DOI:
10.1039/D4RA04745A
(Paper)
RSC Adv., 2024,
14, 25703-25711
Methoxycarbonylation of diisobutylene into methyl isononanoate catalyzed by cobalt complexes dispersed by poly(ionic liquids)
Received
30th June 2024
, Accepted 31st July 2024
First published on 28th August 2024
Abstract
The catalytic performance of cobalt complex catalysts coordinated with various poly(ionic liquids) for the methoxycarbonylation of diisobutene into methyl isononanoate was investigated. The poly(ionic liquids) were synthesized via a solvothermal polymerization method and were characterized using Fourier transform infrared spectroscopy, thermogravimetric analysis, scanning electron microscopy, transmission electron microscopy, N2 adsorption–desorption and elemental analyses. A diisobutene conversion of 88.0% and a methyl isononanoate selectivity of 91.4% were achieved using HVIMI-VPy-DVB (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)@Co2(CO)8 as catalysts at the optimized reaction conditions of 8.0 MPa CO and 150 °C. Furthermore, the catalyst system can be suitable for the methoxycarbonylation of various terminal olefins and exhibits high recoverability and thermostability.
:1)@Co2(CO)8 as catalysts at the optimized reaction conditions of 8.0 MPa CO and 150 °C. Furthermore, the catalyst system can be suitable for the methoxycarbonylation of various terminal olefins and exhibits high recoverability and thermostability.
1. Introduction
Carbonylation is an important means for introducing carbonyl groups into organic molecules, such as those widely used in the chemical industry and organic synthesis.1 Roelen was the first to report the carbonylation reaction process in 1938, and it has more than 80 years history of research and application. Precious metal catalysts of Pd, Ru, and Rh as well as non-precious metal catalysts of Co and Ni have been successfully used in carbonylation reactions.2–8 Since homogeneous catalysts possess the same physical state as the reaction medium, it is difficult to separate and recover them from the reaction system at the end of the reaction;9–11 moreover, complex separation and purification techniques are usually required, which increases production costs. Traditional heterogeneous catalysts are easily separated and recovered from reactants, but their catalytic activities and selectivities are often not ideal. Nowadays, heterogenization of homogeneous catalysts has become a great solution,12–16 which offers both the excellent performance of homogenous catalysts and recyclability of heterogeneous catalysts. However, precious metal catalysts are preferred. For example, Doherty et al.17 reported for the first time that the in situ combination of 2-pyridyldiphenylphosphine anchored on polymers with Pd(OAc)2 forms a heterogeneous complex catalyst, which exhibited excellent catalytic performance for the hydroesterification reaction of phenylacetylene and propargyl acetylene, achieving homogeneous catalytic multiphasing. In 2016, Chen et al.18 synthesized an immobilized Pd/POL-2V-P,N catalyst by anchoring Pd(OAc)2 over a 2-vinyl-functional diphenyl-2-pyridylphosphine polymer (POL-2V-P,N) for the methoxycarbonylation of acetylene. Pd/POL-2V-P,N showed more superior catalytic activity in the catalyzed methoxycarbonylation reaction than the homogeneous catalyst system. Similarly, Ren et al.19 successfully synthesized a porous Rh-based N-containing polymeric materials via an impregnation method, which showed good activity and stability in the catalytic gas-phase carbonylation of methanol to generate acetic acid and methyl acetate and could be operated stably for a much longer time than the corresponding homogeneous catalyst system. In 2016, Yang et al.20 prepared a Y-type molecular sieve-loaded Mo(CO)6 catalyst, which demonstrated excellent activity in the synthesis of propionic acid through the carbonylation of ethylene; the activity of the catalyst was at least 400-fold higher than that of similar homogeneous catalysts. The use of non-precious metal catalysts such as Co is beneficial for reducing the cost of the catalyst, and therefore, it is an important direction for future carbonylation research.
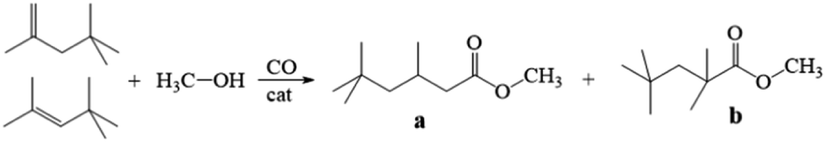
Isononanoic acid and its derivatives are important intermediates of driers for paints and coatings and can be used in the production of paints, lubricants, plasticizers, surfactants, pharmaceutical intermediates, cosmetics, and peroxides. Currently, isononanoic acid is mainly produced from 2-ethylhexanol via a complicated process of dehydration, hydroformylation, and oxidation with low yield, environmental pollution, and high equipment requirements. The preparation of isononanoic acid from the methoxycarbonylation of diisobutene (DIB) is a promising route in the future, at present, a few homogeneous catalytic systems have been applied to the methoxycarbonylation of DIB.21–23 In 2022, our group24 synthesized porous organic polymers bearing pyridine or imidazole complexed Co2(CO)8, which exhibited excellent catalytic activity for the methoxycarbonylation of DIB with methanol and CO. The conversion and selectivity of methyl 3,3,5-trimethylhexanate were up to 89.3% and 92.2% at the reaction conditions of 150 °C and 8.0 MPa for 12 h, respectively. Subsequently, our group25 formed supported cobalt complex catalysts by loading cobalt complex over N-containing ligand-functionalized ZSM-5, in situ, which were used as catalysts. The conversion of 88.3% and selectivity of 93.4% for methyl isononanoate in the methoxycarbonylation of DIB was achieved under solvent-free conditions at 6.0 MPa and 140 °C. Poly(ionic liquids) combines the advantages of ordinary polymers and ionic liquids with a porous structure, such as good thermal and chemical stability, and good solubility for organic substances on its surface, which are widely used in catalytic reactions. Poly(ionic liquids) can not only coordinate with metallic catalytic centers, but also improve the solubility of the substrate on its surface by changing the structure, which could promotes the catalytic reaction.
