DOI:
10.1039/D4RA04643A
(Paper)
RSC Adv., 2024,
14, 27365-27371
Rational design of graphdiyne-based single-atom catalysts for electrochemical CO2 reduction reaction†
Received
26th June 2024
, Accepted 18th August 2024
First published on 28th August 2024
Abstract
Graphdiyne (GDY) has achieved great success in the application of two-dimensional carbon materials in recent years due to its excellent electrochemical catalytic capacity. Considering the unique electronic structure of GDY, transition metal (TM1) (TM = Fe, Ru, Os, Co, Rh, Ir) single-atom catalysts (SACs) with isolated loading on GDY were designed for electrochemical CO2 reduction reaction (CO2RR) with density functional theoretical (DFT) calculations. The charge density difference and projected densities of states have been systematically calculated. The mechanism of electrochemical catalysis and the reaction pathway of CO2RR over Os1/GDY catalysts have also been investigated and high catalytic activity was found for the generation of methane. The calculated results provide a theoretical basis for the design of efficient GDY-based SACs for electrochemical CO2RR.
Introduction
The increasing concentrations of CO2 in the atmosphere has caused a series of environmental problems such as global warming, extreme weather, and land desertification. Therefore, the efficient conversion and utilization of CO2 is a current focus of international research.1–3 Compared to thermal-catalysis, photocatalysis, and biological catalysis, electrocatalysis has certain advantages in terms of economical benefits and the efficiency of electron transfer.4–9 The industrialization of electrochemical carbon dioxide reduction reactions (CO2RR) is a win–win endeavor that can not only reduce carbon dioxide concentrations, but also generate valuable alcohols, ketones, and aldehydes.10–12 The key is to design catalysts with high efficiency, high selectivity, low cost and easy industrialization.13–16
Since Zhang, Li and Liu et al. proposed a new concept of single-atom catalyst (SAC) in 2011,17 the research of which is increasingly extensive in catalysis because of the unique electronic structure and high atomic utilization.18–24 Although the atom utilization of highly dispersed metal SACs achieves 100%, the large surface energy of metal mono-atoms makes it easy to produce agglomeration effects to form nanoparticles. Therefore, the selection of suitable substrates is key to obtaining stable metal SACs.25–28 Compared with conventional metal catalysts, transition metal (TM) atoms have the advantage of controllable coordination geometry of the active center. Due to the geometry, electronic, spin, and magnetic states can be fine-tuned to achieve the desired catalytic activity and selectivity,29–32 TM SACs have been extensively studied in electrochemistry.30,33–36 Usually, single atoms of the same period are chosen to be loaded on the catalyst substrate, and based on this, we would like to study the variation between the group VIII B. Six single-atom catalysts Fe, Ru, Os, Co, Rh and Ir were selected for further computational screening of single-atom catalysts that are more suitable for performing CO2RR. Among carbon materials, GDY is a new isomer of carbon consisting of both sp2 and sp-hybrid carbon atoms, in which sp-hybrid carbon atoms can form strong covalent bonds with metal atoms.37 Metal atoms could exist stably in GDY because of the high electrical conductivity and specific surface area.38–40 GDY-loaded TM SACs have higher catalytic activity than GDY,41,42 which has significance for the activity and selectivity of CO2RR.
Herein, the stability of SACs loaded with TM1 (TM1 = Fe, Ru, Os, Co, Rh, and Ir) on GDY was theoretically investigated by using density functional theoretical (DFT) calculations. Os1/GDY was used as a typical to explore the chemical interaction of TM1 and GDY surface. The electronic structure and electrochemical catalysis properties of Os1/GDY for CO2RR were investigated in detail.
Computational details
The classical calculations are based on the spin polarization DFT implemented in the Vienna ab initio Simulation Package (VASP).43,44 Electron exchange and correlation energy are approximated by Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional.45 Projector augmented wave (PAW) pseudopotential is used to describe the interaction between core and valence electrons,46 projected electronic densities of states (PDOS),47 and charge density difference (CDD).48 GDY cluster is composed of 72 carbon atoms with a cell of 19.6 Å × 19.6 Å × 14 Å, α = β = 90°, γ = 60°. All carbon atoms in the cell were fully relaxed during the structural optimization, and it stopped the optimization when the force was smaller than 0.006 eV Å−1. The energy cut-off of the plane wave base is set to 620 eV, the electron convergence criterion is 1 × 10−5 eV, and the ion relaxation continues until the atomic force is less than 0.05 eV Å−1. The entire calculation is geometrically optimized using a 3 × 3 × 1 k-point density, and the van der Waals (vdW) is evaluated using the DFT-D3 method.49
The binding energy (Eb) was calculated to check the stability of the TM in GDY. The definition equation is as follows
| Eb(TM) = ETM1/GDY − EGDY − ETM, |
where
ETM1/GDY is the total energy of the whole system when the single metal atom is in the best adsorption position,
EGDY is the total energy of GDY substrate,
ETM is the total energy of the single transition metal atom.
