
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHow specific ion effects influence the mechanical behaviors of amide macromolecules? A cross-scale study†
Song Zhang‡
 a,
Mengjia Fang‡b,
Junjun Hea,
Lina Maa,
Xiaohe Miaoc,
Peichuang Li*d,
Shirui Yu*a and
Wanhao Cai*b
a,
Mengjia Fang‡b,
Junjun Hea,
Lina Maa,
Xiaohe Miaoc,
Peichuang Li*d,
Shirui Yu*a and
Wanhao Cai*b
aDepartment of Food Science and Engineering, Moutai Institute, Renhuai 564502, China. E-mail: yushirui@mtxy.edu.cn
bSchool of Food Science and Engineering, Hefei University of Technology, Hefei, Anhui 230009, P.R. China. E-mail: caiwanhao@hfut.edu.cn
cInstrumentation and Service Center for Physical Sciences, Westlake University, Hangzhou 310024, Zhejiang Province, China
dHeze Branch, Qilu University of Technology (Shandong Academy of Sciences), Heze, 274000, China. E-mail: lpc91@qlu.edu.cn
First published on 13th August 2024
Abstract
The mechanisms of specific ion effects on the properties of amide macromolecules is essential to understanding the evolution of life. Because most biological macromolecules contain both complex hydrophilic and hydrophobic structures, it is challenging to accurately identify the contributions of molecular structure to macroscopic behaviors. Herein, we investigated the influence of specific ion effects on the mechanical behaviors of poly(N-isopropylacrylamide) and neutral polyacrylamide (i.e., PNIPAM and NPAM), through a cross-scale study that includes single-molecule force spectroscopy, molecular dynamics simulation and macro mechanical method. The results indicate that the molecular conformation can be markedly influenced by the hydrophilicity (or hydrophobicity) of both macromolecule chain and ions. An extended chain conformation can be obtained when the side groups and ions are relatively hydrophilic, which can also increase the elasticity of a macromolecule chain and film materials. The relatively hydrophobic components promote the collapse of macromolecule chains and reduce the molecular elasticity. It is believed that the hydrogen bonding intensity between a macromolecule chain and aquated ions controls the chain conformation and the elasticity of molecules and films. This study is not only helpful for understanding the self-assembly mechanism of organisms but also provides a way to associate the molecular properties with the macroscopic performance of materials.
1. Introduction
One of the mysteries of life is how macromolecules in aqueous conditions self-assembled into advanced structures; for example, the formation of proteins with multilevel structures.1,2 Ions are critical in catalysing life activities through adjusting the structure of biological macromolecules.3 In 1888, Franz Hofmeister found that the type of ions deeply influences the solubility of egg white protein in aqueous conditions, and the ions were sorted according to their ability to stabilize the protein structure, which is the embryonic form of the well-known Hofmeister series.4–6 During the last century, the Hofmeister effect has been extended to other properties of macromolecules in solution, such as stability of colloidal systems,7 surface tension,8,9 viscosity of resin and condensed-state thermodynamics.6,10 However, it is still a big challenge for scientists to precisely amplify the internal mechanism of the Hofmeister effect since the system involves multiple components including macromolecules, water molecules, and salt ions.11,12At present, several classical theories have been proposed to describe ion-containing solution systems. Debye–Hückel ionic strength theory predicts the activity coefficient of a classical dilute solution,13 and the DLVO theory describes the stability of colloids.14,15 Both the theories consider that the solution properties are only related to the charge and concentration of ions. However, they failed to predict the fact that the same charge ions with different size show different Hofmeister effects, which is called the specific ion effect.16–19 The size of ions determines their charge density, and its introduction in models will improve the existing theory. However, only considering the charge density cannot fully explain the Hofmeister sequence. After all, the role of water cannot be ignored. The intrinsic specific ion effect has been investigated in experiments during recent years with the enrichment of thermodynamic methods.18,20 Single-molecule force spectroscopy (SMFS) allows for the direct manipulation towards a single macromolecule chain, and has been widely applied to investigate the intrachain and intermolecular effects of macromolecules in various environments.21–28 Some examples include detecting the folding and unfolding strength of globulin, and quantifying the influence of specific ion effects on the single-chain elasticity of macromolecules.22,29–33 The lower critical solution temperature (LCST) of poly(N-isopropylacrylamide) (PNIPAM, a thermosensitive macromolecule with both hydrophilic amide group and hydrophobic alkyl group) in a series of sodium salts with a concentration gradient was precisely acquired through a gradient microfluidic device.34–36 Moreover, the action sites of ions on macromolecules can be accurately identified by high-resolution mass spectrometry.37,38 Previous studies have paved the way to characterize the specific ion effect-induced microscopic properties and the macroscopic performance. It is necessary to elucidate the mechanism by which the ion specific effect influences the behaviors of amide macromolecules to better understand the origin of life and the design of high-performance biomaterials.39,40
Herein, by combining SMFS with molecular dynamics (MD) simulations,41,42 and macro-mechanics studies, we investigated the structural and mechanical behaviors of neutral polyacrylamide (NPAM) and PNIPAM (two macromolecules sharing the same backbone but different side groups) induced by the specific ion effect. The results indicate that the single-molecule behaviors of the macromolecules are closely associated with their macroscopic thermodynamics and structural properties in condensed state. The more hydrophilic macromolecule (NPAM) exhibits an obvious extended conformation and outstanding single molecule elasticity in the strong hydrated ion (Ac−) solution. The conformational transition of the relatively hydrophobic macromolecule (PNIPAM) induced by the ion specific effect is much less than that of NPAM. However, the single-molecule elasticity of PNIPAM in the solution of a weakly hydrated ion (SCN−) is drastically weakened. Interestingly, the special extended conformation of NPAM induced by Ac− is proved to be helpful to the formation of a high-strength thin film. The ability to form a hydrogen bond (H-bond) among the molecular chain, water molecules and ions controls the structural and elastic behaviors of macromolecules in salt solutions. Moreover, we found that the single-molecule elasticity shows a positive correlation with the mechanical behavior of film materials.
