
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceα-Terpineol loaded, electron beam crosslinked polyvinyl alcohol/tapioca starch hydrogel sheets; fabrication, characterization and evaluation of wound healing potential on a full thickness acid burn wound†
Maria Khalid‡
a,
Fatima Jameel‡b,
Tooba Jabria,
Abdul Jabbar a,
Asmat Salimb,
Irfan Khanbc and
Muhammad Raza Shah*a
a,
Asmat Salimb,
Irfan Khanbc and
Muhammad Raza Shah*a
aHEJ Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi, 75270, Pakistan. E-mail: mari.talha@gmail.com; toobaasif137@gmail.com; abduljabbar.iccbs@gmail.com; raza.shah@iccs.edu
bDr Panjwani Center for Molecular Medicine and Drug Research, International Center for Chemical and Biological Sciences, University of Karachi, Karachi, 75270, Pakistan. E-mail: fatimajameel43@gmail.com; asmat.salim@iccs.edu
cCenter for Regenerative Medicine and Stem Cell Research, The Aga Khan University, Stadium Road, P. O. Box 3500, Karachi 74800, Pakistan. E-mail: irfankhan.bangash@aku.edu
First published on 3rd September 2024
Abstract
The multifaceted challenges in treating full-thickness acid burn wounds including impaired tissue regeneration, increased risk of infection, and the pursuit of functional and aesthetically pleasing outcomes, highlights the need for innovative therapeutic approaches for their treatment. The exceptional biochemical and mechanical properties of hydrogels, particularly their extracellular matrix-like nature and their potential to incorporate functional ingredients positions them as promising materials for wound dressings, offering a potential solution to the complexities of full-thickness burn wound management. The current study has integrated functional ingredients (starch and α-terpineol), known for their angiogenic, fibroblast-adhesive, and anti-inflammatory properties into an α-terpineol loaded, electron beam crosslinked polyvinyl alcohol/tapioca pearl starch hydrogel. The hydrogel was then explored for its efficacy in treating full-thickness acid burns. The hydrogel sheets, fabricated using a 25 kGy electron beam, were characterized for structural and functional properties. Surface morphology, gel fraction, swelling ratio, moisture retention capacity and thermal stability were also evaluated. PVA/tapioca starch hydrogel demonstrated optimal macroporosity, mechanical strength, thermal stability, water retention, and moisturizing ability, making it ideal for the intended application. In vitro skin compatibility analysis of α-terpineol-loaded hydrogel confirmed its biocompatibility, demonstrating 90% fibroblast viability. In vivo sensitivity testing on normal rat skin showed no inflammatory response. Analysis of the full-thickness rat chemical burn wounds treated with the hydrogels demonstrated that α-terpineol (AT) loaded e-beam crosslinked PVA/tapioca starch hydrogels increased the rate of wound closure, promoted re-epithelialization, facilitated collagen deposition, stimulated angiogenesis, and promoted keratin deposition, ultimately leading to healing of both thick dermal and epidermal tissues, as well as partial restoration of skin appendages over a duration of 30 days as confirmed by the histological and immunohistochemistry staining. Collectively, this study indicates that α-terpineol (AT) loaded e-beam crosslinked PVA/tapioca starch hydrogel holds promise as a cost-effective and efficient wound dressing for expediting full thickness acid burn wound healing, thus expanding the practical applications of the natural polymer based sheet hydrogel dressings.
1 Introduction
Burn therapy poses significant clinical challenges, particularly due to the loss of the skin's barrier, which increases infection risk and impedes the repair process. Burn severity is determined by factors such as size and depth.1 While first-degree burns affect the superficial layer of the epidermis, partial or second-degree burns affect the epidermis as well as the dermis.2 The epidermis, dermis, and subcutaneous tissue are all affected by full thickness or third-degree burns. Burns can also be classified based on the agent causing the burn. Thus, thermal burns refer to the skin injuries resulting from contact with hot liquids, surfaces, steam, or flame. Chemical burns are caused by corrosive acids and alkalis. Treatment strategies for burns include wound management, infection control, escharotomy and debridement of the wound, dressing coverage, skin transplantation, as well as the utilization of skin substitutes. However, as the degree of the burn lesion increases, the number of treatment choices decreases and the treatment becomes more complex.3 Full thickness or third degree wound are particularly challenging in terms of treatment because all three layers of skin are completely destroyed and the wounds pose significant challenges by secondary infection.4 The primary decision regarding a full-thickness wound revolves around whether to opt for surgical repair or allow it to naturally heal through secondary intention.5 Preserving or restoring function is often a primary consideration governing the decision. In most cases involving third-degree burns, surgical intervention is typically necessary to address the damage, often involving the replacement of damaged skin through procedures like skin grafts or the application of skin substitutes. Autologous skin grafts are a widely used surgical practice for third degree burn wound treatments in clinical settings, however in case of severe burns it fails to cover a large surface area due to the unavailability of donor sites. Therefore, there is a need for convenient and efficient way to facilitate and accelerate healing processes for facile restoration of skin integrity and functionality.For full thickness wound healing, the process significantly relies on angiogenesis, neovascularization,6 fibroblast migration, and proliferation,7 along with an optimally regulated inflammatory response.8 Severe burn wounds experience a more significant loss of dermal blood flow compared to superficial burns. As a result, newly formed blood vessels actively contribute to the healing process by providing vital nutrition and oxygen to developing tissues. Stimulating angiogenesis could thus aid in the regeneration of the dermal layers. Moreover, the migration and proliferation of fibroblasts from surrounding dermal tissue play a crucial role in the wound-healing process, serving as essential and rate-limiting steps. These actions are pivotal because dermal fibroblasts hold a central position in the formation granulation tissue, making them critical executors during the healing of full-thickness wounds. Moreover, the inflammatory response plays a crucial role in the healing process, triggering a series of cytokines and growth factors to defend against the risk of infection. However, prolonged inflammation can result in adverse effects, such as scarring and fibrosis. Dressings with multifunctionality, supporting angiogenesis, fibroblast migration, and regulating the inflammatory response, coupled with tunable rheological properties, hemostatic abilities, antibacterial properties, and excellent cytocompatibility, emerge as highly desirable candidates for facilitating the healing of full thickness burn wounds. Hydrogels, characterized by crosslinked polymeric networks containing up to 96% water, stand out as promising dressings suitable for all types of burn wound care, owing to their outstanding features.9 Owing to their structural similarity with the natural extracellular matrix,10 they provide instructive environments for the three-dimensional assembly of vascular networks as well as of the adhesion and proliferation of fibroblasts. A large volume of work on hydrogel-based scaffolds has primarily focused on their applications in wound healing. In addition to serving as scaffolds and extracellular matrix-like dressings, hydrogels exhibit versatility in delivering growth factors,11 antibiotics,12 and cells13 thereby facilitating complete skin regeneration. Hydrogels can be tuned into various shapes and forms using a variety of crosslinking methods including physical chemical and e-beam crosslinking. They are soft, pliable, atraumatic during use, and maintain a moist environment that reduces pain.14 Beyond their intrinsic angiogenetic potential, hydrogels can be customized to incorporate angiogenetic biopolymers15–17 and therapeutic agents.18
In the context of full-thickness wound treatment, whether due to burns or other reasons, previous studies have focused on creating proangiogenic and neovascularization-supportive 3D polymer networks.19,20 A number of other studies have incorporated bioactive ingredients promoting angiogenesis and neovascularization. Examples of utilizing angiogenetic materials within 3D polymeric network hydrogels include development of matrices from polysaccharides for both in vitro and in vivo vascularization. A common material in this regard is hyaluronic acid.21 Alternative polysaccharides, such as dextran and alginate, have also been employed for angiogenic biomaterials. A previous work particularly reports the use of a dextran based acellular hydrogel for the treatment of full thickness burn wounds.6,22 The bioactive ingredients incorporated within these matrices comprise stem cells, extracellular vesicles, growth factors23 and other therapeutic agents.18 The scaffolds and dressings vary enormously in terms of their methods of crosslinking and the morphological forms. Starches have found widespread use as scaffolds in applications requiring accelerated angiogenesis, neovascularization, and cellular adhesion24–26 and there have been instances of using starch-based scaffolds for full-thickness wound treatment.27,28 Their exploration specifically in the form of sheet hydrogels for treating various kinds of full-thickness burn wounds (thermal and chemical), however, remains a relatively unexplored area in the current literature.