Adsorption energy (Ead) is the energy released by the rate reduction during CO2 adsorption. The Ead of CO2 on TM1/GDY-CO2 (TM = Fe, Ru, Os, Co, Rh, Ir) catalyst is calculated to compare the stability of CO2 adsorption on different carriers. The adsorption energy is as follows
| Ead = ETM1/GDY-CO2 − ETM1/GDY − ECO2, |
Here,
ETM1/GDY-CO2 is the energy of CO
2 adsorbed on TM
1/GDY catalyst,
ETM1/GDY is the energy of TM
1/GDY stable structure, and
ECO2 is the energy of CO
2 molecule.
The charge transfer before the formation of TM1/GDY is analyzed by differential charge analysis. The differential charge map is mostly applied to analyze the bonding between atoms of the model after structural optimization in the first nature principle calculations, especially in the interface calculations, where the bonding mode of the interface can be initially visualized by CDD analysis.50 The common definition of differential charge is
| Δρ = ρTM1/GDY − ρGDY − ρTM, |
where
ρTM1/GDY denotes the optimized structural charge density of TM
1/GDY,
ρGDY denotes the charge density of the structurally optimized GDY substrate, and
ρTM denotes the structural charge density of the transition metal single atom.
The free energy (ΔG) distribution of CO2RR is described by the computational hydrogen electrode (CHE) model pioneered by Nørskov et al.51,52 In the CHE method, ΔG is calculated as follows
ΔG = ΔE + ΔEZPE − TΔS + eU + kBT pH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln10 ln10 |
herein Δ
E is the total reaction energy, Δ
EZPE is the correction of zero-point energy (ZPE),
TΔ
S is the entropy contribution, and eU denotes the energy contributed by the applied electrode potential. The pH in our work is set as zero, considering the strong acid environment. The free energy of H
+ + e
− is equal to 1/2H
2 under the standard conditions.
Results and discussion