2. Materials and methods
2.1 Materials and chemicals
PNIPAM and NPAM powders were obtained from Aladdin Corp. (Shanghai, China). V-shaped Si3N4 AFM cantilevers (SNL-10) were purchased from Bruker Corp. (Billerica, MA, USA). The water is ultrapure (UP, >18 MΩ cm). Dimethyl sulfoxide (DMSO), trimethylsiloxane, KAc and KSCN were obtained from Sigma-Aldrich Corp. (St. Louis, MO, USA).2.2 Details of AFM-based SMFS
PNIPAM and NPAM were dissolved in ultrapure water (stirred at 25 °C for 1 h) to form a very dilute solution (10 mg L−1). A quartz slide was first cleaned by ultrasound for 5 min with UP water. Then, the slide was further treated by plasma cleaning in vacuum for 5 min. After that, the slide was immersed into the mixed solution of NaOH solution (1 M) and hydrogen peroxide (30%) at a volume ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 for 30 min (Warning! This process will release a lot of heat and the solution is highly corrosive). After that, the impurity on substrate surface can be thoroughly cleaned, and then be used in SMFS experiments. Finally, the slide was dried by nitrogen. Before the SMFS experiments, about 15 μL PNIPAM or NPAM solution was dropped onto a clean quartz slide for 15 min, then flushed by the surrounding liquid of AFM-based SMFS (Cypher VRS, Asylum Research, Santa Barbara, CA, USA). The moving rate of the AFM tip is 2.0 μm s−1 if not mentioned otherwise, which is a typical value for SMFS. The residence times of the probe on the substrate surface and furthest from the substrate surface are both set as 0.5 s to increase the molecular capture efficiency. The spring constant of the AFM cantilever (about 45 pN nm−1) was calibrated by the thermal noise method. All F–E curves were further analyzed with Igor Pro (Wavemetrics, Portland, OR, USA). In order to prevent the influence of volatilization on the test results, only the first 100 data points were taken for each force experiment under acidic and alkaline conditions, and the test of each sample was completed within 15 min. To ensure the reliability of the experimental results, SMFS experiments were performed on 5 samples for each experimental condition, and the valid data set obtained under each condition is not less than 20.
:4 for 30 min (Warning! This process will release a lot of heat and the solution is highly corrosive). After that, the impurity on substrate surface can be thoroughly cleaned, and then be used in SMFS experiments. Finally, the slide was dried by nitrogen. Before the SMFS experiments, about 15 μL PNIPAM or NPAM solution was dropped onto a clean quartz slide for 15 min, then flushed by the surrounding liquid of AFM-based SMFS (Cypher VRS, Asylum Research, Santa Barbara, CA, USA). The moving rate of the AFM tip is 2.0 μm s−1 if not mentioned otherwise, which is a typical value for SMFS. The residence times of the probe on the substrate surface and furthest from the substrate surface are both set as 0.5 s to increase the molecular capture efficiency. The spring constant of the AFM cantilever (about 45 pN nm−1) was calibrated by the thermal noise method. All F–E curves were further analyzed with Igor Pro (Wavemetrics, Portland, OR, USA). In order to prevent the influence of volatilization on the test results, only the first 100 data points were taken for each force experiment under acidic and alkaline conditions, and the test of each sample was completed within 15 min. To ensure the reliability of the experimental results, SMFS experiments were performed on 5 samples for each experimental condition, and the valid data set obtained under each condition is not less than 20.
2.3 Preparation of PNIPAM and NPAM films, and their water content and mechanical tests
3 g PNIPAM or NPAM particles were dissolved in 10 ml KSCN or KAc solution (0.5 M) under stirring to form a homogeneous viscous fluid, and then the fluid was coated onto an organic glass plate through a four-sided coating device (SZQ, Huaguo, Guang Dong, China). After that, the plate was placed in a fume cupboard for 24 h to dry off the moisture by the forced convection of air. The thickness of both PNIPAM and NPAM films were controlled in 100 ± 10 μm. A 0.5 g film sample was placed in a vacuum drying oven, and the drying temperature was set to 95 °C. After the sample was dried for 1 h, it was taken out, cooled and weighed. The operation was repeated until the mass change of the sample did not exceed 1 mg. The ratio of the mass difference before and after drying to the mass before drying is recorded as the water content. The stress–strain curves of the films were obtained by a microcomputer control electron universal testing machine (BOS-100NW, Boshi, Fujian, China). Samples were prepared into a dumbbell shape, and the length and width were fixed as 100 mm × 25 mm. The stretching rate was set as 0.5 mm min−1.2.4 Details of MD simulations
All MD simulations were performed on a Material Studio software package. The used forcefield is COMPASSII. An amorphous cell that contains a polymer chain (10 repeating units), 1200 water molecules, and other components (KAc or KSCN) with a certain proportion was constructed. The size of the cell (about 30 Å × 30 Å × 50 Å) depends on the van der Waals radius of the system. The electrostatic and van der Waals interactions are chosen as Ewald- and atom-based, respectively. The model cell was geometrically optimized before dynamics. Subsequently, an equalization treatment was performed under the NPT model for 5 ns to obtain the density (ρ) of the cell. Then, dynamics were performed under the NVT model for 20 ns to collect data. The simulations were performed at room temperature (RT). The selected thermostat was Andersen. The electrostatic energy for each system is the statistical results based on the dynamic simulation during data collection.3. Results and discussion
3.1 The inherent single-molecule elasticity
The chemical structures of PNIPAM and NPAM are quite similar. The only difference is that the N atoms of PNIPAM are linked by a hydrophobic isopropyl group, while existing as the primary amine with strong hydrophilicity (Fig. 1A). To accurately quantify the influence of the specific ion effect on the mechanical properties of these two macromolecules, it is necessary to determine the inherent elasticity of the backbone.43,44 It should be noted that either PNIPAM or NPAM may exhibit a strong intrachain H-bond effect.21 Therefore, SMFS studies were firstly carried out in DMSO, a strong H-bond destroyer. The normalized experimental force–extension (F–E) curves of each macromolecule obtained in different stretching events can be superposed perfectly at the elastic elongation region, indicating that the interactions between a macromolecule chain and the surroundings are in equilibrium in dilute solution (see ESI† for more representative curves). When compared together, one can see that the typical experimental F–E curves of PNIPAM and NPAM obtained in DMSO show almost no difference in the entire force region (Fig. 1B). The theoretical F–E curve of the C–C backbone was obtained by Cui et al. by introducing the elastic modulus from quantum-mechanical (QM) calculation into a freely rotating chain (FRC) model, where the effect from the side groups was ignored.45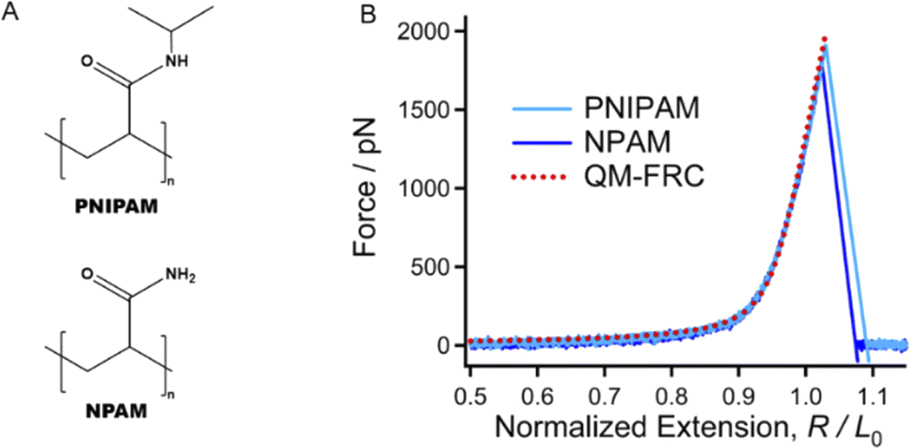 | ||
| Fig. 1 (A) The chemical structures of PNIPAM and PNAM. (B) The typical normalized experimental force–extension (F–E) curves of PNIPAM and NPAM in DMSO (solid curves) compared to the theoretical inherent F–E curve of the C–C backbone. | ||
Cui et al. proposed that the inherent elasticity of a polymer backbone can be described well by introducing the elastic modulus from the quantum-mechanical (QM) calculation into the freely rotating chain (FRC) model, freely jointed chain (FJC) model or wormlike chain (WLC) model.43,46,47 To date, the Cui method has shown outstanding accuracy in describing the inherent molecular elasticity of polymers. The theoretical background of these models can be found in the section “Theoretical Background of Cui method” in ESI.† The QM-FRC model of the C–C backbone proposed by Cui et al. was used to describe the molecular elasticity of PNIPAM and NPAM in DMSO.
