
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrystal structure, spectroscopy, DFT, and thermal studies of 3-cyano-2(1H)-pyridones as potential anticancer agents†
Diana Hurtado-Rodrígueza,
Diana Becerraa,
Hugo Rojasa,
Jovanny A. Gómez Castaño*b,
Mario A. Macías *c and
Juan-Carlos Castillo*a
*c and
Juan-Carlos Castillo*a
aGrupo de Catálisis de la UPTC, Escuela de Ciencias Químicas, Universidad Pedagógica y Tecnológica de Colombia, Avenida Central del Norte 39-115, Tunja 150003, Colombia. E-mail: juan.castillo06@uptc.edu.co
bGrupo Química-Física Molecular y Modelamiento Computacional (QUIMOL), Escuela de Ciencias Químicas, Universidad Pedagógica y Tecnológica de Colombia, Avenida Central del Norte 39-115, Tunja 150003, Colombia. E-mail: jovanny.gomez@uptc.edu.co
cCristalografía y Química de Materiales CrisQuimMat, Departamento de Química, Universidad de los Andes, Carrera 1 No. 18A-10, Bogotá 111711, Colombia. E-mail: ma.maciasl@uniandes.edu.co
First published on 12th August 2024
Abstract
A series of 3-cyano-2(1H)-pyridones 4a–c were efficiently synthesized using an expeditious microwave-assisted multicomponent approach. Single-crystal XRD analysis revealed the presence of six independent molecules in the asymmetric unit cell for all compounds, with the crystal packing stabilized by a network of cyclic dimers formed by N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–H⋯OC intermolecular interactions. Additional supramolecular interactions, including C–H⋯π, C–N⋯π, and π⋯π, and C–H⋯X (for halogenated derivatives, i.e., 4b and 4c), appear crucial for crystal stabilization. Density Functional Theory (DFT) calculations were employed to understand the electronic structures and potential binding affinities. Comprehensive spectroscopic characterization by FT-IR, UV-Vis, NMR, and HMRS techniques confirmed the structures of all synthesized compounds. Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) were employed to evaluate the thermal stability of these compounds. The in vitro anticancer activity was evaluated against a panel of 60 human cancer cell lines, demonstrating promising activity against non-small-cell lung and breast cancer cell lines. Notably, compounds 4a and 4c exhibited the highest anticancer activity against the HOP-92 and MCF7 cell lines, with growth inhibition percentages (GI%) of 54.35 and 40.25, respectively.
C and C–H⋯OC intermolecular interactions. Additional supramolecular interactions, including C–H⋯π, C–N⋯π, and π⋯π, and C–H⋯X (for halogenated derivatives, i.e., 4b and 4c), appear crucial for crystal stabilization. Density Functional Theory (DFT) calculations were employed to understand the electronic structures and potential binding affinities. Comprehensive spectroscopic characterization by FT-IR, UV-Vis, NMR, and HMRS techniques confirmed the structures of all synthesized compounds. Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) were employed to evaluate the thermal stability of these compounds. The in vitro anticancer activity was evaluated against a panel of 60 human cancer cell lines, demonstrating promising activity against non-small-cell lung and breast cancer cell lines. Notably, compounds 4a and 4c exhibited the highest anticancer activity against the HOP-92 and MCF7 cell lines, with growth inhibition percentages (GI%) of 54.35 and 40.25, respectively.
1. Introduction
2(1H)-Pyridone derivatives are six-membered aromatic N-heterocyclic compounds that incorporate carbonyl and amine functionality and have emerged as privileged scaffolds in medicinal chemistry. These molecules exhibit intriguing tautomeric behavior, predominantly existing as the lactam form (2(1H)-pyridone) due to its enhanced thermodynamic stability compared with the lactim form (2-hydroxypyridine) in both solid and solution states (Scheme 1).1 2(1H)-Pyridone derivatives possess a remarkable combination of physicochemical properties, including favorable metabolic stability, water solubility and balanced lipophilicity.2–5 These characteristics, coupled with their ability to act as excellent hydrogen bond donors and acceptors, allow them to mimic nonpeptidic structures and effectively interact with biological targets.2,5 | ||
| Scheme 1 Lactam–lactim tautomerization of 2(1H)-pyridone. | ||
The properties of 2(1H)-pyridones have fueled their exploration in various pharmacological applications (Fig. 1). Naturally occurring examples, such as huperzine A employed for Alzheimer's treatment,6 and fredericamycin A a promising anticancer lead,7 showcase their inherent biological potential. The recent emergence of 2(1H)-pyridone derivatives in FDA-approved drugs further underscores their translational value.8–10 Tazemetostat, a 2020-approved oral inhibitor of enhancer of zeste homolog 2 (EZH2), exemplifies this class.11 Interestingly, its active pharmacophore is the 3-(aminomethyl)-pyridin-2(1H)-one moiety, strategically obtained via chemoselective reduction of a 3-cyano-2(1H)-pyridone precursor. It highlights the continued need for efficient synthetic strategies to access diverse 3-cyano-2(1H)-pyridone derivatives, paving the way for further exploration of their therapeutic potential in various diseases.
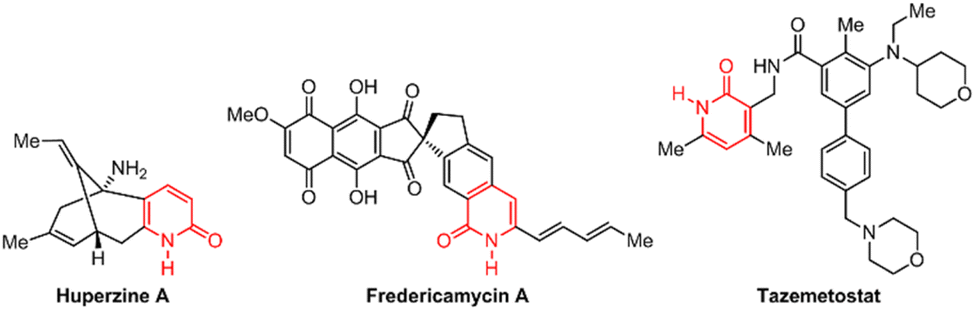 | ||
| Fig. 1 Examples of bioactive 2(1H)-pyridone derivatives. | ||
Traditional synthetic approaches for the preparation of 4,6-diaryl-3-cyano-2(1H)-pyridones often rely on a two-step sequence: (i) Michael-type addition involving enolate and α,β-unsaturated carbonyl acceptors, and (ii) subsequent oxidative intramolecular cyclization.12–15 A diverse range of α,β-unsaturated carbonyl substrates have been effectively employed in this strategy, encompassing ethyl 2-cyano-3-arylacrylates,12,13 2-cyano-3-arylacrylamides,14 and 1,3-diaryl-2-propen-1-ones.15
Multicomponent reactions (MCRs) have emerged as powerful tools for efficiently synthesizing diverse heterocyclic scaffolds, including aza-heterocycles.16,17 However, the application of MCRs to prepare 4,6-diaryl-3-cyano-2(1H)-pyridones 4 remains relatively unexplored.18–21 Pioneering work by Rong et al. described a three-component reaction for the synthesis of compounds 4 with good yields using malononitrile as the active methylene component and sodium hydroxide as the base under both grinding and conventional heating conditions (routes i and ii, Scheme 2a).18,19 This approach was further extended by the same group, replacing malononitrile with cyanoacetamide as the active methylene compound for synthesizing analogous products with good yields (route iii, Scheme 2a).20 El-Sayed et al. subsequently reported a solvent-free four-component reaction utilizing ethyl cyanoacetate as the active methylene compound and ammonium acetate (only 1 equivalent) acting dually as an ammonia surrogate and base, resulting in 4,6-diaryl-3-cyano-2(1H)-pyridones 4 with good yields (route iv, Scheme 2a).21
 | ||
| Scheme 2 Multicomponent approaches for the synthesis of 4,6-diaryl-3-cyano-2(1H)-pyridones 4. | ||
Despite reported synthetic procedures and spectroscopic characterization of 4,6-diaryl-3-cyano-2(1H)-pyridones 4a–c (Scheme 2),18–21 critical knowledge gap persists regarding their comprehensive physicochemical and biological properties. Notably, prior studies have not elucidated their crystal structure, performed detailed computational analysis using DFT, investigated their thermal behavior, or explored their potential as anticancer agents. This study meticulously addresses these shortcomings, offering a multifaceted exploration of compounds 4a–c. Through a combination of crystallographic analysis, DFT calculations, thermal studies, and in vitro anticancer activity assays against a panel of 60 human cancer cell lines, we unveil valuable insights into their potential for future drug development endeavors.