Inspired by the utilization of starch in angiogenetic, cellular adhesion, and tissue engineering scaffolds,29 and considering the potential incorporation of starch into an electron beam crosslinked durable sheet scaffold, this work describes the fabrication and wound-healing efficacy of α-terpineol-loaded polyvinyl alcohol (PVA)/tapioca starch sheet hydrogel dressing for the treatment of third-degree acid burn wounds in rats (Scheme 1). PVA was chosen as a crosslinkable polymer to fabricate hydrogels due to its biocompatibility, non-toxicity, water solubility,30 sheet-forming ability,31 controllable e-beam crosslinking, mechanical strength, high water retention, non-adherence to wounds, and cost-effectiveness32 (considering potential scale-up). These characteristics are particularly desired for treating third-degree acid burn wounds. Tapioca pearl starch, derived from Cassava roots, enhances hydrogel integrity with optimal porosity, mechanical strength, and moisture retention. Its bioadhesive properties support fibroblast adhesion, while Cassava antioxidants may contribute to wound healing.33 α-terpineol (hereafter referred to as AT), with its known wound-healing properties, can be effectively delivered through the hydrogel for a sustained therapeutic impact. The monoterpene demonstrates wound healing,34 antimicrobial35 and antifungal effects.36 AT also inhibits generation of prostaglandin-endoperoxide synthase,37 COX-2 (ref. 38) and IL-6,39 thereby regulating inflammation for appropriate wound healing. It is thus hypothesized that the combining the wound healing properties of tapioca starch and AT within electron beam crosslinked ECM-like sheet hydrogels will lead to a multifunctional and anti-inflammatory ECM like material. The chemical burn wound-healing process is expected to be enhanced by the stimulation of rapid neovascularization through material tissue interaction. Improved wound contraction and skin regeneration in in vivo applications are the anticipated outcomes of this approach. As evidenced, the outcomes validate the potential of the as-fabricated hydrogels as advanced wound dressings, offering a comprehensive solution for the intricate challenges associated with full-thickness burn wounds. The resulting combination effectively harnesses the merits of all components including polyvinyl alcohol, tapioca pearl starch and AT, providing a convenient and versatile ECM like platform for efficient full thickness burn wound management. To the best of our knowledge, this is a first study evaluating the potential of a PVA–tapioca starch-AT based hydrogel sheet dressing for healing a full thickness chemical/acid burn wound.
 | ||
| Scheme 1 Fabrication of PVA–tapioca pearl hydrogels via electron beam crosslinking. | ||
2 Experimental section
2.1 Materials
Poly vinyl alcohol (PVA, MW. 15000 g mol−1), was procured from Sigma-Aldrich. Tapioca starch beads were purchased from local market in Karachi Pakistan. α-Terpineol (AT) was procured from Sigma Aldrich. Distilled water (DW) was utilized for the preparation of all polymer solutions.The details of purchase of various chemicals utilized in biological assays are as follows. MTT dye (M5655, Sigma-Aldrich), DMSO (196055, MP Biomedicals), calcein dye (65-0853-81, Invitrogen eBioscience), DAPI (157574, MP Biomedical Inc.), MEM (12561-056, Gibco Life Technologies), FBS (10438-026, Gibco Life Technologies), sulfuric acid (31350, Fluka, Riedel-dehaen), cell culture grade phosphate buffered saline (70011-044, Gibco Life Technologies), ketamine hydrochloride (K2753-5G, Sigma), xylazine hydrochloride (A148943-5, Ambeed Hts), One-step RNA reagent (BS410A, Bio Basic), penicillin/streptomycin (15140-122, Gibco Life Technologies), sodium pyruvate (11360, Gibco Life Technologies), Triton X-100 (T8787, Sigma), trypsin–EDTA (25200056, Gibco Life Technologies), tween 20 (194724, MP Biomedical, Inc.), Bright green 2× qPCR master mix (G892, Applied Biological Materials Inc.).
2.2 Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 10:4, 11.5:3, 13:2, and 13:4, where the first number represents the concentration of PVA and the second represents the concentration of tapioca pearl starch solution.
:2, 10:4, 11.5:3, 13:2, and 13:4, where the first number represents the concentration of PVA and the second represents the concentration of tapioca pearl starch solution.Subsequently, 40 mL of each formulation was poured into polystyrene plates and exposed to varying electron beam intensities (25–60 kGy) at a conveyor belt speed of 3 m min−1. The resulting sheets were then evaluated for mechanical strength, wearability, water content, swelling ratio, and adhesion to select the best sheet form.
2.3 Characterization
| Swelling ratio (%) = (Ws − Wd)/Wd × 100 | (1) |
The moisture retention capability of the hydrogels was estimated using eqn (2) in which Mn is the moisture content of the material. Ww is the weight of the wet hydrogel and Wd is the weight of the dried sample following a pre-determined period of drying. Hydrogel pieces of almost equal thickness were cut and weighed. These hydrogels were placed at temperatures of 25 °C for 6 h and Wd was recorded at intervals. The moisture retention capacity was calculated as a function of time
| Moisture retention capability (%) = (Wd/Ww) × 100 | (2) |
The hydrogels prepared were dried under vacuum and weighed (Wo). Subsequently, the dried hydrogels were soaked in DW for 24 h to remove soluble components. Following this, the hydrogels were dried once more and re-sweighed (We). The gel fraction percentage was calculated using the following equation:
| Gel fraction (%) = [We/Wo] × 100 |
 000 cells per well were seeded to adhere upon hydrogel surface and incubated for 24 h at 37 °C. MTT dye (0.5 mg mL−1) was added into individual wells followed by a 4 h incubation at 37 °C. Following incubation, dye was removed and DMSO was added for dissolving formazan crystals secreted by metabolically active cells. The suspension was gently collected in a separate 96 well plate and spectrophotometer (SpectraMax, USA) was used to record the absorbance at 590 nm.
000 cells per well were seeded to adhere upon hydrogel surface and incubated for 24 h at 37 °C. MTT dye (0.5 mg mL−1) was added into individual wells followed by a 4 h incubation at 37 °C. Following incubation, dye was removed and DMSO was added for dissolving formazan crystals secreted by metabolically active cells. The suspension was gently collected in a separate 96 well plate and spectrophotometer (SpectraMax, USA) was used to record the absorbance at 590 nm.Skin cells were allowed to grow and culture on the surface of hydrogels (blank and AT loaded gels) and incubate for 24 h at 37 °C. Adherent cells were fixed with 4% paraformaldehyde for 10 min incubation at room temperature. Cells were washed 2–3 times with 1× PBS. DAPI was added and incubated for 10 min, followed with PBS washing. Images were taken at 10× magnification under phase contrast (TE2000 Nikon, Japan) and fluorescence microscopes (NIE, Nikon, Japan) respectively.
000 cells per well were seeded to adhered on the surface of hydrogels and incubated for 24 h at 37 °C. Cells were washed with 1× PBS on the next day. Calcein dye (10 μM) was added for 30 min incubation at 37 °C. 1× PBS was used to wash the cells gently for 15 min. Images of live cells was captured under fluorescence microscope.Hydrogel treatment was similarly applied (5 h per day till one week) for the treatment of burn wound tissues the next day following wound induction. For treatment, hydrogel patch (approximately 1 inch in diameter) was slightly placed on the wound site along with loose gauze bandage. After one-week treatment, gross macroscopic examination was observed at different time points (day 1, 5, 10, 15, 20, 25, and 30).