The experimental F–E curves can be described well by the QM-FRC model from about 0 to 1800 pN (typical breaking strength of the chemical bond). The result indicates that in DMSO, the strength of non-covalent interactions along the chain direction is infinitesimal, and both PNIPAM and NPAM show only the elasticity contributed from the covalent effect of the backbone, i.e., the inherent elasticity of the C–C backbone.
It should be noted that the single-molecule elasticity of polymer chains is not linear, and includes the contribution of conformational change and backbone deformation. Moreover, the length varies for different molecular weights. Therefore, a constant value for the elasticity in the form of elasticity = force/extension seems to not be possible. Nevertheless, a molecular chain adopts an overstretched conformation under high forces, making the conformational change negligible. There, the deformation of the backbone dominates the situation and the force–extension of the molecular chain is almost linear. One can normalize the extension of the molecular chain to the length of its single repeating unit, and perform linear fitting to obtain the elasticity of the segment.48 We find that the segment elasticity is 76.2 ± 0.3 and 77.6 ± 0.3 nN nm−1 for PNIPAM and NPAM, respectively (Fig. S2†). The values are almost the same since they share the same backbone.
3.2 Molecular elasticity in ultrapure water
Next, we performed SMFS experiments on the two macromolecules in aqueous environments. The solid curves in Fig. 2A are the experimental F–E curves of PNIPAM and PNIPAM obtained in ultrapure water. The forces in SMFS can be divided into three regions, where the high, low, and middle force regions are determined only by enthalpy, by entropy, and by both enthalpy and entropy, respectively.49 The curves show a nice consistency with the inherent F–E curve (QM-FRC) at the high forces (more than 600 pN), but obvious deviations can be observed in the lower force region. In UP water, the F–E curves of both PNIPAM and NPAM are above the inherent F–E curve in the low and middle force regions. This means that at the same stretching degree, the force required to stretch these two molecules in water is higher than that in DMSO if the force is not higher than 600 pN; namely, the chains show better elasticity in ultrapure water. The result indicates that both the enthalpic and entropic elasticity were enhanced in ultrapure water, and the elastic increment of NPAM is more obvious than that of PNIPAM. It has been reported that in aqueous conditions, a water bridge can form between the adjacent hydrophilic units of a macromolecule.50 Although amide groups exist in both PNIPAM and NPAM, a more dense water bridge forms along a NPAM chain rather than a PNIPAM chain because PNIPAM is much more hydrophobic.23 Therefore, NPAM shows better single-molecule elasticity than PNIPAM in UP water.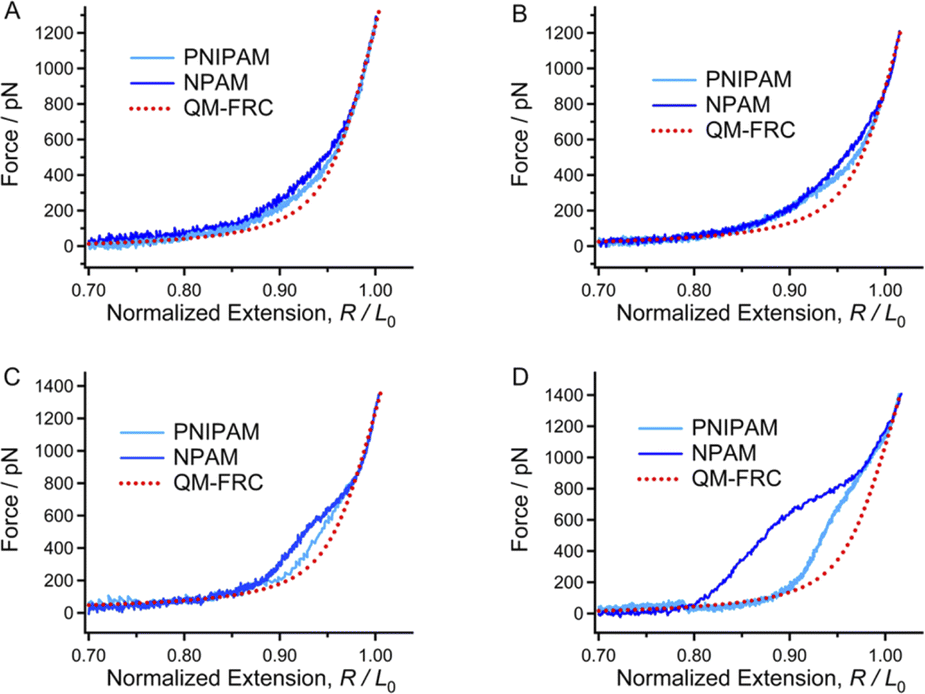 | ||
| Fig. 2 Normalized experimental F–E curves of PNIPAM and NPAM in ultrapure water (A), 0.1 M KAc (B), 0.3 M KAc (C) and 0.5 M KAc (D) in comparison to the inherent F–E curve (red dotted line). | ||
3.3 Huge enhancement in single-molecule elasticity induced by a strong hydrated ion
Many studies have revealed the fact that the ion specificity for most macromolecules to anions is significantly stronger than that of cations.51,52 SMFS studies carried out in KCl solutions (not more than 0.5 M) show that PNIPAM and NPAM almost maintained their single-molecule elasticities compared to that in ultrapure water (Fig. S3†), demonstrating that the existence of K− or Cl− hardly affects the weak interactions along the chain direction since they are in the middle of the Hofmeister sequence. Next, we performed SMFS experiments in KAc solutions with a concentration gradient since Ac− is a strong hydrated monovalent anion (more hydrophilic than Cl−). As shown in Fig. 2B–D, shoulder-like force plateaus can be seen in the F–E curves of both PNIPAM and NPAM obtained in KAc solutions. Moreover, the higher Ac− concentration (not more than 0.5 M) causes a more obvious plateau and a bigger force deviation between the experimental F–E curve and the inherent F–E curve. Ions may be surrounded by water molecules to form a core–shell structure after being dissolved in water.53 For strong hydrated ions, the distance between the ion core and the nearest water shell is relatively shorter than that of weakly hydrated ions. Thus, one can hold that the core–shell structure of Ac− is more stable than that of Cl− and K+. In addition, the strength between the Ac− core–shell bridge and the adjacent units of a macromolecule should be much stronger than the common water bridge, since a large number of water molecules are tightly surrounding Ac−. The density of