
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrtho-quinone methide driven synthesis of kynurenic acid lactams†
Julián Robin Sárik a,
Anasztázia Hetényib,
Róbert Berkeczcd,
István Szatmári*ae and
Bálint Lőrinczi*a
a,
Anasztázia Hetényib,
Róbert Berkeczcd,
István Szatmári*ae and
Bálint Lőrinczi*a
aInstitute of Pharmaceutical Chemistry, University of Szeged, Eötvös u. 6, H-6720 Szeged, Hungary. E-mail: szatmari.istvan@szte.hu; lorinczi.balint@szte.hu
bDepartment of Medical Chemistry, University of Szeged, Dóm tér 8, H-6720 Szeged, Hungary
cInstitute of Pharmaceutical Analysis, University of Szeged, H-6720 Szeged, Somogyi u. 4, Hungary
dDepartment of Forensic Medicine, Albert Szent-Györgyi Health Centre, H-6724 Szeged, Kossuth Lajos sgt. 40, Hungary
eHUN-REN SZTE Stereochemistry Research Group, University of Szeged, Eötvös u. 6, H-6720 Szeged, Hungary
First published on 12th July 2024
Abstract
Lactam formation of different KYNA amides and Mannich bases mediated by ortho-quinone methide has been investigated. The efficiency of the two routes of the cyclization process was revealed and the influence of diverse amide side chains was explored. In this regard compounds bearing a tertiary amine function in the amide side chain resulted in the formation of the lactam product, while the formation of dimer derivatives was observed in the case of other KYNA amides. Furthermore, derivatives bearing different substituents on the KYNA B ring were synthesized and their effects on the ring-closure reaction were investigated.
1 Introduction
Kynurenic acid (KYNA) and 4-hydroxyquinoline-2-carboxylic acid are endogenous substances, produced during tryptophan metabolism (TRP) through the pathway that is also responsible for the production of nicotinamide adenine dinucleotide (NAD+) and NAD phosphate (NADP+).1,2 It is one of the few endogenous excitatory amino acid receptor blockers with a broad spectrum of antagonistic properties in supraphysiological concentrations. KYNA is well established to have high affinity for NMDA receptors and has been shown to have a positive modulatory binding site of higher affinity at the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA).3In recent years there has been increasing interest in the synthesis and pharmacological studies of KYNA derivatives. This is due to their neuroprotective ability as it can prevent neuronal loss following excitotoxic, ischemia-induced, and infectious neuronal injuries.4,5 Several papers have been published on different alterations of the KYNA skeleton. Substitutions at positions 5–8 were achieved by starting from the corresponding anilines using the modified Conrad–Limpach method.6–8 The substituents in positions 2 and 4 have also been investigated in a broad spectrum ranging from alkoxy groups on C-4 or amides on C-2.8–14 Based on the evaluations of previous KYNA amides, a tertiary nitrogen is needed for a biological activity concerning the central nervous system.15–18 Derivatives bearing such functional groups can be synthesized by various methods, such as amidation of the carboxylic function12–14 or the transformation of the 4-hydroxy group.8–10 Another possibility can be the substitution of KYNA using the modified Mannich reaction (mMr). In this one-pot reaction KYNA used as a CH acid can be reacted with different types of secondary amines and aldehydes giving an aminoalkylated derivative with the desired cationic center.19,20
These cationic centers, beside bearing a function possibly required for transport/passage through the blood–brain-barrier, present a new reaction route to synthesize further KYNA derivatives. Depending on the amines used, these groups can function as leaving groups forming an ortho-quinone methide (oQm) intermediate. It is well known that these derivatives bear high reactivity as they contain a methide function21 that may react with a well-selected structural element. Under specific reaction conditions, the aminoalkylated KYNA amides undergo intramolecular ring closure resulting in the formation of pyrroloquinolinone derivatives. Synthesis of similar compounds have already been discussed in the literature: using oxidative methods22 or transforming naturally occurring KYNA derivatives;23 however, this instance is the first to utilize a reaction via oQm (Fig. 1). This structure bears information about the reactivity of specific oQm and they have high resemblance to biologically active compounds, including camptothecin derivatives currently in medical use24 and analogues acting on benzodiazepine receptors.25 Compounds, bearing the 4-quinolone scaffold have also been investigated as potential analgesics, due to their NMDA receptor binding capabilities26 (Fig. 1). Consequently, a suitable synthetic procedure was planned.
 | ||
| Fig. 1 Summary of previous and recent work. | ||
2 Results and discussion
The first KYNA lactam (4) was observed as a side product during the scale-up morpholinomethylation of KYNA amide 1 in mMr. Taking into consideration the present compounds in the reaction media and the possible intermediates, compound 4 could have been formed via two distinct routes: (A) from starting compound 1 and paraformaldehyde with a one-step ring closure or (B) through the aminoalkylation of 1 resulting in a Mannich base (2) that can subsequently act as an intermediate in a two-step reaction route (Scheme 1).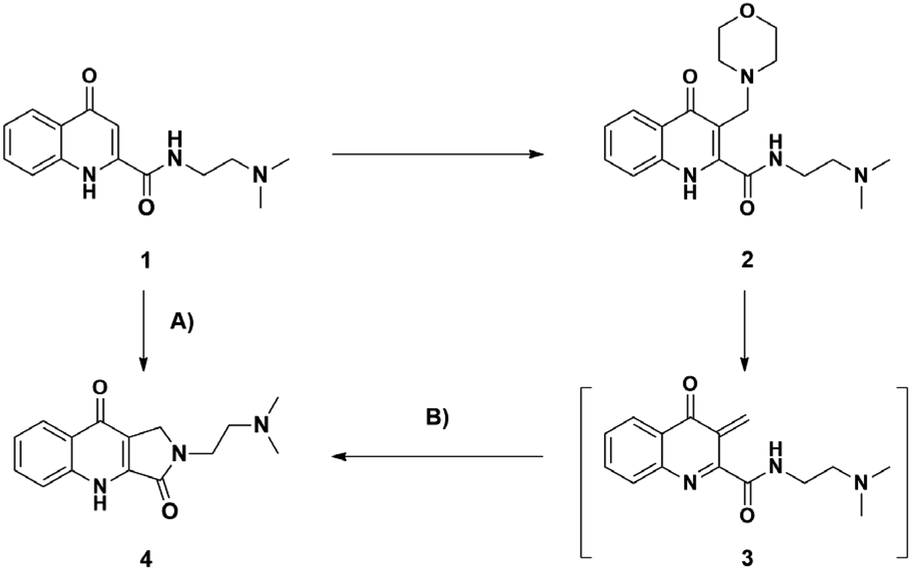 | ||
| Scheme 1 Possible reaction routes toward the formation of lactam derivative 4. (A) direct cyclization of 1 with paraformaldehyde (B) aminoalkylation with paraformaldehyde and morpholine followed by intramolecular cyclization. | ||
Since aminoalkylated KYNA amide 2 was in excess when starting the work-up of the reaction discussed above, route (B) was investigated first. Different solvents (toluene, DCM, 1,4-dioxane, acetonitrile, isopropyl alcohol, EtOH and MeOH) were tested showing the highest conversion in MeOH at 100 °C in 1.5 h. In the next step, the optimization of the reaction was carried out at different temperatures. At lower temperature intervals no conversion was detected (Table 1, entries 8, 9). At higher temperatures increasing conversions were measured (entries 10–12) eventually reaching a plateau at 150 °C after 1.5 h (entry 13). Even though the reaction took place with the same conversion at 160 °C as well (entry 14), 150 °C was chosen as optimal for the preceding reactions. As NMR tracking of the reactions carried out at higher temperatures (entry 13 and 14) showed an almost full transformation of 2 to 4, it is surmised that the reaction fully takes place via oQm 3. However, since the reaction matrix for aminoalkylation also had the starting materials for route (A) as well, a one-pot reaction was planned with entry 13 parameters starting from 1 and utilising morpholine and paraformaldehyde. The aim of this reaction was to decide whether a direct reaction could be achieved via route (B) under the optimized conditions, when starting from the prerequisites of the Mannich base instead of isolated compound 2. According to crude NMR spectra, the reaction showed only minimal formation of the desired lactam, leading to Mannich-base 2 as the main product. Then reactions were carried out via route (A) showing a similar temperature-dependent transformation (Table 1, entries 1–7), but with lower or no conversion at similar temperatures (entries 3 vs. 10, 4 vs. 11, 5 vs. 12, 6 vs. 13) reaching the same plateau at 160 °C (entry 7).