:100. Alexa fluor 488 goat anti-mouse secondary antibody was added to each section at a 1:200 dilution for 1 h at 37 °C. DAPI was used to stain the nuclei. Images were captured at different magnifications under a fluorescence microscope.3 Results and discussion
3.1 Synthesis and optimization
The PVA–tapioca hydrogel sheets were fabricated through a process of free radical polymerization induced by an electron beam. This method generates radicals on both PVA and starch chains, followed by crosslinking, resulting in flexible sheets. To achieve the appropriate sheet form, the polymer concentration and electron beam intensity for irradiating the pre-hydrogel mix were carefully optimized through iterative experiments and analysis. Not all combinations of concentration and electron density resulted in sheet formation; some yielded sheets with inferior mechanical properties or inadequate water content.Fig. 1(a) shows the relationship between total polymer concentration and mechanical strength for some representative combinations. In general, tensile strength increases with increasing polymer content. Therefore, formulations with the highest polymer content (PVA 13%:tapioca 2% and PVA 13%:tapioca 4%) can endure the highest amount of stress before breaking. At the lowest total polymer content, the mechanical strength is so poor that the sheet breaks during the process of lifting it off the plate. The poor mechanical strength at low polymer concentration is due to high water content and fewer crosslinks. However all hydrogel sheets have minimal elongation at break and are relatively brittle, meaning it does not stretch much before it fractures.
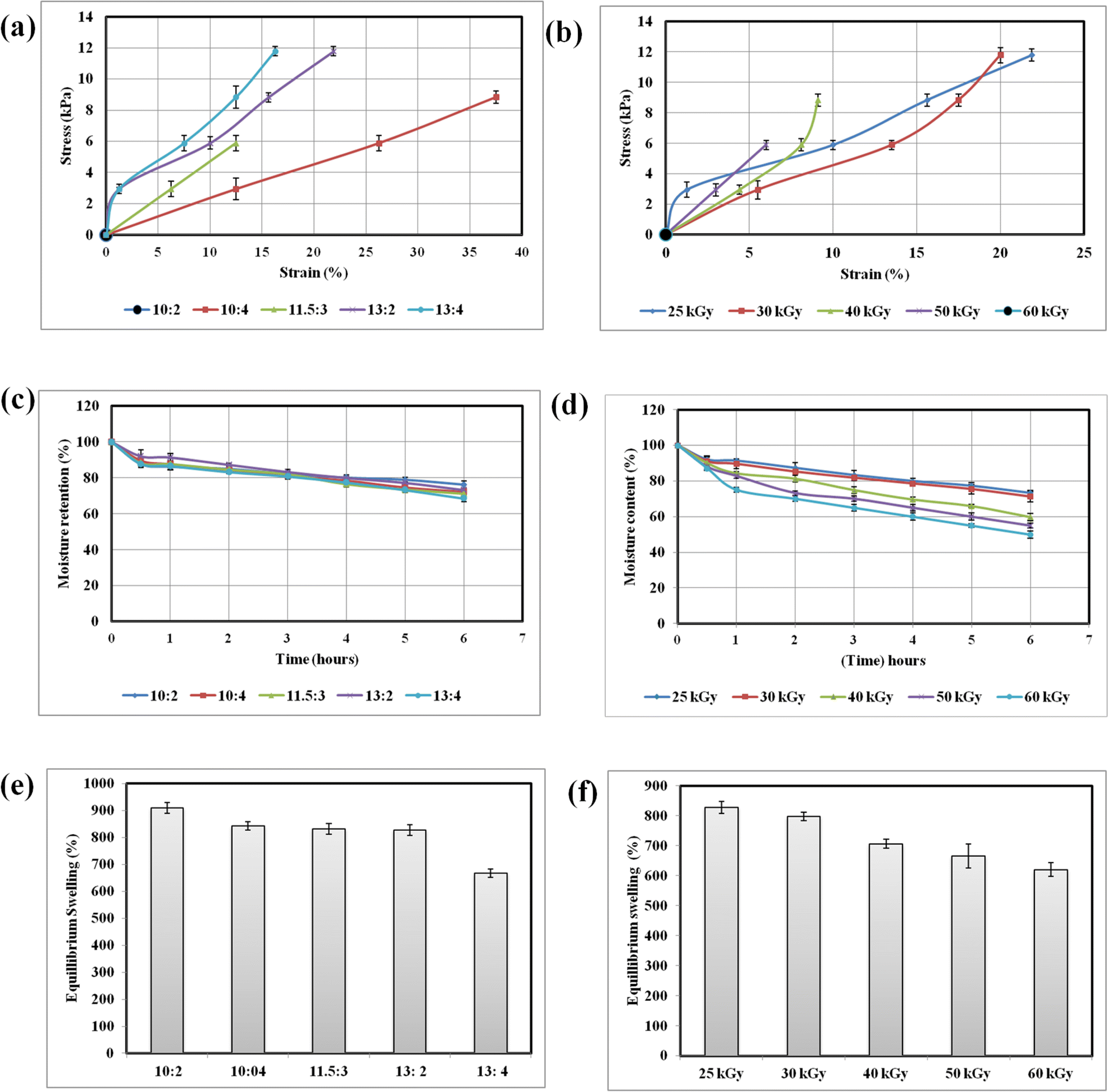 | ||
| Fig. 1 (a) Stress–strain curves at various polymer concentrations. The black dot indicates that tests could not be performed (b) stress–strain curves of PVA:tapioca 13:2 at various electron beam doses. The black dot indicates that tests could not be performed (c) moisture retention of hydrogel sheets fabricated at different polymer concentrations. (d) Moisture retention of PVA:tapioca 13:2 fabricated different electron beam doses. (e) Equilibrium swelling (%) of hydrogel sheets fabricated at different polymer concentrations. (f) Equilibrium swelling (%) of PVA:tapioca 13:2 fabricated at different electron beam doses. | ||
Keeping the polymer concentration constant, the influence of electron beam dose on mechanical strength can be seen in Fig. 1(b). According to literature reports, the mechanical strength increases with radiation dose up to a certain threshold. According to literature reports, 25 kGy is the dose at which PVA hydrogels attain their maximum gel fraction.40 Beyond this point, further increases in the dose lead to a higher crosslink density, making the hydrogel brittle and inflexible. At 60 kGy, the hydrogel breaks under minimal stress, signifying a very rigid structure with low flexibility and increased brittleness. This brittleness may also result from the breakage of polymer chains at higher doses.
Fig. 1(c) and (d) respectively illustrate the impact of increasing polymer concentration and electron beam doses (ranging from 25 kGy to 60 kGy) on the moisture retention of hydrogels. Increased crosslinking density with higher polymer concentration and radiation dose affects the gel's ability to absorb and retain water. Increased crosslink density reduces the hydrogel's water retention capacity. The denser network of polymer chains limits the space available for water molecules, resulting in higher percentage moisture loss. The hydrogels based on PVA:tapioca ratios of 10:2, 10:4: 11.5:3, 13:2 and 13:4 show moisture content of 76.22 ± 1.9%, 72.22 ± 2.0%, 71.1 ± 1.9%, 73.33 ± 0.9% and 68.53 ± 1.9%. It is evident that hydrogels with comparable polymer concentrations (10:4:11.5:3, 13:2) show comparable moisture retention over a period of 6 h. The equilibrium swelling data shown in Fig. 1(e) and (f) also show the same trend. The variation of gel fraction with polymer concentration (Table 1) shows that gel fraction increases only slightly with the increasing polymer concentration and is more or less same for the sheets with comparable polymer concentration.
| PVA:tapioca concentration ratio (w/v%) |
Gel fraction (%) |
|---|---|
| 10:2 |
93.8 ± 1 |
| 10:4 |
94.2 ± 0.8 |
| 11.5:3 |
94.2% ± 0.9 |
| 13:2 |
94.3 ± 0.9 |
| 13:4 |
95.2 ± 0.7 |
The gel fraction data shown in Table 2 indicate that the hydrogel reaches maximum gelation at 25 kGy. Further increase in dose creates multiple crosslinks between the same polymer chains, slightly increasing the gel fraction and causing it to plateau.