the Ac− core–shell bridge along the chain direction of PNIPAM should be lower than that of NPAM because of the steric hindrance and hydrophobic effects from the isopropyl groups. As a result, the increase in the single-molecule elasticity of NPAM is more obvious than that of PNIPAM. Interestingly, the energy to stretch a NPAM chain in 0.5 M KAc is about 2 times greater compared to its inherent elasticity (Fig. S7†), and the energy for PNIPAM in 0.5 M KAc is about 1.5 times greater than the inherent elasticity. The SMFS results in KAc solutions indicate that the single-molecule elasticity of both PNIPAM and NPAM can be improved markedly, giving that the anisotropic elastic NPAM or NPAM materials may be prepared in acetate solutions.3.4 The decrease in single-molecule elasticity induced by a weakly hydrated ion
To further study the influence of the ion hydration ability on the elasticity of a macromolecule chain, SMFS studies were carried out in KSCN solutions with a concentration gradient since SCN− lies on the right side of the Hofmeister series and is considered a weakly hydrated ion. Compared to the F–E curves obtained in ultrapure water shown in Fig. 2A, the F–E curves of PNIPAM and NPAM obtained in 0.1 M KSCN show a smaller deviation with the inherent F–E curve. With the increasing SCN− concentration, the F–E curves of NPAM get closer to the inherent F–E curve, while the F–E curves of PNIPAM are lower than the inherent F–E curve (Fig. 3C and D). Moreover, the deviation between the F–E curves of PNIPAM increase with SCN− concentration. It has been widely accepted that the addition of ions with poor hydratability will destroy the structure of bound water, and this kind of ion tends to go away from the water network and form non-covalent bonding interactions with the hydrophobic groups of other solutes in solution. This progress is the so-called chaotropic effect of hydrophobic ions. It is conceivable that the direct binding of SCN− will compete with that of water molecules on a macromolecule chain. Furthermore, the repulsion between ions along the chain direction will reduce the conformational entropy of a macromolecule chain. As a result, both the enthalpic and entropy elasticity would be reduced by SCN−. For PNIPAM, the hydrophobic isopropyl groups provide ideal binding sites for SCN−. Consequently, the higher SCN− concentration causes a smaller water bridge along the chain. However, the sensitivity of the single-molecule elasticity of NPAM to the SCN− concentration is not obvious since there is no hydrophobic side group. In short, the single-molecule elasticities of PNIPAM and NPAM can both be weakened by introducing SCN− in the aqueous solution, and PNIPAM is much more sensitive to SCN− than NPAM.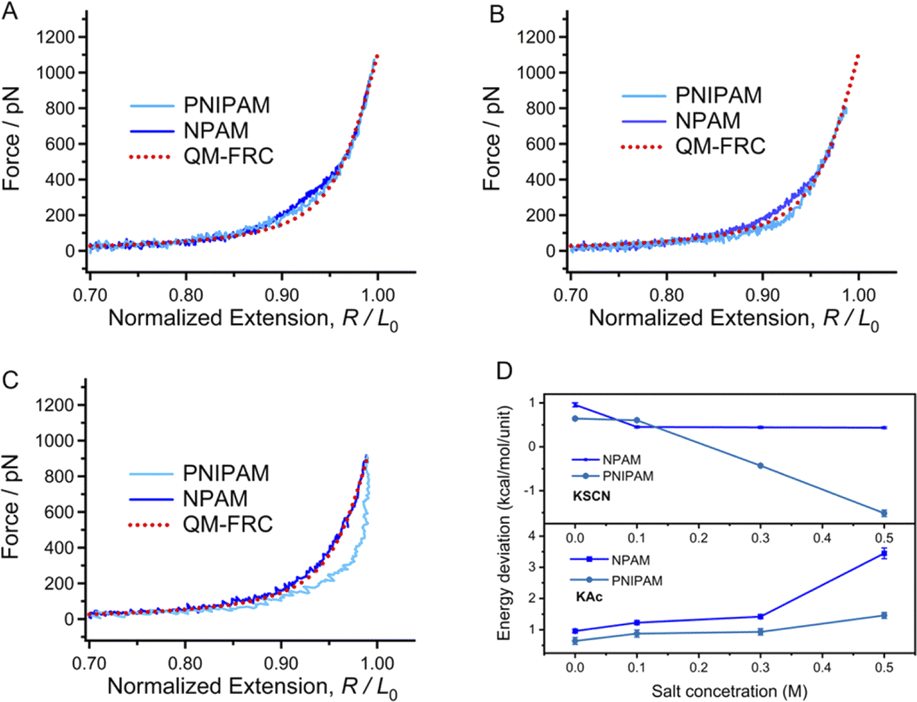 | ||
| Fig. 3 Normalized F–E curves of PNIPAM and NPAM obtained in 0.1 M KSCN (A), 0.3 M KSCN (B), and 0.5 M KSCN (C) in comparison to the inherent F–E curve. (D) The energy difference to stretch PNIPAM and NPAM in solutions with different concentration in comparison to the inherent F–E curve. | ||
3.5 Quantizing the deviation of the stretching energy induced by the specific ion effects
The deviation value of the energy to stretch a PNIPAM or NPAM chain in salt solutions compared to the inherent elasticity, which corresponds to the strength of noncovalent effects along the chain direction, can be obtained by integral methods.44 As shown in Fig. 3D, the energy difference for the two macromolecules shows different trends in KAc and KSCN solutions. Concretely, the energy value is increased with the concentration of Ac−, but decreased with SCN− concentration. Additionally, the Ac−-induced energy increase for NPAM (from 1 to 3.5 kcal per mol per unit) is obviously greater than that for PNIPAM (from 0.6 to 1.3 kcal per mol per unit), while the SCN−-induced energy decrease for PNIPAM (from 1 to −1.5 kcal per mol per unit) is more obvious than that for NPAM (from 0 to 0.5 kcal per mol per unit). Therefore, we can make a conclusion from the aspect of single-molecule elasticity that the specific ion effect for a hydrophilic macromolecule (NPAM) is remarkable for hydrophilic (or kosmotropic) ions (Ac−), and the effect for a relatively hydrophobic macromolecule (PNIPAM) is more sensitive to hydrophobic (or chaotropic) ions (SCN−).3.6 The conformational behaviors of a single chain and its internal driving force from MD simulations