Structure and stability of TM1/GDY
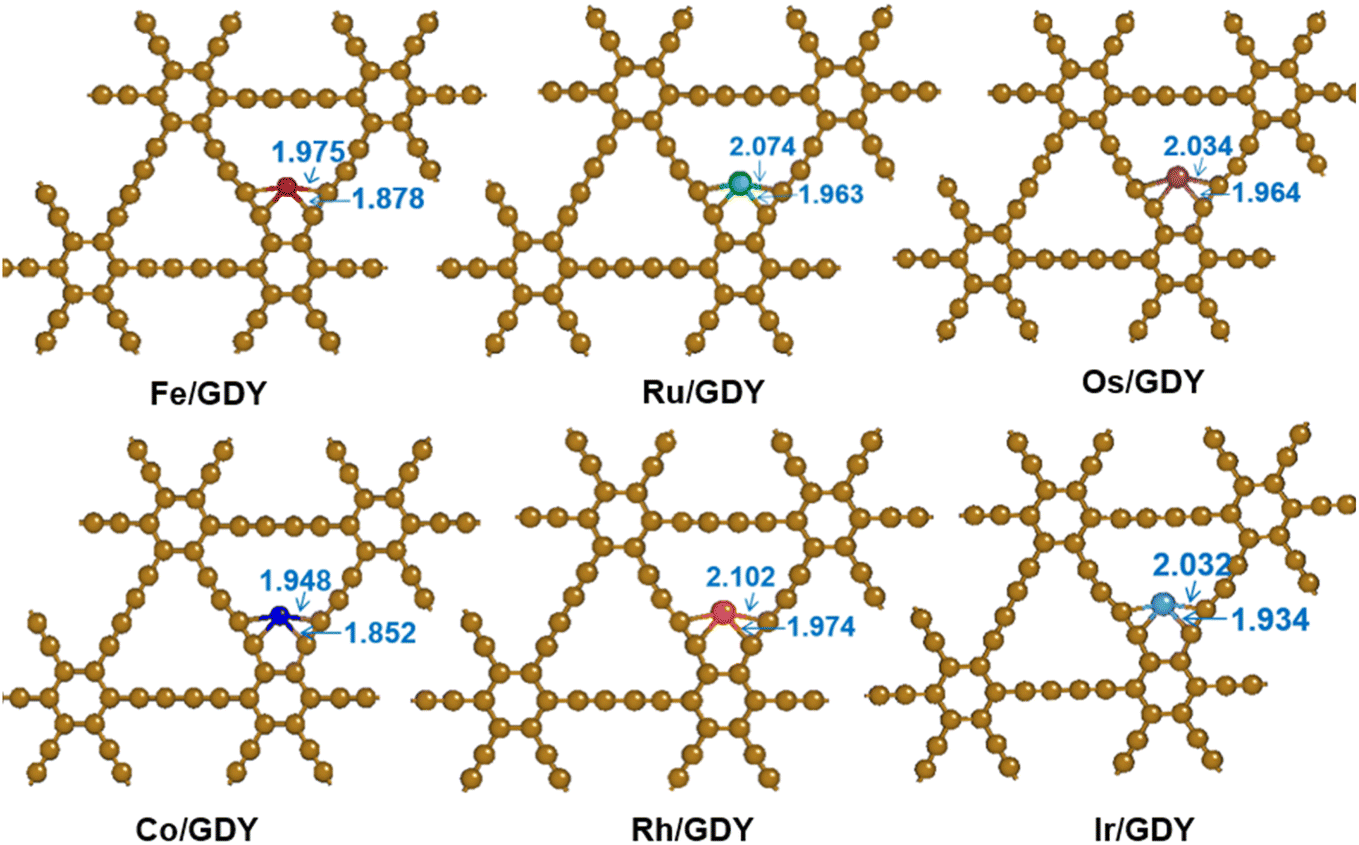
The protocell of GDY is composed of 18 carbon atoms, six of which are located on a hexagonal ring with sp2 hybridization and others on a linear acetylene chain with sp hybridization. The network structure of the graphite alkyne has C6 hexagon consisting of an acetylene bond linking two adjacent sp2-hybridized carbon atoms.53,54 The C–C bond length in the hexagon is 1.43 Å, which is alternately distributed as a single bond, a triple bond, and a single bond with bond lengths of 1.40 Å and 1.23 Å. TM1 SACs (TM = Fe, Ru, Os, Ni, Rh, and Ir) were loaded and optimized on the optimized GDY. The TM1/GDY structure with stable adsorption was obtained by screening three possible binding sites for the TM1 loading in the GDY including the corner site of the large triangular acetylene ring, the central cavity site of the acetylene ring, and the cavity site above the center of the hexagonal ring. As shown in Fig. 1, it can be seen that all transition metals are located within the cavities of the sp bonds in GDY. By measuring the distances between the TM1 and the adjacent C atoms, the results are distributed from 1.85 Å to 2.10 Å. Among them, the Rh atom locates with the largest distance of 2.102 Å and the Co atom with the smallest distance of 1.852 Å. Os, Ru, and Ir exhibit a distance of around 1.93 Å ∼2.07 Å. The distance between Fe and the adjacent carbon atoms is around 1.88 and 1.98 Å, which are shorter than that of Os, Ru, and Ir.
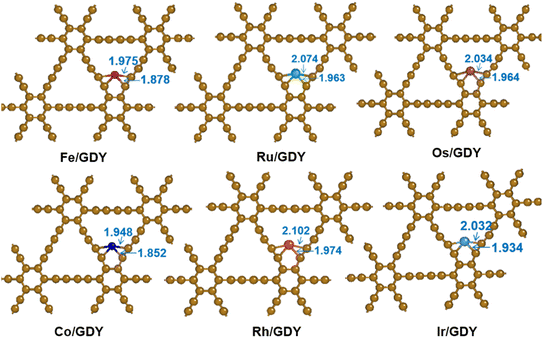 |
| | Fig. 1 Optimized TM1/GDY geometry (TM = Fe, Ru, Os, Ni, Rh, Ir). | |
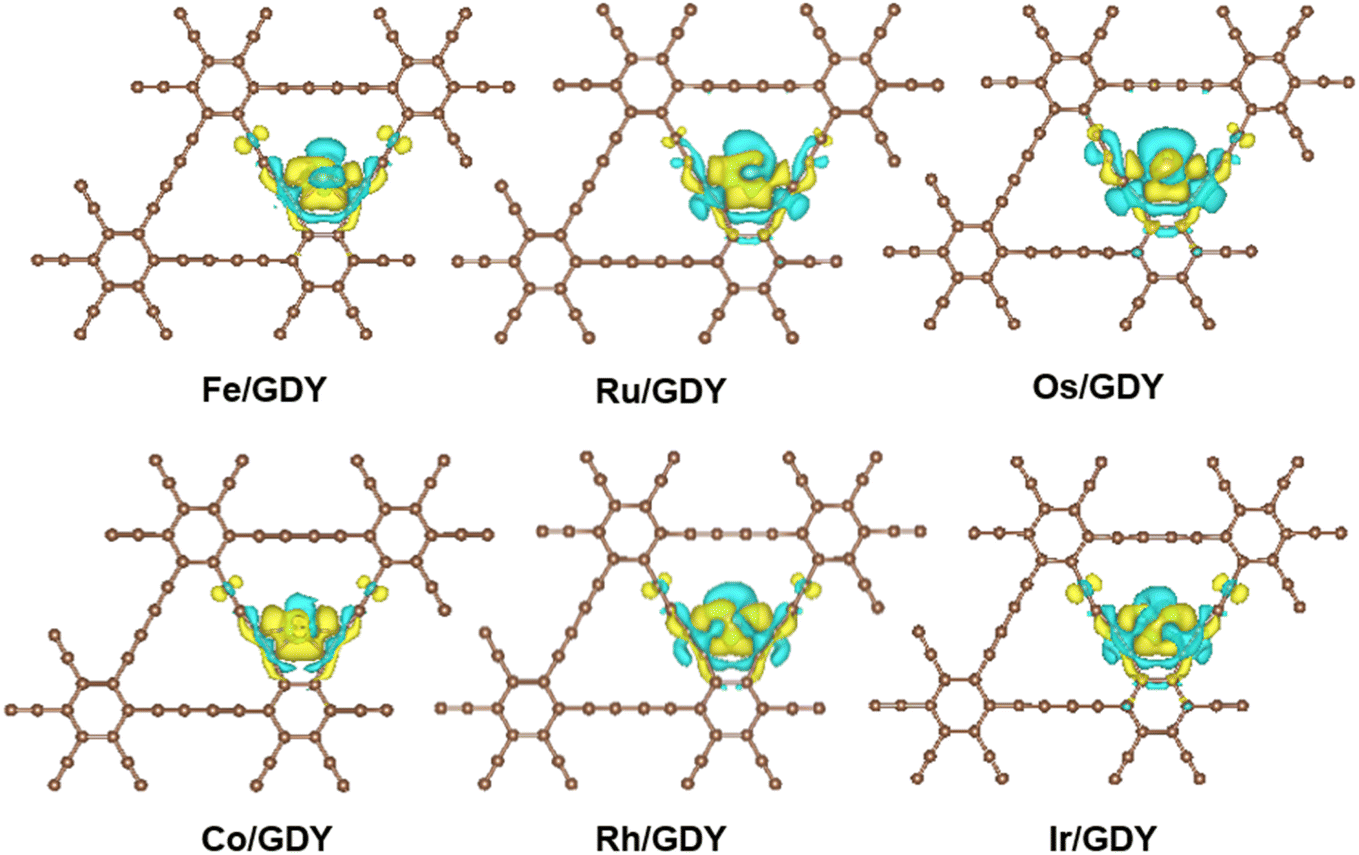