2. Results and discussion
2.1 Chemistry
Regrettably, our attempts to replicate the solvent-free MCR reported by El-Sayed et al., to produce 4,6-diaryl-3-cyano-2(1H)-pyridone 4c at temperatures ranging from 120 °C to 130 °C for 8 min, followed by crystallization in acetic acid, resulted in a yield lower than the reported 83%.21 Therefore, we aimed to improve its efficiency by introducing two modifications: utilizing microwave heating and increasing the amount of ammonium acetate beyond 1 equivalent (Scheme 2b and Table 1). The optimization process evaluated the impact of temperature and reaction time, using 4-chlorobenzaldehyde 1c, acetophenone 2, ethyl cyanoacetate 3, and excess ammonium acetate (3 equivalents) in ethanol (2 mL) under microwave irradiation (Table 1). Analysis of these parameters revealed that heating the reaction mixture at 100 °C for 30 min provided the optimal yield of 4c (entry 1, Table 1). Furthermore, we successfully streamlined the purification process by leveraging the distinct solubility properties of 4c. This approach facilitated the isolation of the target compound in high yield (90%) without column chromatography purification. Using higher temperatures or shorter reaction times resulted in decreased yields of 4c (entries 2–5, Table 1). For comparison, conventional refluxing of the reaction mixture in ethanol for 5 h afforded 4c in a mere 29% (entry 6, Table 1). This stark contrast highlights the superiority of microwave irradiation, which enhances yields and significantly expedites reaction times. The optimized microwave-assisted MCR protocol was further applied to synthesize 3-cyano-2(1H)-pyridones 4a and 4b in 70% and 83% yields, respectively (Scheme 2b).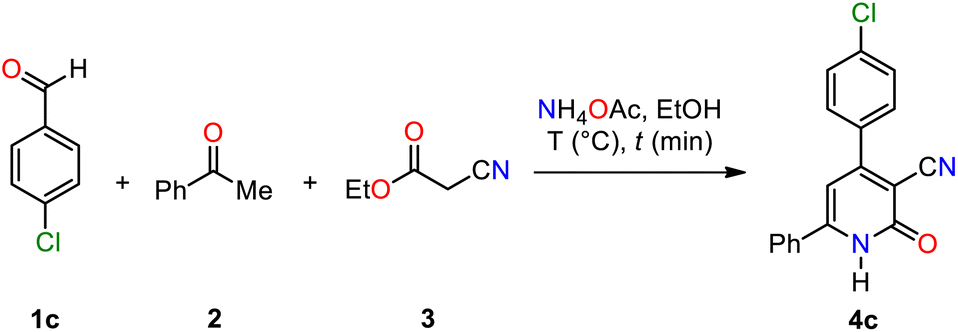
2.2 FT-IR and NMR spectroscopy studies
FT-IR spectroscopy analysis of the synthesized 4,6-diaryl-3-cyano-2(1H)-pyridones 4a–c, revealed characteristic absorption bands consistent with their proposed structures (Scheme 2b). The N–H stretching vibration appeared in the range of 2800–3100 cm−1, while the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N and CO functionalities were identified by their respective stretching vibrations at 2217–2218 cm−1 and 1636–1639 cm−1, respectively (Fig. S1–S3 in ESI†). Further structural elucidation was achieved using 1H-NMR spectroscopy in DMSO-d6 (Fig. S7, S12 and S17 in ESI†). A singlet observed at δ 6.79–6.84 ppm was assigned to the H-5 proton, and a broad singlet at δ 11.65–12.88 ppm corresponded to the exchangeable NH proton of the 2(1H)-pyridone ring. Analysis of the 13C-NMR spectra (Fig. S8, S13 and S18 in ESI†), also recorded in DMSO-d6 with extended acquisition times (up to 6 h), revealed a notable absence of signals for most quaternary carbons within the 2(1H)-pyridone ring. This phenomenon could be attributed to the dynamic equilibrium between lactam and lactim tautomers. While this equilibrium affects the detectability of carbons C-2 to C-6, the absence of signal duplication aligns with the predominant presence of the lactam form in the solution. Moreover, the 13C-NMR spectra successfully confirmed the presence of CO carbon (δ 161.7–162.5 ppm) and the C-5 carbon (δ 105.6–106.1 ppm) within the synthesized compounds.
N and CO functionalities were identified by their respective stretching vibrations at 2217–2218 cm−1 and 1636–1639 cm−1, respectively (Fig. S1–S3 in ESI†). Further structural elucidation was achieved using 1H-NMR spectroscopy in DMSO-d6 (Fig. S7, S12 and S17 in ESI†). A singlet observed at δ 6.79–6.84 ppm was assigned to the H-5 proton, and a broad singlet at δ 11.65–12.88 ppm corresponded to the exchangeable NH proton of the 2(1H)-pyridone ring. Analysis of the 13C-NMR spectra (Fig. S8, S13 and S18 in ESI†), also recorded in DMSO-d6 with extended acquisition times (up to 6 h), revealed a notable absence of signals for most quaternary carbons within the 2(1H)-pyridone ring. This phenomenon could be attributed to the dynamic equilibrium between lactam and lactim tautomers. While this equilibrium affects the detectability of carbons C-2 to C-6, the absence of signal duplication aligns with the predominant presence of the lactam form in the solution. Moreover, the 13C-NMR spectra successfully confirmed the presence of CO carbon (δ 161.7–162.5 ppm) and the C-5 carbon (δ 105.6–106.1 ppm) within the synthesized compounds.
2.3 UV-Vis spectroscopy and solvatochromic studies
UV-Vis spectroscopy was employed to investigate the electronic transitions of the synthesized compounds 4a–c. Initial analyses were conducted in ethanol, a protic and polar solvent, at a concentration of 20 μM, with scans ranging from 200 to 500 nm (Fig. 2).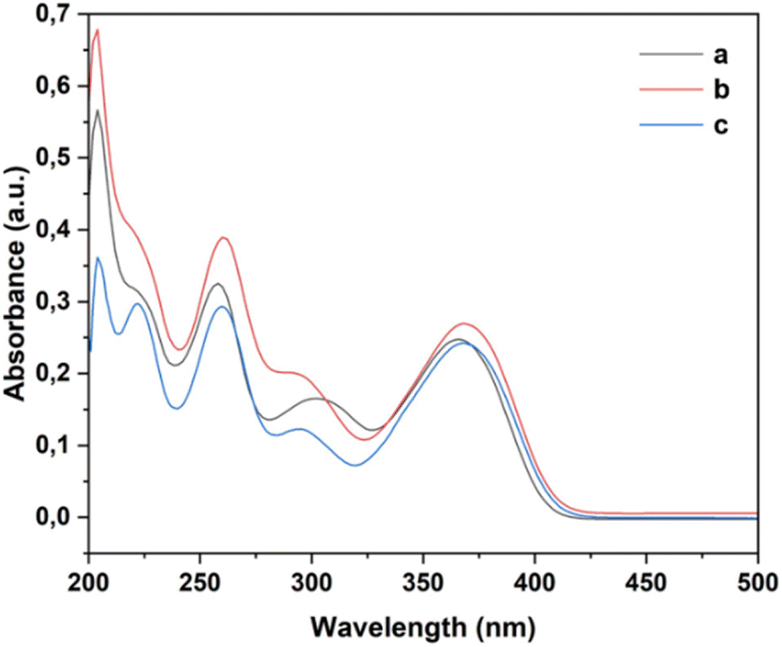 | ||
| Fig. 2 UV-Vis spectra of compounds 4a (R1 = Me), 4b (R1 = Br), and 4c (R1 = Cl) in ethanol (20 μM). | ||
The UV-Vis spectra of compounds 4a–c displayed three distinct absorption bands within the range of 217–275 nm (Table 2). These bands were attributed to various electronic transitions involving both n → π* and π → π* states. In addition, the spectra revealed two broad bands between 275 and 410 nm, which were assigned to the electronic transitions of the HOMO−1 (πPh(C)) and HOMO (nO) to LUMO  orbitals (Table 2). These specific assignments were achieved by a combined approach using vertical electronic and natural bond orbital (NBO) calculations at the B3LYP/6-311++G(2df,2dp) level of theory, in conjunction with previously reported spectra for structurally similar 4,6-diaryl-3-cyano-2(1H)-pyridones.12
orbitals (Table 2). These specific assignments were achieved by a combined approach using vertical electronic and natural bond orbital (NBO) calculations at the B3LYP/6-311++G(2df,2dp) level of theory, in conjunction with previously reported spectra for structurally similar 4,6-diaryl-3-cyano-2(1H)-pyridones.12
| Compound | Solvent | Experimental | Calculated | Δλ (nm) | Transition | Assignmenta | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| λ (nm) | ε × 104 (L mol−1 cm−1) | λ (nm) | f | Normalized coefficient | Transition percentage | |||||
| a For ring labeling (i.e., A, B or C), see Fig. 5. | ||||||||||
| 4a | EtOH | 204 | 2.80 | 241.6 | 0.148 | 0.67332 | 90.7 | −37.6 | HOMO−1–LUMO+1 |  |
| EtOH | 221 | 1.55 | 247.7 | 0.027 | 0.65845 | 86.7 | −26.7 | HOMO–LUMO+2 |  |
|
| EtOH | 257 | 1.60 | 263.0 | 0.212 | 0.68038 | 92.6 | −6.0 | HOMO–LUMO+1 |  |
|
| EtOH | 301 | 0.85 | 328.9 | 0.238 | 0.69747 | 97.3 | −27.9 | HOMO−1–LUMO |  |
|
| EtOH | 365 | 1.20 | 357.4 | 0.493 | 0.69727 | 97.2 | 7.6 | HOMO–LUMO |  |
|
| ACN | 369 | 1.40 | 357.1 | 0.493 | 0.69717 | 97.2 | 11.9 | HOMO–LUMO |  |
|
| DMSO | 376 | 1.25 | 358.1 | 0.509 | 0.69725 | 97.2 | 17.9 | HOMO–LUMO | 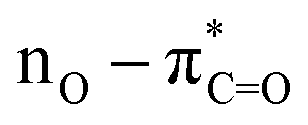 |
|
| 4b | EtOH | 203 | 3.40 | 242.6 | 0.001 | 0.69800 | 97.4 | −39.6 | HOMO−6–LUMO |  |
| EtOH | 217 | 2.05 | 246.7 | 0.012 | 0.65984 | 87.1 | −29.7 | HOMO–LUMO+3 |  |
|
| EtOH | 260 | 1.95 | 264.6 | 0.194 | 0.68574 | 94.0 | −4.6 | HOMO–LUMO+1 |  |
|
| EtOH | 290 | 1.00 | 325.8 | 0.252 | 0.69615 | 96.9 | −35.8 | HOMO−1–LUMO | 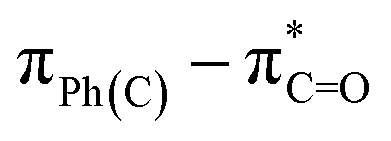 |
|
| EtOH | 368 | 1.35 | 360.9 | 0.515 | 0.69817 | 97.5 | 7.1 | HOMO–LUMO |  |
|
| ACN | 372 | 1.40 | 360.6 | 0.514 | 0.69814 | 97.5 | 11.4 | HOMO–LUMO |  |
|
| DMSO | 378 | 1.25 | 361.6 | 0.530 | 0.69830 | 97.5 | 16.4 | HOMO–LUMO |  |
|
| 4c | EtOH | 204 | 1.80 | 239.5 | 0.031 | 0.51929 | 53.9 | −35.5 | HOMO−1–LUMO+2 |  |
| EtOH | 222 | 1.48 | 246.9 | 0.011 | 0.65532 | 85.9 | −24.9 | HOMO–LUMO+3 |  |
|
| EtOH | 259 | 1.46 | 267.8 | 0.221 | 0.68072 | 92.7 | −8.8 | HOMO–LUMO+1 |  |
|
| EtOH | 295 | 0.60 | 320.3 | 0.246 | 0.69833 | 97.5 | −25.3 | HOMO−1–LUMO |  |
|
| EtOH | 368 | 1.21 | 360.4 | 0.489 | 0.69941 | 97.8 | 7.6 | HOMO–LUMO |  |
|
| ACN | 372 | 1.33 | 360.2 | 0.489 | 0.69938 | 97.8 | 11.8 | HOMO–LUMO |  |
|
| DMSO | 380 | 1.06 | 361.1 | 0.504 | 0.69954 | 97.9 | 18.9 | HOMO–LUMO | 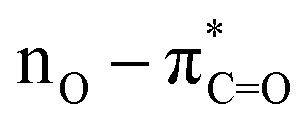 |
|