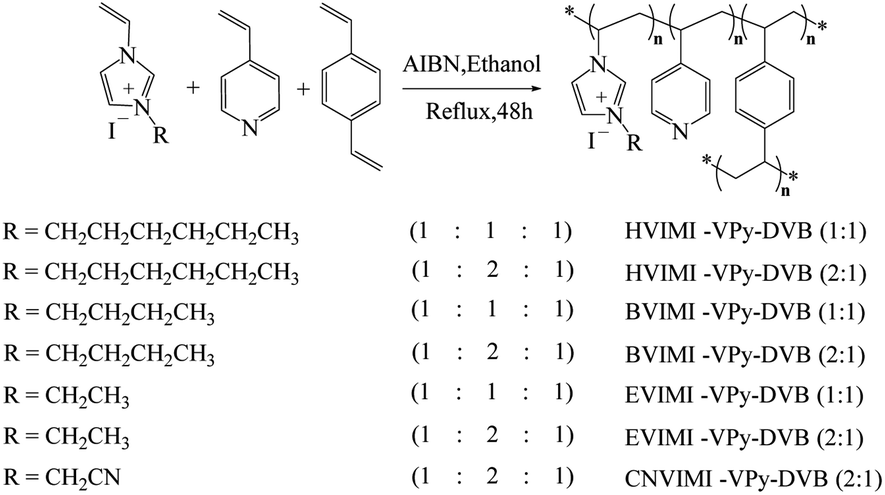
In this study, we synthesized the poly(ionic liquids) supports via co-polymerizing N-vinyl imidazolium iodide containing different side chains with 4-vinylpyridine and p-divinylbenzene. Subsequently, the heterogeneous Co-based catalysts formed in the presence of Co precursor and the poly(ionic liquids) in situ, whose performance was investigated in the methoxycarbonylation of DIB. We selected 1-vinyl-3-hexyl-imidazolium iodide, 1-vinyl-3-butyl-imidazolium iodide, 1-vinyl-3-ethyl-imidazolium iodide, and 1-acetonitrile-3-vinyl-imidazolium iodide as the polymeric monomers, which were polymerized with 4-vinylpyridine and p-divinylbenzene in the presence of an initiator. The structures and properties of the obtained poly(ionic liquids) were comprehensively characterized using Fourier transform infrared spectroscopy (FT-IR), elemental analysis, thermogravimetric analysis (TG), N2 adsorption–desorption (BET), scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Simultaneously, the effects of reaction parameters such as cobalt salt species, catalyst dosage, feedstock ratio, pressure, temperature, and time on the reaction were also investigated to determine the optimal reaction conditions. Besides, a reasonable structure–function relationship was examined. Additionally, catalyst recyclability and substrate scope were also measured.
2. Experimental section
2.1 Materials
Analytically pure Co2(CO)8 (≥99.5%, AR), CoCO3 (98%, Co content: 45–47 wt%, Macklin), Co(acac)3 (98%, Macklin), 1-iodohexane (≥98%, Macklin), 1-iodobutane (98%, Macklin), iodoethane (99%, Macklin), iodoacetonitrile (99%, Macklin), 4-vinylpyridine (96 wt%, Macklin), N-vinyl imidazole (99 wt%, Macklin), p-divinylbenzene (DVB, 80 wt%, Macklin), diisobutylene (DIB, 98%, Macklin), methanol (AR, Rionlon), ethyl alcohol (AR, Chemical reagent), ethyl acetate (AR, Chemical reagent), 1-octene (98%, Macklin), 1-hexene (99%, Macklin), styrene (AR, Mcklin), and azobisi-sobutyronitrile (AIBN, AR, Damao chemical reagent factory) were obtained. The CO used in this study was of 99.999% purity (none of the drugs used in the experiment were purified).
2.2 Catalyst synthesis
2.2.1 Preparation of ionic liquids. The ionic liquid of N-vinyl imidazolium iodide was prepared as follows. Typically, a mixture of 1-iodohexane (0.3 mol) and toluene (50 mL) was slowly added dropwise to a solution of N-vinyl imidazole (0.3 mol) and toluene (200 mL) while stirring vigorously. Subsequently, the mixed solution was gradually heated to 60 °C and refluxed for 48 h. After cooling to room temperature, the lower layers of the mixture were identified as ionic liquids of 1-vinyl-3-hexyl-imidazolium iodide (HVIMI) and were washed with toluene and ethyl acetate (each 100 mL) three times. The obtained ionic liquid was rotary evaporated at 60 °C for 8 h, and then vacuum-dried at 60 °C for 12 h to remove the remaining water and volatiles.26–29 The other ionic liquids of 1-vinyl-3-butyl-imidazolium iodide (BVIMI), 1-vinyl-3-ethyl-imidazolium iodide (HEIMI) and 1-acetonitrile-3-vinyl-imidazolium iodide (CNVIMI) were prepared by the above method, except replacing 1-iodohexane with 1-iodobutane, iodoethane or iodoacetonitrile, respectively.EVIMI. 1H NMR (500 MHz, D2O): δ 7.90 (d, J = 2.2 Hz, 1H), 7.74 (d, J = 2.2 Hz, 1H), 7.28 (t, J = 8.7 Hz, 1H), 5.93 (d, J = 2.8 Hz, 1H), 5.54 (d, J = 2.8 Hz, 1H), 4.41 (q, J = 7.4 Hz, 2H), 1.66 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, D2O): δ 128.43, 122.80, 119.60, 109.63, 45.52, 14.74. Anal. calcd for EVIMI: C, 33.62; H, 4.43; N, 11.20. Found: C, 32.28; H, 3.79; N, 11.53.
BVIMI. 1H NMR (500 MHz, D2O): δ 7.86 (d, J = 2.1 Hz, 1H), 7.68 (d, J = 2.1 Hz, 1H), 7.23 (t, J = 2.9 Hz, 1H), 5.89 (d, J = 3.0 Hz, 1H), 5.50 (d, J = 8.7 Hz, 1H) 4.32 (t, J = 2.3 Hz, 2H), 1.94 (m, 2H), 1.40 (m, 2H), 0.98 (t, J = 1.5 Hz, 3H). 13C NMR (126 MHz, D2O): δ 128.48, 123.07, 119.54, 109.59, 49.88, 31.25, 18.98, 12.99. Anal. calcd for BVIMI: C, 38.87; H, 5.44; N, 10.08. Found: C, 37.69; H, 5.89; N, 10.14.
HVIMI. 1H NMR (500 MHz, D2O): δ 7.82 (d, 1H), 7.63 (d, J = 2.4 Hz, 1H), 7.18 (t, J = 8.7 Hz, 1H), 5.84 (d, J = 2.8 Hz, 1H), 5.46 (d, J = 3.0 Hz, 1H), 4.28 (t, J = 2.7 Hz, 2H), 1.98–1.91 (m, 2H), 1.46–1.23 (m, 6H), 0.88 (t, J = 2.1 Hz, 3H). 13C NMR (126 MHz, D2O): δ 128.26, 122.91, 119.47, 109.39, 50.03, 30.36, 29.06, 25.04, 21.81, 13.32. Anal. calcd for HVIMI: C, 43.15; H, 6.25; N, 9.15. Found: C, 42.87; H, 6.95; N, 9.11.