It can be seen from Fig. 2 that the charge density accumulation between the top of Ru and Co atoms and carbon atoms is enhanced, which is weakened for Os. The charge density at the top is also weakened. Charge transfer on GDY occurs mainly from the p-orbital of carbon atoms to the d-orbital of metal atoms. The charge on the vertical orbitals accumulates on Ru and Co, and is consumed on Fe, Os, Rh, and Ir. The results indicate that the charge transfer occurs mainly on the carbon atoms adjacent to the transition metal atoms on GDY, and also on the sp hybridized carbon atoms where the sp bonds on the benzene rings are involved in the redistribution of the charge density.
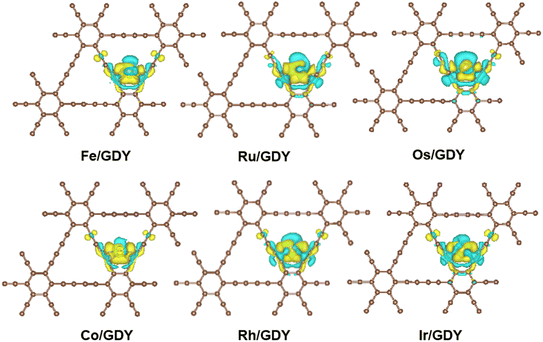 |
| | Fig. 2 The CDD of TM1/GDY. The yellow and blue equivalence surfaces represent charge accumulation and charge depletion, respectively. | |
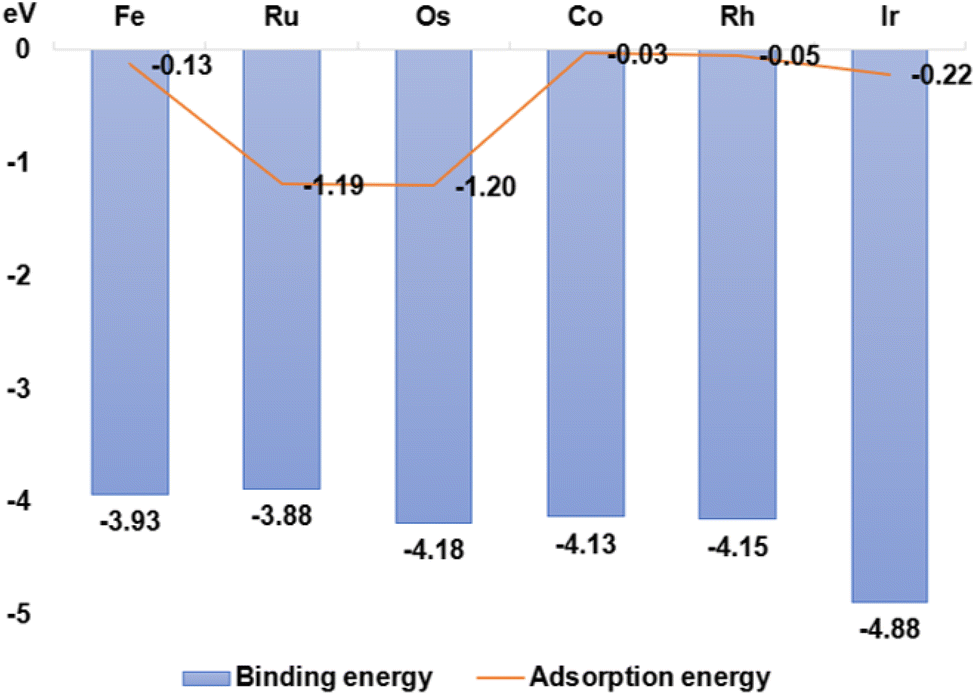
The stability of GDY transition metal composites was quantified by measuring the binding energy of GDY-loaded transition metal SACs. The larger the negative value of the binding energy, the more stable the bonding of TM1 single-atoms with GDY. As seen from Fig. 3, the binding energy of Ru1/GDY is −3.88 eV, which is the highest binding energy among the six TM1 SACs. But Ir1/GDY is more stable with a binding energy of −4.88 eV. First-principles adsorption energy calculation is a quantum mechanics-based measurement method that can be applied to materials design, catalyst development, and gas separation. In the development of catalyst, the adsorption capacity and catalytic activity can be scientifically and rationally predicted by calculating the adsorption energies of different catalyst surfaces or interfaces with molecules. It can also help to design and develop new catalysts and continue to optimize their performance. By calculating and comparing the adsorption energies of TM1/GDY and CO2, the results listed in Fig. 3 showed that Os1/GDY has an adsorption energy of −1.20 eV with CO2 which is the most stable one among the TM1 SACs.