:tapioca 13:2 hydrogel sheets fabricate at different electron beam doses
| Electron beam doses (kGy) | Gel fraction (%) |
|---|---|
| 25 | 94.3 ± 0.9 |
| 30 | 94.6 ± 0.7 |
| 40 | 95.1 ± 0.9 |
| 50 | 95.8 ± 1.2 |
| 60 | 96.4 ± 1.1 |
Thus, higher electron beam intensities enhance the mechanical strength of hydrogels up to a certain point, but they also increase brittleness and reduce moisture retention. In general, mechanical strength increased with higher polymer content and decreased with electron beam intensity in the range of the doses applied during this study. The water content and swelling decreased with the increase in polymer content and radiation doses. The optimized formulation as shown in Table 3, comprised a sheet formed by a 1:1 mixture of 13% w/v PVA and 2% w/v tapioca pearl starch, crosslinked by an electron beam intensity of 25 kGy. The optimized formulation also had moderate adherence to human skin compared to those with higher tapioca content, which is a desirable trait since a strongly adhering dressing might disturb the wound during successive dressing changes, causing trauma, pain, and disrupting the healing process.
| Parameter | Range | Optimal value |
|---|---|---|
| PVA concentration (% w/v) | 10–13 | 13 |
| Tapioca pearl starch concentration (% w/v) | 2–4 | 2 |
| Electron beam intensity (kGy) | 20–60 | 25 |
3.2 Microtopography and porosity of optimized hydrogel sheet
Hydrogel micro-architecture, in particular the porosity of the hydrogel is an important factor determining its usefulness for the intended application.41 In the context of wound healing, the pore size of a hydrogel must be carefully tailored based on the size, particularly the hydrodynamic radius, of the bioactive ingredient or cargo to be delivered. Additionally, the pore size should facilitate the appropriate transmission of gases and nutrients essential for the healing process. The porous structure of the freeze-dried PVA–tapioca pearl starch hydrogels is visible in scanning electron microscopy (SEM) images shown in Fig. 2. The pores are interconnected, with sizes in the micron range, and some tunnel-like pores are also evident. The observed porosity ensures the breathability of the hydrogel sheet as a wound dressing, preventing any surface necrosis of the wound. It is also appropriate for the delivery of small molecular weight AT and for the transfer of gases. | ||
| Fig. 2 Scanning electron micrographs of PVA–tapioca hydrogel at different magnifications. Hydrogels have porous structures with interconnected pore network. Scale bars: (a) 50 μm (b) 20 μm. | ||
3.3 Swelling ratio
The macroporous nature of the PVA/tapioca hydrogel was further confirmed by the swelling characteristics. It was observed that discs with an average weight ∼0.2 g, upon absorption of water at 25 °C were swollen up to ∼2.1–2.2 g during a period of 48 h as shown in Fig. 3(a). This corresponds to weight increase of ∼ 820% by swelling. The hydrogel discs were stable and maintained their swollen weight if stored appropriately in non-drying conditions. The hydrogel discs also resumed their original sheet form following swelling, a characteristic that is important for their drug loading and subsequent application as wound dressing. | ||
| Fig. 3 (a) Swelling ratio of hydrogels as a function of time at 25 °C; each point represents the mean ± SD (n = 3). (b) Moisture content as a function of time at 25 °C. | ||
3.4 Moisture retention
Water retention capacity is a crucial property, especially in the case of hydrogels designed for wound dressing applications. This property is essential to ensure that the hydrogel delivers a cooling effect and effectively maintains a moist environment in the wound bed. Fig. 3(b) shows that optimized hydrogel sheet retains most of its moisture up to 6 h under exposed conditions at room temperature. This slight loss in weight is due to the loss of water attributed to evaporation at room temperature, however it shows that the hydrogel retains sufficient moisture for room temperature wound dressing applications.3.5 Gel fraction
The transition from a system with finite branched polymer to infinite molecules is known as sol–gel transition. After the solution is removed from a hydrogel, the remainder is known as the gel fraction of the hydrogel. Gel fraction is the indication of the degree of crosslinking in the hydrogel. A higher gel fraction corresponds to a higher degree of crosslinking and is indicator of the sheet hydrogel's mechanical strength. The synthesized hydrogel exhibited a gel content of 94.3%, indicating the formation of a stronger hydrogel network. This higher gel fraction is observed because the crosslinking is achieved via exposure to a beam of electrons generating a substantial number of free radicals formed on the polymer chains. These radicals undergo rapid and extensive crosslinking generating a strong hydrogel network.3.6 Powder X-ray diffraction analysis
X-ray diffraction (XRD) pattern (Fig. 4) of the PVA solid shows three typical peaks at 2θ = 19.6°, 22.9°, and 40.8°, corresponding to the (101), (200), and (102) planes of PVA crystallite.42 The X-ray diffractogram of the solid tapioca starch indicate peaks around 17.5°, 19°, 24°, showing a moderate degree of crystallinity which is also consistent with earlier literature reports.43 The dried hydrogel sheet gives a single rather broad peak with a noticeably reduced intensity around 2θ ≈ 20° which shows that hydrogel formation and crosslinking may have disrupted the crystalline structure of PVA as well as starch granules. The XRD pattern of the fabricated hydrogel however was significantly different from the two raw materials indicating the successful synthesis of hydrogels. | ||
| Fig. 4 X-ray diffractograms of PVA, tapioca pearl starch and PVA–tapioca hydrogels. | ||
3.7 Thermogravimetric analysis and differential scanning calorimetry
The TGA thermograms of pure PVA/tapioca pearl starch and the dried hydrogels have been shown in Fig. 5(a). Thermogravimetric analysis was performed to determine the thermal stability. In this study, all examined materials underwent weight loss in two distinct stages during thermal gravimetric analysis; one occurring below 270 °C, attributed to moisture loss, and another above 270 °C, linked to the degradation of the polymeric or hydrogel components. Notably, there are slight variations in the moisture loss pattern between the hydrogels and pure PVA, with the hydrogels exhibiting a lower initial loss compared to PVA. This discrepancy in release kinetics is attributed to differing interactions between water molecules and PVA, tapioca, and the fabricated hydrogels. Additionally, the gradual decrease in weight observed for the hydrogels below 250 °C, in comparison to the pure polymers, underscores the moisture retention capacity of the PVA/tapioca hydrogels. The presence of H-bonded water within the hydrogel 3-D network traps water and prevents its exclusion from crystallizing domains, resulting in slow water release. This characteristic makes the hydrogels ideal for wound healing applications, where a gradual release of moisture is beneficial.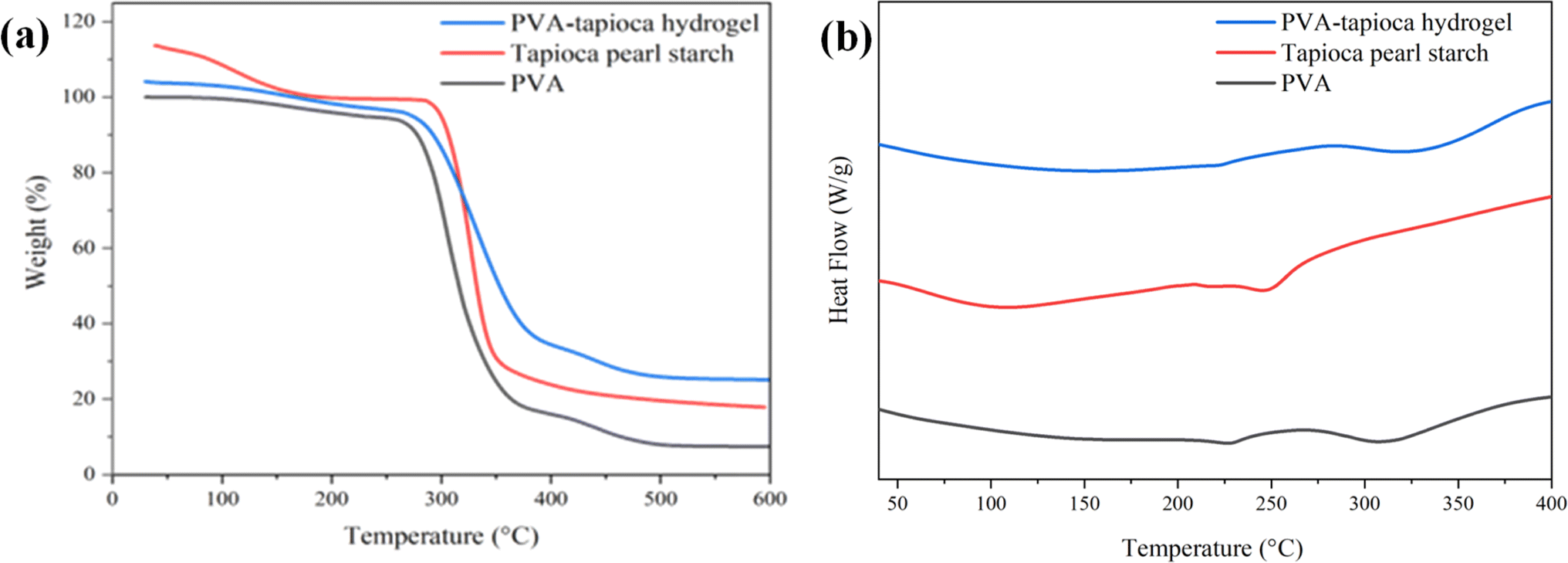 | ||
| Fig. 5 (a) TGA thermograms of PVA, tapioca pearl starch and PVA–tapioca hydrogels. (b) DSC thermograms of PVA, tapioca pearl starch and PVA–tapioca hydrogels. | ||
The hydrogels started to degrade (degradation onset) at temperatures slightly higher (20 °C) than that of pure PVA, indicating an increased thermal stability presumably due to crosslinking. The hydrogel is found to be stable within the temperature range of the intended use or application (wound dressing). This shows that the synthesized hydrogels could be processed at reasonably higher temperature. The as-fabricated hydrogel exhibited a residuum at 600 °C (5%) smaller than either crystalline PVA (7%) or tapioca pearl starch (17%). The TGA analysis provides crucial insights into the thermal properties of the wound dressing hydrogels, offering a comprehensive understanding of their behavior across varying temperature conditions. These findings are particularly valuable as they allow for direct comparisons with similar PVA-based hydrogels44 reported for wound dressing applications.