One can expect that the conformation of a molecular chain may be adjusted by a specific ion effect since the solubility in the macroscopic view can be distinctly influenced. Fig. 4 shows the MD simulation results of PNIPAM and NPAM in pure water, 0.5 M KAc and 0.5 M KSCN, respectively. It is clear that NPAM is more extended and relaxed than PNIPAM in pure water and 0.5 M KAc, while the situation is the opposite in 0.5 M KSCN (Fig. 4A). The conformational behavior of a molecular chain can be largely influenced by the strength of the repulsive effect along the chain when ions are involved in solution. In pure water, the hydrophobic interaction from the side groups promotes PNIPAM to exist as a relatively collapsed conformation compared to NPAM. In 0.5 M KAc, the density of the Ac− hydrated core–shell structure is obviously larger along the chain for NPAM than that of PNIPAM. As a result, the NPAM chain is more extended than PNIPAM in 0.5 M KAc due to a stronger charge repulsion. In 0.5 M KSCN, the density of SCN− along the chain direction of PNIPAM is larger than that of NPAM because its hydrophobic isopropyl side groups provide a lot of binding sites for SCN−. The statistical radius of gyration (Rg) of a macromolecular chain is an important parameter to quantify the conformational behavior, which can reflect the degree of extension of a single macromolecule chain.54 As shown in Fig. 4B, the influence of anion types to the Rg of PNIPAM and NPAM in aqueous conditions shows an inverse tend, corresponding to the result shown in Fig. 4A. It should be noted that the specific ion effect reflected by Rg for NPAM (7.3–9.5 Å) is more obvious than PNIPAM (7.6–7.9 Å). This result is reasonable because the number of binding water molecules and charge repulsion are both favorable to the formation of an extended conformation, which may compete with the hydrophobic effect. In 0.5 M KSCN, the Rg for NPAM is obviously lower than that in pure water because of the reduction of binding water along the chain, while the variation of Rg for PNIPAM is not as sensitive as NPAM because of the competition between the collapse and extension induced by hydrophobicity and charge repulsion, respectively. The situation in 0.5 M KAc is similar to that in 0.5 M KSCN; the difference is that Ac− is hydrophilic.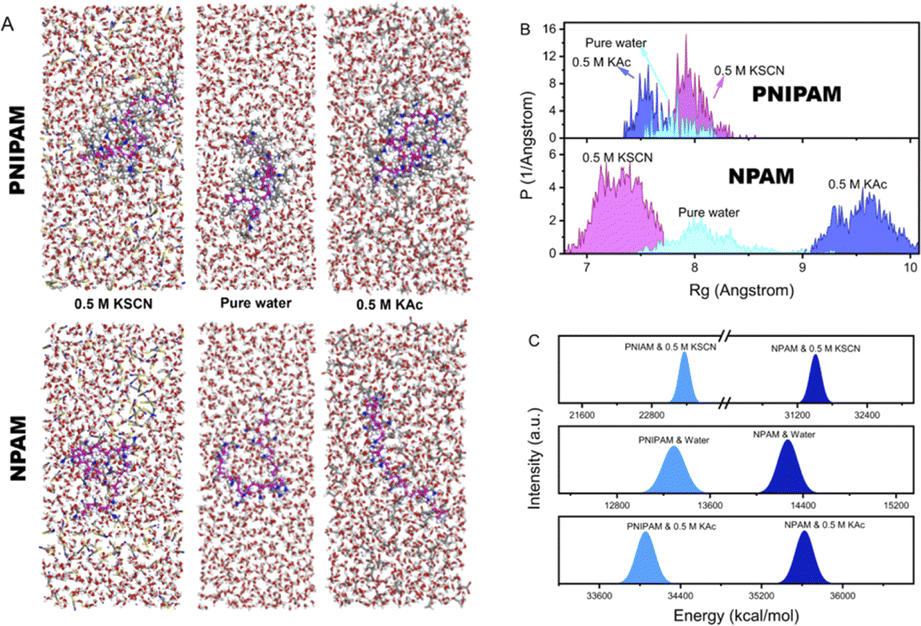 | ||
| Fig. 4 The MD simulation results of PNIPAM and NPAM in different aqueous solutions. (A) The conformational behaviors of a single chain. (B) The Rg distributions of a single chain during MD simulation. (C) The energy between a molecular chain and the surrounding that contributed by electrostatic interactions. | ||
Fig. 4c shows the electrostatic energy between the chain and environment, where NPAM has a much larger value than PNIPAM whether in pure water or ionic solutions. This result indicates that the interactions of NPAM with the water/ions is stronger than that of PNIPAM, and has a better affinity to the local environment. It should be noted that this energy is the total electrostatic interactions between the molecular chain and the surrounding environment. In contrast, the energy obtained from the area difference in SMFS is the energy deviation of molecules under forces in different solutions. This value mainly reflects the consumed energy in breaking the bound water/ion, which is much smaller than that from MD. Therefore, they are not necessarily following the same trend when compared in different environments. For instance, in 0.5 M KSCN, the total electrostatic energy is lower than that in pure water, which differs from the SMFS results. It indicates that although SCN− can weaken the water–chain interactions, the adjustment of molecular conformation and the interacting volume both contribute to the electrostatic energies.
3.7 The mechanics of PNIPAM and NPAM films in the presence of ions
How do the specific ion effects influence the performance of amide macromolecules in a macroscopic scale? Herein, we investigated the mechanical behaviors of PNIPAM and NPAM films prepared in different salt solutions through tensile tests. Typical stress–strain curves of PNIPAM and NPAM prepared in both 0.5 M KAc and 0.5 M KSCN are shown in Fig. 5. Point 1 in the F–E curves correspond to the upper yield point of the film being elongated. Before point 1, the deformation of the films is reversible. Therefore, the stretching energy before point 1 can reflect the elasticity of the films, and it be obtained by integration. Interestingly, for both PNIPAM and NPAM, the stretching energy before yield (Ey) for the film prepared in 0.5 KAc is much larger than that prepared in 0.5 M KSCN. This result is consistent with the fact that the single-molecule elasticity of the two macromolecules in 0.5 M KAc are obviously greater than that in 0.5 M KSCN, indicating that the elasticity of the film materials is positively correlated with the single-molecule elasticity. Point 2 in the stress–strain curves corresponds to the fracture of the films. From the extension value of point 2, the breaking elongation rate relative to the original length (100 mm) can be obtained (ε), which can be calculated from eqn (1).