The electronic absorption assignments observed, alongside FT-IR analysis, support the predominance of the lactam tautomeric form in both solution and solid states. This conclusion is further strengthened by the presence of CO stretching vibrations in the FT-IR spectra (1636–1639 cm−1) and the corresponding CO carbon signals in the 13C-NMR spectra (161.7–162.5 ppm) of compounds 4a–c. The UV-Vis spectroscopy results, coupled with established insights into the lactam–lactim equilibrium, underscore the influence of solvent polarity in determining the predominant tautomeric form. The 2-hydroxypyridine tautomer 5 exhibits lower polarity than the lactam tautomer 4, which can be attributed to the presence of a charge-separated mesomeric form 4′ for the lactam tautomer (Scheme 3).12
 | ||
| Scheme 3 Influence of mesomeric form 4′ on the lactam–lactim tautomeric equilibrium in 4,6-diaryl-3-cyano-2(1H)-pyridones. | ||
Consequently, electron-withdrawing substituents at positions 4 and 6 of the pyridine ring and polar protic solvents significantly stabilize the charge-separated resonance structure 4′, favoring the more polar lactam form 4. The lactam tautomer 4 achieves stability when exposed to polar protic solvents due to its ability to act as a hydrogen-bond donor to carbonyl oxygen. Conversely, the 2-hydroxypyridine tautomer 5 may gain some stabilization in solvents that primarily function as hydrogen-bond acceptors.12 To gain further insight into the n → π* electronic transition of the CO group associated with the lactam–lactim equilibrium in compounds 4a–c, a solvatochromic study was undertaken using a range of polar solvents with varying dielectric constants (ε): ethanol (ε = 24.5), acetonitrile (ε = 37.5), and dimethylsulfoxide (DMSO, ε = 46.7). As illustrated in Fig. 3 and Table 2, all compounds displayed a moderate bathochromic shift (red shift) in the n → π* electronic transition of the CO group with increasing solvent polarity. As mentioned earlier, this phenomenon can be attributed to the enhanced stabilization of the charge-separated mesomeric form 4′ with increasing solvent polarity (Scheme 3).
 | ||
| Fig. 3 UV-Vis spectra and solvatochromic study of compounds 4a (R1 = Me), 4b (R1 = Br), and 4c (R1 = Cl). | ||
Furthermore, we investigated the influence of substituents at the 4-position of the aromatic ring on the electronic transitions. The presence of a more electronegative substituent (R1 = Br and Cl) resulted in a bathochromic shift in the n → π* electronic transition from the HOMO to LUMO orbitals. This observation reveals a correlation between the bathochromic shift and a reduction in the band gap for compounds containing electron-withdrawing substituents (R1 = Br and Cl), which contrasts with the behavior observed for the compound containing an electron-donating group (R1 = Me). While Fig. 3 reveals that compounds 4a–c exhibited their highest absorbance in acetonitrile, interestingly, the n → π* electronic transition displayed a hypochromic effect in DMSO. This observation indicates that the electron-withdrawing substituent (Cl in compound 4c) reduces the absorbance (1.06 × 104 L mol−1 cm−1), which contrasts with the slightly higher absorbance observed for the compound containing an electron-donating substituent (CH3 in compound 4a, 1.25 × 104 L mol−1 cm−1).
2.4 Frontier molecular orbital analysis and molecular reactivity
Frontier Molecular Orbital (FMO) analysis was performed to elucidate the reactivity and chemical stability trends of compounds 4a–c. The HOMO is primarily localized on the oxygen atom of the carbonyl group, while the LUMO is located on the antibonding π* orbital of the same carbonyl group (Fig. 4). These findings are consistent with the previously proposed charge-separated mesomeric form 4' and its corresponding resonance structure, the lactam tautomer 4 (Scheme 3). The calculated HOMO stabilization energies for compounds 4a, 4b, and 4c were 6.44, 6.61, and 6.60 eV, respectively. Similarly, the LUMO energies were 2.60, 2.79, and 2.78 eV, respectively. The HOMO–LUMO energy gaps for compounds 4a, 4b, and 4c were calculated to be 3.84, 3.83, and 3.83 eV, respectively. These results indicate a notable level of chemical stability and reduced reactivity attributable to the enhanced stability of the electron cloud. | ||
| Fig. 4 Calculated frontier molecular orbitals (HOMO and LUMO) and band gap of compounds 4a–c using the B3LYP/6-311++G(2df,2pd) approximation level. | ||
Table 3 presents the calculated global reactivity descriptors for compounds 4a–c, including ionization potential (IP), electron affinity (EA), electrophilicity index (ω), chemical potential (μ), electronegativity (χ), and hardness (η). Koopmans' theorem establishes a correlation between the HOMO and LUMO energy levels of a molecule and its IP and EA, respectively.22 Electronegativity (χ) is calculated as the average of the HOMO and LUMO energies using the equation χ = (IP + EA)/2.23 Hardness (η), a valuable indicator of chemical stability, is inversely proportional to the HOMO–LUMO energy gap.24,25 Parr et al. introduced the electrophilicity index (ω) defined as ω = μ2/2η, where μ is the chemical potential calculated as μ = −(IP + EA)/2.26
| Parameters | 4a | 4b | 4c |
|---|---|---|---|
| HOMO energy | −6.440 | −6.612 | −6.604 |
| LUMO energy | −2.596 | −2.786 | −2.776 |
| HOMO–LUMO energy gap | 3.844 | 3.826 | 3.828 |
| Ionization potential (IP) | 6.440 | 6.612 | 6.604 |
| Electron affinity (EA) | 2.596 | 2.786 | 2.776 |
| Electrophilicity index (ω) | 2.655 | 2.886 | 2.873 |
| Chemical potential (μ) | −4.518 | −4.699 | −4.690 |
| Electronegativity (χ) | 4.518 | 4.699 | 4.690 |
| Hardness (η) | 3.844 | 3.826 | 3.828 |
The calculated electrophilicity index values for 4a (2.66 eV), 4b (2.89 eV), and 4c (2.87 eV) denote moderate electrophilicity toward electron-rich species (Table 3). The chemical potential values (μ) for 4a (−4.52 eV), 4b (−4.70 eV), and 4c (−4.69 eV) imply a moderate ability to attract electrons. The electronegativity values (χ) follow the same trend, ranging from 4.52 to 4.70 eV. The high hardness values (η) calculated from the HOMO–LUMO energy gap ranging from 3.83 to 3.84 eV denote significant resistance to electron transfer for all compounds, resulting in reduced chemical reactivity.
2.5 Crystalline structural analysis
 | ||
| Fig. 5 Molecular structures of compounds (a) 4a, (b) 4b, and (c) 4c show anisotropic thermal vibrations. The ellipsoids represent a 30% probability level, and hydrogen atoms are depicted as spheres with arbitrary radii. Blue arrows denote the free rotation between C7A–C1A and C3A–C13A atoms, resulting in six independent molecules in the asymmetric unit for 4a–c. The dihedral angles between the planes containing rings A–B and A–C for compounds 4a–c were measured using Mercury software (Fig. 4 and Table 4).27 | ||
| Dihedral angles (°) | 4a | 4b | 4c |
|---|---|---|---|
| A–B | 26.8(19) | 23.0(3) | 27.6(13) |
| 32.2(18) | 24.1(3) | 23.1(14) | |
| 30.1(18) | 27.7(3) | 28.2(14) | |
| 32.0(19) | 28.5(2) | 23.9(13) | |
| 27.2(19) | 27.4(3) | 27.9(13) | |
| 29.7(17) | 28.4(3) | 26.8(13) | |
| A–C | 53.0(2) | 49.7(3) | 59.0(14) |
| 57.2(2) | 48.3(3) | 52.1(15) | |
| 48.3(2) | 46.2(3) | 58.9(14) | |
| 57.8(2) | 47.1(3) | 49.3(15) | |
| 53.9(2) | 58.5(3) | 46.1(13) | |
| 49.3(2) | 57.3(3) | 45.6(15) |
Further analysis of the crystal structures of compounds 4a–c revealed the presence of various intermolecular interactions that govern their solid-state packing arrangements (Fig. 6 and Table 5). Independent of the conformational orientation of individual molecules, inversion symmetry-related molecules are linked through a combination of N–H⋯O and C–H⋯O hydrogen bonds. Notably, some of these hydrogen bonds exhibit a split character, where a single oxygen atom acts as a hydrogen bond acceptor for two (N,C)–H donors. As expected, N–H⋯O hydrogen bonds display shorter distances compared to C–H⋯O interactions due to the enhanced acidity of the N–H proton.
