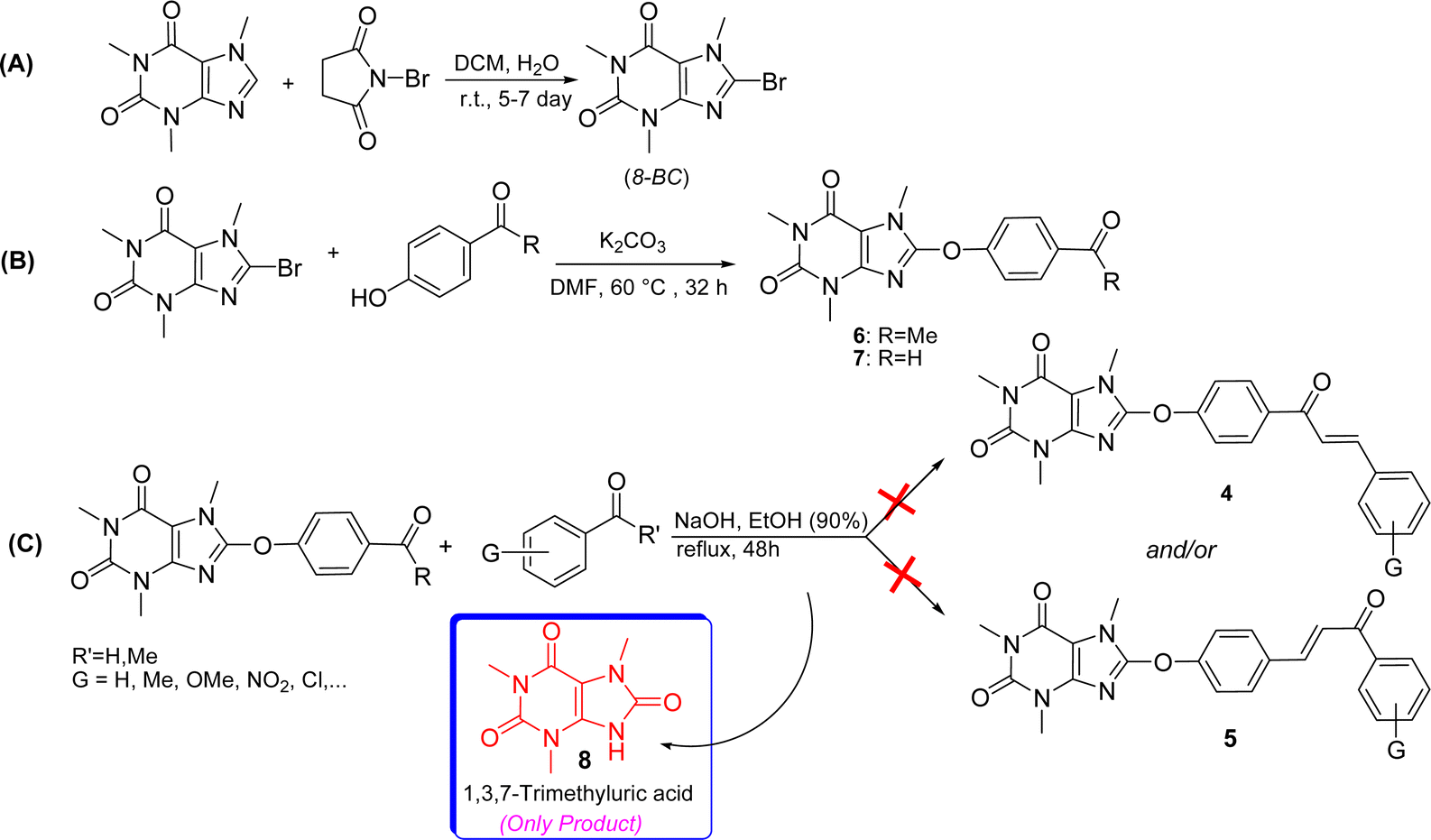 | ||
| Scheme 1 Attempted synthesis of compounds 4 and 5 (A–C) along with the initial generation of 1,3,7-trimethyluric acid. | ||
 | ||
| Scheme 2 A plausible mechanism for generation of 1,3,7-trimethyluric acid. | ||
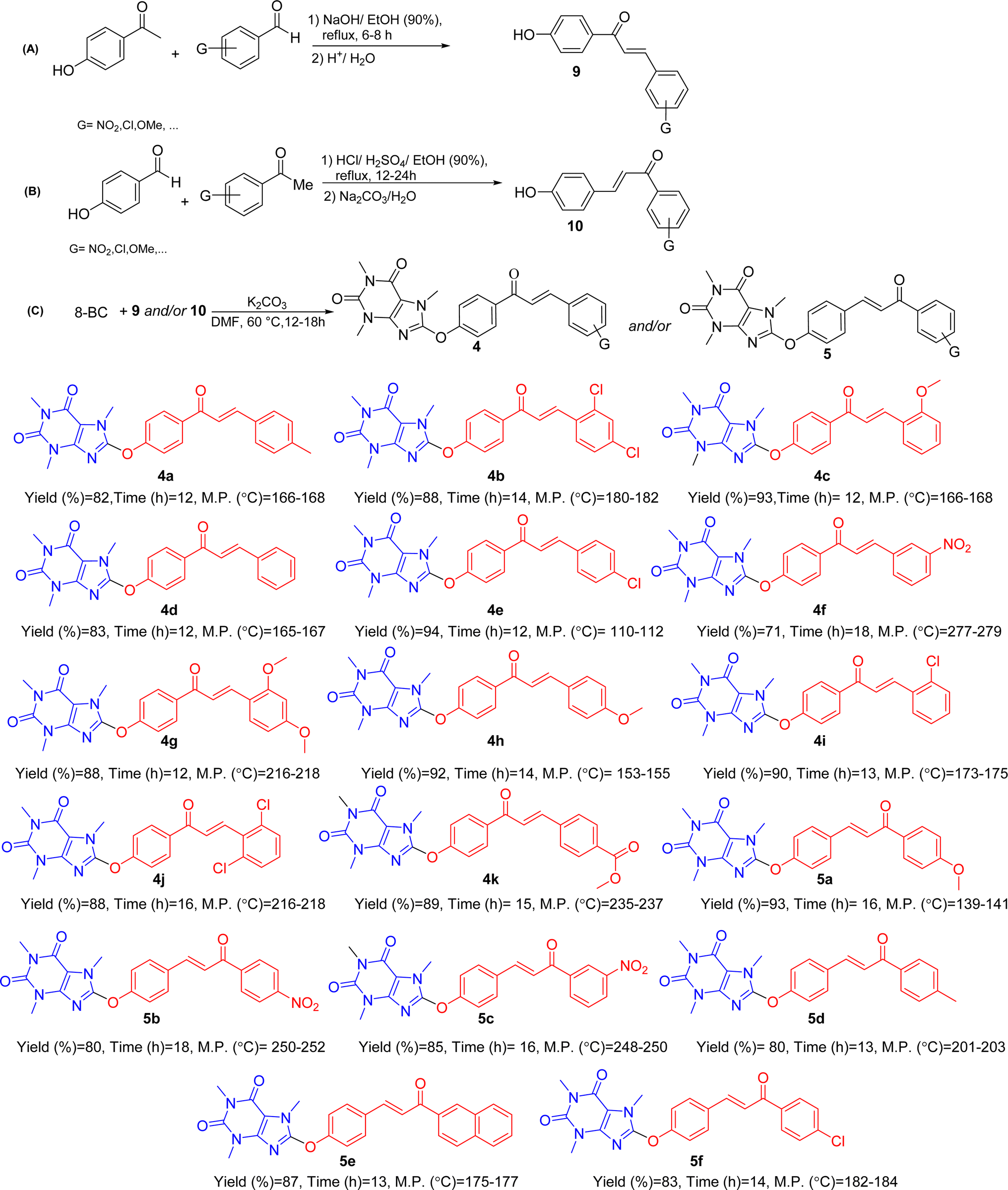 | ||
| Scheme 3 General synthetic pathway (A–C) for preparing novel 8-caffeinyl chalcone hybrid conjugates and the structure of synthesized compounds. | ||
2.2 In vitro anticancer and cytotoxic activity
The A-375 cell line is a well-known human melanoma cell line that originated from a tumor in a 54 year-old woman with malignant melanoma. This cell line is valuable for research in pharmacology, immuno-oncology, and toxicology.27 The MCF-7 cell line, derived from a 69 year-old woman with breast adenocarcinoma in 1973, is among the most widely used human breast cancer cell lines globally. Researchers are drawn to the MCF-7 cell line due to its retention of characteristics resembling those of breast epithelium. It is frequently employed as an in vitro model for studying breast cancer biology. With its various subtypes, this cell line finds extensive applications in developing chemotherapeutic drugs and exploring drug resistance.28 HEK-293, originating from human embryonic kidney cells in 1973, is a normal cell line commonly utilized in fundamental medical and pharmacological research.29 The IC50 values pertaining to toxicity assessments of all synthetic products are outlined in Table 1.| Compound | A375 | MCF-7 | HEK-293 |
|---|---|---|---|
| 4a | 279 ± 2.3 | 38 ± 3.7 | 407 ± 3.1 |
| 4b | 325 ± 3.1 | 230 ± 3.2 | 347 ± 4.2 |
| 4c | 325 ± 3.8 | 269 ± 4.5 | 451 ± 2.4 |
| 4d | 157 ± 4.2 | 18 ± 1.9 | 220 ± 3.4 |
| 4e | 273 ± 3.4 | 35 ± 2.2 | 386 ± 3.2 |
| 4f | >500 | >500 | >500 |
| 4g | 425 ± 3.5 | >500 | >500 |
| 4h | >500 | >500 | >500 |
| 4i | 111 ± 1.9 | 108 ± 3.1 | 162 ± 4.2 |
| 4j | 200 ± 4.3 | 181 ± 3.1 | 214 ± 4.3 |
| 4k | 92 ± 3.2 | 34 ± 3.5 | 99 ± 3 |
| 5a | 249 ± 3.6 | 249 ± 3.2 | >500 |
| 5b | 275 ± 3.7 | 223 ± 3.7 | 419 ± 4.6 |
| 5c | >500 | >500 | >500 |
| 5d | >500 | >500 | >500 |
| 5e | >500 | 333 ± 2.5 | >500 |
| 5f | 368 ± 3.1 | 130 ± 3.4 | 240 ± 3.7 |
| MTX | 418 ± 2.0 | 343 ± 3.6 | 199 ± 2.4 |
Based on the MTT assay results, it was found that, with the exception of compounds 4f, 4h, and 5c–5e, all other entries exhibited stronger anticancer activity compared to the reference drug MTX for the melanoma cancer cell line A375. Among these compounds, 4k (IC50 = 92 ± 3.2) showed the highest potency against the A375 cell line.
In Table 1, it is shown that for the breast cancer cell line MCF-7, the compounds demonstrated greater activity and potency compared to the A375 cell line. For example, compounds 4d (IC50 = 18 ± 1.9), 4e (IC50 = 35 ± 2.2), and 4k (IC50 = 34 ± 3.5) were significantly more potent than MTX, with almost 19, 10, and 10 times greater efficacy, respectively. Except for compounds 4f–4h, 5c, and 5d, all other tested compounds also outperformed MTX against MCF-7.
To assess the toxicity of the compounds, they were tested on the normal cell line HEK-293 (as shown in Table 1). Compounds 4f–4h, 5a, and 5c–5e were found to be non-toxic to HEK-293 cells, while the remaining compounds, including MTX, exhibited varying degrees of toxicity. Among the compounds showing toxicity, 4a–4f, 4j, 5b, and 5f were less toxic than MTX, whereas 4i and 4k were more toxic. Notably, 4k was identified as the most toxic compound, despite displaying the highest potency against all tested cancer cells.