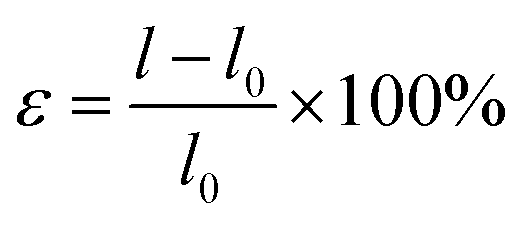 | (1) |
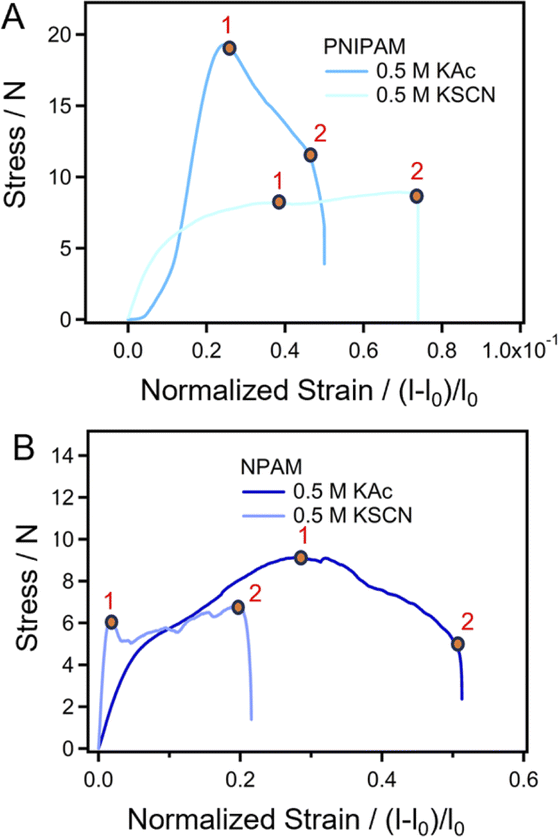 | ||
| Fig. 5 Normalized stress–strain curves of films prepared in different salt solutions. (A) PNIPAM. (B) NPAM. | ||
| Films | Ey/J | ε/% | Water content/% |
|---|---|---|---|
| PNIPAM 0.5 M KAc | 0.186 ± 0.003 | 4.9 ± 0.2 | 5.8 ± 0.4 |
| PNIPAM 0.5 M KSCN | 0.152 ± 0.004 | 2.6 ± 0.3 | 3.1 ± 0.3 |
| NPAM 0.5 M KAc | 1.8 ± 0.01 | 52.1 ± 0.1 | 10.6 ± 0.5 |
| NPAM 0.5 M KSCN | 0.077 ± 0.01 | 22.5 ± 0.6 | 6.7 ± 0.5 |
The results from the SMFS experiments show that the single-molecule elasticity of a polymer in different salt environments is mainly determined by the number of bound waters to the chain. Therefore, one can speculate that the film materials prepared in different salt environments have different water contents under the same moisture condition. In order to verify this conjecture, we used the drying reduction method to determine the water content of different films. The results show that both PNIPAM and NPAM film materials contain a certain amount of bound water (Table 1). The water content of the PNIPAM and NPAM films prepared in 0.5 M KAc was both higher than that in a salt-free environment, and that of the NPAM film in this condition is larger than PNIPAM. However, the water content of the films prepared in 0.5 M KSCN was lower than that in the salt-free environment, indicating that the addition of the SCN− stripped the bound water on the molecular chain. The results are consistent with the conclusion of the SMFS. The elongation rate of the films is determined by the molecular conformation. The more collapsed conformation causes a lower elongation rate, and the more extended chain leads to a higher elongation rate, which is consistent with the MD simulation results.
It should be noted that the mechanics of the single chain and macroscopic films differ from each other, and cannot be compared directly because the structure and stretching conditions are very different. For instance, the elongation rate of the PNIPAM and NPAM film materials involved in this study is relatively low. Nevertheless, the change tendency of mechanical properties (such as the yield strength of film materials prepared in different environments) is consistent with the conclusion of the SMFS experiments. This potential correlation may have the following reasons. (i) The water content deviations in films are associated with single-chain bound water, which plays a role in the energy consumption during elongation. (ii) The molecular chains generally adopt an extended rather than coiled conformation in the film, and are entangled with each other with different orientations. The stretching process is bound to be accompanied by the excessive stretching of a large number of molecules, which directly reflects the molecular elasticity to a certain extent. As a result, the stretching process is bound to be accompanied by the excessive stretching of a large number of molecules, which directly reflects the molecular elasticity to a certain extent. Therefore, it is reasonable that there is a correlation between the macroscopic mechanical properties of the material and the SMFS results. However, we must admit that it is difficult to further derive the quantitative relationship of the results between the two scales, since integrating cross-scale laws has always been a huge challenge. We will further explore this part in our future work.
4. Conclusions
In summary, the influence of specific ion effects on the mechanical properties of amide macromolecules was investigated through a cross-scale study from single-molecule to the macroscopic level. The mechanical and conformational behaviors of two typical macromolecules with different amide side groups (PNIPAM and NPAM) show opposite trends. A more hydrophobic macromolecule (PNIPAM) is more sensitive to a relatively hydrophobic (chaotropic) anion (SCN−), while the macromolecule with prominent hydrophilicity (NPAM) shows more sensitivity to a hydrophilic (kosmotropic) ion (Ac−). The single-molecule elasticity of both PNIPAM and NPAM can be obviously improved by Ac− by forming core-shell-like ion hydration bridges along the chain, and the elastic improvement of NPAM is superior due to the greater bridge number. The existence of SCN− will weaken the backbone elasticity, since the water bridge length along the chain is decreased due to the chaotropic effect of SCN−. The expansion degree of the chain is mainly determined by its ability to bind ions and the hydrophilicity. Moreover, our results initially confirm that the single-molecule elasticity is positively correlated with the macroscopic mechanical performance of materials. Interestingly, the interaction between the strong hydrated macromolecule (NPAM) and kosmotropic ion (Ac−) is conducive to the preparation of materials with high elasticity and toughness. This study may not only be helpful to understand the self-assembly process of macromolecules in salt solutions, but also provides an inspiration for the design of high-performance materials with salt sensitivity.Data availability
The data supporting the findings of this study are available within the article and in the ESI,† and further inquiries can be directed to the corresponding authors.Author contributions
Conceptualization, S. Z. and W. C.; methodology, W. C., S. Y. and S. Z.; validation, S. Z., P. L. and W. C.; investigation, S. Z., W. C., S. Y., M. F., J. H., L. M., P. C., Y. W., X. M., P. L.; data curation, S. Z., P. L. and C. W.; writing—original draft preparation, S. Z., M. F., and W. C.; writing—review and editing, W. C., S. Y., P. L. and S. Z.; funding acquisition, S. Y., W. C. and P. L. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was funded by The Special Funds for Local Scientific and Technological Development Guided by the Central Government ((2019)4006) from Guizhou Science and Technology Department, The Fundamental Research Funds for the Central Universities (JZ2024HGTA0191), Shandong Provincial Natural Science Foundation (ZR2022QE169), Talent Research Project of Qilu University of Technology (2023RCKY062), Innovation Ability Improvement Project of Technologic Minor Enterprise of Shandong Province (2023TSGC0075), Engineering Research Center supported by Guizhou Provincial, and the Education Department (KY(2020)022) and Moutai Institute high-level talents research fund project (mygccrc [2022]087).References
- H. Garcia-Seisdedos, C. Empereur-Mot, N. Elad and E. D. Levy, Proteins evolve on the edge of supramolecular self-assembly, Nature, 2017, 548, 244–247 CrossRef CAS PubMed
.