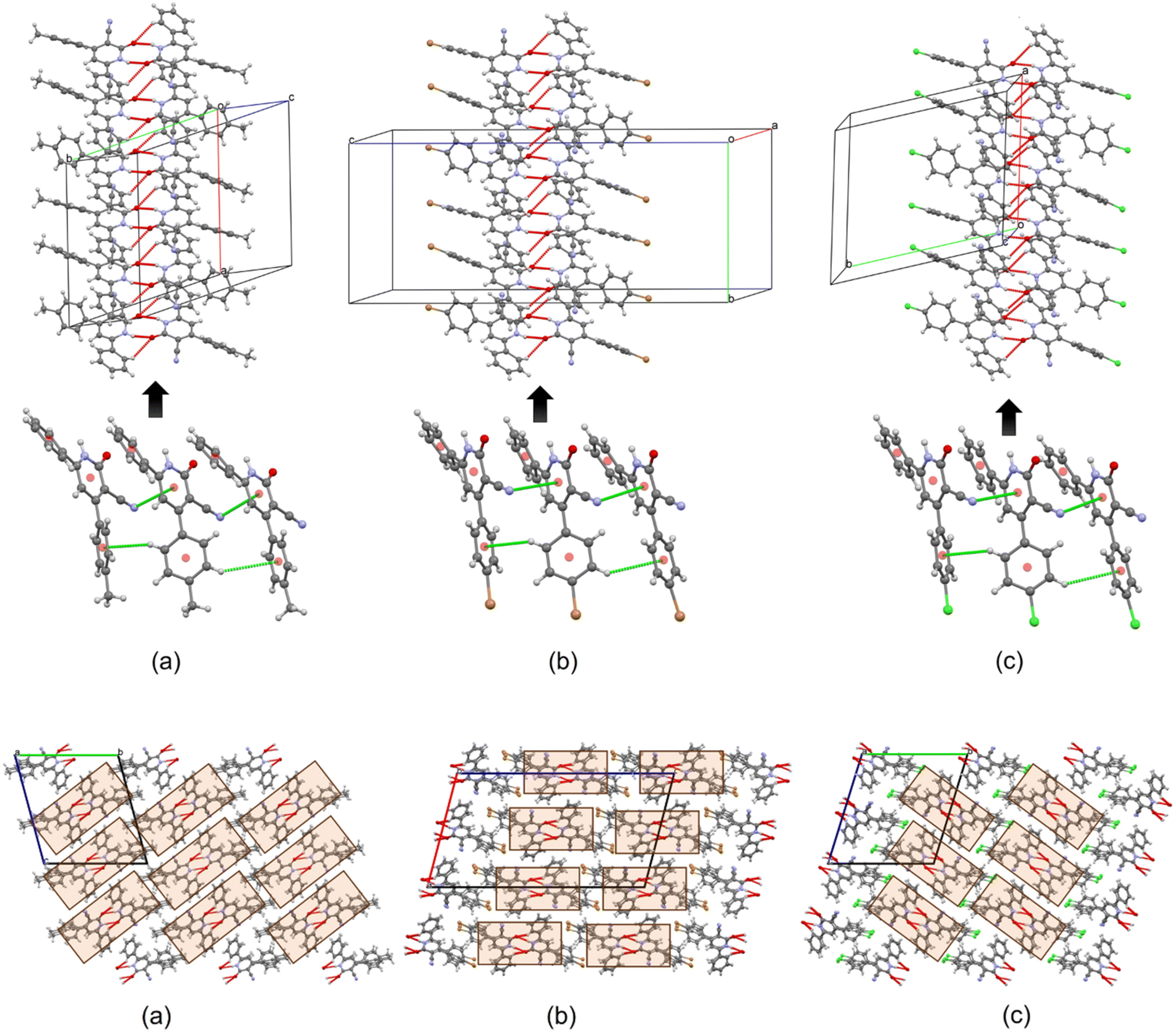 | ||
| Fig. 6 Molecular chains formed by dimers of inversion-related molecules connected by N–H⋯O, C–H⋯O, C–H⋯π, C–N⋯π, and π⋯π interactions for compounds (a) 4a, (b) 4b, and (c) 4c. Brown and green spheres correspond to Br and Cl atoms, respectively. Crystal structures of compounds (a) 4a, (b) 4b, and (c) 4c, showing the molecular packings. | ||
| D–H⋯A | D–H | H⋯A | D⋯A | D–H⋯A | Symmetry code |
|---|---|---|---|---|---|
| Compound 4a | |||||
| N1A–H1A⋯O1D | 0.86 | 1.99 | 2.817(4) | 161 | 1 − x,1 − y,−z |
| N1B–H1B⋯O1F | 0.86 | 1.99 | 2.800(4) | 165 | x,y,z |
| N1C–H1C⋯O1C | 0.86 | 1.99 | 2.788(4) | 161 | 1 − x,1 − y,−z |
| N1D–H1D⋯O1A | 0.86 | 1.99 | 2.827(4) | 164 | 1 − x,1 − y,−z |
| N1E–H1E⋯O1E | 0.86 | 2.00 | 2.819(4) | 158 | −x,1 − y,1 − z |
| N1F–H1F⋯O1B | 0.86 | 1.96 | 2.803(4) | 165 | x,y,z |
| C8C–H8C⋯O1C | 0.93 | 2.57 | 3.189(4) | 125 | 1 − x,1 − y,−z |
| C8D–H8D⋯O1A | 0.93 | 2.57 | 3.202(4) | 126 | 1 − x,1 − y,−z |
| C8F–H8F⋯O1B | 0.93 | 2.55 | 3.205(4) | 128 | x,y,z |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Compound 4b | |||||
| N1A–H1A⋯O1A | 0.86 | 1.98 | 2.797(6) | 158 | 1 − x,−y,1 − z |
| N1B–H1B⋯O1C | 0.86 | 1.97 | 2.812(6) | 166 | 1 − x,1 − y,1 − z |
| N1C–H1C⋯O1B | 0.86 | 1.98 | 2.815(5) | 164 | 1 − x,1 − y,1 − z |
| N1D–H1D⋯O1E | 0.86 | 1.97 | 2.806(6) | 165 | −x,1 − y,1 − z |
| N1E–H1E⋯O1D | 0.86 | 2.01 | 2.834(6) | 161 | −x,1 − y,1 − z |
| N1F–H1F⋯O1F | 0.86 | 1.95 | 2.781(5) | 163 | −x,1 − y,1 − z |
| C8A–H8A⋯O1A | 0.93 | 2.53 | 3.215(7) | 130 | 1 − x,−y,1 − z |
| C8B–H8B⋯O1C | 0.93 | 2.52 | 3.214(6) | 132 | 1 − x,1 − y,1 − z |
| C8C–H8C⋯O1B | 0.93 | 2.45 | 3.148(7) | 132 | 1 − x,1 − y,1 − z |
| C8D–H8D⋯O1E | 0.93 | 2.49 | 3.167(7) | 130 | −x,1 − y,1 − z |
| C8F–H8F⋯O1F | 0.93 | 2.49 | 3.159(7) | 129 | −x,1 − y,1 − z |
|
|||||
| Compound 4c | |||||
| N1A–H1A⋯O1C | 0.86 | 1.97 | 2.816(3) | 166 | x,1 + y,z |
| N1B–H1B⋯O1B | 0.86 | 1.99 | 2.809(3) | 158 | 1 − x,2 − y,1 − z |
| N1C–H1C⋯O1A | 0.86 | 1.97 | 2.815(3) | 165 | x,−1 + y,z |
| N1D–H1D⋯O1F | 0.86 | 1.99 | 2.827(3) | 165 | 1 − x,−y,−z |
| N1E–H1E⋯O1E | 0.86 | 1.94 | 2.777(3) | 163 | −x,−y,−z |
| N1F–H1F⋯O1D | 0.86 | 2.00 | 2.830(3) | 162 | 1 − x,−y,−z |
| C8E–H8E⋯O1E | 0.93 | 2.48 | 3.159(3) | 130 | −x,−y,−z |
The observed free rotation around the C7A–C1A and C3A–C13A bonds, leading to six independent molecules within the asymmetric units of 4a–c, is influenced by packing constraints. Interestingly, the formation of precisely six distinct conformers in each crystal structure is associated with the development of directional molecular chains extending along the [100], [010], and [100] crystallographic directions for 4a, 4b, and 4c, respectively. Within these chains, the 4-substituted aromatic rings (4-MeC6H4, 4-BrC6H4, and 4-ClC6H4) adopt conformations that favor the formation of C–H⋯π interactions, with H⋯π distances of approximately 2.9 Å, contributing to the overall structural stability (Fig. 6). Furthermore, unconventional C–N⋯π contacts (N⋯π distances ranging from 3.4 to 3.6 Å) participate in the assembling of supramolecular chains by facilitating the appropriate molecular orientations necessary for the formation of weak π⋯π interactions.