The viability percentages of the examined cell lines at different concentrations (15.62, 31.25, 62.5, 125, 250, and 500 μM) of 4a–4k, 5a–5f and MTX are depicted in Fig. 6A–C. A p-value less than 0.05 (typically ≤0.05) indicates statistical significance, denoted by stars in Fig. 6A–C. Fig. 6A highlights that for A375, the lowest viability values and thus the highest toxicity are associated with 4i and 4k compared to all synthesized compounds at different concentrations. Furthermore, compounds 4a, 4h, and 5f have shown lower viability and therefore higher toxicity compared to MTX. In the breast cancer cell line MCF-7 (Fig. 6B), 4k was found to have the lowest viability rate compared to the other tested compounds and MTX. Additionally, 4a, 4e, 4g–4i, 5d, and 5f were confirmed to exhibit lower viability than MTX, especially at lower concentrations. Overall, the majority of compounds demonstrated reduced viability to some extent at various concentrations. Compound 4f was found to have the lowest toxicity compared to the other compounds tested. Fig. 6C displays the viability percentage of the normal cell line HEK-293. It is evident from the figure that 4k exhibited the highest toxicity and lowest viability among all examined concentrations compared to the other synthesized compounds for HEK-293. Compounds 4i and 5f were found to be more toxic to HEK-293 compared to MTX, particularly at lower concentrations. Generally, MTX showed the highest toxicity against HEK-293 at 500 μM.
 | ||
| Fig. 6 (A) The viability assessment for 4a–4k, 5a–5f and MTX against A375. (B) The viability assessment for 4a–4k, 5a–5f and MTX against MCF-7. (C) The viability assessment for 4a–4k, 5a–5f and MTX against HEK293. | ||
2.3 Plausible mechanism of anticancer activity and SAR analysis
Folic acid, also known as folacin, is a water-soluble vitamin categorized as vitamin B9. It plays a vital role in various biological processes, including the synthesis and modification of DNA and RNA, the production of methionine, and various chemical reactions within cellular metabolism. Folic acid serves as a precursor for the creation of tetrahydrofolate (coenzyme F) through the enzymatic activity of dihydrofolate reductase (DHFR).30 Through a process involving one-carbon transfer metabolism and the enzymatic function of thymidylate synthase (TS), coenzyme F transfers a methyl group to deoxyuridine monophosphate (dUMP), converting it to deoxythymidine monophosphate (dTMP). This conversion is crucial for DNA synthesis and cell replication.31 Inhibiting DHFR can halt the synthesis of coenzyme F, thereby disrupting DNA synthesis and related biochemical processes. Compounds like methotrexate (MTX) and aminopterin share a structural resemblance to folic acid, comprising three molecular components: pteridine, p-aminobenzoic acid (PABA), and glutamic acid. The primary difference lies in the pteridine core, which is responsible for DHFR inhibition.32 The pteridine core, a key pharmacophoric site in folic acid, MTX, and aminopterin, bears a striking resemblance to xanthine alkaloids. As shown in Fig. 7, the compounds have similarities to folic acid, MTX, and aminopterin. In this regard, the caffeinyl moiety can be considered as a successor of pteridine, while part of chalcone in compound 4 series act as PABA's pharmocophores exist in folic acid and its derivatives. Other part of compound 4 series comprising double bond, aromatic ring “B” and its substituents can be consider as a glutamate residue. In particular, 4k involving hydrolyzable ester moiety in para position is the most potent compound that mimics glutamate residue in folic acid and its antimetabolic derivatives. In general, compound 4 series displayed more reactivity than compound 5 series which can be attributed to more structural resemblance of compound 4 series to folate analogues compare to 5. | ||
| Fig. 7 The possible anticancer mechanism of 8-caffeinyl chalcone hybrid conjugates. | ||
Although there is no apparent trend in the SAR of the title compounds, some insights can be gleaned from the types of substituents in both the compound 4 and 5 series. Generally, the presence of mono chloro or dichloro atoms in both series 4 and 5 led to anticancer activity, as seen in 4b, 4e, 4i, 4j, and 5f. The presence of electron-releasing groups like methoxy, either as a mono or di-substituent, produced varying results in both series 4 and 5. While 4c and 5a exhibited anticancer activity, 4g and 4h did not. Similarly, the presence of nitro as an electron-withdrawing moiety had differing outcomes. Compound 5b showed anticancer activity, while 4f and 5c did not. Similarly, the presence of methyl in 4a confirmed anticancer activity but resulted in the opposite outcome in 5d under the same conditions.
2.4 In silico studies
The significant interactions for stabilization of vemurafenib in the active site of B-RAF V600E kinase enzyme include van der Waals, hydrophobic, π–alkyl, π–π stack, π–sigma, π–cation, and hydrogen bond interactions. The π–π stacking interactions of the heterocyclic ring of vemurafenib were created with Trp531 and Phe583. Six conventional hydrogen bonds were observed between vemurafenib and the residues of Lys483, Gln530, Cys532, Asp594, Phe595, and Gly596. Additionally, one carbon–hydrogen bond was detected between vemurafenib and the residue of Cys532. The side chain of Ala481 and Cys532 was participated in π–alkyl interaction with the heterocyclic ring of vemurafenib. The hydrophobic and van der Waals interactions of vemurafenib were detected with the side chains of Ile463, Val471, Leu505, Ile513, Leu515, Phe516, Ile527, Val528, Thr529, Gly534, Ser535, Ser536, His539, Lys591, and Gly593. The heterocyclic ring of vemurafenib was participated in a π–sigma interaction with Leu514. Also, a π–cation interaction was detected between the residue of Lys483 and the phenyl ring of vemurafenib. The docked conformation of vemurafenib including hydrogen bond interactions and the overlay view of conformations of co-crystallized vemurafenib, re-docked vemurafenib, 4k and 5a in the active site of an enzyme are depicted in Fig. 8. Delightfully, 4k and 5a bind to the identical site similar to vemurafenib and exhibits robust interactions with the B-RAF V600E enzyme as illustrated in Fig. 8.
 | ||
| Fig. 8 Docked conformation of vemurafenib including hydrogen bond interactions [left] and the overlay view of conformations of co-crystallized vemurafenib (yellow), re-docked vemurafenib (green), 4k (red) and 5a (blue) [right] in active site of B-RAF V600E kinase. | ||
The docked conformations of 4k in active site of B-RAF V600E kinase including hydrogen bond interactions is presented in Fig. 9. For 4k, the analysis of the docking protocol revealed that Thr529 is participated in a π–sigma interaction with phenoxy residue. Both rings of caffeine core are involved in π–π interactions with Phe583. Four conventional hydrogen bonds were detected between 4k and the residues of Thr529, Cys532, Asp594, and Gly596. In addition, several carbon–hydrogen bonds were observed between 4k and the side chain of Phe468, Thr529, Gln530, and Cys532. The residue of Val471, Ala481, Leu514, Cys532, and Phe583 was involved in π–alkyl interactions with the caffeine core of 4k. The two phenyl rings of 4k were involved in additional π–alkyl interactions with the side chain of Lys483, Leu485, Leu514, and Ile527. The hydrophobic and van der Waals interactions of 4k are similar to those observed for vemurafenib since both compounds were placed in the same binding pocket.
 | ||
| Fig. 9 Docked conformations of 4k (left) and 5a (right) including hydrogen bond interactions in the active site of B-RAF V600E kinase. | ||
The interactions that play a significant role in stabilizing vemurafenib within the active site of the B-RAF V600E kinase enzyme consist of van der Waals, hydrophobic, π–alkyl, π–π stacking, π–sigma, π–cation, and hydrogen bond interactions. Specifically, the π–π stacking interactions between the heterocyclic ring of vemurafenib and Trp531 and Phe583 were identified. A total of six conventional hydrogen bonds were also noted between vemurafenib and the amino acid residues Lys483, Gln530, Cys532, Asp594, Phe595, and Gly596. Additionally, a carbon–hydrogen bond was observed between vemurafenib and the residue Cys532. The side chains of Ala481 and Cys532 were involved in π–alkyl interactions with the heterocyclic ring of vemurafenib. Furthermore, the hydrophobic and van der Waals interactions of vemurafenib were found to interact with the side chains of Ile463, Val471, Leu505, Ile513, Leu515, Phe516, Ile527, Val528, Thr529, Gly534, Ser535, Ser536, His539, Lys591, and Gly593. The heterocyclic ring of vemurafenib engaged in a π–sigma interaction with Leu514, while a π–cation interaction was observed between Lys483 and the phenyl ring of vemurafenib. The docked conformation of vemurafenib, along with its hydrogen bond interactions, and comparative conformations with other compounds in the active site of the enzyme are depicted in Fig. 8.
Similarly, the docked conformations of 4k within the active site of the B-RAF V600E kinase, including its hydrogen bond interactions, are presented in Fig. 9. Analysis of the docking process for 4k indicated a π–sigma interaction between Thr529 and the phenoxy residue. Both rings of the caffeine core of 4k participated in π–π interactions with Phe583. Four conventional hydrogen bonds were identified between 4k and the residues Thr529, Cys532, Asp594, and Gly596. Additionally, several carbon–hydrogen bonds were observed between 4k and the side chains of Phe468, Thr529, Gln530, and Cys532. The residues Val471, Ala481, Leu514, Cys532, and Phe583 were involved in π–alkyl interactions with the caffeine core of 4k. Moreover, the two phenyl rings of 4k engaged in additional π–alkyl interactions with the side chains of Lys483, Leu485, Leu514, and Ile527. The hydrophobic and van der Waals interactions of 4k were similar to those observed for vemurafenib, given that both compounds occupy the same binding pocket.
The residue from 5a known as anisole engaged in a π–cation interaction with Lys483 (Fig. 9). In addition, it participated in a π–sigma interaction with Gly596. Cys532 formed two π–sulfur interactions with both rings of the caffeine core. Two π–π interactions were observed between the caffeine core of 5a and Phe583. Furthermore, 5a interacted with Thr529 through both π–donor and carbon–hydrogen bonds. Additionally, Glu501, Thr529, Gln530, and Cys532 contributed to other carbon–hydrogen bond interactions with 5a. Three conventional hydrogen bond interactions were noted between 5a and the residues Asp594, Phe595, and Gly596. 5a was also stabilized in the active site of the target enzyme through π–alkyl interactions with the side chains of Val471, Ala481, Phe498, Leu505, Leu514, Ile527, and Phe583. The hydrophobic and van der Waals interactions of 5a showed similarity to those observed for vemurafenib and 4k. The calculated ΔG values for vemurafenib, 4k, and 5a were −154.54, −159.56, and −151.35 kcal mol−1, respectively. The total hydrogen bond energies for vemurafenib, 4k, and 5a were −11.13, −10.90, and −6.13 (kcal mol−1), respectively. These findings suggest that hydrogen bonds, hydrophobic, and van der Waals interactions play a crucial role in stabilizing 4k and 5a in the binding site of the B-RAF V600E kinase enzyme.
Dihydrofolate reductase (DHFR) is essential for folic acid metabolism and the production of purines, thymidylate, and glycine.36 DHFR is a key target for cancer treatment. In this study, the interaction, affinity, binding mode, and orientation of compounds 4k and 5a were investigated in the active site of human DHFR using MVD 6.0 software. The 3D structure of human DHFR in complex with methotrexate (MTX) and nicotinamide adenine dinucleotide phosphate (NADPH) was obtained from the RCSB (PDB code: 1U72). Discovery Studio 2021 software was used for the analysis of results. The docking protocol was validated by redocking MTX in the active site of hDHFR, with a desirable RMSD value of 1.18 Å. Analysis revealed that hydrogen bond interactions are crucial for the stabilization of MTX in the active site. Strong hydrogen bonds were identified between MTX and Ile7, Gln35, Asn64, Arg70, Val115, and Tyr121 residues. NADPH, Arg32, and Ser59 formed carbon–hydrogen bond interactions with MTX. Additionally, Ile7, Ala9, Leu22, and NADPH participated in π–alkyl interactions with the pteridine core of MTX. A π–alkyl interaction was observed between Ile60 and the aryl ring of MTX. Hydrophobic and van der Waals interactions of MTX were detected in a pocket formed by Ile7, Val8, Leu22, TRP24, Phe31, Tyr33, F34, Gln35, Thr38, Thr39, Ser59, Pro61, Leu67, Lys68, Val115, Gly116, Val135, and Phe179. The binding of 4k, 5a, and MTX to the same site in hDHFR with strong interactions is depicted in Fig. 10.
 | ||
| Fig. 10 Docked conformation of MTX including hydrogen bond interactions [left] and the overlay view of conformations of co-crystallized MTX (yellow), re-docked MTX (green), 4k (red) and 5a (blue) [right] in active site of hDHFR. | ||
In Fig. 11, the docked conformation of 4k in the active site of hDHFR, along with its hydrogen bond interactions, is depicted. The ester group of 4k formed two traditional hydrogen bonds with Lys68 and Arg70. Additionally, it engaged in two carbon–hydrogen bond interactions with Asn64 and Lys68. The central caffeine core of 4k participated in multiple carbon–hydrogen bond interactions with the side chains of Ile7, Glu30, Val115, Tyr121, and Thr136. Furthermore, π–π shaped and π–amide stacked interactions were observed between 4k and Phe34 and Val8, respectively. 4k also interacted through π–alkyl interactions with Ala9, Phe31, Fhe34, Ile60, and NADPH. The van der Waals and hydrophobic interactions of 4k in the binding site were similar to those of MTX.
 | ||
| Fig. 11 Docked conformations of 4k (left) and 5a (right) including hydrogen bond interactions in the active site of hDHFR. | ||
On the other hand, in the case of 5a (Fig. 11), there were no conventional hydrogen bond interactions observed. The caffeine core of 5a engaged in four carbon–hydrogen bond interactions with Ile7, Glu30, Val115, and Tyr121. Additionally, a π–donor hydrogen bond formed between Ser59 and the phenoxy ring of 5a. Similar to 4k, π–π shaped and π–amide stacked interactions were detected between 5a and Phe34 and Val8, respectively. The caffeine core of 5a also participated in four π–alkyl interactions with Ala9, Phe34, and NADPH. Other parts of the 5a structure exhibited several π–alkyl interactions with the side chains of Leu22, Pro26, Phe31, Ile60, and Pro61. The hydrophobic and van der Waals interactions of 5a mirrored those observed for MTX and 4k. Calculated ΔG values for MTX, 4k, and 5a were −190.40, −177.89, and −163.62 kcal mol−1, respectively. The total hydrogen bond energies for MTX, 4k, and 5a were −16.33, −4.56, and −7.02 (kcal mol−1), respectively. The docking study results suggest that the stabilization of MTX, 4k, and 5a in the hDHFR enzyme's binding site may be facilitated by strong hydrogen bonds, hydrophobic interactions, and van der Waals forces. The presence of NADPH could also impact the stabilization of ligands in the active site of the enzyme.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) to predict the likelihood of a compound being suitable as a drug.37,38 In the evaluation of drug candidates 4a–4k, 5a–5f, and MTX, in silico pharmacokinetic and physiochemical profiles were analyzed using tools like the preADMET online server,39 OSIRIS Property Explorer,40 and the SwissADME online software,41 with the results summarized in Tables 2 and 3.
P) to predict the likelihood of a compound being suitable as a drug.37,38 In the evaluation of drug candidates 4a–4k, 5a–5f, and MTX, in silico pharmacokinetic and physiochemical profiles were analyzed using tools like the preADMET online server,39 OSIRIS Property Explorer,40 and the SwissADME online software,41 with the results summarized in Tables 2 and 3.
| Compound | Mwa | nRBb | nHBDc | nHBAd | clogPe |
TPSAf | Drug-likeness | Drug score |
|---|---|---|---|---|---|---|---|---|
| a Molecular weight.b Number of rotatable bonds.c Number of hydrogen bond donors.d Number of hydrogen bond acceptors.e Logarithm of octanol–water partition coefficient (logP).f Topological polar surface area (Å2).g Methotrexate. |
||||||||
| 4a | 430.46 | 5 | 0 | 5 | 3.57 | 88.12 | 2.53 | 0.45 |
| 4b | 485.32 | 5 | 0 | 5 | 4.44 | 88.12 | 4.94 | 0.34 |
| 4c | 446.46 | 6 | 0 | 6 | 3.16 | 97.35 | 4.42 | 0.49 |
| 4d | 416.43 | 5 | 0 | 5 | 3.23 | 88.12 | 0.95 | 0.44 |
| 4e | 450.87 | 5 | 0 | 5 | 3.84 | 88.12 | 4.59 | 0.42 |
| 4f | 461.43 | 6 | 0 | 7 | 2.31 | 133.94 | −1.05 | 0.29 |
| 4g | 476.48 | 7 | 0 | 7 | 3.09 | 106.58 | 3.01 | 0.46 |
| 4h | 446.46 | 6 | 0 | 6 | 3.16 | 97.35 | 3.96 | 0.23 |
| 4i | 450.87 | 5 | 0 | 5 | 3.84 | 88.12 | 4.57 | 0.25 |
| 4j | 485.32 | 5 | 0 | 5 | 4.44 | 88.12 | 4.45 | 0.34 |
| 4k | 474.47 | 7 | 0 | 7 | 3.14 | 114.42 | −2.15 | 0.25 |
| 5a | 446.46 | 6 | 0 | 6 | 3.16 | 97.35 | 3.85 | 0.49 |
| 5b | 461.43 | 6 | 0 | 7 | 2.31 | 133.94 | −8.10 | 0.23 |
| 5c | 461.43 | 6 | 0 | 7 | 2.31 | 133.94 | −2.71 | 0.25 |
| 5d | 430.46 | 5 | 0 | 5 | 3.57 | 88.12 | 2.95 | 0.46 |
| 5e | 466.49 | 5 | 0 | 5 | 4.43 | 88.12 | 1.17 | 0.31 |
| 5f | 450.87 | 5 | 0 | 5 | 3.84 | 88.12 | 5.42 | 0.42 |
| MTXg | 454.44 | 10 | 5 | 9 | 1.23 | 210.54 | −7.09 | 0.22 |
| Compound | logS |
HIAa | logKpb |
P-gpc | BBBd | CYP2D6 | CYP3A4 | Carcino.e | hERG inhibitionf |
|---|---|---|---|---|---|---|---|---|---|
| a Human intestinal absorption (%).b Skin permeability (logKp, cm h−1).c P-Glycoprotein.d Blood–brain barrier permeability (C. brain/C. blood).e Carcinogenicity (mouse).f Human ether-a-go-go related gene channel.g Methotrexate. S = substrate, WS = weak-substrate, NS = non-substrate, I = inhibitor, NI = non-inhibitor, NC = non-carcinogenic. |
|||||||||
| 4a | −4.91 | 98.69 | −3.06 | I/NS | 0.29 | NI/NS | NI/NS | NC | Medium |
| 4b | −5.79 | 97.65 | −3.08 | I/NS | 0.49 | NI/NS | NI/WS | NC | Medium |
| 4c | −4.68 | 99.39 | −3.24 | I/NS | 0.40 | NI/NS | NI/NS | NC | Medium |
| 4d | −4.61 | 98.85 | −3.07 | I/NS | 0.27 | NI/NS | NI/WS | NC | Medium |
| 4e | −5.20 | 98.07 | −3.13 | I/NS | 0.33 | NI/NS | NI/WS | NC | Medium |
| 4f | −4.67 | 97.43 | −2.90 | I/NS | 0.22 | NI/NS | NI/WS | C | Medium |
| 4g | −4.75 | 99.73 | −3.48 | I/NS | 0.41 | NI/NS | NI/WS | NC | Medium |
| 4h | −4.68 | 99.39 | −3.27 | I/NS | 0.40 | NI/NS | NI/NS | NC | Medium |
| 4i | −5.20 | 98.07 | −3.10 | I/NS | 0.32 | NI/NS | NI/WS | NC | Medium |
| 4j | −5.20 | 97.65 | −3.06 | I/NS | 0.47 | NI/NS | NI/WS | NC | Medium |
| 4k | −4.69 | 99.65 | −3.27 | I/NS | 0.32 | NI/NS | NI/NS | NC | Medium |
| 5a | −4.68 | 99.29 | −3.30 | I/NS | 0.50 | NI/NS | NI/NS | NC | Medium |
| 5b | −4.67 | 97.43 | −2.90 | I/NS | 0.19 | NI/NS | NI/WS | C | Medium |
| 5c | −4.67 | 96.49 | −2.87 | I/NS | 0.25 | NI/NS | NI/WS | C | Medium |
| 5d | −4.91 | 98.50 | −3.09 | I/NS | 0.55 | NI/NS | NI/NS | NC | Medium |
| 5e | −5.73 | 97.64 | −2.80 | I/NS | 1.12 | NI/NS | NI/WS | NC | Medium |
| 5f | −5.20 | 98.07 | −3.13 | I/NS | 0.56 | NI/NS | NI/WS | NC | Medium |
| MTXg | −1.19 | 36.61 | −4.63 | NI/S | 0.04 | NI/NS | I/WS | NC | High |
From the data presented in Table 2, it can be observed that both MTX and the synthesized compounds adhere to the molecular weight criterion of ≤500 dalton. The number of rotatable bonds (nRB) for MTX and the synthesized compounds falls within the 5–10 range. Additionally, all compounds meet the criteria for hydrogen bond acceptors (nHBA), hydrogen bond donors (nHBD), and the lipophilicity factor indicated by clogP, with values below the specified threshold of 5 as per RO5 criteria.
The total polar surface area (TPSA) is an important parameter in determining a molecule's transport characteristics, influencing factors like intestinal absorption and blood–brain barrier penetration. TPSA values for 4a–4k and 5a–5f are below the accepted limit of 140 Å2 for approved drugs, suggesting favorable membrane permeability or oral absorption. The drug likeness of the synthesized derivatives was evaluated using the OSIRIS Property Explorer software, indicating negative values for compounds 4f, 4k, 5b, and 5c, along with MTX.
These analyses led to the computation of a drug score for each compound, serving as a predictive parameter. The maximum drug score values were observed for compounds 4c and 5a, while the minimum scores belonged to compounds 4h and 5b. Overall, the drug score values for the synthesized compounds surpass that of MTX across the board. Notably, compounds 4k and 5a exhibit promising potential for further research based on these assessments.
The pharmacokinetic profiles of compounds 4a–4k, 5a–5f, and MTX, including their absorption, distribution, metabolism, and toxicity properties, were measured and summarized in Table 3. The analysis focused on assessing these compounds as P-glycoprotein (P-gp) substrates and/or inhibitors, their human intestinal absorption (HIA), aqueous solubility (logS), and skin permeability (logKp) to predict their absorption profiles. Most of the synthesized compounds exhibited moderate solubility in water, while methotrexate (MTX) showed good water solubility. The HIA factor was found to be over 96.49% for 4a–4k and 5a–5f, in contrast to 36.61% for MTX, suggesting ideal intestinal absorption for the former compounds. Skin permeability, crucial for transdermal drug delivery, was comparable among 4a–4k, 5a–5f, and MTX. The compounds were also evaluated for their interaction with the P-gp transporter, which plays a role in drug bioavailability. While all synthesized compounds were found to be non-substrates of P-gp, MTX was predicted to be a substrate; however, it is not an inhibitor. The synthesized compounds were also assessed for their blood–brain barrier (BBB) permeability, with all compounds except 5e showing low permeability, suggesting minimal risk of neurotoxicity. Regarding metabolism, the compounds were found to interact differently with the CYP2D6 and CYP3A4 enzymes. While they were mostly non-inhibitors and non-substrates of CYP2D6, some were substrates of CYP3A4, implicating potential liver metabolism. In terms of carcinogenicity, most compounds were deemed non-carcinogenic in mice. The risk of hERG inhibition, which can lead to cardiac side effects, was found to be high for MTX and moderate for 4a–4k and 5a–5f.