CNVIMI. 1H NMR (500 MHz, D2O): δ 7.98 (d, J = 1.0 Hz, 1H), 7.85 (d, J = 2.0 Hz, 1H), 7.27(t, J = 8.5, 1H), 5.94 (d, J = 3.1 Hz, 1H), 5.60 (d, J = 3.0, Hz, 1H), 4.79 (s, 2H). 13C NMR (126 MHz, D2O) δ 128.07, 123.18, 120.50, 111.13. Anal. calcd for CNVIMI: C, 32.21, H, 3.09; N, 16.10. Found: C, 32.85, H, 3.29; N, 15.46.
2.2.2 Preparation of poly(ionic liquids). As shown in Fig. 1, the poly(ionic liquids) were prepared via the solvothermal polymerization method.30 The ionic liquids of EVIMI, BVIMI, HVIMI, or CNVIMI (0.1 mol) were mixed with 4-vinylpyridine (0.1 or 0.2 mol), p-divinylbenzene (0.1 mol), and azobisisobutyronitrile (AIBN, 3.2 wt%) in 200 mL of ethanol while to form a homogeneous mixture via vigorous stirring. The obtained mixture was then gradually heated to 80 °C and refluxed for 48 h. After the mixture cooled to room temperature and maintained continuously for 12 h, an organic polymer was obtained by centrifugation. After washing with ethanol (100 mL) three times, the solid was dried under vacuum at 60 °C for 12 h to eliminate the residual volatiles and moisture.24,29 Depending on the different monomers, the dried poly(ionic liquids) were denoted as EVIMI-VPy-DVB (x:y), BVIMI-VPy-DVB (x:y), HVIMI-VPy-DVB (x:y), and CNVIMI-VPy-DVB (x:y). Where x:y denotes the molar ratios of 4-vinylpyridine to ionic liquids, respectively. The polymer VPy-DVB (2:1) without any ionic liquid was also prepared as above.
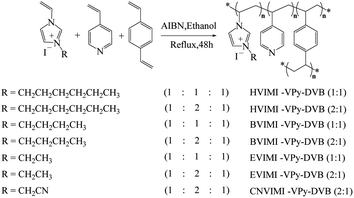 |
| | Fig. 1 Synthetic route of poly(ionic liquids). | |
2.3 Catalysts characterization
FT-IR spectra were obtained by pressing anhydrous potassium bromide with the samples and scanning them in the range of 500–4000 cm−1 using a Nicolet NEXUS 870 system (USA). Thermogravimetric analysis (TG) was performed using a Netzsch STA 449F3 Jupiter instrument operating in an N2 atmosphere in the range of 50–800 °C with a temperature ramping rate of 10°C min−1. The particle size and morphology of the samples were recorded by SEM analysis using a ZEISS Gemini SEM 500 instrument at 10.0 kV. Elemental analysis was performed using an Elementar UNICUBE organic elemental analyzer in the CHNS mode. A Micromeritics ASAP2460 instrument (USA) was used to record the N2 adsorption–desorption isotherms at the liquid nitrogen temperature. The Brunauer–Emmett–Teller (BET) method was used to estimate the surface area. Prior to the measurement, each sample was degassed at 90 °C for 10 h.
2.4 Catalytic performance testing
The methoxycarbonylation reaction of DIB with CO and methanol was carried out in a stainless steel autoclave with a mechanical stirring speed of 500 rpm. Generally, a certain amount of prepared poly(ionic liquids), Co precursor, DIB, and methanol were added to the autoclave. Then, the autoclave was sealed and flushed with CO (2.0 MPa) three times and pressurized to the appropriate pressure. The reaction system was heated to the desired temperature and kept constant during the reaction. After the completion of the reaction, the heating and stirring equipment were turned off, and the reactor was naturally cooled to room temperature. The reaction mixture was subjected to solid–liquid separation by centrifugation. The liquid products and substrates were analyzed using gas chromatography (GC, Agilent 7980) equipped with a SE-54 capillary column and a flame ionization detector (FID) with ethyl acetate as the internal standard. For the recycle experiments, the liquid was pressed out of the reactor through a filter by high pressure, the catalyst was retained at the bottom of the reactor, and the reactants were added directly to the reactor for the next cycle.
3. Results and discussion
3.1 Catalyst characterization
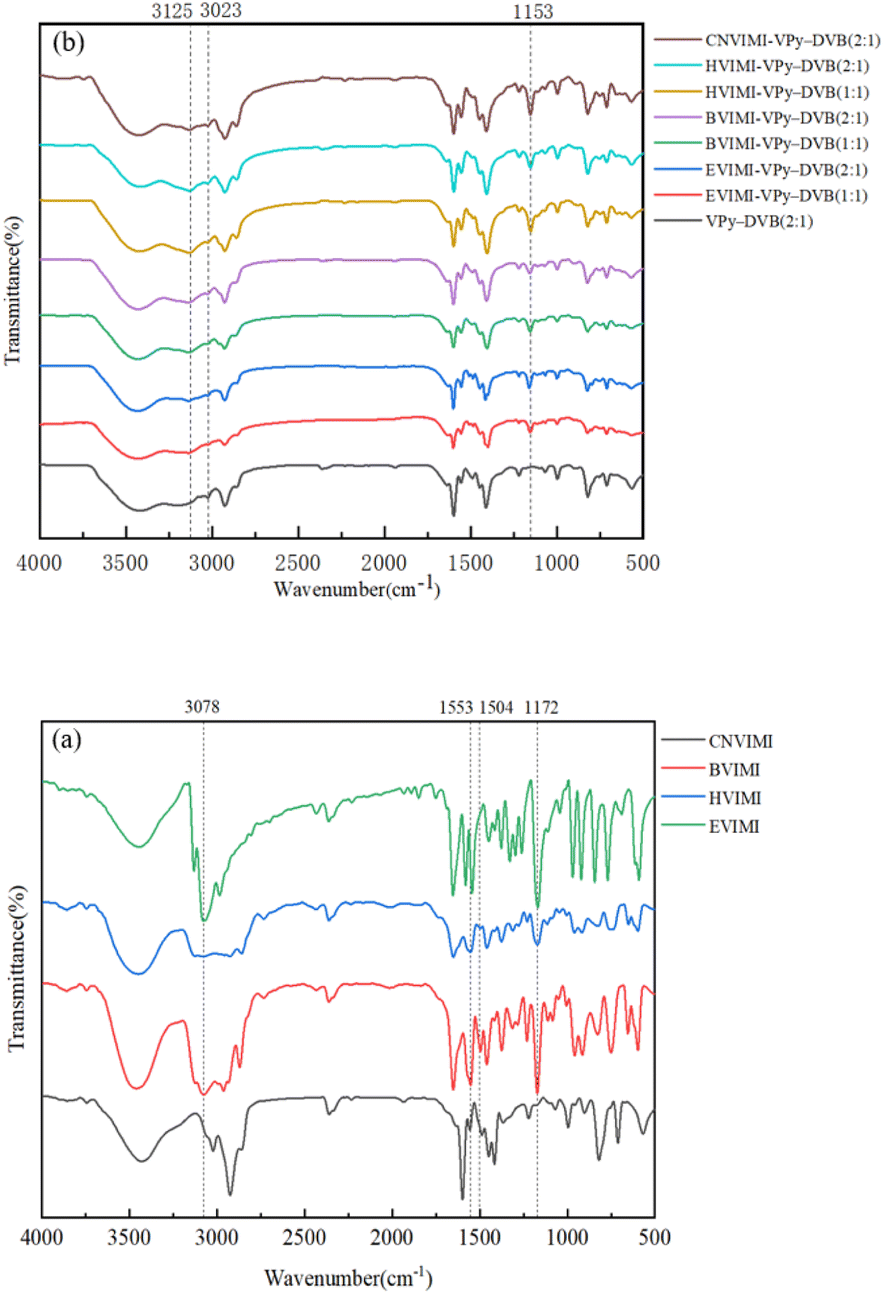
3.1.1 FT-IR spectra. The FT-IR spectra of the ionic liquids of N-vinyl imidazolium iodide with different side chains and poly(ionic liquids) with different monomers and ratios are shown in Fig. 2. As shown in Fig. 2(a), the absorption peaks at 1553 cm−1, 1172 cm−1, and 3078 cm−1 are attributed to the backbone vibrations of the imidazole ring, telescoping vibrations of the ring, and C–H telescoping vibrations in the ring, respectively, and the multiplet peaks in the range of 500–1000 cm−1 are attributed to bending vibrations in the C–H plane.31,32 In Fig. 2(b), the FT-IR absorption peak at 3023 cm−1 is attributed to the stretching vibration of C–H in the pyridine molecule,33 which is observed in all samples. Absorption peaks at 1153 and 3125 cm−1 can be observed in the poly(ionic liquids) spectrum, which are attributed to the stretching vibration of the imidazole ring and the C–H stretching vibration in the poly(ionic liquids), respectively.34 These results indicate that the pyridine and imidazole groups in the monomers are retained and embedded in the poly(ionic liquids) obtained by the solvothermal polymerization method.
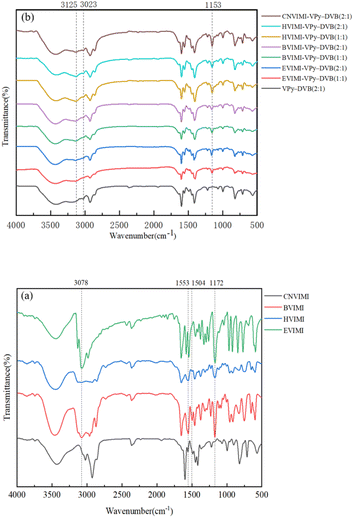 |
| | Fig. 2 FT-IR spectra of (a) ionic liquids and (b) poly(ionic liquids). | |
3.1.2 TG analysis. The thermal stability of the poly(ionic liquids) can be determined by the TG analysis, and the weight loss curves of the samples are shown in Fig. 3. A slight weight loss was observed at 100–280 °C, which is attributed to the release of water, low-boiling solvents, and monomers of polymer inside the pore channels.35 There was a significant weight loss of the poly(ionic liquids) at 280–450 °C, which was attributed to the decomposition of the poly(ionic liquids) formed by the co-polymerization of the ionic liquid, 4-vinylpyridine, and p-divinylbenzene. This indicated that the tested poly(ionic liquids) demonstrated good thermal stability and could be used as catalysts.
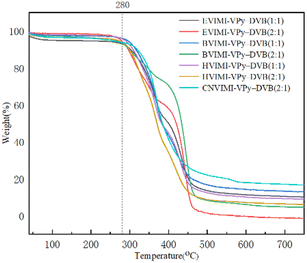 |
| | Fig. 3 TG analysis curves of poly(ionic liquids). | |
3.1.5 SEM and TEM studies. SEM images of different poly(ionic liquids) are illustrated in Fig. 5 and the morphology of the samples varies with the type of monomer. These images show that dried EVIMI-VPy-DVB (1:1), BVIMI-VPy-DVB (1:1), and HVIMI-VPy-DVB (1:1) have a large number of dense pore structures on the surface. This is because poly(ionic liquids) are formed by the polymerization of monomers, and macropores and mesopores may be formed in the interstitial space of the monomers after polymerization. Fig. 6 displays the TEM images of EVIMI-VPy-DVB (1:1), BVIMI-VPy-DVB (1:1), and HVIMI-VPy-DVB (1:1), and it can be seen that this series of poly(ionic liquids) are porous organic polymers.
 |
| | Fig. 5 SEM images of poly(ionic liquids). (a) EVIMI-VPy-DVB (1:1), (b) BVIMI-VPy-DVB (1:1), and (c) HVIMI-VPy-DVB (1:1). | |
 |
| | Fig. 6 TEM images of poly(ionic liquids). (a) EVIMI-VPy-DVB (1:1), (b) BVIMI-VPy-DVB (1:1), and (c) HVIMI-VPy-DVB (1:1). | |
3.2 Performance of catalytic methoxycarbonylation
The methoxycarbonylation of DIB with methanol and CO was chosen as the model reaction to evaluate and compare the catalytic performance of the poly(ionic liquids)@Co catalyst system. The detailed results are presented in Table 3. Firstly, we examined the effect of the N:Co ratio on the methoxycarbonylation under the reaction conditions of CH3OH:DIB = 30:1(mol ratio), 3 wt% Co2(CO)8, 8.0 MPa CO and 150 °C for 12 h. The highest conversion of DIB was observed at N:Co = 1:1(mol ratio) and the selectivity of methyl isononanoate decreased with the increasing of N:Co (Table 3, entries 1–3). Subsequently, the effect of different cobalt precursors, including Co2(CO)8, Co(acac)2 and CoCO3, on the reaction was examined with HVIMI-VPy-DVB (1:1) poly(ionic liquids). Only 34.4% (38.7% or 49.8%) DIB conversion and 47.3% (77.2% or 58.5%) methyl isononanoate selectivity were obtained using the catalyst systems formed with CoCO3 (Co(acac)2 or Co(acac)3) and HVIMI-VPy-DVB (1:1) (Table 3, entries 4–6). Replacing CoCO3 (Co(acac)2 or Co(acac)3) with Co2(CO)8 increased the total DIB conversion to 88.0% and the selectivity of methyl isononanoate to 91.4% (Table 3, entry 7). Co2(CO)8 was selected as the most ideal cobalt precursor.
Table 3 Effect of different catalysts and N:Co on methoxycarbonylation of DIB with methanol and COa
The catalytic performance of different poly(ionic liquids)@Co2(CO)8 for the methoxycarbonylation of DIB to synthesize methyl isononanoate was investigated under 8.0 MPa CO and 150 °C for 12 h. The detailed results are presented in Table 4. The best yield was obtained using HVIMI-VPy-DVB (1:1)@Co2(CO)8 as the catalyst; the conversion of DIB was 88.0%, and the total selectivity of methyl isononanoate was 91.4% (Table 4, entry 1). As the side chain of the ionic liquids grew from ethyl to hexyl at a 1:1 molar ratio of VPy to ionic liquids, DIB conversion increased from 86.0% to 87.0% to 88.0%, and methyl isononanoate selectivity increased from 83.5% to 87.1% to 91.5% (Table 4, entries 1–3). As the side chain of ionic liquids grew from ethyl to hexyl at a 2:1 molar ratio of VPy to ionic liquids, DIB conversion decreased from 86.9% to 85.6% to 81.8%, and methyl isononanoate selectivity increased from 83.6% to 86.5% to 88.2% (Table 4, entries 4–6). This phenomenon indicates that with the growth of the side chain of ionic liquids, the dissolution performance of the support to the substrate increases, and the reactivity is enhanced accordingly. Meanwhile, the reaction conversion and selectivity increased with increasing HVIMI:VPy or BVIMI:VPy, suggesting that the ionic liquids play more role than just as a ligand with Co and that the good solubility of the ionic liquids on the substrate facilitated methoxycarbonylation.
Table 4 Effect of different poly(ionic liquids) on the performance of DIB methoxycarbonylationa
| Entry |
Catalyst |
Conversion (%) |
Selectivity (%) |
| a |
b |
a + b |
| Reaction conditions: CH3OH:DIB = 15:1 (mol ratio), 4 wt% Co2(CO)8, 150 °C, 8 MPa, and 12 h. |
| 1 |
HVIMI-VPy-DVB (1:1)@Co2(CO)8 |
88.0 |
81.0 |
10.4 |
91.4 |
| 2 |
BVIMI-VPy-DVB (1:1)@Co2(CO)8 |
87.0 |
76.5 |
10.6 |
87.1 |
| 3 |
EVIMI-VPy-DVB (1:1)@Co2(CO)8 |
86.0 |
72.2 |
11.3 |
83.5 |
| 4 |
HVIMI-VPy-DVB (2:1)@Co2(CO)8 |
81.8 |
77.6 |
10.6 |
88.2 |
| 5 |
BVIMI-VPy-DVB (2:1)@Co2(CO)8 |
85.6 |
74.2 |
12.3 |
86.5 |
| 6 |
EVIMI-VPy-DVB (2:1)@Co2(CO)8 |
86.9 |
73.2 |
10.5 |
83.6 |
| 7 |
CNVIMI-VPy-DVB (2:1)@Co2(CO)8 |
69.2 |
80.1 |
8.4 |
88.4 |
| 8 |
VPy-DVB (2:1) @Co2(CO)8 |
84.2 |
75.8 |
13.3 |
87.9 |
The reaction parameters, including the molar ratio of methanol to DIB, catalyst concentration, reaction temperature, reaction pressure, and reaction time were investigated in detail. First, the molar ratio of methanol to DIB in the reaction was investigated (Table 5, entries 1–5). With an increase in methanol:DIB (mol ratio), the conversion of DIB and the selectivity of the product increased, and the molar ratio of methanol:DIB = 15:1 was selected for the subsequent reaction. The catalyst concentration had a significant effect on the catalytic performance, as the DIB conversion increased steadily from 73.9% to 91.9% when the Co2(CO)8 loading was increased from 3 wt% to 6 wt% (Table 5, entries 4, 6 and 7). In addition, the selectivity of methyl isononanoate reached a maximum of 91.4% at a Co2(CO)8 loading of 4 wt% (Table 5, entry 6). When the reaction temperature was increased from 140 °C to 160 °C, the conversion and selectivity first increased and then decreased (Table 5, entries 4, 8 and 9), and the optimal reaction temperature was 150 °C (Table 5, entry 6). DIB conversion and methyl isononanoate selectivity gradually increased from 53.4% and 79.4% to 88.0% and 91.4% as CO pressure was increased from 6 to 8 Mpa, respectively (Table 5, entries 4, 10 and 11). The conversion and selectivity increased from 68.0% and 86.6% to 88.0% and 91.4%, respectively, and then decreased to 80.8% and 90.9%, respectively, when the reaction time was gradually increased from 8 h to 14 h, with the best conversion and selectivity at 12 h of reaction.
Table 5 Effect of different reaction conditions of HVIMI-VPy-DVB (1:1)@Co2(CO)8 catalysts on DIB methoxycarbonylation
| Entry |
CH3OH : DIB (mol ratio) |
Catalyst loading (wt%) |
Temp. (°C) |
CO press. (MPa) |
Time (h) |
Conversion (%) |
Selectivity (%) |
| a |
b |
a + b |
| 1 |
50:1 |
3 |
150 |
8 |
12 h |
88.9 |
82.6 |
8.2 |
90.7 |
| 2 |
25:1 |
3 |
150 |
8 |
12 h |
84.2 |
81.9 |
8.6 |
90.5 |
| 3 |
20:1 |
3 |
150 |
8 |
12 h |
84.0 |
81.5 |
8.7 |
90.2 |
| 4 |
15:1 |
3 |
150 |
8 |
12 h |
73.9 |
79.5 |
9.1 |
88.7 |
| 5 |
10:1 |
3 |
150 |
8 |
12 h |
61.8 |
80.4 |
7.3 |
87.7 |
| 6 |
15:1 |
4 |
150 |
8 |
12 h |
88.0 |
81.0 |
10.4 |
91.4 |
| 7 |
15:1 |
6 |
150 |
8 |
12 h |
91.9 |
76.4 |
13.0 |
89.5 |
| 8 |
15:1 |
4 |
140 |
8 |
12 h |
80.5 |
79.0 |
8.7 |
87.7 |
| 9 |
15:1 |
4 |
160 |
8 |
12 h |
66.1 |
71.5 |
5.6 |
77.1 |
| 10 |
15:1 |
4 |
150 |
7 |
12 h |
82.1 |
76.2 |
10.2 |
86.5 |
| 11 |
15:1 |
4 |
150 |
6 |
12 h |
53.4 |
74.0 |
5.4 |
79.4 |
| 12 |
15:1 |
4 |
150 |
8 |
14 h |
80.8 |
82.2 |
8.7 |
90.9 |
| 13 |
15:1 |
4 |
150 |
8 |
10 h |
75.3 |
80.1 |
10.0 |
90.0 |
| 14 |
15:1 |
4 |
150 |
8 |
8 h |
68.0 |
78.0 |
8.6 |
86.6 |
The substrate scope was investigated using HVIMI-VPy-DVB (1:1) @Co2(CO)8 as the catalyst at 150 °C and 8.0 MPa CO. Excellent catalytic performance was achieved using aliphatic terminal olefins as the substrates. The conversions of 1-hexene, 1-octene, and styrene were 98.3%, 97.9%, and 87.1% (Table 6), respectively, whereas the selectivities of the corresponding methoxycarbonylation products were 58.1%, 55.9% and 74.9%, respectively.
Table 6 Methoxycarbonylation of different terminal olefins catalyzed with HVIMI-VPy-DVBa (1:1)@Co2(CO)8
| Entry |
Substrates |
Conversion (%) |
Selectivity (%) |
| Reaction conditions: CH3OH:DIB = 15:1 (mol ratio), 4 wt% Co2(CO)8, 150 °C, 8 MPa, and 12 h. |
| 1 |
1-Octene |
97.9 |
55.9 |
| 2 |
1-Hexene |
98.3 |
58.1 |
| 3 |
Styrene |
87.1 |
74.9 |
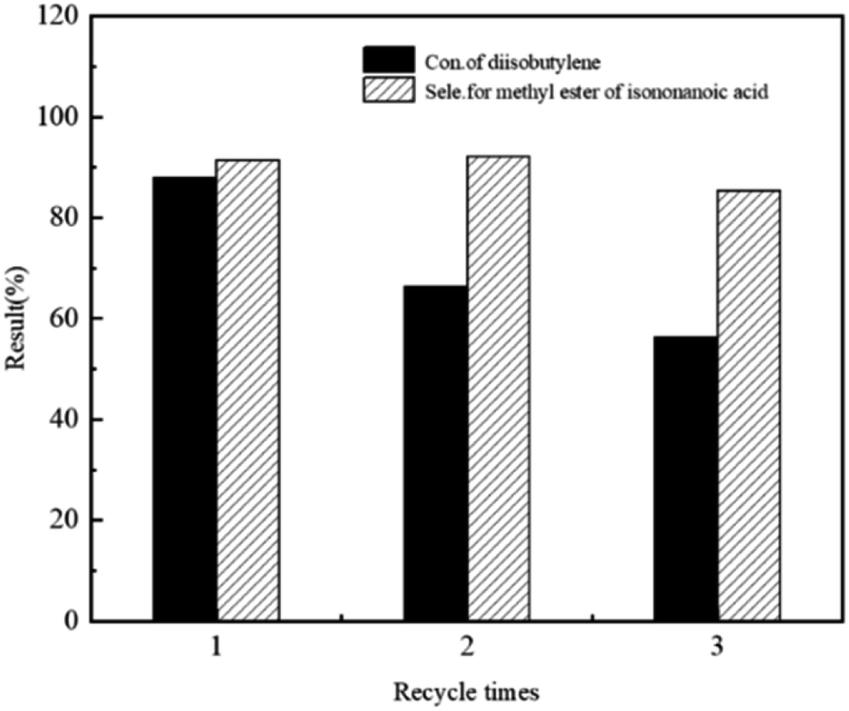
The recyclability of HVIMI-VPy-DVB (1:1)@Co2(CO)8 catalyst was investigated, where the reactor was equipped with a filter through which the liquid at the bottom of the reactor was released, and the catalyst in the mixture was retained at the bottom of the reactor. The catalyst was not treated in any manner during the next cycle. As shown in Fig. 7, after three runs, the DIB conversion and methyl isononanoate selectivity decreased from 88.0% and 91.4% to 56.4% and 85.4%, respectively. The results showed that the poly(ionic liquids)@Co2(CO)8 catalysts were less stable in the DIB methoxycarbonylation reaction. Fig. 8 shows the TEM images before and after use, and it can be seen that there are obvious Co clusters in the TEM images after use. After three runs, Co was gradually agglomerated, the agglomeration of Co would lead to poor dispersion of Co, making it unevenly dispersed on the carrier, thus affecting the catalytic performance of the catalysts and leading to a decrease in conversion and selectivity. Additionally, the textural structure of the catalyst changed significantly during methoxycarbonylation. The BET surface area decreased from 0.783 m2 g−1 to 0.124 m2 g−1 and the total pore volume decreased from 0.012 cm3 g−1 to 0.005 cm3 g−1 (Table 7), respectively. This provides evidence that the channels for the support collapsed after three runs. The apparent changes in the texture and structure of the catalysts can be attributed to the loss of poly(ionic liquids) anions and cations in methanol and the partial dissolution of poly(ionic liquids) resulting in the disruption of its structure. Agglomeration of Co and collapse of the support pores reduced the active sites, thus affecting the catalytic performance of the poly(ionic liquids)@Co2(CO)8 catalysts for the methoxycarbonylation reaction of DIB.
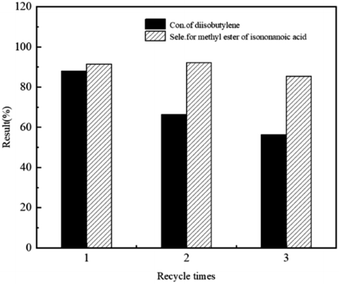 |
| | Fig. 7 Reusability of HVIMI-VPy-DVB (1:1)@Co2(CO)8 in the methoxycarbonylation reaction of DIB and CO. Co2(CO)8: 4 wt%, N:Co = 2:1 (mol ratio), 150 °C, 8.0 MPa, 12 h. | |
 |
| | Fig. 8 The TEM images of HVIMI-VPy-DVB (1:1) @Co2(CO)8 catalyst before and after use. (a) Before use and (b) after use. | |
Table 7 BET surface properties of the HVIMI-VPy-DVB (1:1)@Co2(CO)8 catalyst before and after use
| Sample |
BET surface area (m2 g−1) |
Total pore volume (cm3 g−1) |
Average pore width (nm) |
| Fresh HVIMI-VPy-DVB (1:1)@Co2(CO)8 |
0.783 |
0.012 |
0.793 |
| Used HVIMI-VPy-DVB (1:1)@Co2(CO)8 |
0.125 |
0.005 |
0.781 |
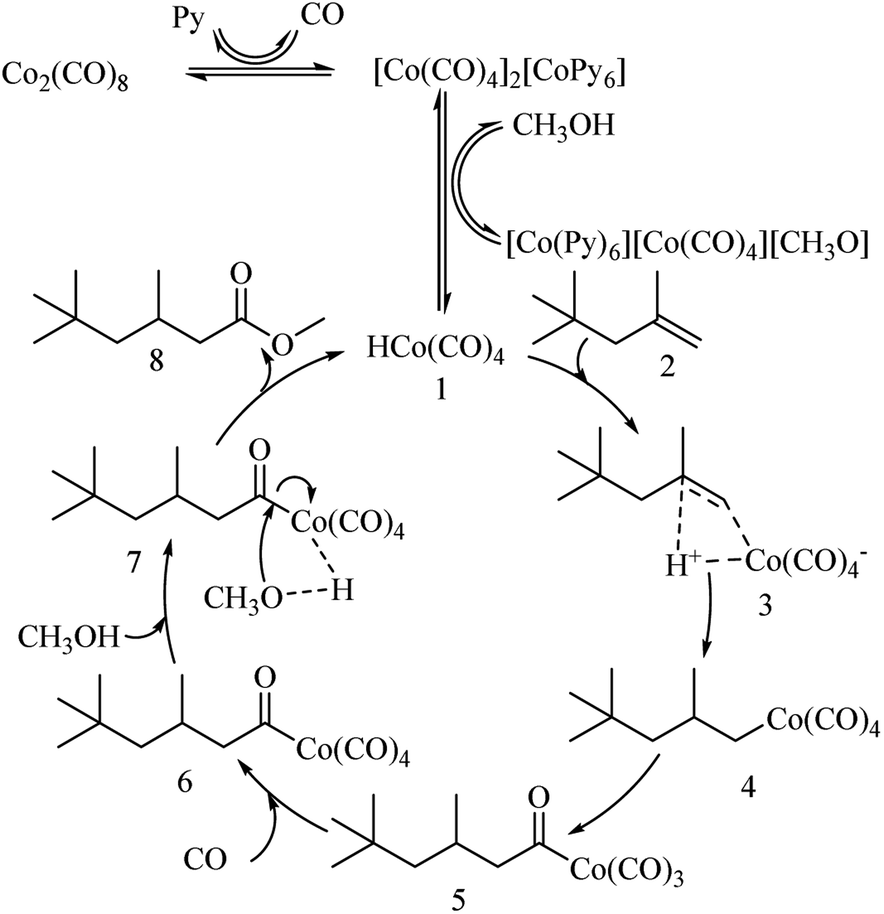
Based on previous studies,37–40 we proposed a reaction mechanism for the methoxycarbonylation of DIB, as shown in Fig. 9 Firstly, in the presence of an N-donors, such as a pyridine compound (Py), Co2(CO)8 is reduced to [CoPy6][Co(CO)4]2, and then HCo(CO)4 (1) is formed. DIB (2) reacts with (1) in an addition reaction to form C8H17Co(CO)4 (4), CO migrates intramolecularly to insert a Co–C bond to form the complex (5). (5) is an active substance that undergoes easy carbonylation in the presence of carbon monoxide to form acyl complex (6), and treatment of acyl complex (6) with methanol gives methyl isononanoate (7).
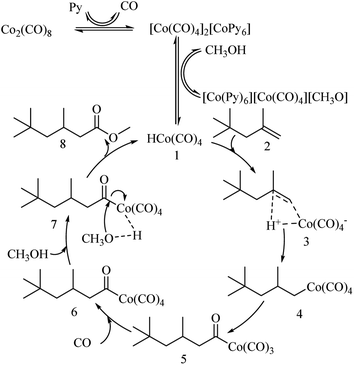 |
| | Fig. 9 Proposed catalytic mechanism for methoxycarbonylation of DIB. | |
4. Conclusions
The heterogeneous catalysts formed by the in situ combination of poly(ionic liquids) with Co2(CO)8 showed good activity and selectivity in the methoxycarbonylation of DIB with CO and methanol. It was found that the catalytic performance depended on the Co precursor, the side chain of ionic liquids, the molar ratio of ionic liquids to VPy, and textural structure. The catalyst HVIMI-VPy-DVB (1:1)@Co2(CO)8 showed 88.0% DIB conversion and 91.4% methyl isononanoate selectivity under the optimum conditions of 8.0 MPa CO and 150 °C for 12 h reaction. However, gradual agglomeration of Co was observed during reuse, and the pore collapse affected its catalytic performance and led to catalyst deactivation. These findings provide an important reference for the preparation of efficient, stable, easily separable and recoverable multiphase homogeneous catalytic systems for the methoxycarbonylation of olefins.
Data availability
All relevant data are within the paper.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the Natural Science Foundation of Gansu Province (22JR5RA349), Gansu Province Key Research and Development Program – Industrial Projects (23YFGA0041) and National Natural Science Foundation of China (22102198).
References
- D. Zhang, S. Sun, Z. Qian, K. Pei, X. Shang, C. Liu and P. Guo, Tetrahedron Lett., 2022, 107, 154104 CrossRef CAS.
- N. Smrečki, B.-M. Kukovec, J. Jaźwiński, Y. Liu, J. Zhang, A.-M. Mikecin and Z. Popović, J. Organomet. Chem., 2014, 760, 224–230 CrossRef.
- M. Jiang, L. Yan, Y. Ding, Q. Sun, J. Liu, H. Zhu, R. Lin, F. Xiao, Z. Jiang and J. Liu, J. Mol. Catal. A: Chem., 2015, 404–405, 211–217 CrossRef CAS.
- T. A. Tshabalala, S. O. Ojwach and M. A. Akerman, J. Mol. Catal. A: Chem., 2015, 406, 178–184 CrossRef CAS.
- I. E. Nifant’ev, N. T. Sevostyanova, S. A. Batashev, A. A. Vinogradov, A. A. Vinogradov, A. V. Churakov and P. V. Ivchenko, Appl. Catal., A, 2019, 581, 123–132 CrossRef.
- Y. Lei, G. Lan, M. Fan and G. Li, Catal. Commun., 2020, 140, 106007 CrossRef CAS.
- N. V. Kolesnichenko, T. I. Batova, A. N. Stashenko, T. K. Obukhova, E. V. Khramov, A. A. Sadovnikov and D. E. Zavelev, Microporous Mesoporous Mater., 2022, 344, 112239 CrossRef CAS.
- J. Zhu, T. Xu, P. Lu, W. Chen and W. Lu, Mol. Catal., 2022, 532, 112736 CrossRef CAS.
- H. Ishii, M. Ueda, K. Takeuchi and M. Asai, J. Mol. Catal. A: Chem., 1999, 144, 369–372 CrossRef CAS.
- A. Vavasori and L. Toniolo, J. Mol. Catal. A: Chem., 2000, 151, 37–45 CrossRef CAS.
- G. Yin, C. Jia, T. Kitamura, T. Yamaji and Y. Fujiwara, J. Organomet. Chem., 2001, 630, 11–16 CrossRef CAS.
- H. Yasuda, K. Watarai, J.-C. Choi and T. Sakakura, J. Mol. Catal. A: Chem., 2005, 236, 149–155 CrossRef CAS.
- G. Fan, J. Huang, Z. Li, T. Li and G. Li, J. Mol. Catal. A: Chem., 2007, 267, 34–40 CrossRef CAS.
- L. Ronchin, A. Vavasori, E. Amadio, G. Cavinato and L. Toniolo, J. Mol. Catal. A: Chem., 2009, 298, 23–30 CrossRef CAS.
- N. Sudheesh, S. K. Sharma, R. S. Shukla and R. V. Jasra, J. Mol. Catal. A: Chem., 2010, 316, 23–29 CrossRef CAS.
- Z. Nairoukh and J. Blum, J. Mol. Catal. A: Chem., 2012, 358, 129–133 CrossRef CAS.
- S. Doherty, J. G. Knight and M. Betham, Chem. Commun., 2006, 88–90, 10.1039/b512556a.
- X. Chen, H. Zhu, T. Wang, C. Li, L. Yan, M. Jiang, J. Liu, X. Sun, Z. Jiang and Y. Ding, J. Mol. Catal. A: Chem., 2016, 414, 37–46 CrossRef CAS.
- Z. Ren, Y. Liu, Y. Lyu, X. Song, C. Zheng, S. Feng, Z. Jiang and Y. Ding, J. Catal., 2019, 369, 249–256 CrossRef CAS.
- C.-C. Yang, S. Yacob, B. A. Kilos, D. G. Barton, E. Weitz and J. M. Notestein, J. Catal., 2016, 338, 313–320 CrossRef CAS.
- J. D. Nobbs, C. H. Low, L. P. Stubbs, C. Wang, E. Drent and M. van Meurs, Organometallics, 2016, 36, 391–398 CrossRef.
- K. Dong, R. Sang, Z. Wei, J. Liu, R. Duhren, A. Spannenberg, H. Jiao, H. Neumann, R. Jackstell, R. Franke and M. Beller, Chem. Sci., 2018, 9, 2510–2516 RSC.
- R. Sang, C. Schneider, R. Razzaq, H. Neumann, R. Jackstell and M. Beller, Org. Chem. Front., 2020, 7, 3681–3685 RSC.
- H. Song, S. Lei, F. Jin and H. Liu, Mol. Catal., 2022, 527, 112408 CrossRef CAS.
- F. Jin, J. Wang, H. Song, Z. Quan, Q. Liu, Y. Yan, M. Kang, H. liu and X. Wang, Arabian J. Chem., 2023, 16, 104907 CrossRef CAS.
- D. Yuan, H. Song, H. Song, M. You, B. Wang, F. Li, Y. Hao and Q. Yu, J. Taiwan Inst. Chem. Eng., 2017, 76, 83–88 CrossRef CAS.
- H. Jia, X. Chen, C.-Y. Liu, X.-J. Liu, X.-C. Zheng, X.-X. Guan and P. Liu, Int. J. Hydrogen Energy, 2018, 43, 12081–12090 CrossRef CAS.
- S. Yousefi, N. Bahri-Laleh, M. Nekoomanesh, M. Emami, S. Sadjadi, S. Amin Mirmohammadi, M. Tomasini, E. Bardají and A. Poater, J. Mol. Liq., 2022, 367, 120381 CrossRef CAS.
- H. Liu, K. Wang, D. Zhang, D. Zhao, J. Zhai and W. Cui, Mater. Sci. Semicond. Process., 2023, 154, 107215 CrossRef CAS.
- Y. Zhang, S. Wei, F. Liu, Y. Du, S. Liu, Y. Ji, T. Yokoi, T. Tatsumi and F.-S. Xiao, Nano Today, 2009, 4, 135–142 CrossRef CAS.
- X. Li, Y. Shen, F. Jin, J. Zhang, Y. Yang and X. Qu, Sep. Purif. Technol., 2024, 335, 126210 CrossRef CAS.
- Y. X. Li, Y. X. Dai, J. Z. Wang, J. Chauvin, X. J. Zhang, S. Cosnier, R. S. Marks and D. Shan, Talanta, 2024, 272, 125779 CrossRef CAS PubMed.
- H. Song, F. Jin, M. Kang and J. Chen, RSC Adv., 2019, 9, 40662–40669 RSC.
- N. K. H. Nguyen, H. H. Dang, L. T. Nguyen, L. M. T. Nguyen, T. T. Truong, H. T. Nguyen, T. Q. Nguyen, C. D. Tran and L.-T. T. Nguyen, Eur. Polym. J., 2023, 199, 112474 CrossRef CAS.
- X. Zhang, M. Han, L. Xu and A. M. AlSofi, Chem. Phys. Lett., 2022, 795, 139538 CrossRef CAS.
- M. Fu, W. Ding, Q. Zhao, Z. Xu, W. Hua, Y. Li, Z. Yang, L. Dong, Q. Su and W. Cheng, Sep. Purif. Technol., 2024, 344, 127174 CrossRef CAS.
- R. b. Tuba, L. s. T. Mika, A. Bodor, Z. n. Pusztai, I. Tóth and I. n. T. Horváth, Organometallics, 2003, 22, 1582–1584 CrossRef CAS.
- J. Liu, J. Chen and C. Xia, J. Mol. Catal. A: Chem., 2006, 250, 232–236 CrossRef CAS.
- Z. Guo, H. Wang, Z. Lv, Z. Wang, T. Nie and W. Zhang, J. Organomet. Chem., 2011, 696, 3668–3672 CrossRef CAS.
- S. Rajendiran, K. Park, K. Lee and S. Yoon, Inorg. Chem., 2017, 56, 7270–7277 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Zhaoxiong Tiana,
Shuangtai Leiab and
Hailong Liu*b
a,
Zhaoxiong Tiana,
Shuangtai Leiab and
Hailong Liu*b