From a supramolecular perspective, the crystal structures of compounds 4a–c can be envisioned as assemblies of structurally similar molecular chains, resembling supramolecular bricks (Fig. 6). In compound 4a, these bricks are connected primarily through weak C–H⋯π interactions and van der Waals forces. In contrast, the packing arrangement in compounds 4b and 4c involves additional intermolecular C–H⋯Cl and C–H⋯Br halogen bonding interactions, with H⋯Cl and H⋯Br distances ranging from 2.8 to 3.1 Å. Furthermore, C–H⋯N–C interactions involving the cyano group (CN), with H⋯N distances of approximately 2.9 Å, contribute to the interconnection of the chains (Fig. 6). It is noteworthy that despite the variation in the 4-substituent on the aromatic ring (4-MeC6H4, 4-BrC6H4, and 4-ClC6H4), the intermolecular interactions governing the assembly between the molecular chains exhibit a high degree of similarity across compounds 4a–c.
As previously mentioned, the carbonyl oxygen atom in compounds 4a–c appears to be crucial for the stability of these crystalline systems. It forms two simultaneous hydrogen bonds: one with the amine's hydrogen and another with aromatic hydrogen from a symmetrically related molecular unit (Fig. 6 and 7-top). To test this hypothesis, we performed QTAIM-C analysis on the unmodified structure of the corresponding dimer extracted from these crystal systems. As shown in Fig. 7-bottom, the topological analysis revealed the presence of two bond critical points (BCPs) and two corresponding bond paths (BPs) connecting each carbonyl oxygen atom with the hydrogen atoms involved in these hydrogen bonding interactions. The topological characterization of these BCPs, such as ρ(r) and ∇2ρ(r), confirms the presence of closed-shell hydrogen bonding interactions, which is consistent with the structural analysis observations. The electron density (ρ) at the BCPs indicates the strength of these interactions, with a higher electron density corresponding to stronger hydrogen bonding. Electron density values at BCPs of the type CO⋯H–Ph HB ranged from 0.007 to 0.009 au in the three systems, indicating weak-to-moderate strength hydrogen bonding interactions (0.002 ≤ ρ ≤ 0.4 au, for neutral systems).28 As shown in Fig. 7, dimer 4a exhibited slightly lower electronic density values for these types of BCPs compared to 4b and 4c. On the other hand, the calculated electron density at the CO⋯H–N BCPs attained values of 0.025 au for system 4a and 0.024 au for systems 4b and 4c, revealing stronger bidirectional dimer interactions between the carbonyl oxygen atom and the amine hydrogen in compounds 4a–c. These findings suggest that a carbonyl oxygen atom in compounds 4a–c is critical for stable crystalline assemblies.
 | ||
| Fig. 7 Dimeric interaction projected from the crystal structures of compounds 4a–c (top) vs. the corresponding critical points (CPs) analysis on the electron density performed using the QTAIM (bottom). Bond paths (pink lines), BCPs (red dots), and RCPs (yellow dots). Electron density (ρ), Laplacian (∇2ρ), and length of contact distances and bond paths (BP) are expressed in atomic units (au). | ||
Our topological analysis unexpectedly revealed a long intermolecular CN⋯H–C bond path (approximately 7.97 au) in halogenated dimers 4b and 4c, connecting the cyano group with an aromatic hydrogen atom from a neighboring molecule (Fig. 7-bottom). The electron densities at these BCPs reached low values of approximately 3.7 × 10−4 au, suggesting weak but potentially significant dispersive interactions contributing to the overall supramolecular organization observed in these molecular crystals. In addition, relatively high ellipticities (ε ≈ 2.2) were observed on these BCPs, indicating that these interactions exhibit partial π-character. The absence of intermolecular CN⋯H–C interactions in compound 4a suggests that the introduction of halogen atoms in compounds 4b and 4c promotes the formation of weak non-covalent interactions, which may play an auxiliary role in stabilizing the molecular packing arrangements.
 | ||
| Fig. 8 Hirshfeld surfaces mapped over dnorm and 2D fingerprint plots including relative contributions (%) to the Hirshfeld surface area for the various close intermolecular contacts in compounds (a) 4a, (b) 4b, and (c) 4c. | ||
The two-dimensional (2D) fingerprint plots in Fig. 8 depict the contributions of various interatomic contacts to the overall HS for each compound. A qualitative comparison of these plots reveals similar patterns for compounds 4b and 4c, which differ from those observed for 4a. These differences can be attributed to the varying 4-substituents on the aromatic ring C (4-MeC6H4, 4-BrC6H4, and 4-ClC6H4), which influence the formation of intermolecular contacts. In all compounds, H⋯H interactions represent the most significant contribution of the HS maps, with the highest percentage observed for compound 4a, as expected. The presence of 4-BrC6H4 and 4-ClC6H4 fragments in compounds 4b and 4c leads to a notable contribution from C–H⋯(Br, Cl) interactions (14.1% and 14.0% for 4b and 4c, respectively), highlighting the importance of these interactions in stabilizing the crystal packing.
Beyond the conventional N–H⋯O, C–H⋯O hydrogen bonds, C–H⋯π and π⋯π interactions, HS analysis revealed the presence of additional, potentially weak C–N⋯π contacts relevant to the supramolecular assembly in all compounds. These C–N⋯π contacts contribute approximately 4.3–5.5% to the total HS map for each compound. Fig. 9 presents the shape index maps, emphasizing the distribution of these C–N⋯π contacts. The presence of red hollows on the shape index maps indicates areas where the surfaces around C–N fragments come into proximity with the surfaces of the aromatic rings in each compound. These hollows, centered on the centroids of the π-systems, further support the existence of these weak C–N⋯π contacts (N⋯π distances: 3.4–3.6 Å).
 | ||
| Fig. 9 Hirshfeld surfaces mapped with shape index showing the hollows (red) on the aromatic rings due to C–N⋯π contacts in compounds (a) 4a, (b) 4b, and (c) 4c. | ||
2.6 UV-Vis diffuse reflectance
UV-Vis diffuse reflectance spectroscopy was used to measure the absorption characteristics of compounds 4a–c (Fig. 10). The estimated absorption wavelengths for these compounds were approximately 350 and 400 nm. Compounds containing electron-withdrawing groups (R1 = Br and Cl) showed higher absorption intensity than those containing an electron-donating group (R1 = Me). The band gap energies of compounds 4a–c were determined by extrapolating the Tauc plot, where the curve intersects the y-axis, assuming a direct band gap. The estimated band gap energies were 2.69, 2.71, and 2.74 eV for compounds 4a, 4b, and 4c, respectively (Fig. 11). However, the band gap energies for compounds 4a, 4b, and 4c were determined to be 3.844, 3.826, and 3.828 eV, respectively, using the B3LYP/6-311++G(2df,2pd) approximation level. In the gas phase, the absence of stabilizing interactions can lead to higher energy transitions, resulting in elevated band gap values. In summary, the disparity between the band gap values obtained from the Tauc plot and those calculated using the DFT method arises from fundamental differences in phase (solid versus gas), methodological approaches (empirical versus theoretical), and environmental effects (presence versus absence of intermolecular interactions). These factors collectively contribute to the observed differences in the band gap energies for compounds 4a–c.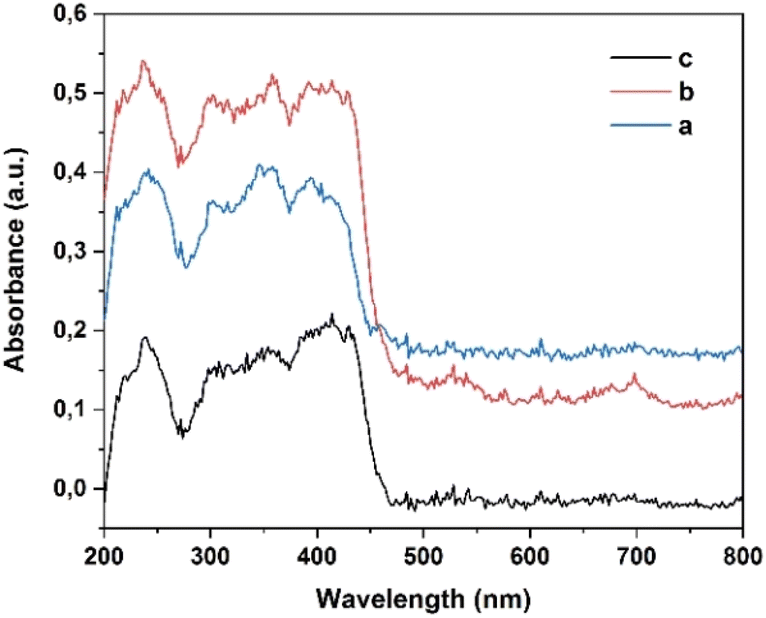 | ||
| Fig. 10 UV-Vis diffuse reflectance spectra of compounds 4a–c. | ||
 | ||
| Fig. 11 Band gap values assessed by a correlated curve of (F(R)hν)2 set against photon energy plots of compounds 4a–c. | ||
2.7 Thermal analysis
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were employed to evaluate the thermal stability of compounds 4a–c. TGA and DSC thermograms for each compound were recorded separately under a controlled nitrogen atmosphere using a heating rate of 10 °C min−1 and a continuous nitrogen flow of 25 mL min−1 over a temperature range of 25 to 400 °C (Fig. 12 and 13).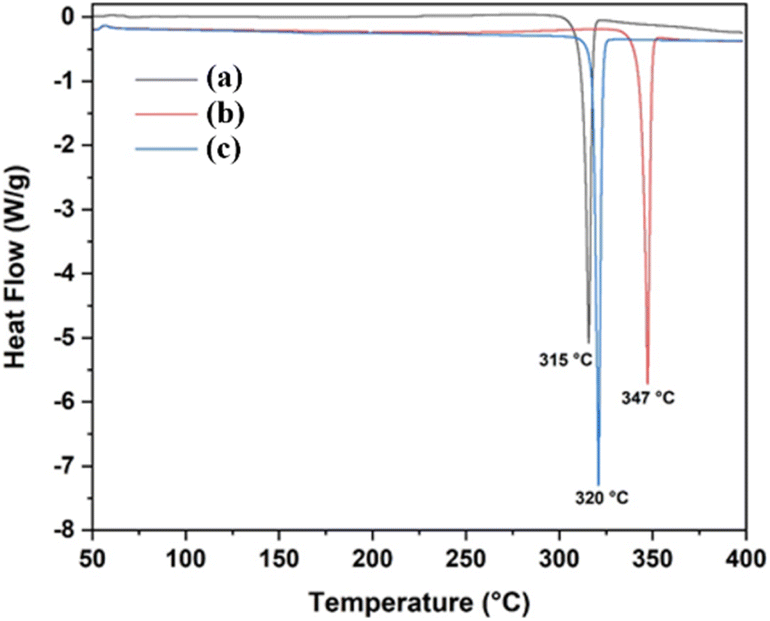 | ||
| Fig. 12 TG thermograms of compounds 4a (R1 = Me), 4b (R1 = Br), and 4c (R1 = Cl). | ||
 | ||
| Fig. 13 DSC thermograms of compounds 4a (R1 = Me), 4b (R1 = Br), and 4c (R1 = Cl). | ||
Analysis of the combined TGA and DSC thermograms for compound 4a (R1 = Me) revealed a melting process within the temperature range of 292–331 °C, characterized by a distinct endothermic peak at 315 °C (ΔHfus = 117 J g−1). Similarly, the TGA and DSC curves for compounds 4b (R1 = Br) and 4c (R1 = Cl) displayed analogous melting transitions within 321–348 °C and 303–337 °C, respectively. These melting processes were accompanied by prominent endothermic peaks observed at 347 °C (ΔHfus = 125 J g−1) for compound 4b and 320 °C (ΔHfus = 132 J g−1) for compound 4c.