2.4.3.1 NBO analysis. In the solution phase, second-order perturbation theory is employed to calculate the stabilization energy Eij(2), for compounds 6, 7, and 8-BC. The donor–acceptor NBO stabilization energy is interpreted as follows:
 | (1) |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Me) and 7 (RH)
Me) and 7 (RH)
| Pairwise charge transfera | E(2), 7 | E(2), 6 | Pairwise charge transferb | E(2), 7 | E(2), 6 | E(2), 8-BC |
|---|---|---|---|---|---|---|
| a Pairwise charge transfer and stabilization energy E(2) (kcal mol−1) for 6 and 7 in ethanol 90%.b Pairwise charge transfer and stabilization energy E(2) (kcal mol−1) for 6, 7 and 8-BC in DMF. | ||||||
| n′ O15 → σ* N7–C8 | 1 | 55 | n N1 → π* C6–O13 | 70 | 70 | 72 |
| n O15 → σ* C8–N9 | 10 | 10 | n N1 → π* C2–O11 | 70 | 69 | 70 |
| n′ O15 → σ* C8–N9 | 47 | — | n N3 → σ* C4–C5 | — | 60 | 59 |
| n N9 → σ* C8–O15 | 4 | 4 | n N3 → σ* C2–O11 | — | 74 | 77 |
| n′ O15 → π* C25–C26 | 22 | 19 | n N9 → π* C4–C5 | — | 92 | 96 |
| n O15 → σ* C25–C30 | 8 | 5 | n N9 → π* N7–C8 | — | 197 | 213 |
| n O32 → σ* C31–C37 | — | 23 | n N9 → σ* C7–C8 | 11 | 11 | 10 |
| n O32 → σ* C31–H37 | 24 | — | n O11 → σ* C2–N1 | 34 | 33 | 33 |
| n O32 → σ* C28–C31 | 23 | 24 | n O11 → σ* C2–N3 | 32 | 32 | 31 |
| σ C4–N9 → σ* C8–O15 | 10 | 10 | n O13 → σ* N1–C6 | 36 | 36 | 35 |
| σ C5–N7 → σ* C8–O15 | 6 | 6 | n O13 → σ* C5–C6 | 22 | 22 | 22 |
| π C25–C26 → π* C29–C30 | 24 | 24 | n O11 → RY C2 | 13 | 13 | 5 |
| π C29–C30 → π* C25–C26 | 33 | 33 | n O13 → RY C6 | 12 | 12 | 4 |
| π C25–C26 → π* C27–C28 | 32 | 32 | n Br15 → σ* N7–C8 | — | — | 29 |
| π C27–C28 → π* C25–C26 | 25 | 26 | π C4–C5 → π* N7–C8 | — | 15 | 20 |
| π C27–C28 → π* C31–O32 | 27 | 24 | σ C4–C5 → π* C6–O13 | — | 40 | 38 |
| π C31–O32 → π* C27–C28 | 6 | 6 | π C5–N7 → π* N3–C4 | 24 | 4 | 3 |
| π C27–C28 → π* C29–C30 | 30 | 30 | π C5–N7 → π* C8–N9 | 51 | 2 | 1 |
| π C29–C30 → π* C27–C28 | 25 | 25 | π N7–C8 → π* C4–C5 | — | 19 | 19 |
| σ C37–H37 → σ* C31–O32 | — | 12 | σ C8–N9 → σ* N3–C4 | 64 | 8 | 8 |
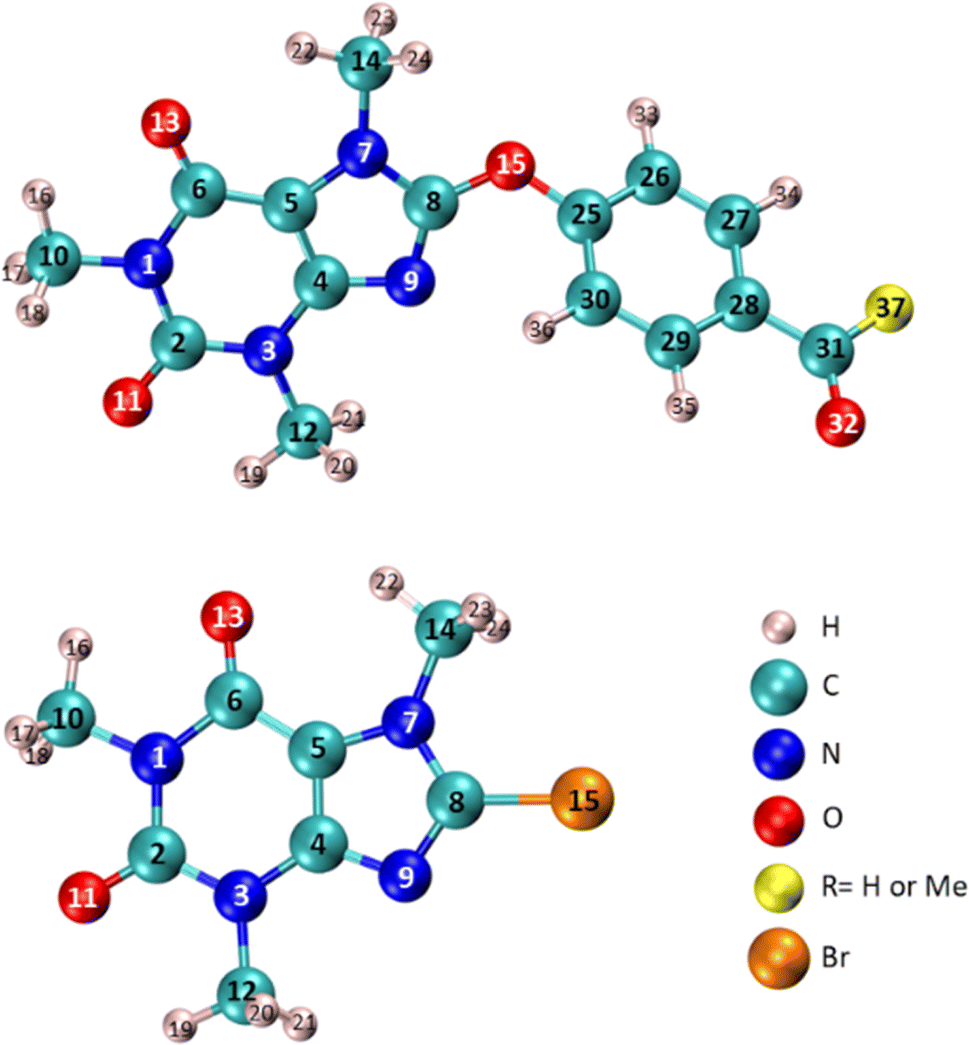 | ||
| Fig. 12 Specification the atom number of compounds 6 (RMe), 7 (RH), and 8-BC used in Table 4. | ||
Table 4 illustrates that the n* NBO of O15 in compounds 6 and 7 displays a more pronounced donor character compared to the corresponding donor role of Br in 8-BC. Additionally, the charge transfer from the lone-pair electrons of O15 in compound 7 to the σ* orbital of C25–C26/C25–C30 (two neighboring C–C bonds) differs slightly from the process observed in compound 6 because of difference in substituent at C28. The significant charge transfers between O15 and the caffeinyl moiety in the molecules originate from the strong interaction between O15 and the caffeinyl moiety. Furthermore, the charge transfer pathway from the atoms surrounding O15 towards the 4-carbonylphenoxy residue signifies an electron flow from the caffeinyl residue of compounds 6 and 7 towards the 4-carbonylphenoxy residue. Hence the charge density is transferred from caffeinyl residue toward 4-carbonylphenoxy group which causes the enhancement of positive charge density on caffeine's C8 and makes it suitable for susceptible attack by a nucleophile like hydroxide ion. These findings support the proposed mechanism depicted in Scheme 2 and are consistent with the experimental observations.
The natural atomic charge is determined by subtracting the nuclear charge from the total natural population of natural atomic orbitals within the atom. The natural atomic charge, referred to as the NBO charge in Fig. 13, is obtained through NBO analysis. Additionally, Fig. 13 illustrates the CHelpG atomic charge derived from Breneman atomic radii, and the ESP atomic charge based on Merz–Kollman atomic radii. In Fig. 13, the atomic charges (NBO, CHelpG, or ESP) of compounds 6, 7, and 8-BC are calculated by dividing the charge data into three parts: (i) the caffeinyl part before atom 15 (O or Br), (ii) atom 15, and (iii) the remaining atoms in both 6 and 7 after atom O15. The analysis reveals significant differences in the behavior of the caffeine moiety when Br is replaced with 4-carbonylphenoxy moiety. According to the NBO analysis results (Table 4), the presence of O15 in 4-carbonylphenoxy group influences the atoms on both sides of the molecule, leading to changes in the behavior of the caffeinyl core in both 6 and 7 compared to 8-BC.
 | ||
| Fig. 13 Charge distribution on three parts of molecules. | ||
Fig. 14 illustrates the electrostatic surface potential (ESP) of solute molecules, with all ESP maps sharing a common color range and isodensity value of 0.02. Additionally, Fig. 14 presents the HOMO and LUMO images of compounds 6, 7, and 8-BC in a solution. While the caffeinyl core influences both the HOMO and LUMO of 8-BC, compounds 6 and 7 exhibit the HOMO primarily on the caffeinyl part and the LUMO on the 4-carbonylphenoxy group, a key element for molecular reactivity. Substituting the bromine atom in 8-BC with 4-carbonylphenoxy moieties (bearing O15) to acquire 6 or 7 shifts the reactive site from caffeinyl core to a linked 4-carbonylphenoxy residue in 6 and 7. The calculated HOMO–LUMO energy gaps for compounds 6, 7, and 8-BC are 6.344, 6.200, and 7.275 eV, respectively, indicating higher reactivity in 6 and 7 compared to 8-BC. This causes the side reaction of 6 and/or 7 with OH− that led to formation of 8 as observed, experimentally.