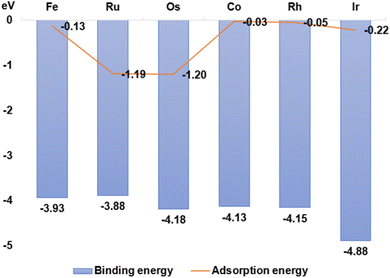 |
| | Fig. 3 The binding energy of GDY-loaded Fe, Ru, Os, Co, Rh, and Ir single atoms and CO2 adsorption energy. | |
Structure of Os1/GDY
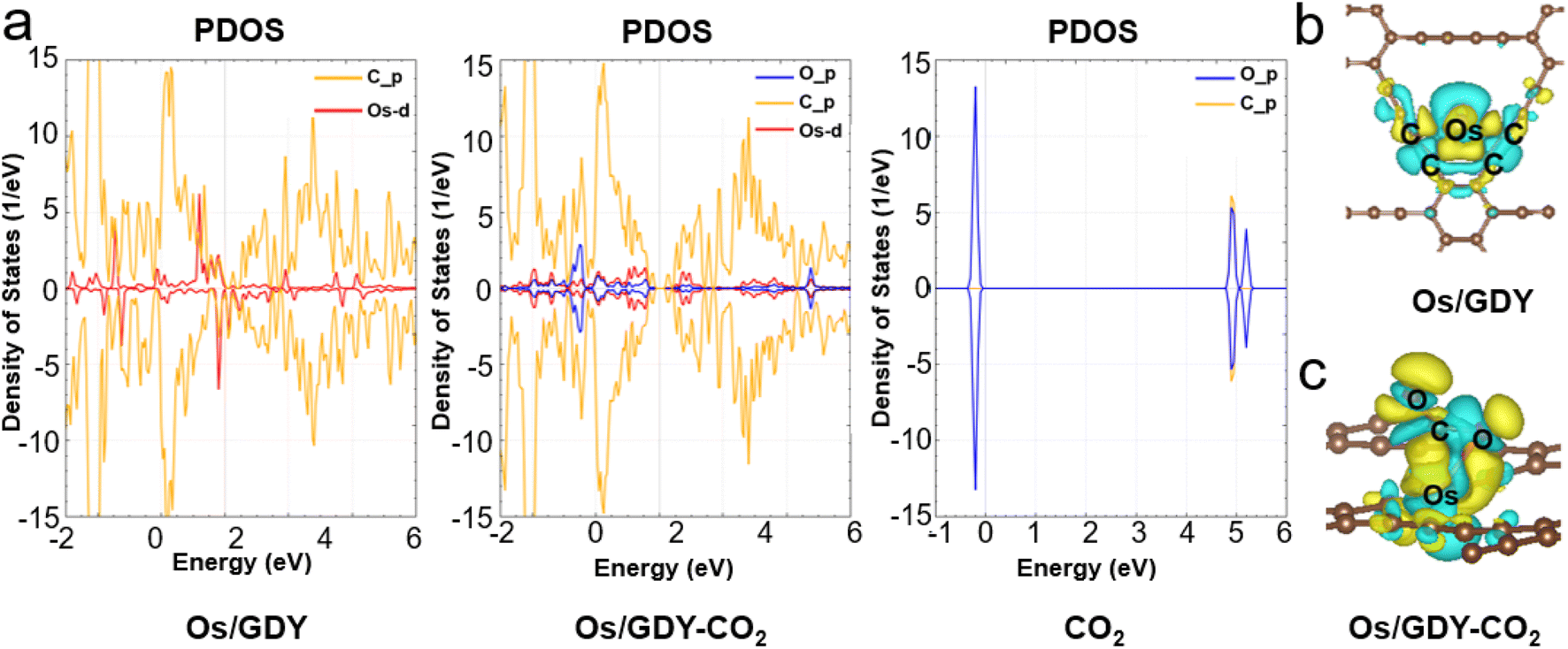
The density of states as a visualization result of energy band structure, which can reflect the distribution of electrons in each orbital and reveal the bonding nature between atomic chemical bonds, is a key parameter to further investigate the electronic structure of SACs.55,56 The spin-polarized PDOS and CDD of Os1/GDY and Os1/GDY adsorbed CO2 are calculated and shown in Fig. 4a, from which we can see that the DOS between the spikes of Os near the Fermi energy level is not zero and the 5d orbital of Os plays a dominant role in bonding with the GDY surface. The pseudopotential gap near the Fermi level due to the p orbitals of C and O in CO2 interact with the d orbitals of Os indicates that the addition of CO2 makes the covalence of Os1/GDY enhanced and facilitates the further of CO2RR. At the same time, the CDD of Os1/GDY adsorbed by the stable configuration Os1/GDY and CO2 was investigated (Fig. 4b and c). Because of the doping of transition metal atoms, the system spin polarization becomes larger to promote the activation of CO2, and also lead electrons to mainly flow from Os atoms to CO2 molecules. When *CO2 is hydrogenated to *COOH, electrons transfer from H to the lone pair of electrons of CO2, which further weakens the C–O bond and makes it more conducive to the subsequent electrocatalysis with H+.