- J. Zhu, N. Avakyan, A. Kakkis, A. M. Hoffnagle, K. Han, Y. Li, Z. Zhang, T. S. Choi, Y. Na and C.-J. Yu, Protein assembly by design, Chem. Rev., 2021, 121, 13701–13796 CrossRef CAS PubMed
- X. Zheng, W. Cheng, C. Ji, J. Zhang and M. Yin, Detection of metal ions in biological systems: A review, Rev. Anal. Chem., 2020, 39, 231–246 CrossRef CAS
- Y. Zhang and P. S. Cremer, Interactions between macromolecules and ions: the Hofmeister series, Curr. Opin. Chem. Biol., 2006, 10, 658–663 CrossRef CAS PubMed
- H. I. Okur, J. Hladílková, K. B. Rembert, Y. Cho, J. Heyda, J. Dzubiella, P. S. Cremer and P. Jungwirth, Beyond the Hofmeister series: Ion-specific effects on proteins and their biological functions, J. Phys. Chem. B, 2017, 121, 1997–2014 CrossRef CAS PubMed
- S. Das, L. Giri and S. Majumdar, Hofmeister series: An insight into its application on gelatin and alginate-based dual-drug biomaterial design, Eur. Polym. J., 2023, 189, 111961 CrossRef CAS
- T. López-León, M. J. Santander-Ortega, J. L. Ortega-Vinuesa and D. Bastos-González, Hofmeister effects in colloidal systems: Influence of the surface nature, J. Phys. Chem. C, 2008, 112, 16060–16069 CrossRef
- L. Musilová, V. Kašpárková, A. Mráček, A. Minařík and M. Minařík, The behaviour of hyaluronan solutions in the presence of Hofmeister ions: A light scattering, viscometry and surface tension study, Carbohydr. Polym., 2019, 212, 395–402 CrossRef PubMed
- L. Ferreira, V. Uversky and B. Zaslavsky, Effects of the Hofmeister series of sodium salts on the solvent properties of water, Phys. Chem. Chem. Phys., 2017, 19, 5254–5261 RSC
- P. Lo Nostro and B. W. Ninham, Hofmeister phenomena: an update on ion specificity in biology, Chem. Rev., 2012, 112, 2286–2322 CrossRef CAS PubMed
- E. E. Bruce, H. I. Okur, S. Stegmaier, C. I. Drexler, B. A. Rogers, N. F. Van Der Vegt, S. Roke and P. S. Cremer, Molecular mechanism for the interactions of Hofmeister cations with macromolecules in aqueous solution, J. Am. Chem. Soc., 2020, 142, 19094–19100 CrossRef CAS PubMed
- B. Rana, D. J. Fairhurst and K. C. Jena, Ion-Specific Water–Macromolecule Interactions at the Air/Aqueous Interface: An Insight into Hofmeister Effect, J. Am. Chem. Soc., 2023, 145, 9646–9654 CrossRef CAS PubMed
- A. Salis and B. W. Ninham, Models and mechanisms of Hofmeister effects in electrolyte solutions, and colloid and protein systems revisited, Chem. Soc. Rev., 2014, 43, 7358–7377 RSC
- K. P. Gregory, G. R. Elliott, H. Robertson, A. Kumar, E. J. Wanless, G. B. Webber, V. S. Craig, G. G. Andersson and A. J. Page, Understanding specific ion effects and the Hofmeister series, Phys. Chem. Chem. Phys., 2022, 24, 12682–12718 RSC
- M. Andreev, A. Chremos, J. De Pablo and J. F. Douglas, Coarse-grained model of the dynamics of electrolyte solutions, J. Phys. Chem. B, 2017, 121, 8195–8202 CrossRef CAS PubMed
- D. J. Tobias and J. C. Hemminger, Getting specific about specific ion effects, Science, 2008, 319, 1197–1198 CrossRef CAS PubMed
- H. Zhao, Biotechnology, Protein stabilization and enzyme activation in ionic liquids: specific ion effects, J. Chem. Technol. Biotechnol., 2016, 91, 25–50 CrossRef CAS PubMed
- S. Z. Moghaddam and E. Thormann, The Hofmeister series: Specific ion effects in aqueous polymer solutions, J. Colloid Interface Sci., 2019, 555, 615–635 CrossRef CAS PubMed
- B. Sarvin, S. Lagziel, N. Sarvin, D. Mukha, P. Kumar, E. Aizenshtein and T. Shlomi, Fast and sensitive flow-injection mass spectrometry metabolomics by analyzing sample-specific ion distributions, Nat. Commun., 2020, 11, 3186 CrossRef CAS PubMed
- L. Moreira and A. J. L. Firoozabadi, Molecular thermodynamic modeling of specific ion effects on micellization of ionic surfactants, Langmuir, 2010, 26, 15177–15191 CrossRef CAS PubMed
- W. Cai, D. Xu, F. Zhang, J. Wei, S. Lu, L. Qian, Z. Lu and S. Cui, Intramolecular hydrogen bonds in a single macromolecule: Strength in high vacuum versus liquid environments, Nano Res., 2022, 15, 1517–1523 CrossRef CAS
- R. Petrosyan, A. Narayan and M. T. Woodside, Single-molecule force spectroscopy of protein folding, J. Mol. Biol., 2021, 433, 167207 CrossRef CAS PubMed
- Y. Bao, Z. Luo and S. Cui, Environment-dependent single-chain mechanics of synthetic polymers and biomacromolecules by atomic force microscopy-based single-molecule force spectroscopy and the implications for advanced polymer materials, Chem. Soc. Rev., 2020, 49, 2799–2827 RSC
- S. Zhang, H. J. Qian, Z. Liu, H. Ju, Z. Y. Lu, H. Zhang, L. Chi and S. Cui, Towards unveiling the exact molecular structure of amorphous red phosphorus by single-molecule studies, Angew. Chem., Int. Ed., 2019, 131, 1673–1677 CrossRef
- W. H. Cai, D. Xu, L. Qian, J. H. Wei, C. Xiao, L. M. Qian, Z. Y. Lu and S. X. Cui, Force-induced transition of pi-pi stacking in a single polystyrene chain, J. Am. Chem. Soc., 2019, 141, 9500–9503 CrossRef CAS PubMed
- W. H. Cai, C. Xiao, L. M. Qian and S. X. Cui, Detecting van der Waals forces between a single polymer repeating unit and a solid surface in high vacuum, Nano Res., 2019, 12, 57–61 CrossRef CAS
- Y. Bao, H. J. Qian, Z. Y. Lu and S. Cui, Revealing the hydrophobicity of natural cellulose by single-molecule experiments, Macromolecules, 2015, 48, 3685–3690 CrossRef CAS