The introduction of halogen substituents (Br and Cl) into compounds 4b and 4c appears to result in a slight increase in the enthalpy of fusion and the melting temperature range compared to compound 4a. This observed trend can be attributed to stronger intermolecular interactions facilitated by halogen incorporation. As evidenced by single-crystal X-ray diffraction analysis (Section 2.5.1), these stronger interactions likely involve C–H⋯Cl and C–H⋯Br hydrogen bonding.
2.8 Anticancer studies
The in vitro anticancer activity of the synthesized 3-cyano-2(1H)-pyridones 4a–c was evaluated against a panel of 60 human cancer cell lines, including leukemia, melanoma, lung, colon, central nervous system, ovarian, renal, prostate, and breast cancers. The National Cancer Institute (NCI) performed the evaluation using a single-dose treatment of 10 μM. Compounds 4a, 4b, and 4c were assigned unique identifiers D-837648/1, D-837640/1, and D-829750/1, respectively, by the NCI. Detailed results, including the mean growth percentage (mean G%) and growth inhibition percentage (GI%) of treated cells relative to untreated controls, are depicted in Tables S2–S4.†31,32 Table 6 summarizes the top five most promising findings, highlighting the highest GI% exhibited by compounds 4a–c against various human cancer cell lines.| Compound | Most sensitive cell lines | GIa (%) |
|---|---|---|
| a GI% = 100 − G%. | ||
| 4a | HOP-92 (non-small cell lung cancer) | 54.35 |
| HCT-15 (colon cancer) | 28.96 | |
| UO-31 (renal cancer) | 26.78 | |
| SR (leukemia) | 26.72 | |
| T-47D (breast cancer) | 26.54 | |
| 4b | MCF7 (breast cancer) | 24.72 |
| HOP-92 (non-small cell lung cancer) | 23.42 | |
| RPMI-8226 (leukemia) | 23.33 | |
| MOLT-4 (leukemia) | 22.34 | |
| NCI-H522 (non-small cell lung cancer) | 21.22 | |
| 4c | MCF7 (breast cancer) | 40.25 |
| HOP-92 (non-small cell lung cancer) | 31.79 | |
| T-47D (breast cancer) | 29.86 | |
| UO-31 (renal cancer) | 28.23 | |
| UACC-62 (melanoma) | 28.18 | |
The 3-cyano-2(1H)-pyridones 4a and 4c exhibited the most promising in vitro anticancer activity. These compounds displayed significant growth inhibition percentages (GI%) against the HOP-92 non-small-cell lung (54.35 for 4a) and MCF7 (40.25 for 4c) breast cancer cell lines. In addition, compounds 4b and 4c demonstrated moderate activity against the HOP-92 cell line with GI% values of 23.42 and 31.79, respectively. Conversely, compounds 4a and 4b showed low activity against the T-47D and MCF7 breast cancer cell lines, with GI% values of 26.54 and 24.72, respectively. Likewise, compounds 4a and 4c demonstrated low activity against the UO-31 renal cancer cell line, with GI% values of 26.78 and 28.23, respectively.
Interestingly, some studies have documented the anticancer activity of 2(1H)-pyridone derivatives against the MCF7 breast cancer cell line,33–36 highlighting the 4-aryl-6-[benzo[f]coumarin-3-yl]-3-cyano-2(1H)-pyridones with GI50 values ranging from 0.003 to >50 μM, as well as 3,4,6-triaryl-2(1H)-pyridones exhibiting higher anticancer activity than the standard drug Nolvadex.34,35 Conversely, limited data exists regarding their efficacy against HOP-92 and UO-31 cancer cell lines. Overall, the promising in vitro anticancer activity of compounds 4a–c against the HOP-92 non-small-cell lung and breast cancer cell lines can be attributed to the presence of the 2(1H)-pyridone scaffold, aligning with findings on the antiproliferative properties of 2-pyridone derivatives.33–36 However, further investigation is warranted to elucidate the underlying mechanisms of action of these compounds and to explore their potential for development as potential anticancer agents.
3. Conclusions
This study presents the multicomponent synthesis, spectroscopic characterization, crystallographic analysis, theoretical investigation, and initial evaluation of the anticancer activity of 4,6-diaryl-3-cyano-2(1H)-pyridones 4a–c. The microwave-assisted protocol furnished the target compounds with good yields. This approach is notable for its simplicity of operation, use of an eco-friendly solvent, straightforward purification process, and simultaneous formation of four chemical bonds in a single step. The structures of 4a–c were confirmed using various analytical techniques (FT-IR, UV-Vis, NMR, HRMS, TG, and DSC) and single-crystal X-ray diffraction analysis.Density functional theory (DFT) calculations at the B3LYP/6-311++G(2df,2pd) level of theory were employed to optimize the molecular geometries of 4a–c in the gas phase and various in silico solvent environments (ethanol, acetonitrile, and DMSO). Time-dependent DFT (TD-DFT) calculations provided insights into the electronic transitions responsible for the UV-Vis spectral bands. Natural bond orbital (NBO) population analysis yielded the frontier molecular orbital surfaces.
The topological analysis of the electron density, conducted using the QTAIM-C approach, highlighted the crucial role of the carbonyl oxygen atom in forming strong bidirectional hydrogen bonding interactions, primarily with the amine hydrogen and secondarily with aromatic hydrogens from neighboring molecules. Furthermore, the analysis revealed the presence of weak intermolecular CN⋯H–C contacts in the halogenated compounds 4b and 4c, which may contribute to the overall stability of the supramolecular assemblies. Hirshfeld surface analysis offered a quantitative and visual representation of these close contacts in real space, providing valuable insights into the nature of the noncovalent interactions that govern the crystalline packing of the 3-cyano-2(1H)-pyridones. These findings can be further utilized to design and synthesize new functional materials with specific properties.
Initial assessment of anticancer activity revealed that all compounds exhibited moderate cytotoxicity against human lung carcinoma (A549) and breast adenocarcinoma (MCF7) cell lines. Compounds 4a and 4c exhibited the most significant anticancer activity against HOP-92 non-small-cell lung and MCF7 breast cancer cell lines, with growth inhibition percentages (GI%) of 54.35 and 40.25, respectively. Further studies are needed to elucidate their mechanisms of action and enhance the anticancer potential of other 3-cyano-2(1H)-pyridones.