Next, the reaction was repeated using different Mannich bases to find the best leaving group. This included two secondary amines (pyrrolidine and piperidine) used for the mMr and subsequently for route (B) (Scheme 2). Based on our previous work concerning C–C bond formations on the KYNA structure starting from Mannich bases, primary amines were omitted from the tests as they proved to be poor leaving groups.27 Reactions were carried out with aminoalkylated derivatives 2 and 5a,b. The results showed that higher conversions could be achieved from 5a,b in only 30 min (99% from 5a,b presumably due to the higher Kb of the amine moieties versus 85% from 2). However, the workup was hindered by the presence of a strong base. Additional steps were necessary for their removal and the overall yields were lower in these cases (56% for 5a, 54% for 5b versus 76% for 2).
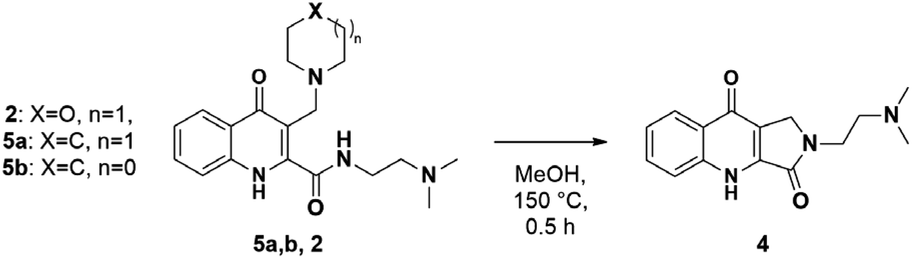 | ||
| Scheme 2 Reaction of alternative Mannich bases. | ||
A further difference between reaction route (A) and (B), beside the possible oQm intermediate, might be in the elimination of morpholine in the case of (B) that can create basic conditions. Based on this information and on results of our previous research, where Mannich bases reacted in a similar manner,27 we planned to test acidic and basic additives in route (A) (Table 2). Reactions applying acidic catalysts (AcOH or para-toluenesulfonic acid [p-TsOH]) showed inhibition of the reaction, similar to the basic N,N-dimethylaminopyridine (DMAP), though the latter had only partial inhibition of the reaction. In the case of triethylamine (TEA), high conversion in shorter reaction (30 min.) was observed. It is notable that TEA promoted the formation of 4 not only in reaction route (A) but also in the case of (B) in a reaction time of 15 min.
The scope and limitations of the reaction were also investigated using different amides bearing alkyl, aryl, and aminoalkyl functions. The relevant amide derivatives and the corresponding Mannich bases were synthesized according to the methods described herein (Scheme 3). The ring-closure reactions were conducted applying the optimized reaction conditions and in the case of route (B) starting from the corresponding Mannich base (Scheme 4, Table 3).
 | ||
| Scheme 3 Synthesis of different alkyl, aryl, and aminoalkyl amide derivatives and their subsequent transformation to Mannich bases. (i) Equimolar diisopropylcarbodiimide (DIC) and 1-hydroxybenzotriazole (1-HOBT) as catalysts at r.t. in DMF (7a–c) (ii) neat conditions, r.t. (7d) (iv) 1,4-Dioxane, reflux (8a–d) (v) acetonitrile, reflux (8e). | ||
 | ||
| Scheme 4 Scope and limitations investigation via amide derivatives. | ||
| Entry | Q- | Reaction route | Conversiona (%) | Product |
|---|---|---|---|---|
| a Starting from 7a–f or 8a–f towards the formation of lactams or dimers (4, 9e–f or 10a–d), determined from the NMR spectra.b Multicomponent reaction mixture.c Conversions could not be determined, because products are highly insoluble in DMSO. | ||||
| 1 | H | (A) | — | — |
| 2 | (B) | 99 | —b | |
| 3 | 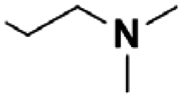 |
(A) | 70 | 4 |
| 4 | (B) | 99 | 4 | |
| 5 | 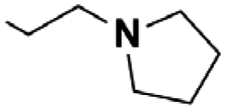 |
(A) | 99 | 9f |
| 6 | (B) | 99 | 9f | |
| 7 | 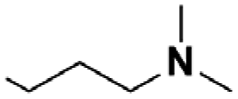 |
(A) | 65 | —b |
| 8 | (B) | 80 | 9e | |
| 9 |  |
(A) | 0 | — |
| 10 | (B) | 65 | 10a | |
| 11 |  |
(A) | 0 | — |
| 12 | (B) | 99 | 10b | |
| 13 |  |
(A) | 0 | — |
| 14 | (B) | 99 | —b | |
| 15 |  |
(A) | —c | 10d |
| 16 | (B) | —c | 10d | |
Based on the experimental results it is hypothesized that an aminoalkyl functional group is crucial for the formation of the lactam ring in both reaction routes. Note, however, that there is still a shift towards route (B) (Table 3, entries 3–8 vs. 15 and 16). Furthermore, a side chain too long is also less favored indicating a possible electrostatic stabilizing effect through a seven-membered ring (entries 7 and 8). Results in entries 15 and 16 are in accordance with this, since n-propylamine is a smaller side-chain in comparison to the aromatic substituents, but also lack a tertiary amine function. It is interesting to note that even though the pyrrolidine function at the end of the alkylamide side chain is bulkier, it facilitates the reaction in route (A) in comparison to the dimethylamine counterpart (entry 5 vs. 3). In the case of aromatic functional groups it is surmised that they have steric hindering effects. Note, that they do not have the tertiary nitrogen atom and, consequently, lactam formation cannot take place (Table 3, entries 9–14). Furthermore, in the case of a directly connected phenyl group, the negative inductive effect of the aromatic system can also be implied (Table 3, entries 9 and 10).
It is interesting to mention that the formation of a dimer derivatives (10a–d) of two KYNA amides connected through a methylene bridge at C-3 was also observed. These compounds were formed in the case of the derivatives bearing no tertiary nitrogen in their amide functions. In case of starting out from amide derivative 7d the formation of such compounds is in accordance with dimers of hydroxy-quinoline derivatives reported previously.28,29 However, in the case of the Mannich derivatives 8a,b,d it is surmised that the reaction may take place via a retro-Mannich pathway. In this, through the elimination of a morpholine the oQm intermediate is formed that reacts through an electrophilic addition with the starting amide that is formed via the elimination of the morpholinomethyl moiety as an iminium cation (Scheme 5). A more complex but also possible pathway was hypothesized by Funk et al. that also depends on the formation of an oQm intermediate and suggests the temporary dearomatisation of the oQm-Mannich base conjugate.30
 | ||
| Scheme 5 Possible reaction mechanism for the formation of dimer derivatives starting out from Mannich derivatives. | ||
To further investigate how different substituents may affect the lactam formation, the reaction was extended to KYNA derivatives substituted at the B ring. Different alkyl, aryl, and halogen analogues with these functional groups being in various positions were synthesized and used (Schemes 6, 7 and Table 4). N1,N1-Dimethylethane-1,2-diamide was chosen to build the amide side chain, because it shows a lesser stabilizing effect compared to 2-(pyrrolidine-1-yl)ethanamine thus giving additional possibility to the modifying effects of the B-ring substituents.
 | ||
| Scheme 6 Synthesis of B-ring-substituted KYNA amides and Mannich bases. (i) N,N-Dimethylethane-1,2-diamine in EtOH, reflux (ii) 1,4-dioxane, reflux. | ||
 | ||
| Scheme 7 Reaction of B-ring-substituted KYNA amides in lactam cyclization. | ||
| Entry | R- | Route | Time (min) | Conversiona (%) | Productsb | Yield (%) |
|---|---|---|---|---|---|---|
| a Starting from 12a–d or 13a–d towards the formation of lactams or dimers (14a,c,d or 15a,c), determined from NMR spectra.b Determined from NMR spectra.c Reaction used for work-up. | ||||||
| 1 | 5-Cl | (A) | 15 | 99 | 14a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15a (1:1) :15a (1:1) |
— |
| 2 | (B) | 15 | 99 | 14a:15a (1:1) |
28:19c |
|
| 3 | 6-Ph | (A) | — | 0 | — | — |
| 4 | (B) | — | 0 | — | — | |
| 5 | 7-Cl | (A) | 15 | 99 | 14c:15c (1:1) |
— |
| 6 | (B) | 15 | 99 | 14c:15c (2:1) |
23:14c |
|
| 7 | 8-Me | (A) | 30 | 99 | 14d:15d (1:0) |
— |
| 8 | (B) | 15 | 99 | 14d:15d (1:0) |
47:–c |
|
In the case of the aryl analogue no conversion was observed in the crude NMR spectra. Consequently, it is surmised that the phenyl substituent of the B ring inhibits the reaction altogether. It is worth mentioning that this inhibition, while not relevant in the case of the ester derivative,19 was observed in the aminoalkylation of the amide derivative, hindering its conversion and altogether inhibiting the subsequent intramolecular ring closure (Table 4, entries 3 and 4). While the methyl substituent showed no difference (entries 7 vs. 8, 30 min. vs. 15 min. until full conversion) compared to the unsubstituted 1 and 2 derivatives, halogen substituents increased the reactivity of the aminoalkylated KYNA derivative and subsequently the reactivity of the oQm intermediate (entries 1, 2, 5, and 6). However, this resulted not only in a faster reaction toward the desired lactam derivatives even from amides, but also increased the quantity of the dimer side product.
3 Conclusions
The synthesis of different KYNA lactam derivatives was investigated. The ring-closing synthesis was explored via two routes: (A) reacting the amide with paraformaldehyde, or (B) using the Mannich-base of the corresponding amide. The latter process led to better conversions, possibly due to the formation of a highly reactive oQm intermediate. Next, different aminoalkylated Mannich bases were investigated to test the role of the leaving group. Despite showing lower conversion, the morpholinomethylated derivative proved to be ideal compared to the pyrrolidino and piperidino analogues, due to the more favorable work-up process. The scope and limitations were further investigated using different alkyl-, aryl-, and tertiary nitrogen-containing amides. During these reactions, the alkylamine derivatives proved to be the most reactive ones, achieving full conversion in 15 min. The cause of this may be a stabilizing interaction mediated by the cationic center in the amide side chain. Alkyl- and arylamides, not bearing the aforementioned tertiary nitrogen functional group in the amide side chain, underwent a different reaction, which resulted in the formation of bis-quinoline-2-carboxamide structured derivatives. To further study the reactivity of the KYNA amides, we investigated how different B-ring substituents affect lactam formation. Using alkyl, aryl, and halogen derivatives we concluded, that chlorination of the B ring has a positive effect on the conversion, promoting the formation of the lactam, but also the dimer derivative. Alkyl substitution of the KYNA amide had little to no effect on the reactivity, while phenyl substitution heavily inhibits the reaction.4 Materials and methods
1H and 13C-NMR spectra were recorded in DMSO-d6 and CDCl3 solutions in 5 mm tubes at room temperature (r.t.) on a Bruker Ascend 500 spectrometer with a 5 mm BBO Prodigy Probe or on a Bruker AVANCE III 600 MHz spectrometer equipped with a 5 mm CP-TCI triple-resonance cryoprobe (Bruker Biospin, Karlsruhe, Baden Württemberg, Germany) at 500 (1H) and 125 (13C) MHz or 600 (1H) and 150 (13C) MHz, with the deuterium signal of the solvent as the lock and TMS as internal standard (1H, 13C).Melting points were determined on a Hinotek X-4 melting point apparatus. Merck Kieselgel 60F254 plates were used for TLC.
Reactions were conducted in a CEM Focused Microwave Synthesis System, Discover SP (CEM, Charlotte, North Carolina, USA).
HRMS flow injection analysis was performed with Thermo Scientific Q Exactive Plus hybrid quadrupole-Orbitrap (Thermo Fischer Scientific, Waltham, MA, USA) mass spectrometer coupled to a Waters Acquity I-Class UPLC™ (Waters, Manchester, UK).
The synthesis of compounds 7e,f and 8f was carried out according to literature method.14,31
4.1. Synthesis
Method II: ethyl 4-hydroxyquinoline-2-carboxylate (6b) (100 mg, 0.460 mmol) was added to an excess of propylamine in a pressure-resistant 10 mL vessel and closed. The reaction was stirred for 1 h and, after the completion of the reaction monitored by TLC (eluent: DCM:MeOH 4:1), it was diluted with 3 mL DCM. The formed precipitate was filtered and washed with 2 × 5 mL DCM.
Method III: the appropriate B-ring-substituted ester (11a–d, 0.340 mmol) and 36 mg, 0.408 mmol N1,N1-dimethylethane-1,2-diamine was added to a round-bottom flask with 20 mL EtOH. The reaction mixture was treated under reflux conditions for 8 h and the solvent was evaporated under vacuum. The residue was crystallized by adding 5 mL of EtOAc and the formed precipitate was filtered and washed with 2 × 5 mL EtOAc.
4.1.1.1 4-Oxo-N-phenyl-1,4-dihydroquinoline-2-carboxamide (7a). Yield: 195 mg, 47%. Mp.: decomposition over 300 °C. HRMS calcd for [M + H+] m/z = 265.0972, found m/z = 265.0965; 1H NMR (500 MHz, DMSO-d6): 6.91 (1H, s); 7.18 (1H, t, J = 7.01 Hz); 7.37–7.42 (3H, m); 7.67–7.76 (1H, m); 7.78–7.88 (2H, m); 7.90–8.00 (1H, m); 8.07–8.15 (1H, m); 10.69 (1H, s); 11.97 (1H, s); 13C NMR (125 MHz, DMSO-d6): 106.4; 108.1; 119.9; 121.2; 124.2; 125.1; 129.3; 132.9; 138.6; 142.3; 161.6; 178.2 (Fig. S1, S2 and S71†).
4.1.1.2 N-Benzyl-4-oxo-1,4-dihydroquinoline-2-carboxamide (7b). Yield: 300 mg, 68%. Mp.: 216.9–217.8 °C. HRMS calcd for [M + H+] m/z = 279.1128, found m/z = 279.1123; 1H NMR (600 MHz, DMSO-d6): 4.53 (2H, d, J = 5.65 Hz); 6.75 (1H, s); 7.26–7.29 (1H, m); 7.36 (5H, d, J = 4.29 Hz); 7.69 (1H, s); 7.94 (1H, d, J = 7.63 Hz); 8.07 (1H, d, J = 7.63 Hz); 9.62 (1H, s); 11.87 (1H, s); 13C NMR (150 MHz, DMSO-d6): 43.4; 107.5; 120.0; 124.2; 125.1; 126.0; 127.5; 127.9; 128.9; 132.8; 139.1; 140.3; 141.9; 162.5; 178.2 (Fig. S3, S4 and S72†).
4.1.1.3 4-Oxo-N-phenethyl-1,4-dihydroquinoline-2-carboxamide (7c). Yield: 191 mg, 41%. Mp.: decomposition over 300 °C. HRMS calcd for [M + H+] m/z = 293.1285, found m/z = 293.1280; 1H NMR (600 MHz, DMSO-d6): 2.89 (2H, t, J = 7.38 Hz); 3.56 (2H, q, J = 6.64 Hz); 6.66 (1H, s); 7.22 (1H, t, J = 6.64 Hz); 7.27 (2H, d, J = 6.64 Hz); 7.29–7.38 (3H, m); 7.64–7.73 (1H, m); 7.94 (1H, d, J = 8.32 Hz); 8.07 (1H, d, J = 6.54 Hz); 9.11 (1H, s); 11.81 (1H, s); 13C NMR (150 MHz, DMSO-d6): 35.1; 41.5; 107.3; 120.0; 124.1; 125.1; 126.0; 126.7; 128.9; 129.2; 132.8; 139.7; 140.3; 142.0; 162.3; 178.2 (Fig. S5, S6 and S73†).
4.1.1.4 4-Oxo-N-propyl-1,4-dihydroquinoline-2-carboxamide (7d). Yield: 92 mg, 87%. Mp.: 161.0–161.8 °C. HRMS calcd for [M + H+] m/z = 231.1128, found m/z = 231.1125; 1H NMR (600 MHz, DMSO-d6): 0.91 (3H, t, J = 7.45 Hz); 1.54–1.60 (2H, m); 3.25–3.30 (2H, q); 6.71 (1H, s); 7.31–7.39 (1H, m); 7.64–7.72 (1H, m); 7.95 (1H, d, J = 8.43 Hz); 8.07 (1H, d, J = 7.49 Hz); 9.01 (1H, s); 11.82 (1H, s); 13C NMR (150 MHz, DMSO-d6): 11.9; 22.5; 41.7; 107.2; 120.0; 124.1; 125.1; 125.9; 132.7; 140.3; 142.1; 162.3; 178.2 (Fig. S7, S8 and S74†).
4.1.1.5 5-Chloro-N-(2-(dimethylamino)ethyl)-4-oxo-1,4-dihydroquinoline-2-carboxamide (12a). Yield: 45 mg, 45%. Mp.: decomposition over 275 °C. HRMS calcd for [M + H+] m/z = 294.1004, found m/z = 294.0999; 1H NMR (600 MHz, DMSO-d6): 2.19 (6H, s); 2.44 (2H, t, J = 6.61 Hz); 3.39 (2H, q, J = 6.09 Hz); 6.71 (1H, brs); 7.31 (1H, s); 7.56 (1H, t, J = 8.17 Hz); 7.90 (1H, d, J = 8.43 Hz); 8.91 (1H, s); 13C NMR (150 MHz, MeOH-d4): 13.7; 36.6; 57.2; 108.1; 121.5; 126.6; 131.5; 132.1; 143.3; 162.2 (Fig. S9–13 and S75†).
4.1.1.6 N-(2-(Dimethylamino)ethyl)-4-oxo-6-phenyl-1,4-dihydroquinoline-2-carboxamide (12b). Yield: 96 mg, 84%. Mp.: decomposition over 260 °C. HRMS calcd for [M + H+] m/z = 336.1707, found m/z = 336.1702; 1H NMR (600 MHz, DMSO-d6): 2.21 (6H, s); 2.45 (2H, t, J = 6.61 Hz); 3.41 (2H, q, J = 6.09 Hz); 6.81 (1H, s); 7.38 (1H, t, J = 7.28 Hz); 7.50 (2H, t, J = 7.82 Hz); 7.75 (1H, d, J = 7.34 Hz); 7.90–7.99 (2H, m); 8.33 (1H, s); 8.84 (1H, brs); 13C NMR (150 MHz, CDCl3): 37.3; 45.1; 56.7; 106.8; 118.9; 123.8; 127.1; 127.7; 128.9; 129.1; 131.8; 132.2; 137.7; 139.5; 160.7 (Fig. S14–18 and S76†).
4.1.1.7 7-Chloro-N-(2-(dimethylamino)ethyl)-4-oxo-1,4-dihydroquinoline-2-carboxamide (12c). Yield: 45 mg, 45%. Mp.: decomposition over 160 °C. HRMS calcd for [M + H+] m/z = 294.1004, found m/z = 294.1000; 1H NMR (600 MHz, MeOH-d4): 2.44 (6H, s); 2.73 (2H, t, J = 6.7 Hz); 3.61 (2H, t, J = 6.52 Hz); 6.90 (1H, s); 7.41 (1H, d, J = 8.77 Hz); 7.88 (1H, s); 8.20 (1H, d, J = 8.76 Hz); 13C NMR (150 MHz, MeOH-d4): 36.4; 43.6; 57.1; 105.9; 120.1; 123.3; 124.6; 126.0; 137.6; 142.6; 145.1; 163.1; 176.8 (Fig. S19–22 and S77†).
4.1.1.8 N-(2-(Dimethylamino)ethyl)-8-methyl-4-oxo-1,4-dihydroquinoline-2-carboxamide (12d). Yield: 57 mg, 61%. Mp.: 158.9–160.3 °C. HRMS calcd for [M + H+] m/z = 274.1550, found m/z = 274.1547; 1H NMR (600 MHz, CDCl3): 2.31 (6H, s); 2.54–2.63 (5H, m); 3.54–3.62 (2H, m); 6.81 (1H, s); 7.28–7.34 (1H, m); 7.49–7.55 (1H, m); 8.18–8.24 (1H, m); 9.46 (1H, brs); 13C NMR (150 MHz, CDCl3): 16.7; 37.3; 45.0; 56.7 106.7; 124.0; 124.1; 125.7; 133.6; 137.5; 137.5; 138.7; 161.3; 180.0 (Fig. S23, S24 and S78†).
Method V: N-(3-(dimethylamino)propyl)-4-hydroxyquinoline-2-carboxamide (7e) (118.9 mg, 0.435 mmol), paraformaldehyde (15.7 mg, 0.522 mmol), and morpholine (45.5 mg, 0.522 mmol) were added to a round-bottom flask. The mixture was treated under reflux conditions in MeCN (10 mL) for 24 h. After completion of the reaction, the solvent was removed under reduced pressure and the desired product was crystallized using EtOAc (5 mL), filtered, and washed with 2 × 5 mL EtOAc.
4.1.2.1 3-(Morpholinomethyl)-4-oxo-N-phenyl-1,4-dihydroquinoline-2-carboxamide (8a). Yield: 104 mg, 66%. Mp.: decomposition over 230 °C. HRMS calcd for [M + H+] m/z = 364.1656, found m/z = 364.1651; 1H NMR (500 MHz, DMSO-d6): 2.47 (4H, s); 3.54 (4H, s); 3.70 (2H, s); 7.19 (1H, t, J = 6.97 Hz); 7.38 (1H, t, J = 8.52 Hz); 7.44 (2H, t, J = 8.52 Hz); 7.70 (1H, t, J = 8.52 Hz); 7.73 (2H, d, J = 7.75 Hz); 7.83 (1H, d, J = 8.52 Hz); 8.13 (1H, d, J = 7.75 Hz); 12.02 (2H, s); 13C NMR (125 MHz, DMSO-d6): 51.1; 52.5; 66.4; 113.5; 120.9; 124.4; 125.1; 125.7; 129.6; 132.7; 138.5; 162.1; 171.4 (Fig. S25, S26 and S79†).
4.1.2.2 N-Benzyl-3-(morpholinomethyl)-4-oxo-1,4-dihydroquinoline-2-carboxamide (8b). Yield: 106 mg, 65%. Mp.: decomposition over 200 °C. HRMS calcd for [M + H+] m/z = 378.1812, found m/z = 378.1808; 1H NMR (500 MHz, DMSO-d6): 2.22–2.28 (4H, m); 3.16–3.24 (4H, m); 3.55 (2H, s); 4.59 (2H, d, J = 5.21 Hz); 7.30–7.35 (2H, m); 7.38 (2H, t, J = 7.42 Hz); 7.43 (2H, d, J = 7.42 Hz); 7.66 (1H, t, J = 7.63 Hz); 7.89 (1H, d, J = 8.48 Hz); 8.08 (1H, d, J = 7.93 Hz); 11.21 (1H, t, J = 5.25 Hz); 11.76 (1H, s); 13C NMR (125 MHz, DMSO-d6): 44.1; 50.5; 51.9; 66.3; 113.4; 119.6; 124.2; 124.4; 125.7; 128.0; 128.7; 129.1; 132.6; 138.6; 139.7; 144.0; 163.2; 177.7 (Fig. S27, S28 and S80†).
4.1.2.3 3-(Morpholinomethyl)-4-oxo-N-phenethyl-1,4-dihydroquinoline-2-carboxamide (8c). Yield: 150 mg, 75%. Mp.: decomposition over 300 °C. HRMS calcd for [M + H+] m/z = 392.1969, found m/z = 392.1960; 1H NMR (500 MHz, MeOH-d4): 2.23–2.32 (4H, m); 3.02 (2H, t, J = 6.51 Hz); 3.36–3.48 (4H, m); 3.56 (2H, s); 3.76 (2H, t, J = 6.51 Hz); 7.25 (1H, t, J = 7.17 Hz); 7.29 (2H, d, J = 7.17 Hz); 7.34 (2H, t, J = 7.17 Hz); 7.44 (1H, t, J = 7.17 Hz); 7.74 (1H, t, J = 7.17 Hz); 7.81 (1H, d, J = 8.76 Hz); 8.24 (1H, d, J = 7.96 Hz); 13C NMR (125 MHz, MeOH-d4): 34.8; 41.8; 49.9; 51.3; 66.4; 113.6; 118.6; 124.1; 124.3; 125.2; 126.3; 128.4; 128.8; 132.7; 139.5; 162.7; 182.0 (Fig. S29, S30 and S81†).
4.1.2.4 3-(Morpholinomethyl)-4-oxo-N-propyl-1,4-dihydroquinoline-2-carboxamide (8d). Yield: 106 mg, 74%. Mp.: decomposition over 190 °C. HRMS calcd for [M + H+] m/z = 330.1812, found m/z = 330.1807; 1H NMR (500 MHz, MeOH-d4): 1.06 (3H, t, J = 7.35 Hz); 1.69–1.77 (2H, m); 2.58–2.70 (4H, m); 3.47 (2H, t, J = 6.97 Hz); 3.67–3.73 (4H, m); 3.78 (2H, s); 7.44 (1H, t, J = 7.43 Hz); 7.73 (1H, t, J = 8.35 Hz); 7.79 (1H, d, J = 8.35 Hz); 8.26 (1H, d, J = 8.15 Hz); 13C NMR (125 MHz, MeOH-d4): 10.7; 22.4; 41.7; 51.7; 66.5; 96.9; 124.3; 125.1; 132.6; 163.4; 178.2 (Fig. S31, S32 and S82†).
4.1.2.5 N-(3-(Dimethylamino)propyl)-3-(morpholinomethyl)-4-oxo-1,4-dihydroquinoline-2-carboxamide (8e). Yield: 126 mg, 78%. Mp.: decomposition over 200 °C. HRMS calcd for [M + H+] m/z = 373.2234, found m/z = 373.2230; 1H NMR (500 MHz, DMSO-d6): 1.70–1.75 (2H, m); 2.15 (6H, s); 2.31 (2H, t, J = 7.04 Hz); 2.44–2.52 (4H, m); 3.38–3.41 (2H, m); 3.53–3.66 (6H, m); 7.34 (1H, t, J = 8.04 Hz); 7.66 (1H, t, J = 8.04 Hz); 7.87 (1H, d, J = 8.31 Hz); 8.09 (1H, d, J = 8.31 Hz); 10.71 (1H, brs); 11.76 (1H, brs); 13C NMR (125 MHz, DMSO-d6): 27.4; 38.3; 45.6; 50.8; 52.2; 57.1; 66.6; 113.3; 119.5; 124.1; 124.3; 125.7; 132.6; 139.7; 144.3; 163.2; 177.6 (Fig. S33, S34 and S83†).
4.1.2.6 5-Chloro-N-(2-(dimethylamino)ethyl)-3-(morpholinomethyl)-4-oxo-1,4-dihydroquinoline-2-carboxamide (13a). Yield: 128 mg, 75%. Mp.: decomposition over 177 °C. HRMS calcd for [M + H+] m/z = 393.1688, found m/z = 393.1684; 1H NMR (600 MHz, DMSO-d6): 2.18 (6H, s); 2.44 (2H, t, J = 6.15 Hz); 2.46–2.50 (4H, m); 3.48 (5.89 Hz); 3.53–3.61 (6H, m); 7.29 (1H, d, J = 7.45 Hz); 7.54 (1H, t, J = 8.19 Hz); 7.84 (1H, d, J = 8.19 Hz); 10.74 (1H, brs); 11.70 (1H, brs); 13C NMR (150 MHz, DMSO-d6): 38.0; 45.6; 50.6; 52.2; 58.7; 66.6; 115.1; 119.1; 120.4; 126.7; 132.3; 132.5; 142.5; 143.3; 162.9; 176.8 (Fig. S35–38 and S84†).
4.1.2.7 N-(2-(Dimethylamino)ethyl)-3-(morpholinomethyl)-4-oxo-6-phenyl-1,4-dihydroquinoline-2-carboxamide (13b). Yield: 51 mg, 27%. Mp.: 179.4–181.3 °C. HRMS calcd for [M + H+] m/z = 435.2391, found m/z = 435.2385; 1H NMR (600 MHz, DMSO-d6): 2.20 (6H, s); 2.46 (2H, t, J = 6.21 Hz); 2.47–2.55 (4H, m); 3.52 (2H, q, J = 5.88 Hz); 3.57–3.61 (4H, m); 3.64 (2H, s); 7.40 (1H, t, J = 6.97 Hz); 7.51 (2H, t, J = 7.96 Hz); 7.74 (2H, d, J = 8.08 Hz); 7.98–8.04 (2H, m); 8.33 (1H, d, J = 2.35 Hz); 10.81 (1H, brs); 11.88 (1H, brs); 13C NMR (150 MHz, DMSO-d6): 38.07; 45.7; 50.6; 52.2; 58.7; 66.6; 113.6; 120.5; 122.9; 124.5; 127.1; 128.0; 129.6; 131.4; 136.0; 139.1; 139.8; 144.0; 163.2; 177.7 (Fig. S39–41 and S85†).
4.1.2.8 7-Chloro-N-(2-(dimethylamino)ethyl)-3-(morpholinomethyl)-4-oxo-1,4-dihydroquinoline-2-carboxamide (13c). Yield: 101 mg, 59%. Mp.: 158.3–160.4 °C. HRMS calcd for [M + H+] m/z = 393.1688, found m/z = 393.1684; 1H NMR (600 MHz, DMSO-d6): 2.18 (6H, s); 2.44 (2H, t, J = 6.31 Hz); 2.46–2.50 (4H, m); 3.49 (2H, q, J = 5.72 Hz); 3.55–3.61 (6H, m); 7.35 (1H, d, J = 8.52 Hz); 7.96 (1H, s); 8.08 (1H, d, J = 8.86 Hz); 10.78 (1H, brs); 11.85 (1H, brs); 13C NMR (150 MHz, DMSO-d6): 38.1; 45.6; 50.5; 52.1; 58.7; 66.5; 114.2; 118.8; 122.9; 124.5; 128.1; 137.3; 140.5; 144.5; 163.0; 177.1 (Fig. S42, S43 and S86†).
4.1.2.9 N-(2-(Dimethylamino)ethyl)-8-methyl-3-(morpholinomethyl)-4-oxo-1,4-dihydroquinoline-2-carboxamide (13d). Yield: 119 mg, 73%. Mp.: 105.1–106.6 °C. HRMS calcd for [M + H+] m/z = 373.2234, found m/z = 373.2230; 1H NMR (600 MHz, CDCl3): 2.28 (6H, s); 2.53–2.59 (5H, m); 2.63–2.73 (4H, m); 3.57–3.87 (8H, m); 7.29 (1H, t, J = 7.51 Hz); 7.50 (1H, d, J = 6.72 Hz); 8.22 (1H, d, J = 8.26 Hz); 9.55 (1H, brs); 11.61 (1H, brs); 13C NMR (150 MHz, CDCl3): 16.4; 38.5; 45.5; 50.5; 51.7; 58.6; 66.8; 114.3; 124.0; 124.3; 124.5; 125.3; 133.4; 136.9; 140.9; 163.3; 179.4 (Fig. S44, S45 and S87†).
:MeOH 4:1 as eluent in SiO2). The fractions were collected and the solvent was removed under reduced pressure. The desired product was crystallized using EtOAc (5 mL), filtered, and washed using 2 × 5 mL EtOAc.Method B: N-(2-(dimethylamino)ethyl)-4-hydroxy-3-(morpholinomethyl)quinoline-2-carboxamide (2) or its derivatives (8a–f and 13a–d) (2.5 mg, 0.070 mmol), triethylamine (72.6 mg, 0.717 mmol) were put in a pressure-resistant 10 mL vessel. The reaction was heated in microwave reactor at 150 °C in MeOH (1.3 mL) for 30 minutes. The solution was filtered in the case of dimer formation (discussed in next paragraph) and the mother liquor was transferred to a 25 mL round-bottom flask and the solvent was removed under reduced pressure. The sample was purified using column chromatography (DCM:MeOH 4:1 as eluent in SiO2). The fractions were collected and a solvent was removed under reduced pressure. The desired product was crystallized using EtOAc (5 mL), filtered, and washed using 2 × 5 mL EtOAc.
4.1.4.1 2-(3-(Dimethylamino)propyl)-1H-pyrrolo[3,4-b]quinoline-3,9(2H,4H)-dione (9e). Yield: 14 mg, 72%. Mp.: decomposition over 240 °C. HRMS calcd for [M + H+] m/z = 298.1550, found m/z = 298.1545; 1H NMR (500 MHz, DMSO-d6): 1.73–1.81 (2H, m); 2.25 (2H, t, J = 6.99 Hz); 3.57 (2H, t, J = 7.30 Hz); 4.37 (2H, s); 7.38 (1H, t, J = 7.44 Hz); 7.71 (1H, t, J = 8.07 Hz); 7.80 (1H, d, J = 8.35 Hz); 8.18 (1H, d, J = 8.07 Hz); 12.59 (1H, brs); 13C NMR (125 MHz, DMSO-d6): 26.2; 41.2; 45.6; 47.4; 56.9; 119.9; 120.5; 124.0; 125.4; 126.7; 132.6; 140.9; 141.3; 164.0; 173.6 (Fig. S48, S49 and S89†).
4.1.4.2 2-(2-(Pyrrolidin-1-yl)ethyl)-1H-pyrrolo[3,4-b]quinoline-3,9(2H,4H)-dione (9f). Yield: 18 mg, 87%. Mp.: decomposition over 220 °C. HRMS calcd for [M + H+] m/z = 286.1550, found m/z = 286.1546; 1H NMR (500 MHz, DMSO-d6): 1.63–1.73 (4H, m); 2.50–2.56 (4H, m); 2.72 (2H, t, J = 6.25 Hz); 3.68 (2H, t, J = 6.10 Hz); 4.43 (2H, s); 7.39 (1H, t, J = 7.62 Hz); 7.72 (1H, t, J = 7.62 Hz); 7.80 (1H, d, J = 7.62 Hz); 8.19 (1H, d, J = 7.62 Hz); 12.61 (1H, brs); 13C NMR (125 MHz, DMSO-d6): 23.6; 41.6; 47.6; 53.9; 54.1; 119.9; 124.0; 125.4; 132.6; 140.8; 141.2; 163.9; 173.6 (Fig. S50, S51 and S90†).
4.1.4.3 8-Chloro-2-(2-(dimethylamino)ethyl)-1H-pyrrolo[3,4-b]quinoline-3,9(2H,4H)-dione (14a). Yield: 6 mg, 28%. Mp.: decomposition over 295 °C. HRMS calcd for [M + H+] m/z = 306.1004, found m/z = 306.0999; 1H NMR (600 MHz, DMSO-d6): 2.22 (6H, s); 2.52–2.58 (2H, m); 3.65 (2H, t, J = 6.05 Hz); 4.37 (2H, s); 7.34 (1H, d, J = 7.44 Hz); 7.60 (1H, t, J = 8.01 Hz); 7.75 (1H, d, J = 8.05 Hz); 12.68 (1H, brs); 13C NMR (150 MHz, DMSO-d6): 40.3; 45.4; 47.6; 57.2; 119.4; 122.7; 126.6; 132.3; 132.7; 140.2; 143.7; 163.7; 173.1 (Fig. S52, S53 and S91†).
4.1.4.4 6-Chloro-2-(2-(dimethylamino)ethyl)-1H-pyrrolo[3,4-b]quinoline-3,9(2H,4H)-dione (14c). Yield: 5 mg, 23%. Mp.: decomposition over 295 °C. HRMS calcd for [M + H+] m/z = 306.1004, found m/z = 306.0999; 1H NMR (600 MHz, DMSO-d6): 2.20 (6H, s); 2.52–2.56 (2H, m); 3.65 (2H, t, J = 5.86 Hz); 4.41 (2H, s); 7.41 (1H, d, J = 8.91 Hz); 7.82 (1H, s); 8.18 (1H, d, J = 8.61 Hz); 12.74 (1H, brs); 13C NMR (150 MHz, DMSO-d6): 40.3; 45.4; 47.5; 57.3; 119.0; 121.4; 124.4; 125.4; 127.8; 137.2; 141.7; 163.7; 173.0 (Fig. S54–56 and S92†).
4.1.4.5 2-(2-(Dimethylamino)ethyl)-5-methyl-1H-pyrrolo[3,4-b]quinoline-3,9(2H,4H)-dione (14d). Yield: 9 mg, 47%. Mp.: decomposition over 230 °C. HRMS calcd for [M + H+] m/z = 286.1550, found m/z = 286.1545; 1H NMR (500 MHz, MeOH-d4): 2.39 (6H, s); 2.68 (3H, s); 2.76 (2H, t, J = 5.98 Hz); 3.85 (2H, t, J = 6.79 Hz); 4.51 (2H, s); 7.39 (1H, t, J = 7.74 Hz); 7.65 (1H, d, J = 7.74 Hz); 8.23 (1H, d, J = 8.45 Hz); 13C NMR (125 MHz, MeOH-d4): 40.2; 44.1; 46.9; 56.4; 120.7; 123.0; 123.9; 133.6; 164.6; 175.0 (Fig. S57, S58 and S93†).
Method B: N-(2-(dimethylamino)ethyl)-4-hydroxy-3-(morpholinomethyl)quinoline-2-carboxamide (2) or its derivatives (8a–f and 13a–d) (2.5 mg, 0.070 mmol), triethylamine (72.6 mg, 0.717 mmol) were put in a pressure-resistant 10 mL vessel. The reaction was heated in microwave reactor at 150 °C in MeOH (1.3 mL) for 2 h. The formed precipitate was filtered and washed with 2 × 2 mL MeOH.
4.1.5.1 3,3′-Methylenebis(4-oxo-N-phenyl-1,4-dihydroquinoline-2-carboxamide) (10a). Yield: 9 mg, 48%. Mp.: decomposition over 300 °C. HRMS calcd for [M + H+] m/z = 541.1870, found m/z = 541.1863; 1H NMR (500 MHz, DMSO-d6): 4.06 (2H, s); 7.17 (2H, t, J = 7.38 Hz); 7.40 (6H, m); 7.75 (2H, t, J = 7.64 Hz); 7.82 (6H, d, J = 8.18 Hz); 8.16 (2H, d, J = 8.18 Hz); 12.70 (2H, s); 13.38 (2H, s); 13C NMR (125 MHz, DMSO-d6): 26.7; 114.7; 120.1; 124.0; 124.7; 125.0; 125.2; 129.6; 133.1; 138.7; 145.2; 145.8; 146.4; 161.9; 178.4 (Fig. S59, S60 and S94†).
4.1.5.2 3,3′-Methylenebis(N-benzyl-4-oxo-1,4-dihydroquinoline-2-carboxamide) (10b). Yield: 10 mg, 49%. Mp.: decomposition over 300 °C. HRMS calcd for [M + H+] m/z = 569.2183, found m/z = 569.2178; 1H NMR (600 MHz, DMSO-d6): 3.94 (2H, s); 4.66 (4H, d, J = 5.90 Hz); 7.26 (2H, t, J = 7.13 Hz); 7.31 (2H, t, J = 7.56 Hz); 7.34 (4H, t, J = 7.56 Hz); 7.43 (4H, d, J = 7.56 Hz); 7.68 (2H, t, J = 7.56 Hz); 7.78 (2H, d, J = 8.64 Hz); 7.96 (2H; d, J = 8.10 Hz); 11.63 (2H, t, J = 5.89 Hz); 12.46 (2H, s); 13C NMR (150 MHz, DMSO-d6): 26.3; 43.2; 115.0; 119.0; 123.9; 124.3; 125.1; 127.6; 128.0; 128.9; 132.8; 138.8; 139.4; 145.0; 163.8; 178.2 (Fig. S61, S62 and S95†).
4.1.5.3 3,3′-Methylenebis(4-oxo-N-propyl-1,4-dihydroquinoline-2-carboxamide) (10d). Yield: 7 mg, 38%. Mp.: decomposition over 300 °C. HRMS calcd for [M + H+] m/z = 473.2183, found m/z = 473.2178; 1H NMR (600 MHz, DMSO-d6): 0.94 (6H, t, J = 6.83 Hz); 1.57–1.63 (4H, m); 3.39 (4H, t, J = 6.07 Hz); 3.94 (2H, s); 7.32 (2H, t, J = 7.83 Hz); 7.68 (2H, t, J = 7.83 Hz); 7.77 (2H, d, J = 7.83 Hz); 8.01 (2H, d, J = 7.83 Hz); 11.13 (2H, s); 12.40 (2H, s); 13C NMR (150 MHz, DMSO-d6): 12.1; 22.7; 29.5; 41.4; 114.6; 119.0; 123.9; 124.2; 125.1; 130.1; 132.8; 139.4; 145.4; 163.6; 178.2 (Fig. S63, S64 and S96†).
4.1.5.4 3,3′-Methylenebis(5-chloro-N-(2-(dimethylamino)ethyl)-4-oxo-1,4-dihydroquinoline-2-carboxamide) (15a). Yield: 4 mg, 19%. Mp.: decomposition over 280 °C. HRMS calcd for [M + H+] m/z = 599.1935, found m/z = 599.1930; 1H NMR (600 MHz, DMSO-d6): 2.10 (12H, s); 2.42–2.50 (4H, m); 3.48 (4H, q, J = 5.70 Hz); 3.97 (2H, s); 7.28 (2H, d, J = 7.64 Hz); 7.56 (2H, t, J = 7.64 Hz); 7.71 (2H, d, J = 8.61 Hz); 10.66 (2H, brs); 13C NMR (150 MHz, DMSO-d6): 26.2; 37.7; 45.3; 57.9; 117.2; 118.6; 120.0; 126.7; 132.1; 132.3; 142.1; 144.1; 163.4; 177.8 (Fig. S65, S66 and S97†).
4.1.5.5 3,3′-Methylenebis(7-chloro-N-(2-(dimethylamino)ethyl)-4-oxo-1,4-dihydroquinoline-2-carboxamide) (15c). Yield: 3 mg, 14%. Mp.: decomposition over 300 °C. HRMS calcd for [M + H+] m/z = 599.1935, found m/z = 599.1933; 1H NMR (600 MHz, DMSO-d6): 2.11 (12H, s); 2.42–2.50 (4H, m); 3.48–3.52 (4H, m); 4.07 (2H, s); 7.33 (2H, d, J = 8.86 Hz); 7.78 (2H, s); 8.01 (2H, d, J = 8.83 Hz); 10.84 (2H, brs); 13C NMR (150 MHz, DMSO-d6): 25.9; 37.7; 45.3; 58.0; 116.1; 118.2; 122.5; 124.6; 127.5; 145.6; 177.8 (Fig. S67–70 and S98†).
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization, I. S., B. L. ; investigation, A. H., R. B., S. J. R.; writing – original draft preparation, B. L., S. J. R.; writing – review and editing, I. S., B. L. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Hungarian Research Foundation (OTKA No. K-138871), the Ministry of Human Capacities, Hungary grant, TKP-2021-EGA-32. L. B. was supported by the ÚNKP-23-4 - SZTE-216 New National Excellence Program of the Ministry for Culture and Innovation from the source of the National Research, Development and Innovation Fund. Furthermore, the scientific work/research and results publicised in this article was reached with the sponsorship of Gedeon Richter Talentum Foundation in framework of Gedeon Richter Excellence PhD Scholarship of Gedeon Richter.Notes and references
- É. Rózsa, H. Robotka, L. Vécsei and J. Toldi, J. Neural. Transm., 2008, 115, 1087–1091 CrossRef.
- T. W. Stone, Expet Opin. Invest. Drugs, 2001, 10, 633–645 CrossRef CAS PubMed.
- K. Sas, H. Robotka, J. Toldi and L. Vécsei, J. Neurol. Sci., 2007, 257, 221–239 CrossRef CAS PubMed.
- G. Gigler, G. Szénási, A. Simó, G. Lévay, L. G. Hársing Jr, K. Sas, L. Vécsei and J. Toldi, Eur. J. Pharmacol., 2007, 564, 116–122 CrossRef CAS.
- E. Luchowska, P. Luchowski, A. Sarnowska, M. Wielosz, W. A. Turski and E. M. Urbańska, Pol. J. Pharmacol., 2003, 55, 443–447 CAS.
- B. L. Harrison, B. M. Baron, D. M. Cousino and I. A. McDonald, J. Med. Chem., 1990, 12, 3130–3132 CrossRef.
- D. Edmont, R. Rocher, C. Plisson and J. Chenault, Bioorg. Med. Chem. Lett., 2000, 16, 1831–1834 Search PubMed.
- F. P. Bonina, L. Arenare, R. Ippolito, G. Boatto, G. Battaglia, V. Bruno and P. de Caprariis, Int. J. Pharm., 2000, 202, 79–88 Search PubMed.
- S. Manfredini, B. Pavan, S. Vertuani, M. Scaglianti, D. Compagnone, C. Biondi, A. Scatturin, S. Tanganelli, L. Ferraro, P. Prasad and A. Dalpiaz, J. Med. Chem., 2002, 45, 559–562 CrossRef CAS PubMed.
- S. Manfredini, S. Vertuani, B. Pavan, F. Vitali, M. Scaglianti, F. Bortolotti, C. Biondi, A. Scatturin, P. Prasad and A. Dalpiaz, Bioorg. Med. Chem., 2004, 12, 5453–5463 CrossRef CAS.
- A. C. Nichols and K. L. Yielding, Mol. Chem. Neuropathol., 1998, 35, 1–12 CrossRef CAS.
- A. Brik, Y. C. Lin, J. Elder and C. H. Wong, Chem. Biol., 2002, 9, 891–896 CrossRef CAS.
- E. Knyihár-Csillik, A. Mihály, B. Krisztin-Péva, H. Robotka, I. Szatmári, F. Fülöp, J. Toldi, B. Csillik and L. Vécsei, Neurosci. Res., 2008, 61, 429–432 Search PubMed.
- F. Fülöp, I. Szatmári, E. Vámos, D. Zádori, J. Toldi and L. Vécsei, Curr. Med. Chem., 2009, 16, 4828–4842 CrossRef.
- D. Zádori, G. Nyíri, A. Szőnyi, I. Szatmári, F. Fülöp, J. Toldi, T. F. Freund, L. Vécsei and P. Klivényi, J. Neural. Transm., 2011, 118, 865–875 Search PubMed.
- D. Zádori, G. Veres, L. Szalárdy, P. Klivényi, J. Toldi and L. Vécsei, J. Alzheim. Dis., 2014, 42, S177–S187 Search PubMed.
- R. Greco, C. Demartini, A. M. Zanaboni, E. Redavide, S. Pampalone, J. Toldi, F. Fülöp, F. Blandini, G. Nappi, G. Sandrini, L. Vécsei and C. Tassorelli, Cephalalgia, 2017, 37, 1272–1284 CrossRef PubMed.
- A. Fejes-Szabó, Z. Bohár, E. Vámos, G. Nagy-Grócz, L. Tar, G. Veres, D. Zádori, M. Szentirmai, J. Tajti, I. Szatmári, F. Fülöp, J. Toldi, A. Paardutz and L. Vecsei, J. Neural. Transm., 2014, 121, 725–738 CrossRef PubMed.
- B. Lőrinczi, A. Csámpai, F. Fülöp and I. Szatmári, Molecules, 2020, 25, 937 CrossRef PubMed.
- B. Lőrinczi, A. Csámpai, F. Fülöp and I. Szatmári, RSC Adv., 2021, 11, 543–554 RSC.
- P. Barta, F. Fülöp and I. Szatmári, Beilstein J. Org. Chem., 2018, 14, 560–575 CrossRef CAS.
- C. Tratrat, S. Giorgi-Renault and H. Henri-Philippe, Synlett, 1998, 10, 1071–1072 CrossRef.
- R. Cincinelli, G. Beretta and S. Dallavalle, Tetrahedron Lett., 2018, 59, 163–166 CrossRef CAS.
- M. Anzini, A. Cappelli, S. Vomero, M. Seeber, M. C. Menziani, T. Langer, B. Hagen, C. Manzoni and J. J. Bourguignon, J. Med. Chem., 2001, 8, 1134 CrossRef.
- Y. Q. Liu, W. Q. Li, S. L. Morris-Natschke, K. Qian, L. Yang, G. X. Zhu, X. B. Wu, A. L. Chen, S. Y. Zhang, X. Nan and K. H. Lee, Med. Res. Rev., 2015, 4, 753 CrossRef.
- A. A. Boteva, I. V. Fefilova, G. A. Triandafilova, V. V. Maslova, S. Y. Solodnikov and O. P. Krasnykh, Pharm. Chem. J., 2019, 53, 616–619 CrossRef CAS.
- B. Lőrinczi, P. Simon and I. Szatmári, Int. J. Mol. Sci., 2022, 13, 7152 CrossRef PubMed.
- J. H. Burckhalter, V. C. Stephens, H. C. Scarborough Jr, W. S. Brinigar and W. H. Edgerton, J. Am. Chem. Soc., 1954, 19, 4902–4906 CrossRef.
- M. Movrin, D. Maysinger and E. Marok, Pharmazie, 1980, 8, 458–460 Search PubMed.
- P. Funk, K. Motyka, M. Soural, M. Malon, H. Koshino, J. Kusz and J. Hlavac, PLoS One, 2017, 12, e0175364 CrossRef PubMed.
- E. Fehér, I. Szatmári, T. Dudás, A. Zalatnai, T. Farkas, B. Lőrinczi, F. Fülöp, L. Vécsei and J. Toldi, Molecules, 2019, 19, 3502 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04341c |
| This journal is © The Royal Society of Chemistry 2024 |