Differential scanning calorimetry (DSC) is a valuable tool for investigating first- and second-order thermal transitions, including glass transition, crystallization, melting, and decomposition processes in polymeric materials. These transitions occur as the sample temperature increases, following a predefined temperature program.45 PVA exhibits an endotherm with the lowest point on the dip at 227 °C, corresponding to the crystalline melting point (Tm) of PVA as shown in Fig. 5(b).46 The decomposition endotherm (TD) was observed at 307 °C. Tapioca pearl starch exhibits its melting endotherm around (Tm) 160 °C whereas the corresponding decomposition endotherm (TD) was observed around 250 °C. For the hydrogel, the melting endotherm is observed at 220 °C and is rather broad. The incorporation of tapioca starch and subsequent crosslinking reaction within the PVA matrix results in the broadening and shifting of the melting endotherm of PVA to lower temperatures. Moreover, in comparison to pure PVA, the decomposition endotherm of PVA/tapioca hydrogels was observed at a higher temperature indicating the improved thermal stability of hydrogels.
3.8 Hydrogel degradation analysis
Burn wounds present a variety of pH levels, depending on various factors such as the wound healing stages, the presence or absence of infection, and bacterial load.47 The degradation of PVA–tapioca pearl starch hydrogels was investigated at different pH values corresponding to those observed in burn wounds. The data shows that these hydrogels are remarkably stable across the pH range investigated, exhibiting only minimal weight loss, which aligns with the gel fraction data (Fig. 6). This implies that the crosslinked fraction, approximately 94%, remains structurally and functionally intact, enabling it to perform its therapeutic function effectively. | ||
| Fig. 6 Hydrogel degradation analysis at pH 6, 7, 8 and 9. | ||
This stability is particularly beneficial for moist wound healing, which requires the gel to retain therapeutic moisture levels, providing the advantages of a moist healing environment. The fabricated hydrogel patch is stable and does not contain any pH-sensitive moieties or crosslinks that could compromise its structural and functional integrity.
3.9 Determination of non-cytotoxic dose of AT and evaluation of its wound healing potential
Using MTT assay, the non-cytotoxic dose of AT was determined to be 10 μM (Fig. 7(a)). Using the same dose, scratch assay can be used to evaluate the wound healing potential of AT. In scratch assay analysis, enhanced cell migration or significant reduction of wound closure was observed in AT treated cells as compared to untreated control. Blue arrows indicate fibroblasts migration towards the scratch area (Fig. 7(b)). The quantitative scratch analysis showed wound closure at 24 h (68%), 48 h (86%), 72 h (90%) in the AT treated group as compared to untreated control (Fig. 7(c)). Furthermore, IL-1β, IL-6, BAX were downregulated and IL-13, BCL-2, VEGF, FGF, PRDX-4, GPX-7, and SOD-1 were upregulated in treated group as compared to control (Fig. 7(d)). These findings collectively suggest a positive influence of AT treatment on wound healing, potentially mediated through the modulation of relevant paracrine factors and enhanced cell migration, further verifying their role in loaded PVA/tapioca pearl starch hydrogels. | ||
| Fig. 7 (a) 10 μM dose of α-terpineol (AT) was determined non-cytotoxic via MTT assay. Cytotoxicity analysis was performed in rat skin cells (fibroblasts). (b) Cell migration was observed via scratch assay analysis. (c) Bar graph representing statistical analysis of percentage scratch wound closure. (d) Transcriptional analysis of wound healing mediator following complete wound closure. IBM SPSS 21 software was used for one-way ANOVA and Bonferroni post hoc test; p-value ≤ 0.05 was considered statistically significant (*** = p ≤ 0.001, ** = p ≤ 0.01 and * = p ≤ 0.05). | ||
3.10 Drug loading experiments and characterization of loaded hydrogels
A 10 μM non cytotoxic concentration of AT was determined via MTT assay as shown in Fig. 7(a). Wet Hydrogel patches each with 1 inch diameter were dried to constant weight of approximately 0.2 g and immersed in a 10 μM solution of AT (approximately 8 mL/0.2 g hydrogel disc). The hydrogel patches were allowed to swell up to their equilibrium swelling ratio. This absorbed amount was thus available for the therapeutic effect on the third degree burn wound. The molarity of the left over solution (20 mL) was determined by making use of the calibration curve and was determined to be 6.7 μM. Each hydrogel patch thus encapsulated a 590 μg of AT with a loading efficiency of 46%.The IR spectra of the AT-loaded and unloaded hydrogel formulations are very similar as shown in Fig. 8(a), there are only minor peak shifts which signify the interaction between the hydrogel network and AT. All the representative peaks of the drug-free hydrogel are retained in the AT-loaded hydrogels, and no new peaks were observed in the AT loaded hydrogels, indicating the integrity and stability of AT within the hydrogel network structure. The SEM micrograph of loaded hydrogel is shown in Fig. 8(b).
 | ||
| Fig. 8 (a) Infrared spectra of AT, blank PVA/tapioca pearl starch hydrogel and AT-loaded hydrogel. (b) SEM micrograph of AT-loaded hydrogel. | ||
3.11 In vitro drug release assay
Fig. 9 shows the release profile of AT in aqueous solution. The hydrogel patch exhibited an initial burst release within the first 30 min, followed by a slower release over the next 5 h, eventually reaching a plateau. The initial burst release can be attributed to the drug adsorbed on the surface of the hydrogel. Following this phase, the drug within the hydrogel matrix diffused through the porous structure into the release medium. This release behavior is consistent with that observed in other PVA-based hydrogels. The initial burst release is advantageous for providing an immediate therapeutic effect, particularly beneficial for treating third-degree acid burn wounds. The overall cumulative release amounted to nearly 50% within 5–6 h, indicating a significant release of the active compound over a relatively short period. | ||
| Fig. 9 In vitro release curve of AT. | ||
3.12 In vitro skin compatibility of hydrogels and morphological characteristics of skin cells
Cultured fibroblasts adhered on the surface of hydrogels and showed proliferation with spindle shaped fibroblast like morphology under phase contrast microscope. The surface of AT loaded gel exhibited increased number of proliferated cells in contrast to blank gel (Fig. 10(a)). Both hydrogels were found to be non-cytotoxic for skin cells. For confirmation, DAPI was used to stain nuclei of proliferated cells (Fig. 10(b)). However, statistical analysis showed 90% cell viability in AT loaded gel as compared to blank gel (Fig. 10(c)). It was further witnessed by live cell imaging under fluorescence microscope as shown in Fig. 10(d). Overall, these results collectively provide a comprehensive understanding of the impact of AT-loaded hydrogels on fibroblast behavior, including morphology, proliferation, and cell viability, supporting the potential efficacy of the AT-loaded gel in cellular or wound healing applications. | ||
| Fig. 10 (a) Image showing cell seeding and morphological characteristics of skin cells on the surface of blank & AT loaded hydrogels. All images were taken at 10× magnification under phase contrast. (b) In vitro skin compatibility was performed to evaluate non cytotoxic effect of these hydrogels on skin cells. DAPI was used to stain nuclei of adhered cells and images were taken at 10× magnification under fluorescence microscope (c), quantitative analysis showed more than 90% cell viability in AT loaded gel as compared to the blank gel. IBM SPSS 21 software was used for one-way ANOVA and Bonferroni post hoc test; p-value ≤ 0.05 was considered statistically significant (*** = p ≤ 0.001, ** = p ≤ 0.01 and * = p ≤ 0.05) (C), (d) calcein was used to stain live cells. Image exhibiting increase number of live cells in AT loaded gel in contrast to the blank gel. (e) Quantitative analysis of cell survival rate by calcein AM/PI staining. | ||
3.13 In vivo skin compatibility of hydrogels
To verify the specificity of the hydrogels for injured tissues while preserving the integrity of surrounding healthy tissues, skin sensitivity tests were conducted. Application of both hydrogels onto a single rat's skin (5 h per day for 5 days) was followed by thorough macroscopic examinations for up to 15 days post-application. No observable inflammatory reactions such as erythema or edema was detected, affirming the in vivo skin compatibility of the hydrogels (Fig. 11(a)). | ||
| Fig. 11 (a) Skin sensitivity against hydrogels was tested on normal rat skin. Both hydrogels were placed on a single rat's skin. Macroscopic examination was observed till day 15 after hydrogel application. No inflammatory response (erythema & edema) was observed on the skin. (b) Wound model development and hydrogel application at the wound site for 5 h per day along with gauze. (c) Temporal healing pattern was observed in burn wound (control), blank gel treated wound, AT loaded gel treated wound. (d) Graphical representation of wound area over time. | ||
3.14 Macroscopic examination of wound tissues
Continual gross macroscopic observations of third-degree acid burn wound healing were conducted up to day 30, as depicted in Fig. 11(c). Initial observations on day 1 revealed no significant differences between the groups. However, at later time points, the control group exhibited extensive tissue necrosis, while wounds treated with the blank gel displayed crust/scab formation without tissue destruction—indicative of the initiation of the recovery phase. Meanwhile, wounds treated with AT-loaded gel showcased notable wound contraction at the same time point (Fig. 11(d)). The wound site also exhibited the development of skin appendages, notably the emergence of new hair follicles, providing compelling evidence of healing.The dressing demonstrated accelerated wound closure rates and enhanced tissue re-epithelialization. Although this study marks a novel approach in treating third-degree chemical burn wounds using hydrogels. An analysis of macroscopic wound healing characteristics between the AT loaded tapioca pearl hydrogel dressings and similar dressings reported in literature (such as those employed by Mohamad et al., for partial thickness burn wounds48 or by Jiji et al. for third degree burn wound49) showcased superior performance of the dressings fabricated in this work. While dressings demonstrated healing patterns similar to those observed in other studies,48,49 the treatment durations were notably shorter, signaling an enhanced overall efficacy.
3.15 Histological analysis
The histological analysis of burn wound tissues (Fig. 12) at day 30 further corroborated our macroscopic findings. In the case of control burn wound tissues at day 30, damage to both the epidermal and dermal layers, along with inflamed hair follicles and a disorganized collagen network was evident. | ||
| Fig. 12 Histological analysis of control untreated third degree acid burn wound and blank gel and AT loaded hydrogel treated burn wound tissues stained with H & E and Masson trichrome on day 30. Images: ×10 magnification; bright field microscope. | ||
In contrast, burn wounds treated with blank hydrogels continued to exhibit a devastated extracellular matrix (ECM) at day 30. Additionally, there was evidence of cellular infiltration, although re-epithelialization remained incomplete.
However, in the treatment group, significant improvements were observed. By day 30, there was complete re-epithelialization, with regenerated skin adnexa and a restored epidermal layer. Furthermore, the ECM appeared relatively healthy, suggesting improved tissue remodeling and regeneration in response to AT loaded hydrogel treatment (Fig. 12).
3.16 Immunohistochemistry
A comprehensive analysis of protein expression was conducted, focusing on alpha smooth muscle actin (α-SMA) and keratin 5 (KRT5), in burn wound tissues treated with control, blank hydrogel, and AT-loaded formulations (Fig. 13).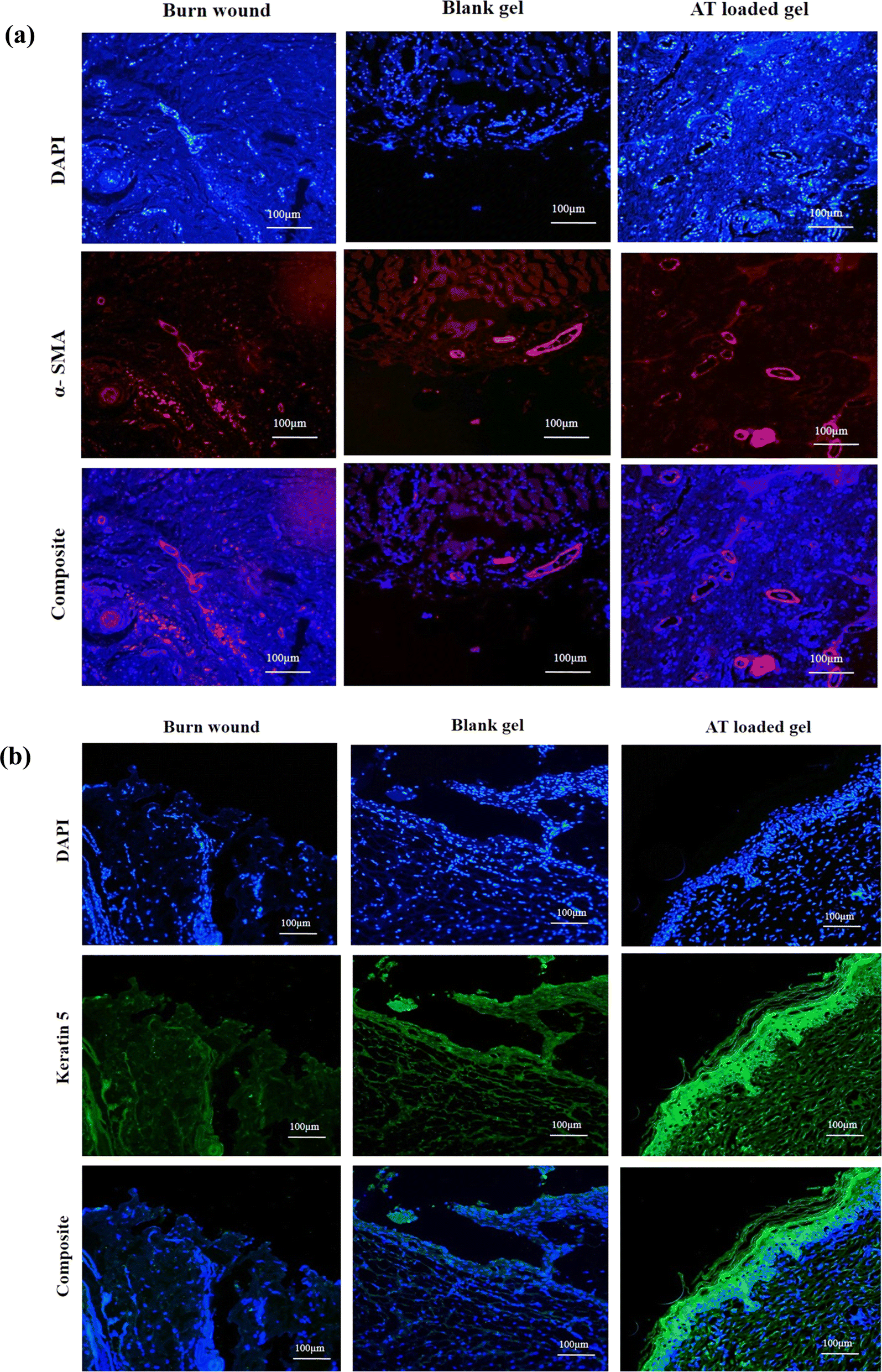 | ||
| Fig. 13 Analysis of neovascularization and re-epithelization: immunohistochemical analysis of (a) α-SMA, (b) keratin 5 at day 30. Secondary antibody: Alexa fluor 488 goat anti-mouse; DAPI for nuclei staining. Images: ×10, ×20, and ×50 magnifications; fluorescence microscope. | ||
α-SMA, a marker expressed in various cell types including myofibroblasts, blood vessels, endothelial cells, and hair follicles, is known to play a crucial role in wound healing dynamics. Myofibroblasts, particularly during the proliferation phase, are essential for wound contraction and are indicative of healthy wound healing. To assess the angiogenic effects of AT-loaded hydrogels, the expression levels α-SMA were evaluated using immunohistochemistry staining. An upregulation of α-SMA expression was observed in the AT-loaded hydrogel-treated burn wounds at day 30, signifying increased blood vessel formation during the remodeling phase. This was evidenced by the migration of stained infiltrating cells to the deep wound and enhanced neovascularization, with the presence of larger blood vessels, compared to the untreated control group. Similarly, the expression of KRT5, a key intermediate filament protein predominantly found in epithelial tissues, was investigated. KRT5, along with KRT14, forms keratin intermediate filaments crucial for maintaining the structural integrity of the epidermis. Findings revealed a positive or enhanced expression of KRT5 during the complete maturation of the treated group, suggesting robust epithelial regeneration. While positive KRT5 expression was also exhibited by the control group, it was notably lower compared to the blank and AT hydrogel-treated groups, underscoring the superior efficacy of the treatment in promoting epithelial repair and regeneration. Overall, the results highlight the beneficial effects of AT-loaded hydrogel treatment in promoting wound healing, as evidenced by the enhanced expression of α-SMA and KRT5, indicative of improved tissue remodeling and epithelial regeneration.
4 Conclusion
The PVA/tapioca pearl hydrogel synthesized in this work demonstrated an optimal combination of macroporosity, mechanical strength, thermal stability, water retention, and moisturizing ability, making it ideal for the full thickness burn wound dressing applications (Table 4). α-Terpineol (AT) loaded hydrogels manifested optimum degree of biocompatibility and demonstrated a 90% cell viability. In vivo sensitivity testing on normal rat skin revealed no signs of inflammatory response, erythema or edema. Macroscopic analysis of full-thickness acid burn wounds following treatment with the α-terpineol hydrogels revealed enhanced wound contraction compared to both unloaded hydrogels as well as untreated wounds. The work thus demonstrates the potential of electron beam crosslinked PVA/tapioca pearl starch hydrogels loaded with α-terpineol for the effective treatment of full-thickness acid burn wounds. In summary, our findings clearly demonstrate that the hydrogel actively promotes rapid neovascularization and complete skin regeneration, even without the addition of growth factors or cytokines. This remarkable potential positions it as a unique and promising device for acid burn wound treatment in clinical applications. Overall, the ease of fabrication, scalability potential, and promising therapeutic outcomes position α-terpineol loaded electron beam crosslinked PVA/tapioca starch hydrogel sheets for further advancement into an “off-the-shelf” treatment, highlighting their commercial viability and substantial impact in the field. Future research directions include evaluating the antimicrobial potential of the dressing on infected wounds and exploring its use in combination with stem cells to enhance regenerative wound healing.| Feature/criteria | Tapioca starch-based hydrogel (current study) | Hydrogel based wound dressings in literature |
|---|---|---|
| Ease of fabrication | Simple polymer solubilization and radiation exposure for sheet formation | Varies; some may be complex or costly |
| Cost of materials | Low | Varies; often higher for advanced materials |
| Hydration capacity | High, retains 73% of the original moisture content upto 5 h | Varies from 0–80% (ref. 50) |
| Tensile strength | Low (12 kPa approx) | Ranges between 1–100 kPa for hydrogels51 |
| Moderately high (830%) | Ranges upto 1200% for PVA hydrogels52,53 | |
| Biocompatibility | High | Generally high, varies by material |
| Therapeutic release | 50% cumulative release over 5–6 h | Varies for PVA based hydrogels, some studies report upto 90% release54 |
| Degradability | Nondegradable | Both degradable and nondegradable hydrogel wound dressings have been reported55 |
| Wound healing potential | Enhanced wound healing observed for a third degree acid burn over shorter treatment duration48,49 | No reports of hydrogels on third-degree acid burns; varying efficacies reported for hydrogels for burn healing |
Ethical approval
All studies involving animals were conducted according to approved guidelines and following formal approval from the institutional committee under relevant animal study protocol.Data availability
Raw data was generated at different departments and institutes in the University of Karachi, Karachi, Pakistan. Derived data supporting the findings of this study are available from the corresponding author Dr Muhammad Raza Shah upon reasonable request.Author contributions
Maria Khalid: conceptualization, methodology, validation, investigation, visualization and writing original draft; Fatima Jameel: conceptualization, methodology, validation, investigation, visualization and writing – original draft; Tooba Jabri: methodology, investigation, validation. Abdul Jabbar: methodology investigation, revision. Asmat Salim: resources, supervision; Irfan Khan: project administration and supervision; Muhammad Raza Shah: resources, supervision.Conflicts of interest
The authors declare that they have no competing financial or other interests that might influence the publication of this work.Acknowledgements
The authors are grateful for the support of Pak Electron Beam Irradiation (Pvt.) Limited (PEBL) for providing the electron beam usage facility to the researchers for the fabrication of sheet hydrogel.References
- M. G. Jeschke, M. E. van Baar, M. A. Choudhry, K. K. Chung, N. S. Gibran and S. Logsetty, Nat. Rev. Dis. Prim., 2020, 6, 11 CrossRef PubMed.
- W. Żwierełło, K. Piorun, M. Skórka-Majewicz, A. Maruszewska, J. Antoniewski and I. Gutowska, Int. J. Mol. Sci., 2023, 24, 3749 CrossRef.
- H. Kim, S. Shin and D. Han, Medicina, 2022, 58, 400 CrossRef PubMed.
- Y. Wang, J. Beekman, J. Hew, S. Jackson, A. C. Issler-Fisher, R. Parungao, S. S. Lajevardi, Z. Li and P. K. Maitz, Adv. Drug Delivery Rev., 2018, 123, 3–17 CrossRef CAS PubMed.
- A. E. Rivera and J. M. Spencer, Clin. Dermatol., 2007, 25, 39–48 CrossRef PubMed.
- G. Sun, X. Zhang, Y.-I. Shen, R. Sebastian, L. E. Dickinson, K. Fox-Talbot, M. Reinblatt, C. Steenbergen, J. W. Harmon and S. Gerecht, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20976–20981 CrossRef CAS.
- R. Cen, L. Wang, Y. He, C. Yue, Y. Tan, L. Li and X. Lei, Front. Pharmacol, 2022, 12, 781282 CrossRef PubMed.
- X. L. Strudwick and A. J. Cowin, Hot Topics in Burn Injuries, Intechopen, Adelaide, South Australia, Australia, 2018, pp. 37–57 Search PubMed.
- H. M. Nguyen, T. T. N. Le, A. T. Nguyen, H. N. T. Le and T. T. Pham, RSC Adv., 2023, 13, 5509–5528 RSC.
- H. Geckil, F. Xu, X. Zhang, S. Moon and U. Demirci, Nanomedicine, 2010, 5, 469–484 CrossRef CAS PubMed.
- F. Wang, Y. Gao, H. Li, L. Zhou, H. Shi, S. Feng, J. Chen and Z. Mei, Nanotechnol. Rev., 2022, 11, 2493–2512 CrossRef CAS.
- A. T. Zafalon, V. J. dos Santos, F. Esposito, N. Lincopan, V. Rangari, A. B. Lugão, and D. F. Parra, Synthesis of polymeric hydrogel loaded with antibiotic drug for wound healing applications, in Characterization of Minerals, Metals, and Materials 2018, ed. S. Banerjee, S. R. H. M. Rahman, and A. S. E. Gali, Springer International Publishing, Cham, 2018, pp. 165–176 Search PubMed.
- Q. Li, D. Wang, Z. Jiang, R. Li, T. Xue, C. Lin, Y. Deng, Y. Jin and B. Sun, Front. Chem., 2022, 10, 1038839 CrossRef CAS.
- W. Shu, Y. Wang, X. Zhang, C. Li, H. Le and F. Chang, Front. Bioeng. Biotechnol., 2021, 9, 788461 CrossRef PubMed.
- M. T. Ngo and B. A. Harley, Biomaterials, 2020, 255, 120207 CrossRef CAS.
- A. Pal, B. L. Vernon and M. Nikkhah, Bioact. Mater., 2018, 3, 389–400 Search PubMed.
- L. A. Wells, M. S. Valic, A. Lisovsky and M. V. Sefton, Isr. J. Chem., 2013, 53, 637–645 CrossRef CAS.
- I. Firlar, M. Altunbek, C. McCarthy, M. Ramalingam and G. Camci-Unal, Gels, 2022, 8, 127 CrossRef CAS PubMed.
- J. J. He, C. McCarthy and G. Camci-Unal, Adv. NanoBiomed Res., 2021, 1, 2100004 CrossRef CAS.
- D. Chouhan, T. U. Lohe, P. K. Samudrala and B. B. Mandal, Adv. Healthcare Mater., 2018, 7, 1801092 CrossRef.
- L. Hong, M. Shen, J. Fang, Y. Wang, Z. Bao, S. Bu and Y. Zhu, J. Mater. Sci.: Mater. Med., 2018, 29, 1–11 CrossRef CAS.
- Y.-I. Shen, H.-H. G. Song, A. E. Papa, J. A. Burke, S. W. Volk and S. Gerecht, J. Invest. Dermatol., 2015, 135, 2519–2529 CrossRef CAS.
- L. Wei, J. Tan, L. Li, H. Wang, S. Liu, J. Chen, Y. Weng and T. Liu, Int. J. Mol. Sci., 2022, 23, 1249 CrossRef CAS PubMed.
- M. R. Roslan, N. M. Nasir, E. M. Cheng, and N. A. M. Amin, Tissue engineering scaffold based on starch: a review, 2016 International Conference on Electrical, Electronics, and Optimization Techniques (ICEEOT), IEEE, Chennai, India, 2016, pp. 1857–1860 Search PubMed.
- V. S. Waghmare, P. R. Wadke, S. Dyawanapelly, A. Deshpande, R. Jain and P. Dandekar, Bioact. Mater., 2018, 3, 255–266 Search PubMed.
- M. Qamruzzaman, F. Ahmed and M. I. H. Mondal, J. Polym. Environ., 2022, 30, 19–50 CrossRef CAS.
- R. Chhabra, V. Peshattiwar, T. Pant, A. Deshpande, D. Modi, S. Sathaye, A. Tibrewala, S. Dyawanapelly, R. Jain and P. Dandekar, ACS Appl. Bio Mater., 2020, 3, 2920–2929 CrossRef CAS.
- H. Salehi, M. Mehrasa, B. Nasri-Nasrabadi, M. Doostmohammadi, R. Seyedebrahimi, N. Davari, M. Rafienia, M. E. Hosseinabadi, M. Agheb and M. Siavash, J. Res. Med. Sci., 2017, 22(1), 110 CrossRef PubMed.
- C. P. Palanisamy, B. Cui, H. Zhang, V. P. Gunasekaran, A. L. Ariyo, S. Jayaraman, P. Rajagopal and Q. Long, Int. J. Biol. Macromol., 2022, 222, 1852–1860 CrossRef CAS PubMed.
- P. Uttayarat, R. Chiangnoon, T. Thongnopkoon, K. Noiruksa, J. Trakanrungsie, W. Phattanaphakdee, C. Chittasupho and S. Athikomkulchai, Gels, 2023, 9, 52 CrossRef CAS PubMed.
- B. Gajra, S. S. Pandya, G. Vidyasagar, H. Rabari, R. R. Dedania and S. Rao, Int. J. Pharm. Res., 2012, 4, 2026 Search PubMed.
- J. Liao, Z. Liu, J. Wang and Z. Ye, ACS Omega, 2020, 5, 8272–8282 CrossRef CAS PubMed.
- S. R. N. S. P. Mohidin, S. Moshawih, A. Hermansyah, M. I. Asmuni, N. Shafqat and L. C. Ming, J. Evidence-Based Integr. Med., 2023, 28, 2515690X231206227 CrossRef CAS PubMed.
- E. Dagne, D. Bisrat, M. Alemayehu and T. Worku, J. Essent. Oil Res., 2000, 12, 467–470 CrossRef CAS.
- S.-N. Park, Y. K. Lim, M. O. Freire, E. Cho, D. Jin and J.-K. Kook, Anaerobe, 2012, 18, 369–372 CrossRef CAS.
- M. Park, K. Gwak, I. Yang, K. Kim, E. Jeung, J. Chang and I. Choi, Fitoterapia, 2009, 80, 290–296 CrossRef CAS PubMed.
- Z. Khalil, A. L. Pearce, N. Satkunanathan, E. Storer, J. J. Finlay-Jones and P. H. Hart, J. Invest. Dermatol., 2004, 123, 683–690 CrossRef CAS.
- J. Kawata, M. Kameda and M. Miyazawa, Int. J. Essent. Oil Ther., 2008, 2, 145–148 CAS.
- S. Held, P. Schieberle and V. Somoza, J. Agric. Food Chem., 2007, 55, 8040–8046 CrossRef CAS.
- F. Gulenoor, P. Poddar, M. Bossunia, N. Dafader and S. Chowdhury, Chem. Sci. J., 2016, 7, 1000125 Search PubMed.
- V. Chimisso, M. A. Aleman Garcia, S. Yorulmaz Avsar, I. A. Dinu and C. G. Palivan, Molecules, 2020, 25, 4090 CrossRef CAS.
- Y.-N. Chen, C. Jiao, Y. Zhao, J. Zhang and H. Wang, ACS Omega, 2018, 3, 11788–11795 CrossRef CAS PubMed.
- D. A. Ajiya, S. S. B. Jikan, B. Talip, H. Matiasperalta, N. Badarulzaman and S. Yahya, Int. J. Curr. Res. Sci. Eng. Technol., 2018, 1, 2410–2415 Search PubMed.
- K. H. Hong, Polym. Bull., 2017, 74, 2861–2872 CrossRef CAS.
- P. Gill, T. T. Moghadam and B. Ranjbar, J. Biomol. Tech., 2010, 21, 167 Search PubMed.
- D. Thomas, E. Zhuravlev, A. Wurm, C. Schick and P. Cebe, Polymer, 2018, 137, 145–155 CrossRef CAS.
- H. Richards and S. Falder, Burns, 2018, 44, 2104–2105 CrossRef PubMed.
- N. Mohamad, M. C. I. M. Amin, M. Pandey, N. Ahmad and N. F. Rajab, Carbohydr. Polym., 2014, 114, 312–320 CrossRef CAS PubMed.
- S. Jiji, S. Udhayakumar, C. Rose, C. Muralidharan and K. Kadirvelu, Int. J. Biol. Macromol., 2019, 122, 452–460 CrossRef CAS PubMed.
- T. Maver, S. Hribernik, T. Mohan, D. M. Smrke, U. Maver and K. Stana-Kleinschek, RSC Adv., 2015, 5, 77873–77884 RSC.
- S.-Q. Chen, Q. Liao, O. W. Meldrum, L. Guo, K. Wang, S. Zhang, Y. Liu, X. Chen, J. Zhu and L. Li, Carbohydr. Polym., 2023, 321, 121268 CrossRef CAS PubMed.
- T. M. Tamer, M. M. Sabet, Z. A. Alhalili, A. M. Ismail, M. S. Mohy-Eldin and M. A. Hassan, Pharmaceutics, 2022, 14, 2649 CrossRef CAS.
- E. E. Doğan, P. Tokcan, M. E. Diken, B. Yilmaz, B. Kizilduman and P. Sabaz, Adv. Mater. Sci., 2019, 19, 32–45 CrossRef.
- M. Afshar, G. Dini, S. Vaezifar, M. Mehdikhani and B. Movahedi, J. Drug Delivery Sci. Technol., 2020, 56, 101530 CrossRef CAS.
- P. Mahalakshmi, G. Reshma, C. Arthi, M. Másson and J. Rangasamy, Eur. Polym. J., 2023, 112390 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04572f |
| ‡ Maria Khalid and Fatima Jameel contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2024 |