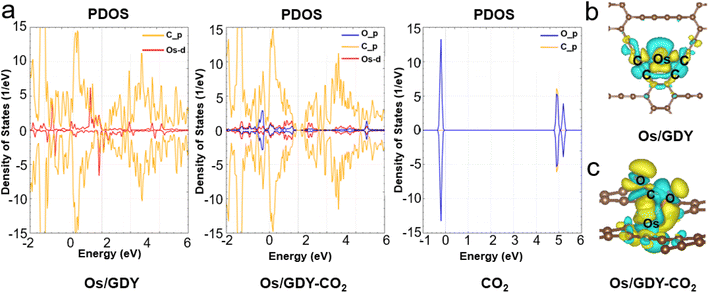 |
| | Fig. 4 (a) The schematic diagram of projected electronic densities of states of Os1/GDY, Os1/GDY-CO2, and CO2. CDD plots of (b) Os1/GDY and (c) Os1/GDY-CO2. | |
Hydrogen evolution reaction
The competition between the hydrogen evolution reaction (HER) and CO2RR is crucial in aqueous electrolytes because the first step in CO2RR is the protonation of CO2 to *COOH or *OCOH, consuming *H and electrons. And the HER starts with the synthesis of *H with a catalyst, where *H and electrons are essential. On TM1/GDY, the transition metal interacts with *H. The predicted overpotential of HER is lower than that of Fe1/GDY and Co1/GDY. When U = 0 V, Fe1/GDY acts as a great HER catalyst candidate (Fig. 5a). As shown in Fig. 5b, we found that the lower free energy changes showed higher selectivity through comparing the adsorption free energy changes (ΔG) of *CO2 and *H. Among them, Fe, Ru, Os, Co, Rh, and Ir are all located below the parity line which defines the selectivity between CO2RR and HER. Therefore, the resistance of TM1/GDY to HER can be effectively used as CO2RR catalysts. The loading of transition metal single-atoms improved the activity and selectivity of CO2 initial reduction. For Os1/GDY, the adsorption of *CO2 is better than *H, which means that it favors to become a catalyst for CO2RR. Because the active site is easily occupied by *CO2 and *CO2 is more negative than *H, the Os1/GDY catalyst can effectively adsorb and further catalyze CO2.
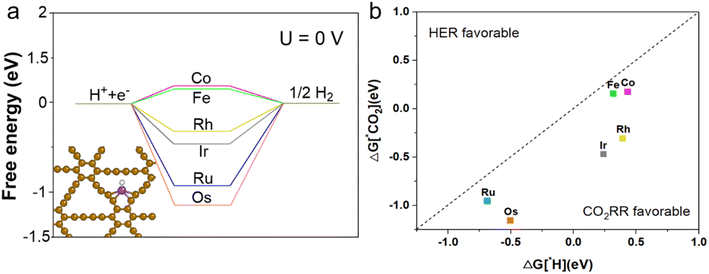 |
| | Fig. 5 (a) The free energy diagrams for HER at equilibrium U = 0 V on TM1/GDY. (b) The change of ΔG in the first protonation step of CO2RR adsorption and HER on TM1/GDY catalysts. The region below/above the parity test represents CO2RR/HER selectivity, respectively. | |
Electrochemical CO2RR
In the CO2RR, the C1 products including CO, HCOOH, CH4, and CH3OH are frequently generated. CO2 is first reduced over a catalyst into *COOH or *OCOH, which forms *CO or *HCOOH by further electrochemical reduction.57 Followed by desorption of *CO and *HCOOH from the SACs, CO and HCOOH is generated, respectively.58,59
| CO2 → *OCOH → *HCOOH → HCOOH |
The basic reaction step of further protonation of *CO intermediates on the catalyst surface is currently the rate-determining step for most electrocatalysts, and this process may form either *CHO or *COH. Peterson et al. have concluded that the process of *CO protonated to form *CHO would more efficient when the catalyst surface bind both *CO and *CHO strongly.52 The research group of Nie proposed that the final product is strongly dependent on two key intermediates *CHO and *COH.60 If *CO is protonated to form *CHO, then *CHO continues to be protonated to form *OCH2 and *OCH3, and the location of further hydrogenation of *OCH3 may affect the selectivity of the final product, which eventually forms CH4 on the catalyst surface.61 There are usually two pathways to generate CH4. Based on studies of the initial reduction of CO2, CO2RR involves more steps of proton–electron pair transfer, leading to an end product that is often not monolithic.
| *CHO/*COH → *CHOH → *CH → *CH2 → *CH3 → *CH4 |
| *CHO/*COH → *OCH2 → *OCH3 → *OCH4 → *OH + CH4 → H2O |
| *CHO/*COH → * CHOH → * CH2OH → CH3OH |
One process of CH4 formation is the continuous protonation of C into *O and CH4 and the further hydrogenation of *O to H2O. In another process, the O is protonated to form H2O and *C, and then *C is deoxidized to form CH4.62,63 However, hydrogenation reactions should also be considered for processes that occur between C and O. The free energy pathways involving the protonation of *COH and *CHO were shown in Fig. 6. It can be seen that the protonation of *CO to *COH on the surface of the Os1/GDY catalyst is the step with the maximum free energy variation (ΔG = 0.61), and the protonation of *CHO to *CH2O is the reaction step with the optimal energy. Subsequently, *CH2O is more energetically inclined to form *CH3O relative to *CH2OH. Eventually, *CH3O was further protonated to form *OCH4 intermediates, which further produce CH4. Synthesizing the previous discussion on the electronic structure of Os1/GDY, doping Os on GDY changes the charge density and state density, which is more conducive to the adsorption of CO2, and thus has better performance for electrocatalytic CO2RR to generate CH4.
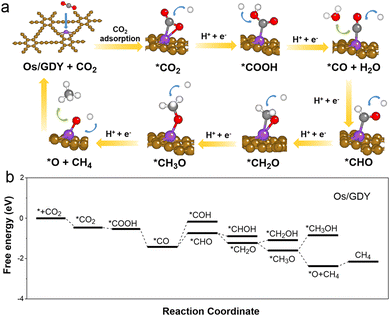 |
| | Fig. 6 Reaction mechanism of CO2RR to CH4 on Os1/GDY. (a) The structure of the reaction intermediates; (b) different energy profiles CHE model and the DFT method of PBE functional are used to calculate the path. | |
Conclusions
In this paper, DFT calculations were used to investigate the selectivity of CO2RR activity of GDY-loaded transition metal SACs by screening TM1/GDY (TM = Fe, Ru, Os, Co, Rh, and Ir) monatomic catalysts. By studying the electronic structure including DOS and CDD, it was found that the sp-hybridized carbon atoms on GDY have a great potential to tune the electronic structure of SACs and participate in the adsorption process of TM. The CO2RR of the Os1/GDY catalysts showed high performance towards CH4 with an energy potential barrier of 0.61 eV. Therefore, the GDY-loaded transition metal SAC may be a promising catalyst for application in other reduction reactions.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Author contributions
L. J. and M. Z. performed first-principles calculations, data curation, formal analysis and wrote the original draft. Q. Y. obtained funding support, participated in the discussion of the electronic structure calculations used in the paper and reviewed the manuscript.
Conflicts of interest
The authors declare no competing financial or non-financial interests.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 92061109), Natural Science Basic Research Program of Shaanxi (Program No. 2021JCW-20 and 2022KJXX-18). We gratefully acknowledge HZWTECH for providing computation facilities.
References
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- Z. Chen, Z. Liu and X. Xu, Nat. Commun., 2023, 14, 936 CrossRef CAS PubMed.
- T. Theerathanagorn, A. Vidal-Lopez, A. Comas-Vives, A. Poater and V. D’Elia, Green Chem., 2023, 4336–4349 RSC.
- M. D. Hossain, Q. Zhang, T. Cheng, W. A. Goddard and Z. Luo, Carbon, 2021, 183, 940–947 CrossRef CAS.
- K. Liu, J. Li, Y. Liu, M. Wang and H. Cui, J. Energy Chem., 2023, 515–534 CrossRef.
- Q. Yu, R. Lin, L. Jiang, J. Wan and C. Chen, Sci. China Mater., 2018, 61, 1007–1011 CrossRef CAS.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 CrossRef CAS PubMed.
- Y. Dai and Y. Xiong, Nano Res. Energy, 2022, 1, e9120006 CrossRef.
- J. Yuan, H. Li, Q. Wang, Q. Yu, X. Zhang, H. Yu and Y. Xie, Mater. Lett., 2012, 81, 123–126 CrossRef CAS.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS PubMed.
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani and X. Wang, Science, 2020, 367, 661–666 CrossRef PubMed.
- T. Zheng, K. Jiang and H. Wang, Adv. Mater., 2018, 30, 1802066 CrossRef PubMed.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- T. Ahmad, S. Liu, M. Sajid, K. Li, M. Ali, L. Liu and W. Chen, Nano Res. Energy, 2022,(1), e9120021 CrossRef.
- Q. Zeng, G. Yang, J. Chen, Q. Zhang, Z. Liu, B. Qin and F. Peng, Carbon, 2023, 202, 1–11 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- Q. Yang, Y. Jiang, H. Zhuo, E. M. Mitchell and Q. Yu, Nano Energy, 2023, 108404 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- Q. Zhang and J. Guan, Adv. Funct. Mater., 2020, 30, 2000768 CrossRef CAS.
- M. B. Gawande, P. Fornasiero and R. Zbořil, ACS Catal., 2020, 10, 2231–2259 CrossRef CAS.
- S. Ren, Q. Yu, X. Yu, P. Rong, L. Jiang and J. Jiang, Sci. China Mater., 2020, 63, 903–920 CrossRef CAS.
- Q. Yu, H. Li, Q. Wang, S. Cheng, L. Jiang, Y. Zhang, T. Ai and C. Guo, Mater. Lett., 2014, 128, 284–286 CrossRef CAS.
- C. Ye, Y. Zhou, H. Li and Y. Shen, Green Chem., 2023, 3931–3939 RSC.
- A. Tripathi and R. Thapa, Carbon, 2023, 208, 330–337 CrossRef CAS.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong and Y. Wang, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- V. Giulimondi, S. Mitchell and J. Pérez-Ramírez, ACS Catal., 2023, 13, 2981–2997 CrossRef CAS PubMed.
- L. Li, S. Cheng, H. Li, Q. Yu, J. Liu and X. Lv, Nano-Micro Lett., 2010, 2, 154–159 CrossRef CAS.
- H.-Y. Zhuo, X. Zhang, J.-X. Liang, Q. Yu, H. Xiao and J. Li, Chem. Rev., 2020, 120, 12315–12341 CrossRef CAS PubMed.
- C.-X. Zhao, J.-N. Liu, J. Wang, C. Wang, X. Guo, X.-Y. Li, X. Chen, L. Song, B.-Q. Li and Q. Zhang, Sci. Adv., 2022, 8, eabn5091 CrossRef CAS PubMed.
- Y.-N. Gong, W. Zhong, Y. Li, Y. Qiu, L. Zheng, J. Jiang and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 16723–16731 CrossRef CAS PubMed.
- P. Rong, Y.-F. Jiang, Q. Wang, M. Gu, X.-L. Jiang and Q. Yu, J. Mater. Chem. A, 2022, 10, 6231–6241 RSC.
- C. Xu, A. Vasileff, Y. Zheng and S. Z. Qiao, Adv. Mater. Interfaces, 2021, 8, 2001904 CrossRef CAS.
- Q. Yu, Sci. China Mater., 2023, 66, 1079–1088 CrossRef CAS.
- J.-C. Jiang, J.-C. Chen, M.-d. Zhao, Q. Yu, Y.-G. Wang and J. Li, Nano Res., 2022, 15, 7116–7123 CrossRef CAS.
- Q. An, S. Bo, J. Jiang, C. Gong, H. Su, W. Cheng and Q. Liu, Advanced Science, 2023, 10, 2205031 CrossRef CAS PubMed.
- X. Gao, J. Li and Z. Zuo, Nano Res. Energy, 2022, 1, e9120036 CrossRef.
- L. Hui, Y. Xue, H. Yu, Y. Liu, Y. Fang, C. Xing, B. Huang and Y. Li, J. Am. Chem. Soc., 2019, 141, 10677–10683 CrossRef CAS PubMed.
- C. Huang, Y. Li, N. Wang, Y. Xue, Z. Zuo, H. Liu and Y. Li, Chem. Rev., 2018, 118, 7744–7803 CrossRef CAS PubMed.
- P. Jiao, D. Ye, C. Zhu, S. Wu, C. Qin, C. An, N. Hu and Q. Deng, Nanoscale, 2022, 14, 14322–14340 RSC.
- X. Li and D. Xing, J. Phys. Chem. C, 2019, 123, 8843–8850 CrossRef CAS.
- T. He, S. K. Matta, G. Will and A. Du, Small Methods, 2019, 3, 1800419 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- H. Försterling and H. Kuhn, Int. J. Quantum Chem., 1968, 2, 413–430 CrossRef.
- M. D. Kostin, J. Math. Phys., 1991, 32, 1341–1343 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- J. Ortiz-Medina, F. López-Urías, H. Terrones, F. Rodríguez-Macías, M. Endo and M. Terrones, J. Phys. Chem. C, 2015, 119, 13972–13978 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- X. Fu, X. Zhao, T. B. Lu, M. Yuan and M. Wang, Angew. Chem., Int. Ed., 2023, 62, e202219242 CrossRef CAS PubMed.
- C. Huang, Y. Zhao and Y. Li, Adv. Mater., 2019, 31, 1904885 CrossRef CAS PubMed.
- J. Gao, D. Nandi and M. Gupta, J. Appl. Phys., 2018, 124, 014502 CrossRef.
- V. Fung, G. Hu, P. Ganesh and B. G. Sumpter, Nat. Commun., 2021, 12, 88 CrossRef CAS PubMed.
- S. Back and Y. Jung, ACS Energy Lett., 2017, 2, 969–975 CrossRef CAS.
- J.-H. Jeoung and H. Dobbek, Science, 2007, 318, 1461–1464 CrossRef CAS PubMed.
- S. Liu, J. Xiao, X. F. Lu, J. Wang, X. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2019, 58, 8499–8503 CrossRef CAS PubMed.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., 2013, 125, 2519–2522 CrossRef.
- X. Nie, W. Luo, M. J. Janik and A. Asthagiri, J. Catal., 2014, 312, 108–122 CrossRef CAS.
- G. Shi, Y. Xie, L. Du, X. Fu, X. Chen, W. Xie, T. B. Lu, M. Yuan and M. Wang, Angew. Chem., Int. Ed., 2022, 61, e202203569 CrossRef CAS PubMed.
- T. Jitwatanasirikul, T. Roongcharoen, P. Sikam, K. Takahashi, T. Rungrotmongkol and S. Namuangruk, Adv. Mater. Interfaces, 2023, 2201904 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Mengdie Zhao‡
a,
Mengdie Zhao‡