4. Experimental
4.1 Materials and methods
All chemical reactions were performed under a dry nitrogen atmosphere using oven-dried glassware. The reaction progress was monitored by thin-layer chromatography (TLC) on precoated silica gel (Merck Kieselgel 60 F254). Visualization was achieved using UV light at 254 and 365 nm. Solvents and commercially available reagents were purchased from Sigma-Aldrich and used without further purification unless otherwise stated. Microwave-assisted reactions were conducted using an Anton Paar Monowave 400 microwave reactor with 10 mL glass reaction vials (G30). Infrared spectra were recorded using a Thermo Scientific Nicolet 1550 Infrared spectrophotometer. 1H and 13C NMR spectra were acquired on a Bruker Avance 400 MHz spectrometer (for compounds 4a and 4c) and a Varian UNITYPLUS 500 MHz spectrometer (for compound 4b), using deuterated dimethylsulfoxide (DMSO-d6). Chemical shifts (δ) are reported in ppm relative to the residual solvent peak (δ = 2.50 ppm for 1H and 39.52 ppm for 13C) and coupling constants (J) are reported in Hz. Multiplicity was abbreviated as s (singlet), d (doublet), t (triplet), and m (multiplet). High-resolution mass spectra (HRMS) were recorded using a Q-TOF spectrometer equipped with electrospray ionization (ESI, 4000 V) in positive mode. The UV-Vis spectra were recorded using a Thermo Scientific Evolution 300 spectrophotometer over the range of 200–800 nm using quartz cuvettes of 1 cm path-length. Solvatochromic studies were conducted at a concentration of 20 μM in ethanol, acetonitrile, and dimethylsulfoxide. Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) were performed using an STA7200 differential thermogravimetric analyzer under a nitrogen atmosphere. The band gap energies of compounds were determined using a Cary 100 spectrophotometer equipped with a diffuse reflectance accessory.4.2 Microwave-assisted multicomponent synthesis of 4,6-diaryl-3-cyano-2(1H)-pyridones 4
A mixture of the corresponding aromatic aldehyde 1a–c (3 mmol), acetophenone 2 (3 mmol, 350 μL), ethyl cyanoacetate 3 (3 mmol, 319 μL), and ammonium acetate (9 mmol, 694 mg) was prepared in ethanol (2 mL). The reaction mixture was irradiated in a sealed vessel using a microwave reactor (Anton Paar Monowave 400) at 100 °C for 30 min. After irradiation, the solvent was removed under reduced pressure using a rotary evaporator. The crude product was then purified by consecutive washes. The first wash involved a mixture of n-hexane and dichloromethane (5:1, v/v), which was repeated three times. This was followed by three washes with a mixture of n-hexane and ethanol (5:1, v/v). The resulting solid was transferred to a Büchner funnel and washed with a small amount of cold ethanol (1 mL) to remove any residual solvent. Finally, the product was dried under a vacuum to afford 3-cyano-2(1H)-pyridones 4a–c. Crystals of compounds 4a–c suitable for single-crystal X-ray diffraction analysis were grown by slow evaporation from a solution of compounds in a mixture of methanol and N,N-dimethylformamide (1:5, v/v) under ambient temperature and pressure conditions for two weeks.
N) 2218, ν(CO) 1639, ν(CC) 1607, 1497, 1472, 1227, 1078, 918, 816, 768, 694, 637, 558, and 540 cm−1. UV-Vis (20 μM in ethanol): 221 (λmax, ε = 1.55 × 104 L mol−1 cm−1, π → π*), 257 (ε = 1.60 × 104 L mol−1 cm−1, π → π*), 301 (ε = 0.85 × 104 L mol−1 cm−1, π → π*), and 365 (ε = 1.20 × 104 L mol−1 cm−1, n → π*(CO)) nm. 1H-NMR (400 MHz, DMSO-d6): 2.39 (s, 3H, CH3), 6.79 (s, 1H, H-5), 7.37 (d, J = 8.0 Hz, 2H, ArH), 7.49–7.57 (m, 3H, ArH), 7.64 (d, J = 8.0 Hz, 2H, ArH), 7.89 (dd, J = 7.8, 1.8 Hz, 2H, ArH), and 11.65 (br s, 1H, NH) ppm. 13C-NMR (100 MHz, DMSO-d6): 20.9 (CH3), 97.8 (Cq, CN), 106.0 (CH, C-5), 116.8 (Cq, C-3), 127.7 (2CH, Ar), 128.2 (2CH, Ar), 128.9 (2CH, Ar), 129.3 (2CH, Ar), 131.1 (CH, Ar), 132.6 (Cq, Ar), 133.2 (Cq, Ar), 140.4 (Cq, Ar), 151.6 (Cq, C-6), 159.5 (Cq, C-4), and 162.5 (CO, C-2) ppm. HRMS (ESI+): m/z calculated for C19H15N2O+ = 287.1179 [M + H]+; found 287.1185.N) 2217, ν(CO) 1636, ν(CC) 1604, 1499, 1471, 1226, 1071, 1009, 910, 821, 767, ν(C–Br) 689, 609, and 547 cm−1. UV-Vis (20 μM in ethanol): 217 (λmax, ε = 2.05 × 104, L mol−1 cm−1, π → π*), 260 (ε = 1.95 × 104 L mol−1 cm−1, π → π*), 290 (ε = 1.00 × 104 L mol−1 cm−1, π → π*), and 368 (ε = 1.35 × 104 L mol−1 cm−1, n → π*(CO)) ppm. 1H-NMR (500 MHz, DMSO-d6): 6.83 (s, 1H, H-5), 7.51–7.59 (m, 3H, ArH), 7.70 (d, J = 9.0 Hz, 2H, ArH), 7.79 (d, J = 8.5 Hz, 2H, ArH), 7.90 (d, J = 6.5 Hz, 2H, ArH), and 12.87 (br s, 1H, NH) ppm. 13C-NMR (125 MHz, DMSO-d6): 98.8 (Cq, CN), 105.6 (CH, C-5), 116.3 (Cq, C-3), 124.1 (Cq, Cp), 127.8 (2CH, Co′), 128.9 (2CH, Cm′), 130.3 (Cq, Ci′), 130.4 (2CH, Cm), 131.2 (CH, Cp′), 131.8 (2CH, Co), 135.2 (Cq, Ci), 151.4 (Cq, C-6), 158.6 (Cq, C-4), and 161.7 (CO, C-2) ppm. HRMS (ESI+): m/z calculated for C18H1279BrN2O+ = 351.0128 [M + H]+; found 351.0134.N) 2217, ν(CO) 1637, ν(CC) 1603, 1490, 1338, 1226, 1179, 1092, 1013, 962, 908, 822, ν(C–Cl) 766 (ν C–Cl), 689, 624, and 547 cm−1. UV-Vis (20 μM in ethanol): 222 (λmax, ε = 1.38 × 104 L mol−1 cm−1, π → π*), 259 (ε = 1.46 × 104, L mol−1 cm−1, π → π*), 295 (ε = 0.60 × 104 L mol−1 cm−1, π → π*), and 368 (ε = 1.21 × 104, n → π*(CO)) nm. 1H-NMR (400 MHz, DMSO-d6): 6.84 (s, 1H, H-5), 7.51–7.59 (m, 3H, Hm′, Hp′), 7.64 (d, J = 8.4 Hz, 2H, Hm), 7.77 (d, J = 8.8 Hz, 2H, Ho), 7.90 (d, J = 7.2 Hz, 2H, Ho′), and 12.88 (s, 1H, NH) ppm. 13C-NMR (100 MHz, DMSO-d6): 96.6 (Cq, CN), 106.1 (CH, C-5), 116.4 (Cq, C-3), 127.8 (2CH, Co′), 128.9 (2CH, Cm), 129.0 (2CH, Cm′), 130.3 (2CH, Co), 131.3 (CH, Cp′), 132.3 (Cq, Ci′), 134.8 (Cq, Ci), 135.4 (Cq, Cp), 151.3 (Cq, C-6), 158.5 (Cq, C-4), and 162.0 (CO, C-2) ppm. HRMS (ESI+): m/z calculated for C18H1235ClN2O+ = 307.0633 [M + H]+; found 307.0636.4.3 Single-crystal X-ray diffraction analysis
Single-crystal X-ray diffraction data for compounds 4a–c were collected at 298 K using an Agilent SuperNova, dual, Cu at zero, Atlas four-circle diffractometer equipped with a CCD plate detector. CuKα radiation (λ = 1.54184 Å) was employed. The data collection, integration, and absorption correction were performed using the CrysAlis PRO software package.39 Crystal structures of compounds 4a–c were solved through an iterative algorithm,40 followed by a difference Fourier map. Refinement of these structures was performed using SHELXL2018/3 software,41 with all non-hydrogen atoms refined anisotropically. Hydrogen atoms were geometrically generated and placed at calculated positions using a rigid model (C–H = 0.93–0.96 Å, N–H = 0.86 Å), and constrained with Uiso values set to 1.2 times (for methyl groups) or 1.5 times (for other hydrogens) the equivalent Ueq value of the attached atom. During refinements, substitutional disorder was refined over Br and Cl atoms in compounds 4b and 4c. Molecular and supramolecular graphics were generated using the graphical software Mercury.274.4 Computational methods
All electronic structure calculations for compounds 4a–c were performed using the Gaussian 09 software package.42 Density functional theory (DFT) calculations were employed at the B3LYP/6-311++G(2df,2pd) level of theory. The molecular geometries of compounds 4a–c were optimized in the gas phase and in silico solvent environments using the implicit polarizable continuum model (PCM) to represent ethanol, acetonitrile, and DMSO. All atomic coordinates were relaxed during the optimization process. The optimized geometries were confirmed as local minima on the potential energy surface (PES) by harmonic vibrational frequency calculations, ensuring the absence of imaginary frequencies. To aid in the assignment of UV-Vis spectral bands, vertical excitation energies and oscillator strength values (f) were calculated in the gas phase, and the simulated solvents using the time-dependent DFT (TD-DFT) method at the B3LYP/6-311++G(2df,2pd) level of theory. A total of five excited states were included in these calculations (NStates = 5). Natural bond orbital (NBO) population analyses were performed on the optimized gas-phase geometries to generate the frontier molecular orbital (FMO) surfaces. The GaussView graphical interface was used for the visualization of molecular structures,43 the generation of simulated IR and UV-Vis spectra, and the depiction of molecular surfaces such as frontier orbitals.Topological analysis of the electron density (ρ) within the crystalline packing of compounds 4a–c was performed using the quantum theory of atoms in molecules and crystals (QTAIM-C) approach developed by Bader.44 To represent the crystalline environment, a dimeric model was considered for each compound by extracting two symmetrically related molecules from the corresponding unit cell. The wavefunctions of these dimers were calculated using Gaussian 09 software at the B3LYP/6-311++G(2df,p) level of theory. These wavefunctions were then used as input for the AIM2000 program45 to perform the QTAIM-C analysis. Critical points on the gradient vector field of the electron density (∇ρ) were located using Newton's method with a maximum of 120 iterations and a step size factor of 0.5. The nature of each CP was determined by analyzing its Laplacian (∇2ρ) and eigenvalue properties. The molecular graphs were constructed following the established QTAIM-C protocol. This involved tracing the paths of steepest descent (downhill paths) from bond critical points (BCPs, (3,+1) CPs) and paths of steepest ascent (uphill paths) from ring critical points (RCPs, (3,−1) CPs). In addition, bond paths connecting neighboring BCPs were identified.
Intermolecular interactions in the crystal packing of compounds 4a–c were investigated using Hirshfeld surface (HS) analysis.29 CrystalExplorer21.5 software30 was employed to generate the HS and associated two-dimensional fingerprint plots. The three-dimensional dnorm (normalized contact distance) HS map was generated for each compound. A standard surface resolution was used for HS generation, and a fixed color scale of −0.2023 to +1.3588 au was applied to represent the range of normalized contact distances.
Data availability
The data supporting this article have been included as part of the ESI.† The crystallographic data have been deposited with the Cambridge Crystallographic Data Center, CCDC, and are assigned the deposition codes 2335843 (4a), 2335844 (4b), and 2335845 (4c).Author contributions
DH-R: investigation, methodology; DB: conceptualization, writing – original draft; HR: writing – review & editing; JAGC: computational analyses, methodology, writing – original draft; MAM: methodology, writing – original draft; J-CC: conceptualization, methodology, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Universidad Pedagógica y Tecnológica de Colombia and Universidad de los Andes. This work was funded by the Dirección de Investigaciones of Universidad Pedagógica y Tecnológica de Colombia through project SGI-3470 and the young researcher encouragement project SGI-3650. We express our gratitude to the National Cancer Institute (NCI) for conducting the anticancer screening of the compounds, to Universidad de Alcalá (Spain) for providing the NMR spectra, and to Dr Claudia Castañeda for acquiring the diffuse reflectance spectra.References
- A. Loppinet-Serani, F. Charbonnier, C. Rolando and I. Huc, J. Chem. Soc., Perkin Trans. 2, 1998, 937–942 RSC.
- D. Hurtado-Rodríguez, A. Salinas-Torres, H. Rojas, D. Becerra and J.-C. Castillo, RSC Adv., 2022, 12, 35158–35176 RSC.
- M. M. K. Amer, M. A. Aziz, W. S. Shehab, M. H. Abdellattif and S. M. Mouneir, J. Saudi Chem. Soc., 2021, 25, 101259 CrossRef CAS.
- S. Sangwan, N. Yadav, R. Kumar, S. Chauhan, V. Dhanda, P. Walia and A. Duhan, Eur. J. Med. Chem., 2022, 232, 114199 CrossRef CAS PubMed.
- Y. Zhang and A. Pike, Bioorg. Med. Chem. Lett., 2021, 38, 127849 CrossRef CAS PubMed.
- G. Yang, Y. Wang, J. Tian and J.-P. Liu, PLoS One, 2013, 8, e74916 CrossRef CAS PubMed.
- M. D. Latham, C. K. King, P. Gorycki, T. L. Macdonald and W. E. Ross, Cancer Chemother. Pharmacol., 1989, 24, 167–171 CrossRef CAS PubMed.
- S. Bauer, M. C. Heinrich, S. George, J. R. Zalcberg, C. Serrano, H. Gelderblom, R. L. Jones, S. Attia, G. D'Amato, P. Chi, P. Reichardt, J. Meade, Y. Su, R. Ruiz-Soto, J.-Y. Blay, M. Mehren and P. Schöffski, Clin. Cancer Res., 2021, 27, 6333–6342 CrossRef CAS PubMed.
- S. J. Smith, G. T. Pauly, A. Akram, K. Melody, Z. Ambrose, J. P. Schneider and S. H. Hughes, J. Acquired Immune Defic. Syndr., 2016, 72, 485–491 CrossRef CAS PubMed.
- I. W. Flinn, S. O'Brien, B. Kahl, M. Patel, Y. Oki, F. F. Foss, P. Porcu, J. Jones, J. A. Burger, N. Jain, V. M. Kelly, K. Allen, M. Douglas, J. Sweeney, P. Kelly and S. Horwitz, Blood, 2018, 131, 877–887 CrossRef CAS PubMed.
- M. G. Zauderer, P. W. Szlosarek, S. L. Moulec, S. Popat, P. Taylor, D. Planchard, A. Scherpereel, M. Koczywas, M. Forster, R. B. Cameron, T. Peikert, E. K. Argon, N. R. Michaud, A. Szanto, J. Yang, Y. Chen, V. Kansra, S. Agarwal and D. A. Fennell, Lancet, 2022, 23, 758–767 CrossRef CAS PubMed.
- A. S. A. Alimmari, B. D. Božić, A. D. Marinković, D. Ž. Mijin and G. S. Ušćumlić, J. Solution Chem., 2012, 41, 1825–1835 CrossRef CAS.
- A. Marinković, D. Mijin, J. Mirković, V. Maslak and C. O. Kappe, J. Serb. Chem. Soc., 2014, 79, 759–765 CrossRef.
- S. K. Rai, S. Khanam, R. S. Khanna and A. K. Tewari, RSC Adv., 2014, 4, 44141–44145 RSC.
- K. Salamanca-Perdigón, D. Hurtado-Rodríguez, J. Portilla, I. Iriepa, H. Rojas, D. Becerra and J.-C. Castillo, ChemPlusChem, 2024, e202400172 CrossRef PubMed.
- D. Becerra, R. Abonia and J.-C. Castillo, Molecules, 2022, 27, 4723 CrossRef CAS PubMed.
- C. Simon, T. Constantieux and J. Rodriguez, Eur. J. Org Chem., 2004, 24, 4957–4980 CrossRef.
- L. Rong, X. Li, H. Wang, D. Shi and S. Tu, Chem. Lett., 2006, 35, 1314c1315 CrossRef.
- L. Rong, H. Wang, J. Shi, F. Yang, H. Yao, S. Tu and D. Shi, J. Heterocycl. Chem., 2007, 44, 1505–1508 CrossRef CAS.
- L. Rong, H. Han, H. Jiang, D. Shi and S. Tu, Synth. Commun., 2008, 38, 217–224 CrossRef CAS.
- H. A. El-Sayed, N. H. Ouf and A. H. Moustafa, Res. Chem. Intermed., 2014, 40, 407–412 CrossRef CAS.
- I. S. Antonijevic, G. V. Janjic, M. Milcic and S. D. Zaric, Cryst. Growth Des., 2016, 16, 632–639 CrossRef CAS.
- A. Nataraj, V. Balachandran and T. Karthick, J. Mol. Struct., 2013, 1031, 221–233 CrossRef CAS.
- P. Politzer and F. Abu-Awwad, Theor. Chem. Acc., 1998, 99, 83–87 Search PubMed.
- R. S. Mulliken, J. Chem. Phys., 1934, 2, 782 CrossRef CAS.
- R. G. Parr, L. von Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- P. L. A. Popelier, Atoms in Molecules: An Introduction, Pearson Education, Harlow, Great Britain, 2000 Search PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman. CrystalExplorer17, University of Western Australia, 2017, available at: https://hirshfeldsurface.net/ Search PubMed.
- J.-C. Castillo, D. Becerra and M. A. Macías, Crystals, 2023, 13, 694 CrossRef CAS.
- C. Serrano-Sterling, D. Becerra, J. Portilla, H. Rojas, M. Macías and J.-C. Castillo, J. Mol. Struct., 2021, 1244, 130944 CrossRef CAS.
- I. V. Magedov, M. Manpadi, M. A. Ogasawara, A. S. Dhawan, S. Rogelj, S. V. slambrouck, W. F. A. Steelant, N. M. Evdokimov, P. Y. Uglinskii, E. M. Elias, E. J. Knee, P. Tongwa, M. Y. Antipin and A. Kornienko, J. Med. Chem., 2008, 51, 2561–2570 CrossRef CAS PubMed.
- A. A. Fadda, K. S. Mohamed, H. M. Refat and E. E. El-Bialy, Heterocycles, 2015, 91, 134–148 CrossRef CAS.
- M. I. Ansari, A. Arun, M. K. Hussain, R. Konwar and K. Hajela, ChemistrySelect, 2016, 1, 4255–4264 CrossRef CAS.
- Y. Tangella, K. L. Manasa, V. L. Nayak, M. Sathish, B. Sridhar, A. Alarifi, N. Nagesh and A. Kamal, Org. Biomol. Chem., 2017, 15, 6837–6853 RSC.
- R. Mekheimer, Pharmazie, 1994, 49, 322–324 CAS.
- L. Rong, H. Wang, J. Shi, F. Yang, H. Yao, S. Tu and D. Shi, J. Heterocycl. Chem., 2009, 44, 1505 CrossRef.
- CrysAlisPro 1.171.39.46e, Rigaku Oxford Diffraction, 2018 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView 6.0.16, Semichem, Inc., Shawnee Mission, 2016 Search PubMed.
- R. F. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- F. Biegler-Konig, J. Schonbohm and D. J. Bayles, AIM2000 – a program to analyze and visualize atoms in molecules, J. Comput. Chem., 2001, 22, 545–559 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2335843–2335845. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra04563g |
| This journal is © The Royal Society of Chemistry 2024 |