 | ||
| Fig. 14 ESP, HOMO, and LUMO of 8-BC, compounds 6 and 7. | ||
3. Conclusion
In this paper, we conducted a comprehensive evaluation of the design, synthesis, characterization, anticancer properties, toxicity, and in silico analysis of novel 8-caffeinyl chalcone hybrid conjugates. These compounds were prepared by combining 8-BC with chalcones having diverse substituents. The synthesis process involved three stages: bromination of caffeine to produce 8-BC, synthesis of chalcones, and subsequent coupling of chalcones with 8-BC. The anticancer efficacy of these compounds was evaluated in vitro against two important cancer cell lines, breast cancer MCF-7 (ATCC HTB-22) and melanoma cancer A-375 (ATCC CRL-1619), with methotrexate (MTX) used as a reference drug. Among the compounds tested, 4k showed the highest potency against A-375 (IC50 = 92 ± 3.2), while 4d, 4k, 4e, and 4a exhibited significant inhibitory effects on MCF-7 cell growth, with their activities surpassing that of MTX. Toxicity assessments on the normal cell line HEK-293 (ATCC CRL-11268) revealed varying degrees of toxicity in the compounds, with exceptions noted for 4f, 4g, 5a, and 5c–5e, which demonstrated minimal toxicity. Molecular docking studies on 4k and 5a indicated strong binding affinities to B-RAF kinase and hDHFR enzymes. Furthermore, in silico pharmacokinetic and physiochemical profiling of the compounds showed that most adhered to Lipinski's rule of five (RO5). Additionally, density functional theory (DFT) calculations at the M06-2X-D3/6-31+G(d,p) level were employed to analyze atom charges, HOMO–LUMO energies, electrostatic potential (ESP), and other parameters of the intermediates, providing insights to support and elucidate the underlying mechanisms.4. Experimental
4.1 Materials and cell lines
Cancer cell lines MCF-7 (ATCC HTB-22) derived from breast cancer, A-375 (ATCC CRL-1619) from melanoma, and the normal cell line HEK-293 (ATCC CRL-11268) were sourced from the Pasteur Institute of Iran. Fetal bovine serum (FBS) was procured from Gibco (USA). Phosphate-buffered saline (PBS) tablets, tetrazolium salt, and MTT (3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide) were obtained from Sigma-Aldrich (USA). Dimethyl sulfoxide (DMSO), trypsin, penicillin/streptomycin, and Dulbecco's Modified Eagle's Media (DMEM) cell culture medium were supplied by Shellmax (China).4.2 Evaluation of anticancer activity
The anticancer activity of the title compounds and MTX43 was assessed using the MTT assay. All compounds were dissolved in a PBS solution containing 0.5% DMSO, and necessary dilutions were made using the same solvent.The cell lines (MCF-7, A-375, and HEK-293) were cultured in culture flasks in DMEM complete medium (comprising 10% FBS and 1% penicillin–streptomycin) and then incubated at 37 °C in a CO2 (5%) and air (95%) environment. Following separation using trypsin, the cell lines were seeded in 96-well plates and incubated for 48 hours. Subsequently, the culture media was removed, and each well was replenished with 75 μL of fresh complete medium. The compounds at concentrations of 15.62, 31.25, 62.5, 125, 250, and 500 μM were added to the wells. After another incubation period of 48 hours at 37 °C in a 95% air and 5% CO2 atmosphere, the contents of the plates were discarded, and the wells were washed with 100 μL of PBS. Then, 100 μL of MTT solution (0.5 mg mL−1) was added to each well and incubated for an additional 4 hours. Following this, 100 μL of DMSO was added to each well to dissolve the formazan crystals that were formed. In each plate, six wells served as the control group, containing a PBS solution with 0.5% DMSO (25 μL) and DMEM with the cells (75 μL). Finally, the absorbance (A) of each well was measured at 570 nm using an ELISA plate reader. The cell viability at each concentration was calculated using eqn (2).
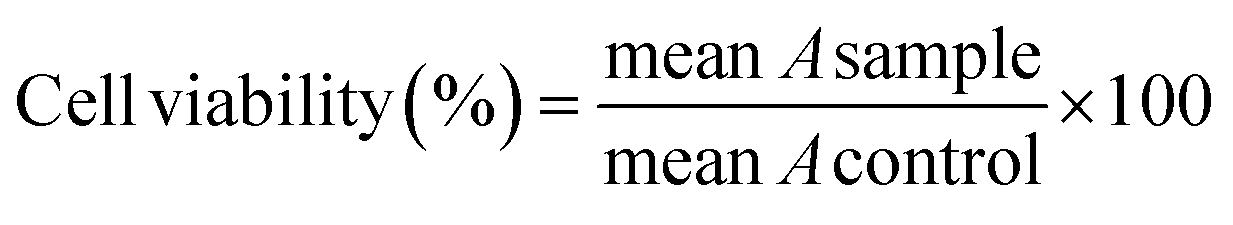 | (2) |
4.3 Molecular docking studies procedure
The molecular docking investigation was conducted utilizing Molegro Virtual Docker (MVD 6.0) software with its default settings.34 For this study, the crystal structures of B-RAF V600E bound to vemurafenib (PDB code: 3OG7) and human DHFR bound to MTX and NADPH (PDB code: 1U72) were retrieved from the RCSB website. The enzyme structures were prepared by removing water molecules and ligands, adjusting hydrogen atoms, and ensuring accurate bond and atom assignments. Specifically, in the case of B-RAF V600E, chain B was excluded, and only chain A was used for docking. For hDHFR, the docking was performed with NADPH present. The docking simulations were run 50 times for each system. The active site of the enzymes was defined as residues within a 7 Å radius around the inhibitor ligand. The structures of vemurafenib, methotrexate, 4k, and 5a were optimized using the PM6 method in Gaussian09.44,45 Initially, the docking was validated by re-docking vemurafenib and methotrexate into the active sites of B-RAF V600E and hDHFR, respectively. Subsequently, the optimized structures of ligands 4k and 5a were introduced into the analysis. The interactions were examined and assessed based on the docking outcomes using the Discovery Studio 2021 visualizer.4.4 Chemistry general
All chemicals were bought from Merck and other chemical suppliers and directly used without additional purifications. The reactions were observed using TLC on SILG/UV 254 silica-gel plates. Purifications through short column chromatography were carried out using silica gel 60 (mesh size 0.063–0.200 mm, 70–230 mesh; ASTM). Melting points were taken using the Electrothermal IA 9000 in open capillary tubes without correction. Elemental analyses, GC/MS, and IR spectra were obtained using the PerkinElmer 240-B micro-analyzer, Shimadzu GC/MS-QP 1000-EX apparatus (m/z; rel.%), and the Shimadzu FT-IR-8300 spectrophotometer, respectively. 1H- and 13C-NMR spectra were recorded on the Brüker Avance-DPX-300 spectrometer operating at 300/75 MHz, respectively. Chemical shifts are referenced in δ relative to tetramethylsilane (TMS) as an internal standard, and coupling constants J are given in Hz. Abbreviations used for 1H-NMR signals include s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad, etc.4.4.5.1 (E)-1,3,7-Trimethyl-8-(4-(3-p-tolylacryloyl)phenoxy)-1H-purine-2,6(3H,7H)-dione (4a). Compound 4a was obtained according to the general procedure A from a mixture of 8-BC (2.73 g, 10 mmol), (E)-1-(4-hydroxyphenyl)-3-p-tolylprop-2-en-1-one (2.38 g, 10 mmol), and K2CO3 (1.38, 10 mmol) in anhydrous DMF (20 mL) after 12 h as pale orange solid (3.52 g, 82%); m.p.: 166–168 °C, 1H NMR (CDCl3, 400 MHz): δppm = 8.13 (d, J = 8.8 Hz, 2H, aryl), 7.85 (d, J = 15.6 Hz, 1H, C(O)–CH), 7.57–7.46 (complex, 5H, aryl, C(O)–CH
CH), 7.25 (d, J = 8.0 Hz, 2H, aryl), 3.91 (s, 3H, N(7)-CH3), 3.48 (s, 3H, N(3)-CH3), 3.42 (s, 3H, N(1)-CH3), 2.41 (s, 3H, PhCH3). 13C NMR (CDCl3, 100 MHz): δppm = 188.61, 156.87, 154.81, 152.03, 151.31, 145.64, 144.58, 134.29, 133.11, 130.79, 129.85, 129.03, 128.21, 121.41, 119.82, 104.03, 30.69, 29.53, 27.58, 21.81. IR (KBr): 3050, 2986, 1705, 1697, 1652, 1553, 1484, 1275, 1056 cm−1. MS (EI): m/z (%) = 430 (27.5) [M+]. Anal. calc. for C24H22N4O4: C, 66.97; H, 5.15; N, 13.02; found: C, 66.85; H, 5.29; N, 13.14.
4.4.5.2 (E)-8-(4-(3-(2,4-Dichlorophenyl)acryloyl)phenoxy)-1,3,7-trimethyl-1H-purine-2,6(3H,7H)-dione (4b). Compound 4b was obtained according to the general procedure A from a mixture of 8-BC (2.73 g, 10 mmol), (E)-3-(2,4-dichlorophenyl)-1-(4-hydroxyphenyl)prop-2-en-1-one (2.93 g, 10 mmol), and K2CO3 (1.38, 10 mmol) in anhydrous DMF (20 mL) after 14 h as creamy solid (4.27 g, 88%); m.p.: 180–182 °C, 1H NMR (CDCl3, 400 MHz): δppm = 8.16–8.11 (m, 3H, aryl), 7.71 (d, J = 15.6 Hz, 1H, C(O)–CH), 7.50–7.47 (complex, 4H, aryl, C(O)–CH
CH), 7.33 (d, J = 10.4 Hz, 1H, aryl), 3.92 (s, 3H, N(7)-CH3), 3.49 (s, 3H, N(3)-CH3), 3.42 (s, 3H, N(1)-CH3). 13C NMR (CDCl3, 100 MHz): δppm = 188.65, 156.62, 154.58, 152.46, 151.34, 145.72, 141.17, 135.97, 135.01, 133.25, 131.49, 130.38, 129.46, 127.76, 126.91, 124.38, 119.49, 104.07, 30.51, 29.37, 27.82. IR (KBr): 3028, 2972, 1706, 1695, 1650, 1561, 1480, 1268, 1050, 870 cm−1. MS (EI): m/z (%) = 484 (34.8) [M+]. Anal. calc. for C23H18Cl2N4O4: C, 56.92; H, 3.74; N, 11.54; found: C, 57.11; H, 3.91; N, 11.70.
4.4.5.3 (E)-8-(4-(3-(2-Methoxyphenyl)acryloyl)phenoxy)-1,3,7-trimethyl-1H-purine-2,6(3H,7H)-dione (4c). Compound 4c was obtained according to the general procedure A from a mixture of 8-BC (2.73 g, 10 mmol), (E)-1-(4-hydroxyphenyl)-3-(2-methoxyphenyl)prop-2-en-1-one (2.54 g, 10 mmol), and K2CO3 (1.38, 10 mmol) in anhydrous DMF (20 mL) after 12 h as yellow solid (4.15 g, 93%); m.p.: 166–168 °C, 1H NMR (CDCl3, 400 MHz): δppm = 8.17 (br s, 1H, C(O)–CH), 8.13 (d, J = 8.4 Hz, 2H, aryl), 7.66–7.62 (complex, 2H, C(O)–CH
CH, aryl), 7.47 (d, J = 8.8 Hz, 2H, aryl), 7.43 (t, J = 7.6 Hz, 1H, aryl), 7.04–6.96 (m, 2H, aryl), 3.94 (s, 3H, N(7)-CH3), 3.91 (s, 3H, N(3)-CH3), 3.49 (s, 3H, N(1)-CH3), 3.43 (s, 3H, OCH3). 13C NMR (CDCl3, 100 MHz): δppm = 188.57, 156.51, 155.07, 152.43, 151.49, 150.75, 149.35, 145.85, 144.98, 135.83, 130.58, 127.70, 123.34, 120.02, 119.33, 114.32, 112.53, 103.96, 55.76, 30.69, 29.90, 27.58. IR (KBr): 3075, 2959, 1703, 1692, 1657, 1552, 1465, 1268, 1059 cm−1. MS (EI): m/z (%) = 446 (29.2) [M+]. Anal. calc. for C24H22N4O5: C, 64.57; H, 4.97; N, 12.55; found: C, 64.40; H, 5.06; N, 12.42.
4.4.5.4 (E)-8-(4-Cinnamoylphenoxy)-1,3,7-trimethyl-1H-purine-2,6(3H,7H) dione (4d). Compound 4d was obtained according to the general procedure A from a mixture of 8-BC (2.73 g, 10 mmol), (E)-1-(4-hydroxyphenyl)-3-phenylprop-2-en-1-one (2.24 g, 10 mmol), and K2CO3 (1.38, 10 mmol) in anhydrous DMF (20 mL) after 12 h as pale creamy solid (3.45 g, 83%); m.p.: 165–167 °C, 1H NMR (CDCl3, 400 MHz): δppm = 8.14 (d, J = 8.8 Hz, 2H, aryl), 7.87 (d, J = 15.6 Hz, 1H, C(O)–CH), 7.68–7.66 (m, 2H, aryl), 7.57 (d, J = 15.6 Hz, 1H, C(O)–CH
CH), 7.49–7.44 (m, 5H, aryl), 3.92 (s, 3H, N(7)-CH3), 3.49 (s, 3H, N(3)-CH3), 3.42 (s, 3H, N(1)-CH3). 13C NMR (CDCl3, 100 MHz): δppm = 188.83, 156.58, 154.95, 152.43, 151.59, 145.66, 145.25, 135.48, 134.72, 130.75, 130.56, 129.02, 128.50, 121.51, 119.25, 103.97, 30.58, 29.91, 27.88. IR (KBr): 3068, 2971, 1706, 1697, 1662, 1541, 1460, 1260, 1051 cm−1. MS (EI): m/z (%) = 416 (23.9) [M+]. Anal. calc. for C23H20N4O4: C, 66.34; H, 4.84; N, 13.45; found: C, 66.53; H, 5.01; N, 13.63.
4.4.5.5 (E)-8-(4-(3-(4-Chlorophenyl)acryloyl)phenoxy)-1,3,7-trimethyl-1H-purine-2,6(3H,7H)-dione (4e). Compound 4e was obtained according to the general procedure A from a mixture of 8-BC (2.73 g, 10 mmol), (E)-3-(4-chlorophenyl)-1-(4-hydroxyphenyl)prop-2-en-1-one (2.58 g, 10 mmol), and K2CO3 (1.38, 10 mmol) in anhydrous DMF (20 mL) after 12 h as pale orange solid (4.23 g, 94%); m.p.: 110–112 °C, 1H NMR (CDCl3, 400 MHz): δppm = 8.14 (d, J = 8.8 Hz, 2H, aryl), 7.82 (d, J = 15.6 Hz, 1H, C(O)–CH), 7.61 (d, J = 8.4 Hz, 2H, aryl), 7.54–7.41 (complex, 5H, C(O)–CH
CH, aryl), 3.92 (s, 3H, N(7)-CH3), 3.49 (s, 3H, N(3)-CH3), 3.43 (s, 3H, N(1)-CH3). 13C NMR (CDCl3, 100 MHz): δppm = 188.53, 156.67, 154.96, 152.39, 151.59, 145.65, 143.73, 136.66, 135.28, 133.20, 130.57, 129.65, 129.32, 121.87, 119.30, 104.01, 30.59, 29.92, 27.90. IR (KBr): 3100, 2980, 1707, 1694, 1658, 1550, 1459, 1280, 1057, 840 cm−1. MS (EI): m/z (%) = 450 (30.1) [M+]. Anal. calc. for C23H19ClN4O4: C, 61.27; H, 4.25; N, 12.43; found: C, 61.09; H, 4.06; N, 12.27.
4.4.5.6 (E)-1,3,7-Trimethyl-8-(4-(3-(3-nitrophenyl)acryloyl)phenoxy)-1H-purine-2,6(3H,7H)-dione (4f). Compound 4f was obtained according to the general procedure A from a mixture of 8-BC (2.73 g, 10 mmol), (E)-1-(4-hydroxyphenyl)-3-(3-nitrophenyl)prop-2-en-1-one (2.69 g, 10 mmol), and K2CO3 (1.38, 10 mmol) in anhydrous DMF (20 mL) after 18 h as yellow solid (3.27 g, 71%); m.p.: 277–279 °C, 1H NMR (CDCl3, 400 MHz): δppm = 8.31 (br s, 1H, C(O)–CH), 8.06–7.63 (complex, 5H, aryl, C(O)–CH
CH), 7.46–7.29 (complex, 3H, aryl), 7.04 (s, 1H, aryl), 3.69 (s, 3H, N(7)-CH3), 3.25 (s, 3H, N(3)-CH3), 3.19 (s, 3H, N(1)-CH3). 13C NMR (CDCl3, 100 MHz): δppm = 187.99, 156.96, 154.96, 151.59, 148.65, 145.63, 141.93, 140.84, 137.37, 134.78, 130.71, 129.01, 125.15, 124.27, 123.55, 119.45, 115.70, 104.05, 30.61, 29.92, 27.92. IR (KBr): 3050, 2970, 1705, 1698, 1663, 1565, 1548, 1449, 1351, 1260, 1053 cm−1. MS (EI): m/z (%) = 461 (21.4) [M+]. Anal. calc. for C23H19N5O6: C, 59.87; H, 4.15; N, 15.18; found: C, 60.02; H, 4.33; N, 15.31.
4.4.5.7 (E)-8-(4-(3-(2,4-Dimethoxyphenyl)acryloyl)phenoxy)-1,3,7-trimethyl-1H-purine-2,6(3H,7H)-dione (4g). Compound 4g was obtained according to the general procedure A from a mixture of 8-BC (2.73 g, 10 mmol), (E)-3-(2,4-dimethoxyphenyl)-1-(4-hydroxyphenyl)prop-2-en-1-one (2.84 g, 10 mmol), and K2CO3 (1.38, 10 mmol) in anhydrous DMF (20 mL) after 12 h as orange solid (4.19 g, 88%); m.p.: 216–218 °C, 1H NMR (CDCl3, 400 MHz): δppm = 8.13 (d, J = 8.8 Hz, 2H, aryl), 7.82 (d, J = 15.6 Hz, 1H, C(O)–CH), 7.48 (d, J = 15.2 Hz, 2H, aryl), 7.42 (d, J = 15.2 Hz, 1H, C(O)–CH
CH), 7.27 (d, J = 8.4 Hz, 1H, aryl), 7.17 (br s, 1H, aryl), 6.93 (d, J = 8.4 Hz, 1H, aryl), 3.96 (s, 3H, OCH3), 3.95 (s, 3H, OCH3), 3.91 (s, 3H, N(7)-CH3), 3.48 (s, 3H, N(3)-CH3), 3.42 (s, 3H, N(1)-CH3). 13C NMR (CDCl3, 100 MHz): δppm = 188.90, 156.43, 154.95, 152.47, 151.61, 151.58, 149.27, 145.66, 145.46, 135.75, 130.47, 127.67, 123.32, 119.42, 119.20, 111.12, 110.09, 103.96, 56.04, 55.99, 30.58, 29.91, 27.88. IR (KBr): 3035, 2968, 1704, 1694, 1660, 1550, 1462, 1270, 1049 cm−1. MS (EI): m/z (%) = 476 (28.3) [M+]. Anal. calc. for C25H24N4O6: C, 63.02; H, 5.08; N, 11.76; found: C, 62.86; H, 5.21; N, 11.90.
4.4.5.8 (E)-8-(4-(3-(4-Methoxyphenyl)acryloyl)phenoxy)-1,3,7-trimethyl-1H-purine-2,6(3H,7H)-dione (4h). Compound 4h was obtained according to the general procedure A from a mixture of 8-BC (2.73 g, 10 mmol), (E)-1-(4-hydroxyphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (2.54 g, 10 mmol), and K2CO3 (1.38, 10 mmol) in anhydrous DMF (20 mL) after 14 h as creamy solid (4.10 g, 92%); m.p.: 153–155 °C, 1H NMR (CDCl3, 400 MHz): δppm = 8.13 (d, J = 8.8 Hz, 2H, aryl), 7.85 (d, J = 15.6 Hz, 1H, C(O)–CH), 7.64 (d, J = 8.8 Hz, 2H, aryl), 7.48–7.41 (complex, 3H, C(O)–CH
CH, aryl), 6.97 (d, J = 8.8 Hz, 2H, aryl), 3.91 (s, 3H, OCH3), 3.88 (s, 3H, N(7)-CH3), 3.49 (s, 3H, N(3)-CH3), 3.42 (s, 3H, N(1)-CH3). 13C NMR (CDCl3, 100 MHz): δppm = 188.87, 161.84, 156.41, 154.96, 152.50, 151.60, 145.68, 145.12, 135.79, 130.45, 130.33, 127.44, 119.20, 119.15, 114.48, 103.97, 55.46, 30.58, 29.92, 27.89. IR (KBr): 3085, 2947, 1706, 1699, 1656, 1550, 1472, 1283, 1060 cm−1. MS (EI): m/z (%) = 446 (24.9) [M+]. Anal. calc. for C24H22N4O5: C, 64.57; H, 4.97; N, 12.55; found: C, 64.76; H, 5.12; N, 12.69.
4.4.5.9 (E)-8-(4-(3-(2-Chlorophenyl)acryloyl)phenoxy)-1,3,7-trimethyl-1H-purine-2,6(3H,7H)-dione (4i). Compound 4i was obtained according to the general procedure A from a mixture of 8-BC (2.73 g, 10 mmol), (E)-3-(2-chlorophenyl)-1-(4-hydroxyphenyl)prop-2-en-1-one (2.58 g, 10 mmol), and K2CO3 (1.38, 10 mmol) in anhydrous DMF (20 mL) after 13 h as yellow solid (4.05 g, 90%); m.p.: 173–175 °C, 1H NMR (CDCl3, 400 MHz): δppm = 8.22 (d, J = 15.6 Hz, 1H, C(O)–CH), 8.12 (d, J = 8.8 Hz, 2H, aryl), 7.76–7.74 (m, 1H, C(O)–CH
CH) 7.51–7.44 (m, 4H, aryl), 7.37–7.30 (m, 2H, aryl), 3.90 (s, 3H, N(7)-CH3), 3.47 (s, 3H, N(3)-CH3), 3.41 (s, 3H, N(1)-CH3). 13C NMR (CDCl3, 100 MHz): δppm = 188.80, 156.67, 154.93, 152.40, 151.57, 145.64, 141.00, 135.54, 135.15, 133.04, 131.38, 130.69, 130.36, 127.79, 127.13, 124.26, 119.30, 103.97, 30.58, 29.92, 27.89. IR (KBr): 3037, 2962, 1705, 1696, 1658, 1557, 1479, 1270, 1054, 857 cm−1. MS (EI): m/z (%) = 450 (26.7) [M+]. Anal. calc. for C23H19ClN4O4: C, 61.27; H, 4.25; N, 12.43; found: C, 61.41; H, 4.09; N, 12.26.
4.4.5.10 (E)-8-(4-(3-(2,6-Dichlorophenyl)acryloyl)phenoxy)-1,3,7-trimethyl-1H-purine-2,6(3H,7H)-dione (4j). Compound 4j was obtained according to the general procedure A from a mixture of 8-BC (2.73 g, 10 mmol), (E)-3-(2,6-dichlorophenyl)-1-(4-hydroxyphenyl)prop-2-en-1-one (2.93 g, 10 mmol), and K2CO3 (1.38, 10 mmol) in anhydrous DMF (20 mL) after 16 h as green solid (4.27 g, 88%); m.p.: 216–218 °C, 1H NMR (CDCl3, 400 MHz): δppm = 8.13 (d, J = 8.8 Hz, 2H, aryl), 7.91 (d, J = 16.0 Hz, 1H, C(O)–CH), 7.70 (d, J = 16.0 Hz, 1H, C(O)–CH
CH), 7.49 (d, J = 8.4 Hz, 2H, aryl), 7.41 (d, J = 8.0 Hz, 2H, aryl), 7.25 (t, J = 8.0 Hz, 1H, aryl), 3.91 (s, 3H, N(7)-CH3), 3.48 (s, 3H, N(3)-CH3), 3.41 (s, 3H, N(1)-CH3). 13C NMR (CDCl3, 100 MHz): δppm = 188.56, 156.81, 154.94, 152.36, 151.58, 145.64, 138.19, 135.19, 134.90, 132.43, 130.82, 130.02, 129.99, 128.92, 119.34, 103.97, 30.59, 29.93, 27.89. IR (KBr): 3070, 2978, 1703, 1695, 1648, 1559, 1467, 1281, 1053, 859 cm−1. MS (EI): m/z (%) = 484 (19.4) [M+]. Anal. calc. for C23H18Cl2N4O4: C, 56.92; H, 3.74; N, 11.54; found: C, 57.10; H, 3.90; N, 11.67.
4.4.5.11 (E)-Methyl 4-(3-oxo-3-(4-(1,3,7-trimethyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yloxy)phenyl)prop-1-enyl)benzoate (4k). Compound 4k was obtained according to the general procedure A from a mixture of 8-BC (2.73 g, 10 mmol), (E)-methyl 4-(3-(4-hydroxyphenyl)-3-oxoprop-1-enyl)benzoate (2.82 g, 10 mmol), and K2CO3 (1.38, 10 mmol) in anhydrous DMF (20 mL) after 15 h as pale yellow solid (4.22 g, 89%); m.p.: 235–237 °C, 1H NMR (DMSO-d6, 400 MHz): δppm = 8.30 (d, J = 8.4 Hz, 2H, aryl), 8.03–7.95 (complex, 3H, C(O)–CH, aryl), 7.88 (d, J = 3.2 Hz, 2H, aryl), 7.79 (d, J = 15.6 Hz, 1H, C(O)–CH
CH), 7.58 (d, J = 8.8 Hz, 2H, aryl), 3.80 (s, 3H, N(7)-CH3), 3.30 (s, 3H, N(3)-CH3), 3.23 (s, 3H, N(1)-CH3), 2.09 (s, 3H, OCH3). 13C NMR (DMSO-d6, 100 MHz): δppm = 188.82, 170.34, 161.86, 156.85, 154.89, 152.61, 151.12, 145.95, 144.90, 135.81, 130.74, 129.60, 127.37, 119.94, 118.95, 114.71, 103.93, 54.02, 30.60, 29.90, 27.82. IR (KBr): 3067, 2958, 1730, 1705, 1697, 1659, 1550, 1477, 1279, 1180, 1061 cm−1. MS (EI): m/z (%) = 474 (21.7) [M+]. Anal. calc. for C25H22N4O6: C, 63.29; H, 4.67; N, 11.81; found: C, 63.41; H, 4.82; N, 11.65.
4.4.7.1 (E)-8-(4-(3-(4-Methoxyphenyl)-3-oxoprop-1-enyl)phenoxy)-1,3,7-trimethyl-1H-purine-2,6(3H,7H)-dione (5a). Compound 5a was obtained according to the general procedure B from a mixture of 8-BC (2.73 g, 10 mmol), (E)-3-(4-hydroxyphenyl)-1-(4-methoxyphenyl)prop-2-en-1-one (2.54 g, 10 mmol), and K2CO3 (1.38, 10 mmol) in anhydrous DMF (20 mL) after 16 h as brown solid (4.15 g, 93%); m.p.: 139–141 °C, 1H NMR (CDCl3, 400 MHz): δppm = 8.07–8.02 (complex, 3H, C(O)–CH
CH, aryl), 7.83–7.70 (complex, 3H, C(O)–CH, aryl), 7.56–7.52 (m, 2H, aryl), 7.43–7.38 (m, 2H, aryl), 3.91 (s, 3H, OCH3), 3.89 (s, 3H, N(7)-CH3), 3.48 (s, 3H, N(3)-CH3), 3.43 (s, 3H, N(1)-CH3). 13C NMR (CDCl3, 100 MHz): δppm = 188.57, 163.61, 159.01, 154.58, 151.64, 144.33, 142.57, 132.63, 130.87, 129.86, 127.16, 122.07, 119.84, 116.07, 113.79, 104.02, 55.50, 30.53, 29.95, 27.92. IR (KBr): 3045, 2981, 1704, 1695, 1657, 1548, 1460, 1273, 1039 cm−1. MS (EI): m/z (%) = 446 (20.2) [M+]. Anal. calc. for C24H22N4O5: C, 64.57; H, 4.97; N, 12.55; found: C, 64.38; H, 4.81; N, 12.43.
4.4.7.2 (E)-1,3,7-Trimethyl-8-(4-(3-(4-nitrophenyl)-3-oxoprop-1-enyl)phenoxy)-1H-purine-2,6(3H,7H)-dione (5b). Compound 5b was obtained according to the general procedure B from a mixture of 8-BC (2.73 g, 10 mmol), (E)-3-(4-hydroxyphenyl)-1-(4-nitrophenyl)prop-2-en-1-one (2.69 g, 10 mmol), and K2CO3 (1.38, 10 mmol) in anhydrous DMF (20 mL) after 18 h as pale orange solid (3.69 g, 80%); m.p.: 250–252 °C, 1H NMR (CDCl3, 400 MHz): δppm = 8.40 (d, J = 8.8 Hz, 2H, aryl), 8.18 (d, J = 8.4 Hz, 2H, aryl), 7.90 (d, J = 16.0 Hz, 1H, C(O)–CH
CH), 7.77 (d, J = 8.8 Hz, 2H, aryl), 7.51–7.43 (complex, 3H, C(O)–CH, aryl), 3.92 (s, 3H, N(7)-CH3), 3.50 (s, 3H, N(3)-CH3), 3.43 (s, 3H, N(1)-CH3). 13C NMR (CDCl3, 100 MHz): δppm = 188.74, 155.25, 152.59, 151.61, 150.13, 145.27, 143.20, 142.88, 131.78, 130.86, 130.27, 129.42, 123.93, 121.40, 119.97, 106.21, 30.56, 29.92, 27.90. IR (KBr): 3036, 2975, 1706, 1698, 1656, 1559, 1530, 1450, 1341, 1267, 1052 cm−1. MS (EI): m/z (%) = 461 (25.8) [M+]. Anal. calc. for C23H19N5O6: C, 59.87; H, 4.15; N, 15.18; found: C, 59.69; H, 4.03; N, 15.01.
4.4.7.3 (E)-1,3,7-Trimethyl-8-(4-(3-(3-nitrophenyl)-3-oxoprop-1-enyl)phenoxy)-1H-purine-2,6(3H,7H)-dione (5c). Compound 5c was obtained according to the general procedure B from a mixture of 8-BC (2.73 g, 10 mmol), (E)-3-(4-hydroxyphenyl)-1-(3-nitrophenyl)prop-2-en-1-one (2.69 g, 10 mmol), and K2CO3 (1.38, 10 mmol) in anhydrous DMF (20 mL) after 16 h as yellow solid (3.92 g, 85%); m.p.: 248–250 °C, 1H NMR (CDCl3, 400 MHz): δppm = 8.87 (br s, 1H, C(O)–CH
CH), 8.49–8.39 (m, 2H, aryl), 7.94–7.78 (complex, 4H, C(O)–CH, aryl), 7.56–7.46 (m, 3H, aryl), 3.92 (s, 3H, N(7)-CH3), 3.50 (s, 3H, N(3)-CH3), 3.43 (s, 3H, N(1)-CH3). 13C NMR (CDCl3, 100 MHz): δppm = 188.93, 155.67, 154.86, 152.75, 151.65, 145.82, 143.75, 142.64, 135.68, 132.64, 130.17, 129.46, 128.26, 127.65, 126.97, 122.57, 119.98, 104.04, 30.69, 29.81, 27.89. IR (KBr): 3050, 2968, 1705, 1693, 1658, 1564, 1545, 1479, 1350, 1282, 1051 cm−1. MS (EI): m/z (%) = 461 (30.6) [M+]. Anal. calc. for C23H19N5O6: C, 59.87; H, 4.15; N, 15.18; found: C, 60.04; H, 4.29; N, 15.37.
4.4.7.4 (E)-1,3,7-Trimethyl-8-(4-(3-oxo-3-p-tolylprop-1-enyl)phenoxy)-1H-purine-2,6(3H,7H)-dione (5d). Compound 5d was obtained according to the general procedure B from a mixture of 8-BC (2.73 g, 10 mmol), (E)-3-(4-hydroxyphenyl)-1-p-tolylprop-2-en-1-one (2.38 g, 10 mmol), and K2CO3 (1.38, 10 mmol) in anhydrous DMF (20 mL) after 13 h as creamy solid (3.44 g, 80%); m.p.: 201–203 °C, 1H NMR (CDCl3, 400 MHz): δppm = 7.96 (d, J = 8.0 Hz, 2H, aryl), 7.83 (d, J = 16.0 Hz, 1H, C(O)–CH
CH), 7.73 (d, J = 8.8 Hz, 2H, aryl), 7.55 (d, J = 15.6 Hz, 1H, C(O)–CH), 7.41 (d, J = 8.8 Hz, 2H, aryl), 7.33 (d, J = 8.0 Hz, 2H, aryl), 3.90 (s, 3H, N(7)-CH3), 3.48 (s, 3H, N(3)-CH3), 3.42 (s, 3H, N(1)-CH3), 2.45 (s, 3H, PhCH3). 13C NMR (CDCl3, 100 MHz): δppm = 189.65, 154.94, 154.65, 152.79, 151.60, 145.72, 143.86, 142.87, 135.47, 132.53, 129.89, 129.40, 128.65, 122.24, 119.82, 103.87, 30.52, 29.91, 27.87, 21.72. IR (KBr): 3072, 2980, 1707, 1696, 1645, 1566, 1457, 1270, 1063 cm−1. MS (EI): m/z (%) = 430 (18.2) [M+]. Anal. calc. for C24H22N4O4: C, 66.97; H, 5.15; N, 13.02; found: C, 66.85; H, 5.34; N, 13.16.
4.4.7.5 (E)-1,3,7-Trimethyl-8-(4-(3-(naphthalen-2-yl)-3-oxoprop-1-enyl)phenoxy)-1H-purine-2,6(3H,7H)-dione (5e). Compound 5e was obtained according to the general procedure B from a mixture of 8-BC (2.73 g, 10 mmol), (E)-3-(4-hydroxyphenyl)-1-(naphthalen-2-yl)prop-2-en-1-one (2.74 g, 10 mmol), and K2CO3 (1.38, 10 mmol) in anhydrous DMF (20 mL) after 13 h as brown solid (4.05 g, 87%); m.p.: 175–177 °C, 1H NMR (CDCl3, 400 MHz): δppm = 8.13 (d, J = 15.6 Hz, 1H, C(O)–CH
CH), 8.02–7.87 (complex, 5H, C(O)–CH, aryl), 7.78 (d, J = 8.4 Hz, 2H, aryl), 7.72–7.61 (m, 3H, aryl), 7.44 (d, J = 8.8 Hz, 2H, aryl), 3.91 (s, 3H, N(7)-CH3), 3.49 (s, 3H, N(3)-CH3), 3.42 (s, 3H, N(1)-CH3). 13C NMR (CDCl3, 100 MHz): δppm = 190.03, 154.92, 154.74, 152.79, 143.30, 135.54, 135.38, 132.53, 132.49, 130.02, 129.52, 128.68, 128.54, 127.87, 127.80, 126.90, 124.41, 122.22, 119.88, 103.87, 30.54, 29.94, 27.90. IR (KBr): 3046, 2968, 1705, 1697, 1660, 1559, 1474, 1283, 1050 cm−1. MS (EI): m/z (%) = 466 (35.3) [M+]. Anal. calc. for C27H22N4O4: C, 69.52; H, 4.75; N, 12.01; found: C, 69.64; H, 4.91; N, 12.16.
4.4.7.6 (E)-8-(4-(3-(4-Chlorophenyl)-3-oxoprop-1-enyl)phenoxy)-1,3,7-trimethyl-1H-purine-2,6(3H,7H)-dione (5f). Compound 5f was obtained according to the general procedure B from a mixture of 8-BC (2.73 g, 10 mmol), (E)-1-(4-chlorophenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (2.58 g, 10 mmol), and K2CO3 (1.38, 10 mmol) in anhydrous DMF (20 mL) after 14 h as pale brown solid (3.74 g, 83%); m.p.: 182–184 °C, 1H NMR (CDCl3, 400 MHz): δppm = 7.98 (d, J = 8.4 Hz, 2H, aryl), 7.84 (d, J = 15.6 Hz, 1H, C(O)–CH
CH), 7.73 (d, J = 8.4 Hz, 2H, aryl), 7.50–7.485(complex, 3H, C(O)–CHCH, aryl), 7.42 (d, J = 8.4 Hz, 2H, aryl), 3.90 (s, 3H, N(7)-CH3), 3.47 (s, 3H, N(3)-CH3), 3.41 (s, 3H, N(1)-CH3). 13C NMR (CDCl3, 100 MHz): δppm = 188.89, 154.87, 152.73, 151.59, 145.71, 143.83, 139.39, 136.32, 132.20, 130.03, 129.90, 129.00, 121.63, 119.88, 116.10, 103.92, 30.53, 29.91, 27.89. IR (KBr): 3070, 2974, 1703, 1694, 1656, 1563, 1471, 1269, 1065, 850 cm−1. MS (EI): m/z (%) = 450 (28.7) [M+]. Anal. calc. for C23H19ClN4O4: C, 61.27; H, 4.25; N, 12.43; found: C, 61.02; H, 4.38; N, 12.27.
Data availability
The data pertaining to this manuscript are available upon request.Conflicts of interest
The authors have declared no conflict of interest.Acknowledgements
The authors wish to thank the Shiraz University of Technology research council for partial support of this work.References
- C. de Martel, D. Georges, F. Bray, J. Ferlay and G. M. Clifford, Global burden of cancer attributable to infections in 2018: a worldwide incidence analysis, Lancet Glob. Health, 2020, 8, e180–e190 CrossRef PubMed.
- E. Bidram, Y. Esmaeili, H. Ranji-Burachaloo, N. Al-Zaubai, A. Zarrabi, A. Stewart and D. E. Dunstan, A concise review on cancer treatment methods and delivery systems, J. Drug Deliv. Technol., 2019, 54, 101350 CrossRef CAS.
- S. Akkın, G. Varan and E. Bilensoy, A review on cancer immunotherapy and applications of nanotechnology to chemoimmunotherapy of different cancers, Molecules, 2021, 26, 3382 CrossRef PubMed.
- J. Zugazagoitia, C. Guedes, S. Ponce, I. Ferrer, S. Molina-Pinelo and L. Paz-Ares, Current challenges in cancer treatment, Clin. Ther., 2016, 38, 1551–1566 CrossRef PubMed.
- S. h. Wang, Y. Liu, Y. Feng, J. Zhang, J. Swinnen, Y. Li and Y. Ni, A review on curability of cancers: more efforts for novel therapeutic options are needed, Cancers, 2019, 11, 1782 CrossRef CAS PubMed.
- M. A. Shalaby, S. A. Rizk and A. M. Fahim, Synthesis, reactions and application of chalcones: a systematic review, Org. Biomol. Chem., 2023, 21, 5317–5346 RSC.
- C. Zhuang, W. Zhang, C. Sheng, W. Zhang, C. Xing and Z. Miao, Chalcone: a privileged structure in medicinal chemistry, Chem. Rev., 2017, 117, 7762–7810 CrossRef CAS PubMed.
- S. Narwal, B. Devi, T. Dhanda, S. Kumar and S. Tahlan, Exploring chalcone derivatives: synthesis and their therapeutic potential, J. Mol. Struct., 2024, 1303, 137554 CrossRef CAS.
- M. N. Gomes, E. N. Muratov, M. Pereira and J. C. Peixoto, Chalcone derivatives: promising starting points for drug design, Molecules, 2017, 22, 1210 CrossRef PubMed.
- K. Mezgebe, Y. Melaku and E. Mulugeta, Synthesis and pharmacological activities of chalcone and its derivatives bearing N-heterocyclic scaffolds: a review, ACS Omega, 2023, 8, 19194–19211 CrossRef CAS PubMed.
- K. Dzobo, The role of natural products as sources of therapeutic agents for innovative drug discovery, Compr. Pharm., 2022, 2, 408–422 Search PubMed.
- B. G. Katzung, Basic & Clinical Pharmacology, Lange Medical Publications, McGraw-Hill Companies, 14 edn, 2018 Search PubMed.
- T. M. McLellan, J. A. Caldwell and H. R. Lieberman, A review of caffeine's effects on cognitive, physical and occupational performance, Neurosci. Biobehav. Rev., 2016, 71, 294–312 CrossRef CAS PubMed.
- N. Singh, A. K. Shreshtha, M. S. Thakur and S. Patra, Xanthine scaffold: scope and potential in drug development, Heliyon, 2018, 4, e00829 CrossRef PubMed.
- M. N. Soltani Rad, S. Behrouz, S. Aghajani, M. Behrouz, E. Zarenezhad and A. Ghanbariasad, Design, synthesis, anticancer and in silico assessment of 8-caffeinyl-triazolylmethoxy hybrid conjugate, RSC Adv., 2023, 13, 3056–3070 RSC.
- B. Strydom, J. J. Bergh and J. P. Petzer, 8-Aryl- and alkyloxycaffeine analogues as inhibitors of monoamine oxidase, Eur. J. Med. Chem., 2011, 46, 3474–3485 CrossRef CAS PubMed.
- M. M. van der Walt and G. Terre'Blanche, Selected C8 two-chain linkers enhance the adenosine A1/A2A receptor affinity and selectivity of caffeine, Eur. J. Med. Chem., 2017, 125, 652–656 CrossRef CAS PubMed.
- A. A. Kadi, K. E. H. El-Tahir, Y. Jahng and A. F. M. Motiur Rahman, Synthesis, biological evaluation and structure activity relationships (SARs) study of 8-(substituted)aryloxycaffeine, Arab. J. Chem., 2019, 12, 2356–2364 CrossRef CAS.
- A. H. Alkhzem, T. J. Woodman and I. S. Blagbrough, Design and synthesis of hybrid compounds as novel drugs and medicines, RSC Adv., 2022, 12, 19470–19484 RSC.
- B. Meunier, Hybrid molecules with a dual mode of action: dream or reality?, Acc. Chem. Res., 2008, 41, 69–77 CrossRef CAS PubMed.
- M. d. O. Pedrosa, R. M. D. d. Cruz, J. d. O. Viana, R. O. d. Moura, H. M. Ishiki, J. M. B. Filho, M. F. F. M. Diniz, M. T. Scotti, L. Scotti and F. J. B. Mendonca, Hybrid compounds as direct multitarget ligands: a review, Curr. Top. Med. Chem., 2017, 17, 1044–1079 CrossRef CAS PubMed.
- M. N. Soltani Rad and S. Maghsoudi, Two-step three-component process for one-pot synthesis of 8-alkylmercaptocaffeine derivatives, RSC Adv., 2016, 6, 70335–70342 RSC.
- M. N. Soltani Rad, S. Behrouz, K. Zokaei, M. Behrouz, A. Ghanbariasad and E. Zarenezhad, Synthesis of some novel 8-(4-alkylpiperazinyl) caffeine derivatives as potent anti-leishmania agents, Bioorg. Chem., 2022, 128, 106062 CrossRef PubMed.
- M. N. Soltani Rad, S. Behrouz, K. Shahbazkhani, M. Behrouz, E. Zarenezhad and A. Ghanbariasad, Design, synthesis, anticancer and in silico assessment of 8-piperazinyl caffeinyl-triazolylmethyl hybrid conjugates, RSC Adv., 2023, 13, 24656–24673 RSC.
- L. Claisen and A. Claparède, Condensationen von ketonen mit aldehyden, Ber. Dtsch. Chem. Ges., 1881, 14, 2460–2468 CrossRef.
- X. Zhang, Y. Q. Li, C. Qian, L. An, W. Wang, X. F. Li, X. Z. Shao and Z. Lia, Research progress of catalysts for aldol condensation of biomass based compounds, RSC Adv., 2023, 13, 9466–9478 RSC.
- E. Caputo, L. Maiorana, V. Vasta, F. M. Pezzino, S. Sunkara, K. Wynne, G. Elia, F. M. Marincola, J. A. McCubrey, M. Libra, S. Travali and M. Kane, Characterization of human melanoma cell lines and melanocytes by proteome analysis, Cell Cycle, 2011, 10, 2924–2936 CrossRef CAS PubMed.
- Ş. Comşa, A. M. Cimpean and M. Raica, The story of MCF-7 breast cancer cell line: 40 years of experience in research, Anticancer Res., 2015, 35, 3147–3154 Search PubMed.
- Y.-C. Lin, M. Boone, L. Meuris, I. Lemmens, N. Van Roy, A. Soete, J. Reumers, M. Moisse, S. Plaisance, R. Drmanac, J. Chen, F. Speleman, D. Lambrechts, Y. Van de Peer, J. Tavernier and N. Callewaert, Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations, Nat. Commun., 2014, 5, 4767 CrossRef CAS PubMed.
- https://en.wikipedia.org/wiki/Folate, accessed 11 June 2024.
- B. Chabner, and D. L. Longo, Cancer Chemotherapy and Biotherapy Principles and Practice, Oxford University press, Wolters Kluwer Health/Lippincott Williams & Wilkins, 5th edn, 2011 Search PubMed.
- L. Genestier, R. Paillot, L. Quemeneur, K. Izeradjene and J. P. Revillard, Mechanisms of action of methotrexate, Immunopharmacology, 2000, 47, 247–257 CrossRef CAS PubMed.
- I. Puzanov, R. K. Amaravadi, G. A. McArthur, K. T. Flaherty, P. B. Chapman, J. A. Sosman, A. Ribas, M. Shackleton, P. Hwu, B. Chmielowski, K. B. Nolop, P. S. Lin and K. B. Kim, Long-term outcome in BRAF (V600E) melanoma patients treated with vemurafenib: patterns of disease progression and clinical management of limited progression, Eur. J. Cancer, 2015, 51, 1435–1443 CrossRef CAS PubMed.
- Molegro Virtual Docker, Version 6.0.0, CLC Bio, 8200, Aarhus N, Denmark, 2012 Search PubMed.
- G. Marcou and D. Rognan, Optimizing fragment and scaffold docking by use of molecular interaction fingerprints, J. Chem. Inform. Model, 2007, 47, 195–207 CrossRef CAS PubMed.
- V. Cody, J. R. Luft and W. Pangborn, Understanding the role of Leu22 variants in methotrexate resistance: comparison of wild-type and Leu22Arg variant mouse and human dihydrofolate reductase ternary crystal complexes with methotrexate and NADPH, Acta Cryst., 2005, D61, 147–155 CrossRef CAS PubMed.
- R. J. Bienstock, Library Design, Search Methods, and Applications of Fragment-Based Drug Design, A.C.S., Washington, 2011 Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliv. Rev., 2001, 46, 3–25 CrossRef CAS PubMed.
- PreADMET online server, https://preadmet.webservice.bmdrc.org/adme, accessed 2 June 2024 Search PubMed.
- OSIRIS Property Explorer, https://www.organic-chemistry.org/prog/peo, accessed 2 June 2024 Search PubMed.
- SwissADME online software, https://www.swissadme.ch/index.php, accessed 2 June 2024 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- J. J. P. Stewart, Application of the PM6 method to modeling the solid state, J. Mol. Model., 2008, 14, 499–535 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04787g |
| This journal is © The Royal Society of Chemistry 2024 |