- S. Zhang, Z. Li, Y. Bao, S. Lu, Z. Gong, H.-J. Qian, Z.-Y. Lu and S. Cui, Nanoscopic Characterization Reveals that Bulk Amorphous Elementary Boron Is Composed of a Ladder-like Polymer with B4 as the Structural Unit, ACS Nano, 2023, 17, 10958–10964 CrossRef CAS PubMed
- G. M. Nam and D. E. Makarov, Extracting intrinsic dynamic parameters of biomolecular folding from single-molecule force spectroscopy experiments, Protein Sci., 2016, 25, 123–134 CrossRef CAS PubMed
- C. Bustamante, L. Alexander, K. Maciuba and C. M. Kaiser, Single-molecule studies of protein folding with optical tweezers, Annu. Rev. Biochem., 2020, 89, 443–470 CrossRef CAS PubMed
- S. Howorka and Z. Siwy, Nanopore analytics: sensing of single molecules, Chem. Soc. Rev., 2009, 38, 2360–2384 RSC
- Z. L. Hu, M. Z. Huo, Y. L. Ying and Y. T. Long, Biological nanopore approach for single-molecule protein sequencing, Angew. Chem., Int. Ed., 2021, 133, 14862–14873 CrossRef
- W. Cai, J. L. Trefs, T. Hugel and B. N. Balzer, Anisotropy of π–π Stacking as Basis for Superlubricity, ACS Mater. Lett., 2022, 5, 172–179 CrossRef
- P. Xue, J. Nan, T. Wang, S. Wang, S. Ye, J. Zhang, Z. Cui and B. Yang, Ordered micro/nanostructures with geometric gradient: from integrated wettability “library” to anisotropic wetting surface, Small, 2017, 13, 1601807 CrossRef PubMed
- Y. Zhang, S. Furyk, L. B. Sagle, Y. Cho, D. E. Bergbreiter and P. S. Cremer, Effects of Hofmeister anions on the LCST of PNIPAM as a function of molecular weight, J. Phys. Chem. C, 2007, 111, 8916–8924 CrossRef CAS PubMed
- Y. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, Specific ion effects on the water solubility of macromolecules: PNIPAM and the Hofmeister series, J. Am. Chem. Soc., 2005, 127, 14505–14510 CrossRef CAS PubMed
- C. Wesdemiotis, Multidimensional mass spectrometry of synthetic polymers and advanced materials, Angew. Chem., Int. Ed., 2017, 56, 1452–1464 CrossRef CAS PubMed
- N. P. Lockyer, S. Aoyagi, J. S. Fletcher, I. S. Gilmore, P. A. Van Der Heide, K. L. Moore, B. J. Tyler and L.-T. Weng, Secondary ion mass spectrometry, Nat. Rev. Methods Primers, 2024, 4, 32 CrossRef CAS
- P. G. Higgs and N. Lehman, The RNA World: molecular cooperation at the origins of life, Nat. Rev. Genet., 2015, 16, 7–17 CrossRef CAS PubMed
- A. B. Grommet, M. Feller and R. Klajn, Chemical reactivity under nanoconfinement, Nat. Nanotechnol., 2020, 15, 256–271 CrossRef CAS PubMed
- S. Wolf, B. Lickert, S. Bray and G. Stock, Multisecond ligand dissociation dynamics from atomistic simulations, Nat. Commun., 2020, 11, 2918 CrossRef CAS PubMed
- B. Sohmen, C. Beck, V. Frank, T. Seydel, I. Hoffmann, B. Hermann, M. Nüesch, M. Grimaldo, F. Schreiber and S. Wolf, The Onset of Molecule-Spanning Dynamics in Heat Shock Protein Hsp90, Adv. Sci., 2023, 10, 2304262 CrossRef CAS PubMed
- S. Cui, Y. Yu and Z. Lin, Modeling single chain elasticity of single-stranded DNA: A comparison of three models, Polymer, 2009, 50, 930–935 CrossRef CAS
- Y. Bao, X. Huang, J. Xu and S. Cui, Effect of intramolecular hydrogen bonds on the single-chain elasticity of poly (vinyl alcohol): Evidencing the synergistic enhancement effect at the single-molecule level, Macromolecules, 2021, 54, 7314–7320 CrossRef CAS
- W. Cai, S. Lu, J. Wei and S. Cui, Single-chain polymer models incorporating the effects of side groups: An approach to general polymer models, Macromolecules, 2019, 52, 7324–7330 CrossRef CAS
- S. X. Cui, C. Albrecht, F. Kuhner and H. E. Gaub, Weakly bound water molecules shorten single-stranded DNA, J. Am. Chem. Soc., 2006, 128, 6636–6639 CrossRef CAS PubMed
- S. X. Cui, J. Yu, F. Kuhner, K. Schulten and H. E. Gaub, Double-stranded DNA dissociates into single strands when dragged into a poor solvent, J. Am. Chem. Soc., 2007, 129, 14710–14716 CrossRef CAS PubMed
- Y. Liu, Y. Yu, J. Gao, Z. Wang and X. Zhang, Water-soluble supramolecular polymerization driven by multiple host-stabilized charge-transfer interactions, Angew. Chem., Int. Ed., 2010, 49, 6576–6579 CrossRef CAS PubMed
- Z. Luo, A. Zhang, Y. Chen, Z. Shen and S. Cui, How big is big enough? Effect of length and shape of side chains on the single-chain enthalpic elasticity of a macromolecule, Macromolecules, 2016, 49, 3559–3565 CrossRef CAS
- N. Cao, Y. Zhao, H. Chen, J. Huang, M. Yu, Y. Bao, D. Wang and S. Cui, Poly (ethylene glycol) becomes a supra-polyelectrolyte by capturing hydronium ions in water, Macromolecules, 2022, 55, 4656–4664 CrossRef CAS
- V. Mazzini and V. S. Craig, What is the fundamental ion-specific series for anions and cations? Ion specificity in standard partial molar volumes of electrolytes and electrostriction in water and non-aqueous solvents, Chem. Sci., 2017, 8, 7052–7065 RSC
- E. M. Wright and J. M. Diamond, Anion selectivity in biological systems, Physiol. Rev., 1977, 57, 109–156 CrossRef CAS PubMed
- M. Ma, Z. Gan and Z. Xu, Ion structure near a core-shell dielectric nanoparticle, Phys. Rev. Lett., 2017, 118, 076102 CrossRef PubMed
- E. Yamamoto, T. Akimoto, A. Mitsutake and R. Metzler, Universal relation between instantaneous diffusivity and radius of gyration of proteins in aqueous solution, Phys. Rev. Lett., 2021, 126, 128101 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04360j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |