
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUpgrading furfural to bio-fuels using supported molybdenum carbides: study of the support effect†
Leticia F. Sosa a,
Priscilla M. de Souzab,
Raphaela A. Rafaelb,
Eric Marceaub,
Valérie Brioisc,
Fabio S. Tonioloa,
Fabio B. Noronhabd,
Franck Dumeignilb and
Sébastien Paul*b
a,
Priscilla M. de Souzab,
Raphaela A. Rafaelb,
Eric Marceaub,
Valérie Brioisc,
Fabio S. Tonioloa,
Fabio B. Noronhabd,
Franck Dumeignilb and
Sébastien Paul*b
aChemical Engineering Program of COPPE/UFRJ, Federal University of Rio Janeiro, P. O. Box 68502, Rio de Janeiro, CEP 21941-972, Brazil
bUniv. Lille, CNRS, Centrale Lille, Univ. Artois, UMR 8181 – UCCS – Unité de Catalyse et Chimie du Solide, F-59000 Lille, France. E-mail: sebastien.paul@centralelille.fr
cSynchrotron SOLEIL, L'Orme des Merisiers, Saint-Aubin, BP 48, 91192 Gif-sur-Yvette Cedex, France
dNational Institute of Technology, Catalysis, Biocatalysis and Chemical Processes Division, Av. Venezuela 82, Rio de Janeiro, 20081-312, RJ, Brazil
First published on 27th August 2024
Abstract
Materials exhibiting different textural and surface properties (SiO2, TiO2, ZrO2 and ZSM-5) were investigated as supports for Mo carbides in the upgrading of furfural (FF) in liquid phase to produce 2-methylfuran (2MF). The state of the catalysts after carburization, passivation, and reactivation under a hydrogen atmosphere was investigated by XAS analysis. The effect of the supports was observed in the first step of the reaction, i.e., the hydrogenation of FF to furfuryl acid and related to Lewis acidic and basic sites. The nature of the supports was also relevant to the final state of the Mo carbides after carburization, passivation, and reactivation. The comparison of the materials showed that Mo2C/SiO2 was the least decarburized catalyst after reactivation, and the most active in converting furfural, while the Mo2C/TiO2 system presented smaller carbide particles after carburization and more disorganized particles after reactivation. Mo carbide supported on SiO2 and on TiO2 were found to be suitable catalysts for producing a mixture containing 2-methylfuran and C10 compounds with potential application as biofuel.
1 Introduction
Lignocellulosic biomass is an inexpensive and abundant non-edible biomass suitable for the production of biofuels and biochemicals.1,2 Processing this feedstock by hydrolysis generates platform molecules such as furfural (FF), which can be used to produce several compounds of industrial interest such as furfuryl alcohol, tetrahydrofurfuryl alcohol (THFA), 2-methylfuran (2MF), 2-methyltetrahydrofuran. Among them, 2MF stands out due to its application as a biofuel and fuel additive.3–5 2-Methylfuran is produced from FF via furfuryl alcohol (FA) as an intermediate. The aldehyde function in furfural is firstly hydrogenated forming FA, which then undergoes hydrodeoxygenation (HDO) yielding 2MF.3,4 It has been reported that two types of active sites, metallic and Lewis acidic sites, are needed to promote this reaction.6–8 Lewis acidic sites attract and weaken the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in FF, which is hydrogenated by hydrogen species provided by the metallic sites.
O bond in FF, which is hydrogenated by hydrogen species provided by the metallic sites.
In the literature, both functionalities are found in catalysts containing a mixture of a reduced metal (M) and its respective metal oxide (MOx).9–12 These species are metallic sites, and Lewis acidic sites found as coordinatively unsaturated cations in metal oxides, respectively.13 To improve the activity and selectivity to 2MF it is necessary then to achieve an ideal M/MOx ratio. The M/MOx ratio is controlled by varying parameters related to the synthesis of the materials, such as the preparation method, calcination temperature, and reduction conditions.
Gong et al.8 tested Cu supported on activated carbon (AC) catalysts for the HDO of FF using 2-propanol at 170 °C and 30 bar of hydrogen for 4 hours. According to the authors, a full FF conversion and selectivity to 2MF were obtained due to the presence of different Cu species in the Cu/AC catalyst calcined at 400 °C for 2 hours. Electron-deficient copper oxide species (Cu+ and Cu2+) acted as Lewis acidic sites attracting and weakening the CO bond in FF, which was hydrogenated by activated hydrogen species formed on Cu0 centers. Finally, the C–O bond in furfuryl alcohol was dissociated on Cu+ sites forming 2MF. Notwithstanding, the recyclability tests of the Cu/AC catalyst showed a reduction in its catalytic performance, which was attributed to the reduction of CuOx to the metallic state Cu0. The drawback of using catalysts with this design is that they are easily susceptible to structural changes, either through metal oxidation or oxide reduction, which causes the catalyst to lose functionality or ideal M/MOx ratio. To overcome this, Lewis acidic oxides such as Al2O3, TiO2, ZrO2 are preferable, as they are less prone to structural changes and can be use as supports for species containing metallic properties.
The metals most used to upgrade furfural are Ni, Cu, Co, Pd, Pt, Ru11,14–18 and alternative materials such as carbides, nitrides, and phosphides.19–21 Among these, transition metal carbides, especially Mo carbide, are very promising since they are highly selective for breaking CO bonds, favoring the formation of deoxygenated products.22 In addition, Mo carbide exhibits activity comparable to noble metals.23,24 In this sense, molybdenum carbides would be a promising candidate for the upgrading of furfural to 2-methylfuran.
To the best of our knowledge, up to now, the influence of the support of the catalyst on the product distribution in the HDO of furfural is still not clear. In the work of Liu et al.,7 the production of 2MF was directly related to the amount of Lewis acidic sites in the supports (SiO2, TiO2, Al2O3 and CeO2) for Co-based catalysts exhibiting similar metal dispersion. The Co/Al2O3 catalyst was the most active and selective to 2MF (93%) at 180 °C and 10 bar of hydrogen. On the other hand, the formation of 2MF was not influenced by the nature of the support for ReOx catalysts supported on SiO2, Al2O3, and SiO2–Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25 or bimetallic catalysts containing Cu and Pd supported on SiO2, TiO2, ZrO2 and Al2O3.26
25 or bimetallic catalysts containing Cu and Pd supported on SiO2, TiO2, ZrO2 and Al2O3.26
Nevertheless, it is well known that the physicochemical properties of a material used as support for catalysts can play an important role in both substrate conversion and product distribution depending on the reaction. The nature of the supports can significantly influence the dispersion of the active phases, thereby modifying the adsorption/desorption of the reactants and products and, consequently, the catalytic performance. In addition, the acidic properties can influence the type of products formed.
Monometallic and bimetallic Cu–Ni catalysts supported on Al2O3 and TiO2 were studied by Seemala et al.27 for the hydrodeoxygenation of FF. When supported on Al2O3, Ni showed minimal interaction with Cu, while on TiO2 these metals were found as alloys. These configurations led the higher dispersed Ni on Cu–Ni/TiO2 catalyst to be unable to adsorb the furan ring of furfural preferentially producing 2MF, while the opposite occurred over the Cu–Ni/Al2O3 catalyst, which produced ring hydrogenation products, such as THFA. Finally, the difference in the activities of the monometallic Cu catalysts was attributed to the greater dispersion of this metal in alumina compared to titania, while the acidity of the supports did not seem to influence the reactivity of the catalyst. A similar effect was observed for bimetallic Cu–Co catalysts supported on SiO2, Al2O3, and ZSM-5 tested for the HDO of furfural.28 The performance of the catalysts was mainly associated with different metal dispersion over the supports and not related to their acidic nature.
The nature of the support is more relevant when the reaction is performed in the absence of external hydrogen. Higher yields of 2MF were observed over Cu–Pd catalysts supported on ZrO2 and TiO2 in comparison with SiO2 and Al2O3, which was associated to the acid–base sites of the former materials, that assisted the adsorption and dissociation of the alcohols to produce hydrogen in catalytic transfer hydrogenation (CTH) reactions.26
Aiming to get more insight about the support effect, this work seeks to study SiO2, TiO2, ZrO2 and ZSM-5 as supports for Mo carbides in the upgrading of furfural to biofuels, especially 2-methylfuran. For this, the acidic properties of the materials, such as the total amount and nature of the acidic sites was considered. Silica has scarcely acidic properties, while TiO2 and ZrO2 exhibit Lewis acidic sites and ZSM-5 possesses both Lewis and Brønsted acidic sites. In addition, the effect of the support's nature for the synthesis, passivation and reactivation of Mo carbides was investigated by in situ X-ray absorption spectroscopy (XAS) analyses.
2 Experimental
2.1 Catalyst preparation
Before preparation of the catalysts, the supports, i.e., silica (SiO2, Aerosil 200, Evonik Industries), titania (TiO2, Aeroxide P25, Degussa Brasil LTDA), monoclinic zirconia (ZrO2, Saint-Gobain) and an ammonium zeolite (ZSM-5, FCC S.A.) were heat treated. Titania and zirconia were dried under static air at 110 °C for 12 h to eliminate water. Due to the low density of silica, this material was moistened with deionized water, dried for 3 h at 120 °C, and heated at 500 °C (10 °C min−1) for 6 h. The zeolite ZSM-5 was first heated to 110 °C (10 °C min−1) for 30 min and then at 500 °C (10 °C min−1) for 3 h to obtain its protonic form. The precursors of supported Mo carbides were prepared by incipient wetness impregnation using an appropriate amount of aqueous solution of ammonium heptamolybdate ((NH4)6Mo7O24, Sigma-Aldrich, 0.173 g mL−1) to achieve 20% (w/w) Mo2C. After impregnation, the samples were calcined at 500 °C (5 °C min−1) for 3 h under static air to decompose the molybdenum salt. The carbides were then synthesized by temperature-programmed carburization of the calcined precursors (1 g) at 650 °C for 2 h using a heating ramp of 2.5 °C min−1 under 20% (v/v) CH4/H2 (200 mL min−1 STP).29 Due to the pyrophoric nature of the carbides, the samples were passivated at room temperature using a mixture of 0.5% (v/v) O2/N2 (30 mL min−1 STP) for 14 h before being exposed to the atmosphere. The catalysts were designated as Mo2C/S where S informs on the support used (S = SiO2, TiO2, ZrO2, or ZSM-5).2.2 Catalyst characterization
X-ray diffraction (XRD) analysis was performed using a Bruker AXS D8 diffractometer employing a Cu Kα radiation (λ = 0.1538 nm). The diffractograms were obtained over a 2θ range of 5–80° at a scan rate of 0.02° per step and a scan time of 1 s per step. The identification of the phases was carried out by comparison with the JCPDS standard spectra software. When possible, the crystallite diameters (Dc) of Mo carbides were estimated by the Scherrer equation using the line corresponding to the (110) plane at 2θ = 61.5° for β-Mo2C, considering that the particles have a spherical geometry (shape factor equal to 0.9).The amounts of Mo in the carbides were determined by inductively coupled plasma optical emission spectrometry using a 720-ES ICP-OES spectrometer (Agilent) with axial viewing and simultaneous CCD detection. Meanwhile, elemental analysis (EA) was performed to estimate the C content in the catalysts using a Thermo Scientific FlashSmart automated analyzer.
Textural properties were measured by nitrogen adsorption at −196 °C using a Micromeritics TriStar II Plus analyzer and a Micromeritics Accelerated Surface Area and Porosity (ASAP) depending on the porous type in the materials. The samples were previously outgassed under vacuum firstly at 75 °C for 1 h and then at 300 °C for up to 24 h. Specific surface areas were estimated using the Brunauer–Emmett–Teller (BET) method and the total pore volume was measured using t-plot or Barrett–Joyner–Halenda (BJH) methods whether the sample was micro or mesoporous, respectively.
The synthesis of carbides was followed by temperature-programmed carburization (TPC) in a multipurpose unit coupled to a Pfeiffer Vacuum mass spectrometer (MS) model QME 200. Before analysis, the calcined precursors (0.1 g) were treated under He (50 mL min−1 STP) at 200 °C (10 °C min−1) for 1 h to eliminate water and then cooled down to 30 °C. Then, He was replaced by 20% (v/v) CH4/H2 (100 mL min−1 STP) and then, the temperature was increased up to 800 °C (2.5 °C min−1). The signals of the ions m/z = 18 (H2O), m/z = 15 (CH4) and m/z = 28 (CO) were continuously monitored on the mass spectrometer.
X-ray absorption spectroscopy (XAS) was carried out in the transmission mode at the ROCK Quick-EXAFS beamline at the French synchrotron radiation facility SOLEIL.30 The beamline benefits from a 2.81 Tesla Super-Bend source which delivers nearly 1012 ph per s between 8 to 20 keV. Spectra were acquired in in situ conditions at the Mo K-edge (20000 eV) during carburization under 20% (v/v) CH4/H2. The monochromator used is based on a Si (111) channel-cut installed on a tilt table allowed to oscillate around the Bragg angle characteristic of the element of interest, i.e., 5.6550° for Mo, with an amplitude of 0.5°. The Si (111) channel-cut oscillation frequency was set to 2 Hz and recorded two quick-EXAFS spectra every 0.5 s. Every 10 acquired spectra were merged in order to improve the signal/noise ratio. Ionization chambers were filled with a mixture 50:50 of nitrogen and argon for the Mo K edge measurements. The beam size at the sample position was ∼500 μm (H) × 300 μm (V).
Experiments were performed using a dedicated gas-feeding set-up installed on the ROCK beamline.31 A quartz capillary (1.5 mm × 80 mm × 0.04 mm) was used as sample holder. The powder catalyst bed (length ∼8–9 mm) was maintained at the center of the capillary between two pieces of quartz wool and heated using a gas blower. After recording XAS spectra at room temperature, a carburization of the calcined precursors of the Mo2C/SiO2 and Mo2C/TiO2 catalysts was performed by heating the cell from room temperature to 650 °C (2.5 °C min−1) under a 20% (v/v) CH4/H2 flow (5 mL min−1). Spectra of the carburized catalysts were recorded back at room temperature. In a separate set of measurements, reactivation was performed on catalysts that had been previously passivated (Mo2C, Mo2C/SiO2, Mo2C/TiO2 and Mo2C/ZSM-5). Mo2C/ZrO2 was not tested because of its strong X-ray absorption. Samples were heated up to 400 °C (5 °C min−1) under a H2 flow (5 mL min−1), and spectra of the reactivated catalysts were recorded at room temperature.
Energy calibration with respect to the reference metal foil (Mo) and a XAS data-normalization procedure were first carried out using the Python normal_gui graphical interface developed at SOLEIL for the fast handling of Quick-XAS data.32 The EXAFS signal extraction and Fourier transform of the EXAFS spectra were done using the Athena graphical interface software.33 EXAFS fitting of coordination numbers N, Debye–Waller factors σ2 and interatomic distances R was simultaneously performed on k-, k2- and k3-weighted χ(k) functions with the Artemis interface to IFeFFIT using least-squares refinements.34 Fits were first performed on the metallic foil reference for the determination of the S02 factor. Fourier-transformed EXAFS signals are presented as k3-χ(k) functions and Fourier transforms shown without phase correction. In parallel with the XAS analyses, Raman spectra were measured using a commercial RXN1 Raman spectrometer (Kaiser Optical Systems, Inc.).
The total acidity of the supports and catalysts was estimated by temperature-programmed desorption of NH3 (TPD-NH3). Before analysis, the supports (100 mg) were treated at 200 °C (10 °C min−1) for 1 h in He (50 mL min−1 STP) and the catalysts at 450 °C (10 °C min−1) for 1 h in H2 (50 mL min−1 STP). After treatment, all materials were cooled down to 100 °C in He (50 mL min−1 STP) and purged for 15 min. The NH3 adsorption was performed at 100 °C for 30 min using 10% (v/v) NH3/He (30 mL min−1 STP) and then the material surface was purged in He (50 mL min−1 STP) for 2 h. The NH3 desorption was performed up to 500 °C (10 °C min−1) in He (30 mL min−1 STP) and kept isothermal until the NH3 signal returned to the baseline (around 2 h). The experiments were conducted in an AutoChem II equipment from Micromeritics equipped with a thermal conductivity detector and a mass spectrometer, in which the signals of NH3 (m/z = 17, 16, and 15) and H2O (m/z = 18, 17, and 16) ions were monitored.
2.3 Catalytic experiments
The hydrodeoxygenation reaction of furfural in the liquid phase was performed in a 30 mL batch Hastelloy Parr autoclave equipped with a thermocouple and mechanical stirring. Before the reaction, the passivated Mo carbides were formerly reactivated at 400 °C (5 °C min−1) with pure H2 (50 mL min−1 STP) for 1 h under 30 bar H2 to eliminate the passivation layer on the catalyst surface in a Screening Pressure Reactor (SPR) system from Unchained Labs equipped with 24 parallel stainless-steel batch reactors of 6 mL each (REALCAT platform). Then, the hermetically sealed reactor vessel was transferred to a glovebox (MBRAUN), in which all catalysts were stored to avoid reoxidation.Inside the glovebox, the Parr autoclave was charged with the desired amount of catalyst and the reaction mixture (15 mL, 0.25 mol L−1 of furfural in 2BuOH) without exposure to air. Then the system was purged 3 times and 30 bar of H2 was added. The reaction mixture was heated at 5 °C min−1 to 200 °C (45 bar total pressure) and then the stirring was started (600 rpm); at this moment the time was set to zero. After 4 h of reaction, the stirring was stopped and the autoclave was cooled in an ice bath until reaching a temperature lower than 30 °C. Around 2 mL was taken from the liquid mixture and 1 mL aliquots were analyzed after filtration.
Reaction products were analyzed using a gas chromatograph (Shimadzu GC-2010 Plus) equipped with a ZB-5MS column (30 m × 0.25 mm × 0.25 μm) and a flame ionization detector (FID). The compounds were identified by gas chromatography coupled to a mass spectrometry GC-MS (Agilent Technologies, 7890B GC System) using CP-Wax 52 CB column (30 m × 0.25 mm × 0.25 μm).
The furfural conversion (XFF), product yield (Y), and carbon balance (CB) were calculated using the following eqn (1)–(3):
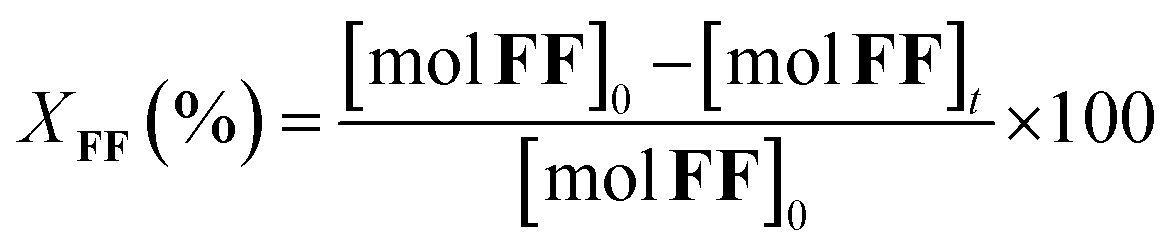 | (1) |
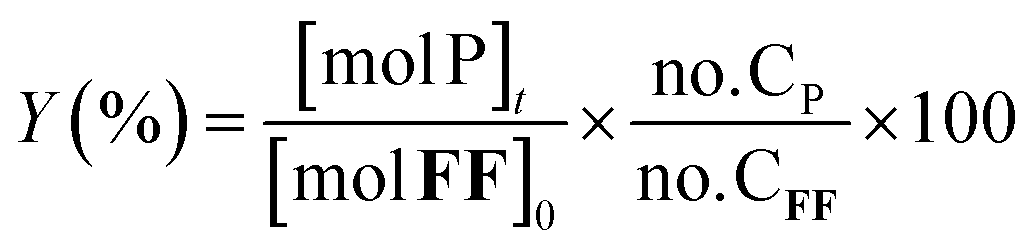 | (2) |
 | (3) |
3 Results and discussion
3.1 Catalyst characterization
The diffractograms of the supports and passivated Mo carbide catalysts are displayed in Fig. 1. The broad peak at 2θ = 21.9° corresponds to the classical pattern of amorphous SiO2 (JCPDS 39-1425). For titania P25, two crystalline phases could be identified: anatase (a-TiO2, JCPDS 21-1272) and rutile (r-TiO2, JCPDS 21-1276). The diffractogram of zirconia exhibits the lines characteristic of the monoclinic phase (m-ZrO2, JCPDS 37-1484) and the diffraction pattern that matches the ZSM-5 diffractogram is JCPDS 44-0002. The molybdenum species formed in all catalysts after calcination corresponds to the MoO3 phase (JCPDS 05-0508) (Fig. S1†). The high-intensity diffraction lines related to the MoO3 phase in the Mo2C/ZSM-5 catalyst's diffractogram indicate that by the size of the particles, they are mainly deposited in the outer surface of the zeolite.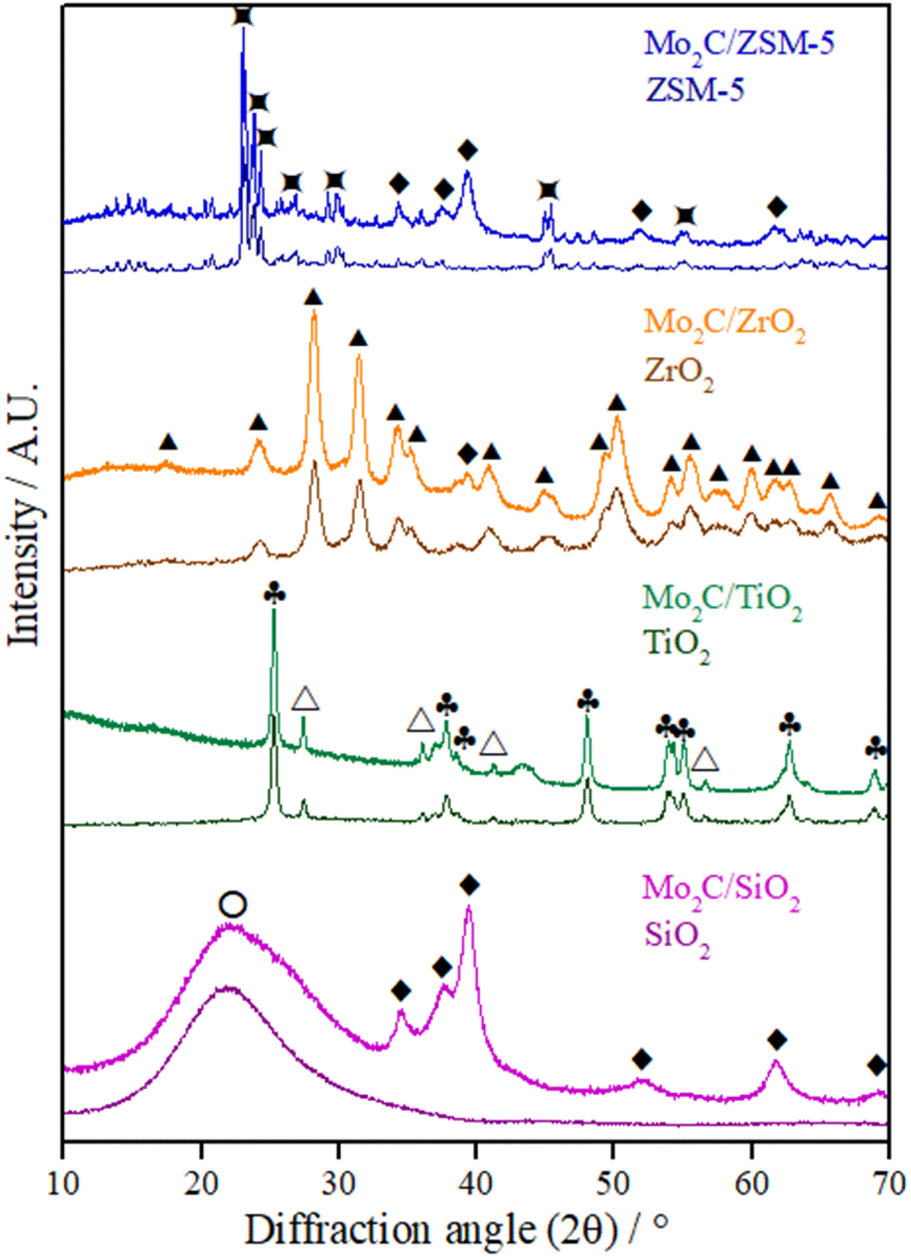 | ||
Fig. 1 Diffractograms of supports and passivated Mo carbides supported on different materials. ♦ β-Mo2C,  ZSM-5, ▲ m-ZrO2, ♣ a-TiO2, △ r-TiO2, ○ SiO2. ZSM-5, ▲ m-ZrO2, ♣ a-TiO2, △ r-TiO2, ○ SiO2. | ||
All supports appeared to be unchanged after the carburization and passivation stages since their crystalline structure was preserved and no new phase resulting from a reaction between the support and Mo oxide or carbide was identified.35
In the diffractograms of the passivated samples (Fig. 1), the diffraction lines corresponding to the hexagonal β-Mo2C phase (JCPDS 35-0787) at 2θ = 34.4, 37.8, 39.4, 61.5, and 74.6° could be easily identified for the carbides supported on SiO2 and ZSM-5. The mean crystallite diameter (Dc) of the Mo2C/SiO2 and Mo2C/ZSM-5 catalysts was estimated by the Scherrer equation to be 5 nm (Table 1).
| Material | Content (wt%) | SSAa (m2 g−1) | Pvb (cm3 g−1) | Dc (nm) | ||
|---|---|---|---|---|---|---|
| Mo | C | Mo2C | ||||
| a Determined by BET method.b Determined by BJH method.c Determined by t-plot method. | ||||||
| SiO2 | — | — | — | 198 | 1.13 | — |
| TiO2 | — | — | — | 41 | 0.15 | — |
| ZrO2 | — | — | — | 98 | 0.29 | — |
| ZSM-5 | — | — | — | 515 | 0.32 | — |
| Mo2C/SiO2 | 17.6 | 0.8 | 18.4 | 140 | 0.53 | 5 |
| Mo2C/TiO2 | 18.4 | 1.1 | 19.5 | 44 | 0.17 | — |
| Mo2C/ZrO2 | 17.3 | 1.4 | 18.7 | 70 | 0.18 | — |
| Mo2C/ZSM-5 | 17.1 | 1.3 | 18.4 | 318 | 0.04c | 5 |
Meanwhile, only the most intense characteristic line of the β-Mo2C phase (2θ = 39.4°) could be detected on ZrO2.36 For the carbide supported on TiO2, it is not possible to identify any carbide phase, as previously reported by Boullosa-Eiras et al.37 for the same catalyst with a carbide content of 15 wt%, which was associated with the high dispersion of the Mo carbide phase or the passivation treatment. Only a non-identified broad peak at 2θ = 43.4° could be seen.
The lack of diffraction lines corresponding to Mo carbide in the diffractograms of Mo2C/TiO2 and Mo2C/ZrO2 catalysts might be an indication of a better Mo2C dispersion on these supports or a deeper passivation with the formation of poorly crystalline species.
The Mo and C contents in the passivated catalysts estimated by ICP-OES and elemental analysis are reported in Table 1. The theoretical value of the C/Mo molar ratio is 0.5 according to the chemical formula of the Mo carbide (Mo2C). The values obtained for Mo2C/SiO2 (0.41) and Mo2C/TiO2 (0.52) catalysts were close to the expected value, while an excess of carbon was observed for both Mo2C/ZrO2 (0.60) and Mo2C/ZSM-5 (0.61) catalysts.
The textural properties of the supports and passivated catalysts determined by N2 physisorption are shown in Table 1. Except for the Mo2C/TiO2 catalyst, which did not change, a decrease in the specific surface area (SSA) and pore volume (Pv) was observed for all materials after the synthesis of carbides in comparison with the bare supports, which is consistent with the formation of 20 wt% Mo2C, a nonporous material with low surface area. This effect was more pronounced for the Mo2C/ZSM-5 catalyst since almost no pore volume is observed after the addition of the Mo carbide, which might be clogging the pores of the zeolite.
All catalysts showed similar N2 adsorption–desorption isotherms to their respective supports (Fig. S2†). According to the IUPAC classification, the adsorption curves on SiO2, TiO2, and ZrO2 supports and supported catalysts exhibited a type IV isotherm with different hysteresis loops: Mo2C/SiO2 and Mo2C/TiO2: H3 hysteresis loop, which is associated to the capillary condensation in meso and micropores; Mo2C/TiO2: H4 hysteresis loop, which is found on aggregated crystals of mesoporous materials. Finally, ZSM-5 and Mo2C/ZSM-5 materials exhibited type I isotherms, typical of microporous materials, in which there is a rapid increase in the adsorbed amount and a long and nearly flat region at higher pressures.38
To better understand the formation of the carbides, the carburization process was accompanied by TPC, following the water and CO formation (Fig. 2). The TPC profiles of all catalysts showed two main regions in the range between 300–450 °C and above 500 °C. According to the literature, these regions are associated with the reduction of MoO3 to MoO2, and carburization of MoO2 to β-Mo2C, respectively.39–41
 | ||
| Fig. 2 Water and CO formation profiles during TPC of the calcined precursors of Mo2C/SiO2, Mo2C/TiO2, Mo2C/ZrO2, and Mo2C/ZSM-5. | ||
The carburization process of the catalysts supported on SiO2 and ZSM-5 are rather similar with two main peaks of water formation at 396 °C and 550 °C. In comparison with the literature, a similar profile was observed during the carburization of 10MoOx/HZSM-5 in a mixture containing more diluted methane (10% CH4/H2). In this case, however, the transformations took place at higher temperatures. The reduction of MoO3 to MoO2 occurred at 450 °C, then MoO2 was reduced to Mo0 at 650 °C, and above 665 °C was observed the carbide formation accompanied by the consumption of CH4 and release of CO and CO2.42
For the Mo2C/TiO2 and Mo2C/ZrO2 catalysts, the first step corresponding to the reduction of Mo oxides takes place at 335 and 360 °C, respectively, i.e., at lower temperatures in comparison with the Mo carbides supported on SiO2 and ZSM-5. On the contrary, the carburization occurred at higher temperatures for these catalysts in comparison with the ones supported on SiO2 and ZSM-5.
The m/z = 28 signal that accompanies the signal of water is ascribed to the CO release from the carburization of MoO3. However, the undefined increase of the m/z = 28 signal observed for all the samples at higher temperatures may be associated with the formation of ethene according to the following reaction that occurs on the surface of molybdenum carbide: 4CH4s → 2CH2s˙ + 4Hs → C2H4 + 2H2.39
The molybdenum transformations during the synthesis of the Mo2C/SiO2 and Mo2C/TiO2 catalysts were investigated by Raman and XAS analyses.
XAS analysis at the Mo K-edge was performed for the calcined precursors of Mo2C/SiO2 and Mo2C/TiO2 catalysts (Fig. S3–S5†). For both precursors, the shape of the XANES spectra and position in energy are similar to those of the MoO3 standard (Fig. S3†). Bands at 994, 819, 665, 337, 287, and 243 cm−1 in the Raman spectra of the calcined precursors (Fig. S6†) confirm the presence of MoO343 in a well-crystallized form, which is in agreement with the XRD results (Fig. S2†). In addition, in the spectrum of the precursor of the Mo2C/TiO2 catalyst, the bands at 636, 514, and 397 cm−1 are ascribed to the anatase phase of the TiO2 support.44
After carburization, the XANES spectra, EXAFS oscillations, and Fourier transforms of Mo2C/SiO2 and Mo2C/TiO2 catalysts are rather similar and resemble that of reference bulk β-Mo2C (an unsupported standard whose structure was identified by XRD after passivation, and that was reactivated under H2 at 400 °C before recording the XAS spectrum) (Fig. S7–S9†). However, the shape of the XANES spectrum (Fig. S7†) after the edge is slightly different. The two features between 20020 and 20040 eV are better defined for Mo2C supported on TiO2. In fact, EXAFS oscillations (Fig. S8†) are much less intense for the Mo2C/TiO2 catalyst above k = 5 Å−1 (where the oscillations are dominated by the contribution of the heavier atoms), which is reflected by a peak of Mo neighbors significantly less intense than for Mo2C/SiO2 on the Fourier transform (Fig. S9†).
The results of the fits for the first two shells of neighbors (C and Mo atoms) are presented in Fig. 3 and Table 2. They are consistent with the formation of Mo carbides. The number of Mo neighbors found by EXAFS fitting is 5 in Mo2C/TiO2, against 7 on unsupported β-Mo2C and Mo2C/SiO2: possibly smaller particles of Mo carbide exist on TiO2 after carburization.
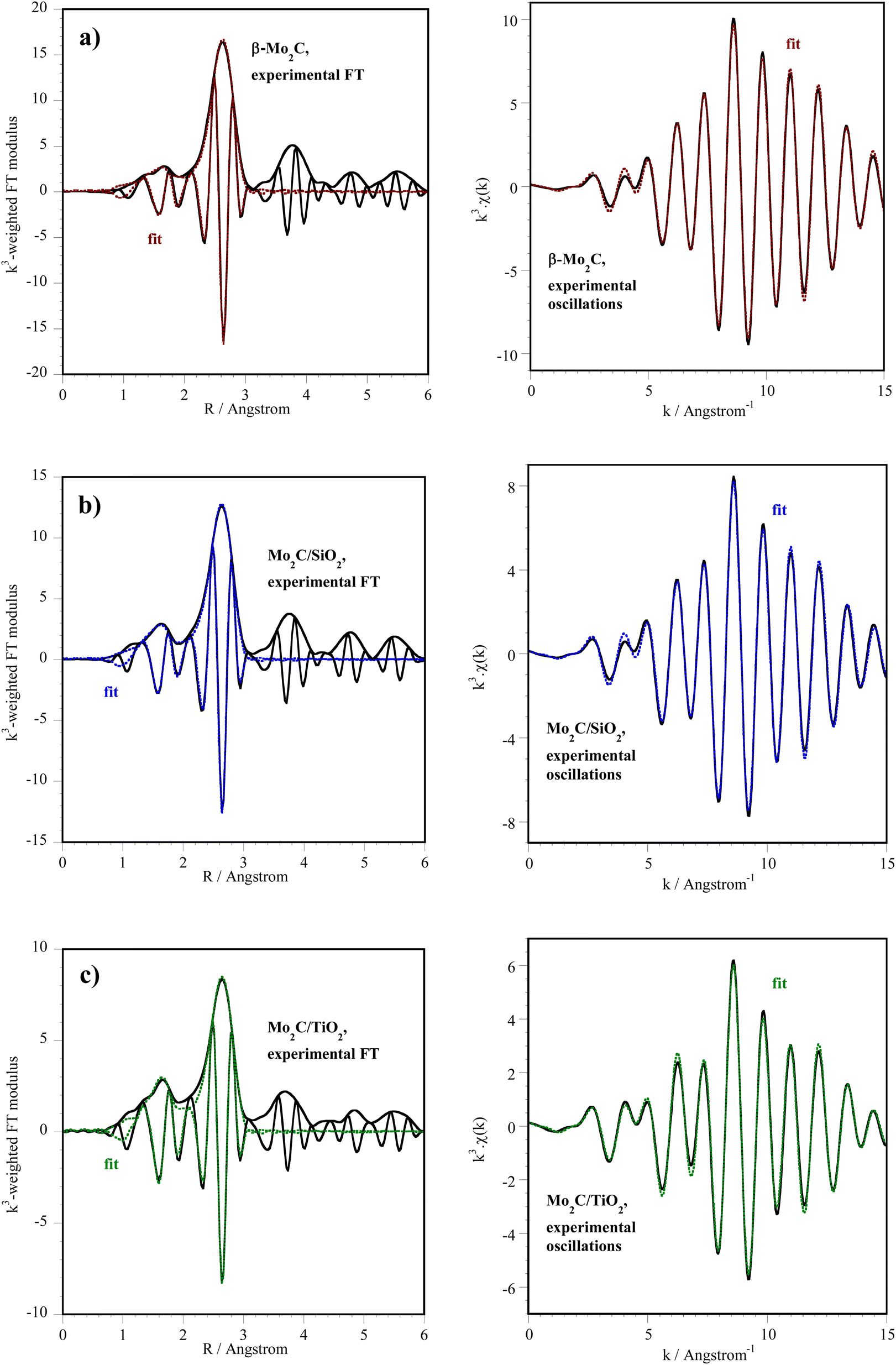 | ||
| Fig. 3 XAS data at the Mo K-edge of (a) β-Mo2C (reactivated in H2 at 400 °C) and after carburization of (b) Mo2C/SiO2 and (c) Mo2C/TiO2 (spectra recorded at room temperature). Fit of the first and second shells of neighbors: Fourier transform (left) and EXAFS oscillations (right). k = 3.5–15 Å−1. | ||
013 eV, S02 = 0.98) determined from the EXAFS analysis of spectra recorded at room temperature on carburized catalysts. k = 3.5–15 Å−1. Fit of the first peak(s) from the Fourier transform between 1 and 3 Å
| Catalyst | Backscatter | N | σ2 (Å2) × 103 | R (Å) |
|---|---|---|---|---|
| a After reactivation in H2 at 400 °C. | ||||
| β-Mo2Ca | C | 2.7 ± 0.9 | 4.1 ± 0.3 | 2.08 ± 0.02 |
| Mo | 7.3 ± 0.8 | 5.7 ± 0.5 | 2.966 ± 0.005 | |
| ΔE0 = −5.5 eV, r-factor = 0.01575, χ2 = 592, Nind = 13, Nvar = 7 | ||||
| Mo2C/SiO2 | C | 3.5 ± 0.9 | 6 ± 2 | 2.10 ± 0.01 |
| Mo | 7.3 ± 0.8 | 7.3 ± 0.6 | 2.978 ± 0.005 | |
| ΔE0 = −3.8 eV, r-factor = 0.01462, χ2 = 441, Nind = 13, Nvar = 7 | ||||
| Mo2C/TiO2 | C | 3.1 ± 0.6 | 5 ± 2 | 2.11 ± 0.01 |
| Mo | 5.1 ± 0.7 | 7.5 ± 0.8 | 2.984 ± 0.005 | |
| ΔE0 = −2.9 eV, r-factor = 0.01036, χ2 = 121, Nind = 13, Nvar = 7 | ||||
After carburization, the catalysts were passivated under diluted O2. The passivated Mo carbides supported on SiO2, TiO2, and ZSM-5 were evaluated by XAS analysis at the Mo K-edge, and the data are reported in Fig. S10–S12.† The XANES spectra (Fig. S10†) reveal that all three catalysts were oxidized to similar degrees. The EXAFS oscillations (Fig. S11†) of Mo carbide are still recognizable for Mo2C/SiO2 and Mo2C/ZSM-5, while for the Mo2C/TiO2 catalyst they are quite different. This is also reflected in the poorly intense peaks on the Fourier transform (Fig. S12†). One can suggest that on TiO2, the initially more dispersed carbide became more disorganized upon passivation than the carbides on the other supports.
The fits for the three supported passivated catalysts are still based on Mo carbide but they are all improved when one adds a small O contribution to the first shell of neighbors (Table S1 and Fig. S13†). The large error bar on N(C) does not allow for a precise discussion on the degree of carburization. Much more significant is the strong decrease of N(Mo). Compared with the just-carburized state, this decrease is marginal for bulk β-Mo2C, but N(Mo) falls to 4 for SiO2 and ZSM-5 (initially 7 after carburization on SiO2), and to 2 on TiO2 (initially 5). The passivation of the catalysts not only oxidized the carbide, but also disorganized the particles into smaller entities, or left crystallized Mo carbide as a minor fraction. Moreover, for TiO2, the quality of the fit is improved when one adds a Mo contribution at 2.52 Å (corresponding to the small peak at 2.1 Å on the Fourier transform), which can be interpreted as a Mo–Mo distance in MoO2. It thus clearly appears that passivation strongly alters the nature of the carbide phases on the supported systems, with a more pronounced effect when the Mo carbide was already poorly organized after carburization.
After reactivation of the passivated catalysts under hydrogen (Fig. S14†), the carbide spectrum reappears, but the spectrum of Mo2C/TiO2 is still quite different as can be seen in the relative intensity of the two features after the edge in the zoomed figure.
The EXAFS oscillations (Fig. S15†) extracted for the three reactivated catalysts are different: the oscillations measured for Mo2C/SiO2 look like those from the β-Mo2C standard; the shape is the same for Mo2C/ZSM-5 but the oscillations are quite damped; the shape is different for Mo2C/TiO2, which is not restored to its carbide state. These differences mostly come from the Mo shell, as seen when one compares the intensity of the Mo peak in the Fourier transforms (Fig. S16†): it decreases in the order SiO2, ZSM-5, and TiO2.
The oxidic fraction disappears from the fit, but the number of Mo neighbors remains more or less the same as on the passivated systems (Table S2 and Fig. S17†). If passivation contributed to break particles apart compared to the fresh carburized state, the reduction would then lead to a status quo in terms of particle size, and the reactivated state of Mo2C/ZSM-5 and especially Mo2C/TiO2 is indeed different from the initial carbide. Moreover, the fits are improved upon the addition of a minor contribution of Mo at 2.49 Å for Mo2C/TiO2 (assigned to traces of MoO2 above) or at 2.69 Å for Mo2C/ZSM-5 (possibly attributed to traces of metallic Mo).
A rough assessment of the degree of carburization of the catalysts after reactivation was performed using linear combinations of standard XANES and EXAFS spectra, following a previously described procedure.45 While the results for Mo2C/ZSM-5 were not very far from those found for Mo2C/SiO2 (76% of Mo carbide, 19% of oxycarbide MoO2−xCx and 5% of metallic Mo), to be compared with 80% of Mo carbide and 20% of oxycarbide for the silica-supported system,45 a composition of 63% of Mo carbide only and 37% of oxycarbide was found for Mo2C/TiO2. Even if the addition of the spectrum of MoO2 in the linear combination does not improve the fit, and thus does not ascertain the presence of oxide suggested by the EXAFS analysis, these results clearly point to a larger extent of decarburization for Mo2C/TiO2.
The disruption of the carbide particles caused by passivation and maintained upon reactivation could be accompanied by the persistence of minor oxidic and decarburized Mo phases that either remain unreduced, or that are reduced to metallic Mo upon reactivation.
The NH3 desorption profiles of supports and catalysts are displayed in Fig. 4. While SiO2 showed almost no acidity, two NH3 desorption peaks below 300 °C related to weak and moderate acidic sites are usually observed for TiO2 and ZrO2.46 In the obtained profiles, these peaks seem to have overlapped forming a broad curve. The maximum temperature for TiO2 (290 °C) was higher compared to ZrO2 (233 °C), which is in agreement with the literature.47
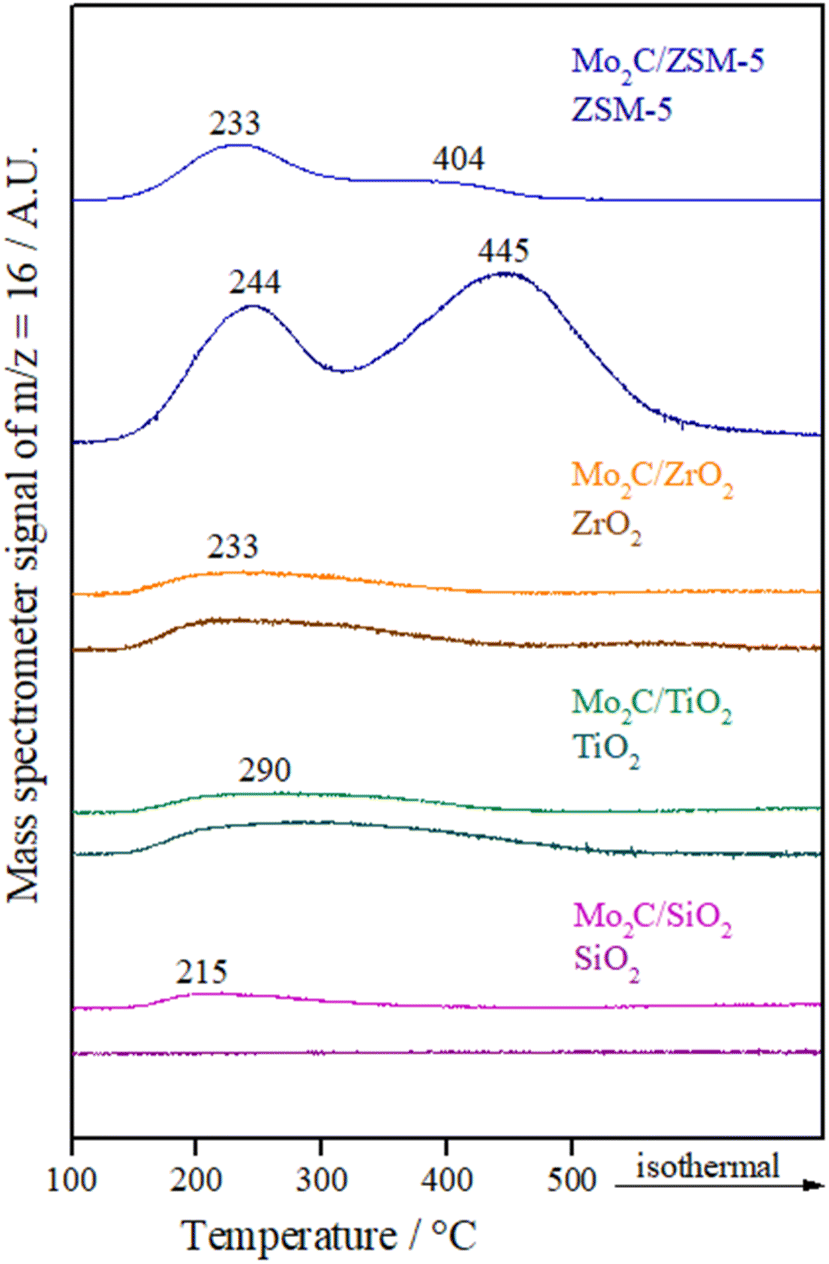 | ||
| Fig. 4 NH3 desorption profiles of supports and reactivated Mo carbides. | ||
ZSM-5 profile presents two main peaks, one located at low temperature (<300 °C) and one at high temperature (around 500 °C). In general, the low-temperature peak is associated with ammonia adsorbed on weak acidic sites and the high-temperature peak is ascribed to the strong adsorption of ammonia on acidic sites.48 Acidic sites of weak strength are ascribed to silanol groups or extra framework aluminum species, while acidic sites with strong strength are attributed to bridging hydroxyl groups (Si–OH–Al).49
Regarding the NH3 desorption profiles of catalysts, a broad peak in a low-temperature region can be seen for the Mo2C supported on SiO2, which indicates the presence of weak acidic sites on this material due to Mo carbide. Meanwhile, the Mo2C/TiO2 and Mo2C/ZrO2 catalysts showed a similar NH3 desorption profile as their supports. On the other hand, it was observed a change related to the strength of acidic sites for Mo2C/ZSM-5 catalyst. When Mo2C is supported on ZSM-5 more weak acidic sites appear compared with the bare support, which shows mainly strong acidic sites.
In comparison with the bare supports, the amount of NH3 desorbed from the materials (Table 3) shows that the total acidity is reduced after impregnation and synthesis of Mo2C with exception of Mo2C/SiO2 catalyst, which showed a slight increase, indicating that pure Mo2C already has acidity.50 This loss of acidity for most materials is ascribed to the coverage or neutralization of the acid sites of the support by the active phase.
| Material | Ammonia desorbed (μmol gcat−1) | Ammonia desorbed (μmol m−2) |
|---|---|---|
| SiO2 | 13 | 0.07 |
| TiO2 | 162 | 3.95 |
| ZrO2 | 125 | 1.28 |
| ZSM-5 | 2061 | 4.00 |
| Mo2C/SiO2 | 41 | 0.29 |
| Mo2C/TiO2 | 82 | 1.86 |
| Mo2C/ZrO2 | 86 | 1.23 |
| Mo2C/ZSM-5 | 632 | 1.99 |
Although TiO2 has more acidic sites than ZrO2, which is in agreement with the literature,47 similar values were obtained when Mo2C was supported on these materials. The Mo2C/ZSM-5 catalyst showed the highest number of total acidic sites.
3.2 HDO of furfural over supported Mo carbides-based catalysts
The supported Mo carbides were evaluated for the HDO of furfural using 2BuOH as solvent at 200 °C, 30 bar of H2 for 4 h. The activity of the supports and catalysts was compared at the same furfural and active phase molar ratio (R = 32) and the results are reported in Table 4.| Entry | Material | R | XFF (%) | Yield (%) | CB (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
Othera | |||||
| a 2-(sec-Butoxymethyl)furan (SBMF), 2-(dibutoxymethyl)furan (FDA), sec-butyl 4-oxopentanoate (SBOP). | |||||||||
| 1 | SiO2 | 32 | 2 | 1 | 0 | 0 | 0 | 2 | 100 |
| 2 | TiO2 | 32 | 20 | 12 | 0 | 0 | 0 | 1 | 94 |
| 4 | ZrO2 | 32 | 93 | 90 | 0 | 0 | 0 | 0 | 97 |
| 5 | ZSM-5 | 32 | 0 | 0 | 0 | 0 | 0 | 0 | — |
| 6 | Mo2C/SiO2 | 32 | 77 | 5 | 16 | 6 | 18 | 2 | 69 |
| 7 | Mo2C/TiO2 | 32 | 55 | 8 | 16 | 2 | 9 | 2 | 81 |
| 8 | Mo2C/ZrO2 | 32 | 46 | 11 | 6 | 1 | 1 | 3 | 79 |
| 9 | Mo2C/ZSM-5 | 26 | 54 | 4 | 8 | 2 | 10 | 6 | 76 |
| 10 | Mo2C/SiO2 | 19 | 100 | 0 | 34 | 7 | 11 | 4 | 57 |
| 11 | Mo2C/SiO2 | 48 | 62 | 7 | 12 | 4 | 14 | 0 | 76 |
| 12 | Mo2C/SiO2 | 127 | 30 | 7 | 2 | 1 | 8 | 2 | 91 |
| 13 | Mo2C/TiO2 | 16 | 81 | 7 | 34 | 3 | 10 | 1 | 75 |
| 14 | Mo2C/TiO2 | 21 | 68 | 7 | 22 | 3 | 7 | 3 | 74 |
| 15 | Mo2C/TiO2 | 64 | 34 | 9 | 5 | 1 | 2 | 5 | 89 |

The results show that SiO2 support does not show any activity for the HDO reaction of furfural. On the other hand, ZrO2 showed almost full conversion of FF producing FA with a high yield (90%). In fact, the excellent performance of this material in the MPV reduction involving furfural and a secondary alcohol has been observed previously and it was attributed to a variety of catalytic active sites found in ZrO2. The presence of hydroxyl species in this material can act as weak basic sites and assists the formation of alkoxide species with acidic sites by the deprotonation of alcohol molecules.47,51 The solvent used in this work, 2-butanol, is adsorbed as an alkoxide on ZrO2, which strongly favors the hydrogen transfer step.52,53 On the other hand, the lower FF conversion and consequently lower yield to FA over TiO2 might be related to the lower number of basic sites in this material.47 In any case, these results reveals that the reduction of furfural is catalyzed not only by acidic, but also basic sites.
Meanwhile, for the ZSM-5 support, the high acidity of this material promoted the dehydration of the solvent 2-butanol to 2-butene, which was detected in the gas phase by the increase in system pressure and dissolved in the liquid phase as confirmed by GCMS. In this sense, this reaction caused part of the solvent to go into the gas phase as 2-butene, concentrating furfural, which had a higher concentration than the reaction mixture at the start of the reaction, compromising the quantification of the substrate. Therefore, the use of a highly acidic support such as ZSM-5 in an alcoholic medium is not recommended.
Most of the catalysts were more active in converting FF compared to their respective supports (entries 1–9). In the case of the Mo2C/SiO2 catalyst, the significant gain in activity is attributed to the Mo carbide phase, since the support SiO2 exhibits no activity (entries 1 and 6). On the contrary, a different trend was observed for the Mo2C/ZrO2 catalyst, which was much less active than its corresponding support (entries 4 and 8). This effect has already been observed in the hydrogen transfer reduction of FF when using bare ZrO2 and Cu/ZrO2 catalyst and it was attributed to the reduction of the specific surface area of the catalyst and consequent loss of basic active sites.53
The comparison of the supported Mo carbides activity (entries 6–9) shows that a higher FF conversion is obtained when Mo2C is supported on SiO2 (77%), while lower conversions were observed over TiO2 (55%), ZrO2 (46%) and ZSM-5 (54%, even when using a higher amount of catalyst, R = 26). These results seem to be related to the final state of the carbides after passivation and reactivation procedures. As observed by XAS, the particles of Mo2C were disordered after passivation for the catalysts supported on TiO2 and ZSM-5, and the carburization degree was not the same for these materials after reactivation. On the other hand, the Mo2C/SiO2 catalyst was less de-carburized and, for this reason, presents a higher activity. These results show that the type of support used influences the final state of the carbide after reactivation.
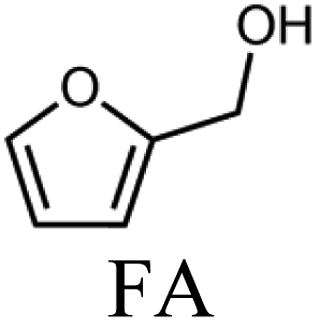
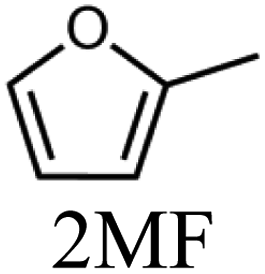
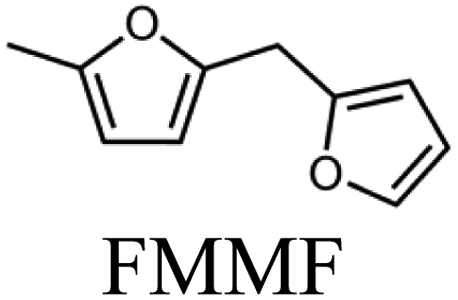
To evaluate the product distribution, all catalysts were compared in conditions close to iso-conversion (entries 7, 8, 9, and 11) by modification of the furfural and active phase molar ratio. As reported in our previous work,54 the main compounds produced over supported Mo carbides in the presence of 2-butanol were 2-methylfuran (2MF), furfuryl alcohol (FA), 2-(furan-2-ylmethyl)-5-methylfuran (FMMF) and 2,2-(1,2-ethenediyl)bis-furan (EBF).
Furfuryl alcohol is obtained by the hydrogenation of the carbonyl group in FF, followed by the deoxygenation of FA to produce 2MF.3,4 C10 compounds such as EBF and FMMF are produced by condensation reactions favoured in acidic sites. EBF is formed from the condensation of two molecules of furfural, followed by hydrogenation and dehydration reactions,19 while FMMF can be obtained by three main routes: FA dimerization, hydroxyalkylation/alkylation of 2MF or by the reaction between FA and 2MF, which is the most likely to occur.14
The highest yield of 2MF was observed over the Mo carbide supported on TiO2 (16%) followed by SiO2 (12%). Meanwhile, the Mo2C/ZrO2 and Mo2C/ZSM-5 catalysts were less effective in obtaining 2MF. A higher FA yield was observed over Mo2C/ZrO2, which agrees with the high selectivity of the bare ZrO2 support to promote the conversion of furfural to furfuryl alcohol. This also limits the FF condensation, which was reflected by the reduction of the EBF and FMMF yields. In contrast, a higher production of these products was observed for the other catalysts. Condensation reactions are reported to occur in acidic sites,55–58 which are present in all materials, as demonstrated by TPD-NH3. The main route followed by each catalyst is shown in Scheme 1.
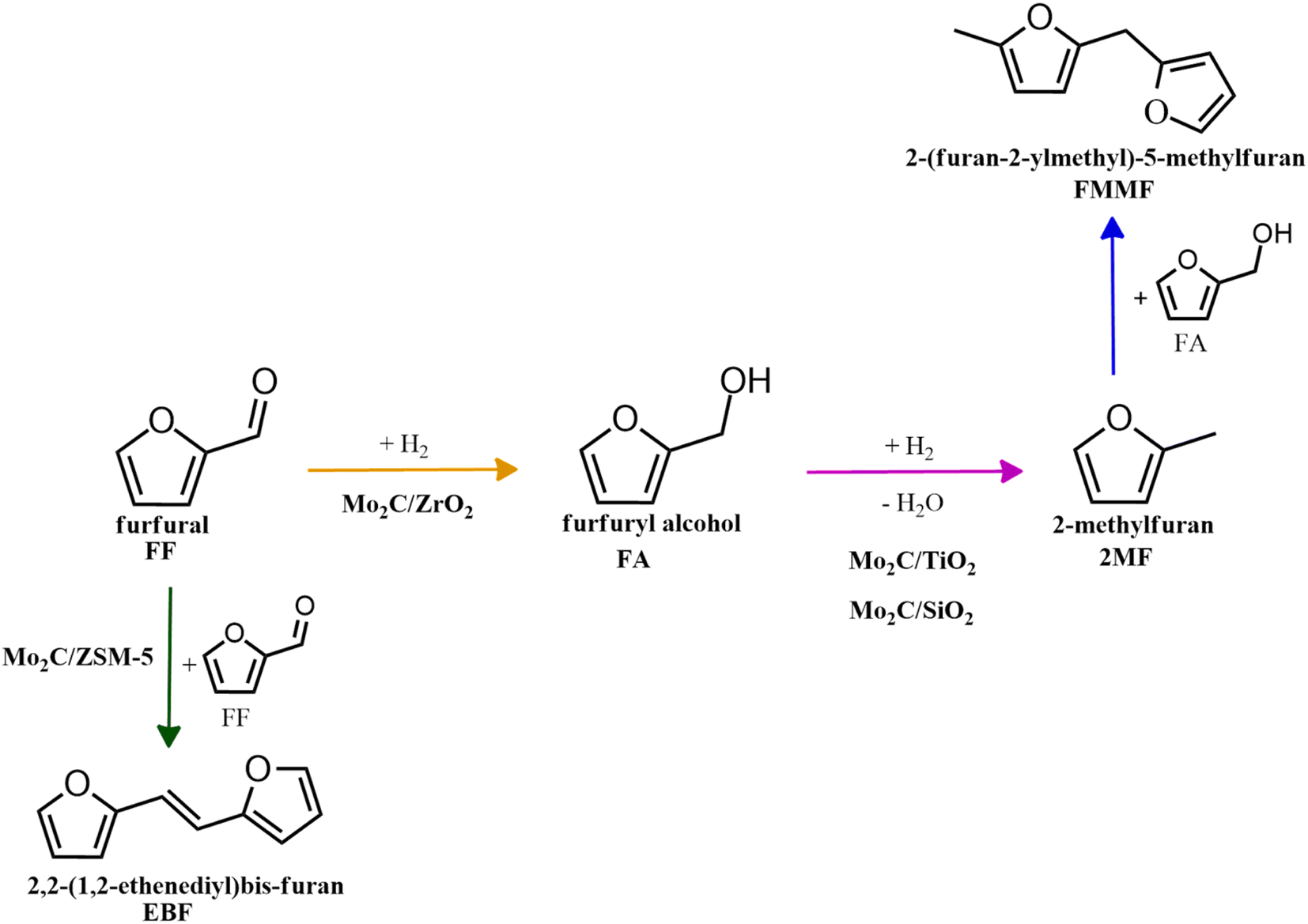 | ||
| Scheme 1 Main routes for the FF conversion over Mo carbides supported on SiO2, TiO2, ZrO2 and ZSM-5. | ||
The variation in the yield of products as a function of the FF conversion was evaluated for the Mo2C/SiO2 and Mo2C/TiO2 catalysts (Fig. 5). The increase in the conversion of FF was accompanied by the increase in the yield to 2MF and decrease in the FA yield, confirming that furfuryl alcohol is a reaction intermediate. Both catalysts showed similar trends regarding the evolution of the products. Over the Mo2C/TiO2 catalyst, lower yields of EBF were observed in comparison with Mo2C/SiO2. This is ascribed to the higher initial reaction rate of the former catalyst, which converts FF faster avoiding the formation of EBF. The FMMF yield remained below 7% for both catalysts.
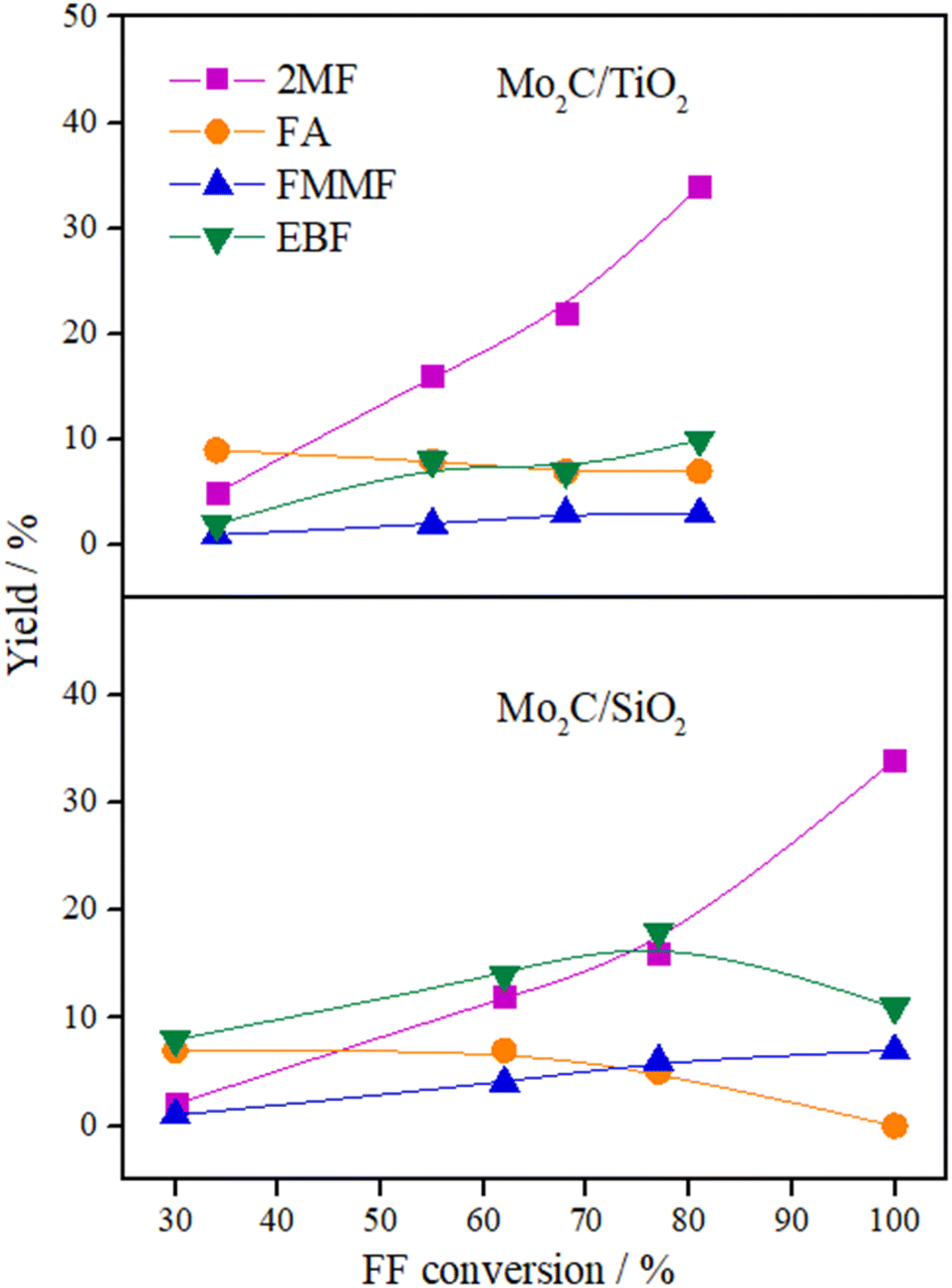 | ||
| Fig. 5 Product yields versus FF conversion for Mo2C/SiO2 and Mo2C/TiO2 catalysts (reaction conditions: 200 °C, 30 bar of H2, 4 h, 2-butanol, Parr reactor). | ||
In summary, Mo2C/TiO2 is a suitable catalyst to produce 2MF, while a mixture containing both 2MF and C10 compounds are yielded over the Mo2C/SiO2 catalyst.
4 Conclusion
The effect of the support for Mo carbides (SiO2, TiO2, ZrO2, and ZSM-5) was evaluated for the HDO of furfural in liquid phase at 200 °C, 30 bar of H2 for 4 hours using 2-butanol as solvent. The carburization, passivation, and reactivation of the supported Mo carbides were evaluated by XAS analysis. The Mo carbide supported on TiO2 was the most affected by these treatments. Smaller Mo2C particles formed during carburization were disorganized after passivation and a minor fraction of crystallized Mo carbide remained in the Mo2C/TiO2 catalyst. After reactivation, the Mo2C/SiO2 catalyst was the least de-carburized among all, while traces of MoO2 and metallic Mo were observed for the Mo2C/TiO2 and Mo2C/ZSM-5 catalysts, respectively.SiO2 exhibited lack in activity to convert FF, while higher conversions were observed over the supports presenting acidic properties (TiO2, ZrO2). On the other hand, a too elevated acidity, such as that observed for ZSM-5 caused the degradation of the solvent 2-butanol to 2-butene. The activity of the supports was not directly related to the total number or nature of the acidic sites but to the presence of both Lewis acidic sites and basic sites that promote the conversion of FF to FA by the MPV reaction, in which ZrO2 exhibited the highest conversion of FF and yield to FA.
Nevertheless, the presence of acidic and basic sites in the supports was not relevant after impregnation and synthesis of the Mo carbides. In this case, the supports influenced the final state of the Mo carbides after carburization, passivation, and reactivation. The highest activity was observed for the least de-carburized catalyst, Mo2C/SiO2, even though SiO2 was not active in the reaction, therefore the activity is mainly attributed to the Mo carbide.
Although the Mo2C/SiO2 catalyst is more active than the Mo2C/TiO2 catalyst, at similar conversions (around 80%), the latter produced 2MF in higher yields (34%). In any case, a mixture containing 2MF and C10 compounds can be achieved using Mo2C/SiO2 as catalyst.
Data availability
The data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Leticia F. Sosa thanks Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior (CAPES – Finance code 001) and CAPES – COFECUB program (88881.142911/2017-01) for the scholarship. This study was supported by the French government through the Programme Investissement d’Avenir (I-SITE ULNE/ANR-16-IDEX-0004 ULNE) managed by the Agence Nationale de la Recherche, CNRS, Métropole Européen de Lille (MEL) and Region Hauts-de-France for “CatBioInnov” project are also acknowledged. Fabio B. Noronha thanks Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ – E-26/202.783/2017; 200.966/2021) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq – 303667/2018-4; 305046/2015-2; 302469/2020-6; 310116/2019-82) for financial support. We also thank the French synchrotron radiation facility SOLEIL for the assigned beamtime at ROCK (project 20210494) to perform the XAS experiments. The REALCAT platform benefits from a state subsidy administered by the French National Research Agency (ANR) within the frame of the ‘Future Investments’ program (PIA) with the contractual reference ANR-11-EQPX-0037. The European Union, through the ERDF funding administered by the Hauts-de-France Region, has co-financed the platform. Centrale Lille, the CNRS, and Lille University, as well as the Centrale Initiatives Foundation, are thanked for their financial contributions to the acquisition and implementation of the equipment of the REALCAT platform.References
- J. A. Okolie, S. Nanda, A. K. Dalai and J. A. Kozinski, Chemistry and Specialty Industrial Applications of Lignocellulosic Biomass, Waste Biomass Valorization, 2021, 12, 2145–2169, DOI:10.1007/s12649-020-01123-0.
- Y. Liu, Y. Nie, X. Lu, X. Zhang, H. He, F. Pan, L. Zhou, X. Liu, X. Ji and S. Zhang, Cascade utilization of lignocellulosic biomass to high-value products, Green Chem., 2019, 21, 3499–3535, 10.1039/C9GC00473D.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, How Catalysts and Experimental Conditions Determine the Selective Hydroconversion of Furfural and 5-Hydroxymethylfurfural, Chem. Rev., 2018, 118, 11023–11117, DOI:10.1021/acs.chemrev.8b00134.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. López Granados, Furfural: a renewable and versatile platform molecule for the synthesis of chemicals and fuels, Energy Environ. Sci., 2016, 9, 1144–1189, 10.1039/C5EE02666K.
- P. Khemthong, C. Yimsukanan, T. Narkkun, A. Srifa, T. Witoon, S. Pongchaiphol, S. Kiatphuengporn and K. Faungnawakij, Advances in catalytic production of value-added biochemicals and biofuels via furfural platform derived lignocellulosic biomass, Biomass Bioenergy, 2021, 148, 106033, DOI:10.1016/j.biombioe.2021.106033.
- A. Tuan Hoang and V. Viet Pham, 2-Methylfuran (MF) as a potential biofuel: A thorough review on the production pathway from biomass, combustion progress, and application in engines, Renewable Sustainable Energy Rev., 2021, 148, 111265, DOI:10.1016/j.rser.2021.111265.
- P. Liu, L. Sun, X. Jia, C. Zhang, W. Zhang, Y. Song, H. Wang and C. Li, Efficient one-pot conversion of furfural into 2-methyltetrahydrofuran using non-precious metal catalysts, Mol. Catal., 2020, 490, 110951, DOI:10.1016/j.mcat.2020.110951.
- W. Gong, C. Chen, H. Zhang, G. Wang and H. Zhao, Efficient Synthesis of 2-Methylfuran from Bio-Derived Furfural over Supported Copper Catalyst: The Synergistic Effect of CuO x and Cu, ChemistrySelect, 2017, 2, 9984–9991, DOI:10.1002/slct.201702206.
- W. Gong, C. Chen, Y. Zhang, H. Zhou, H. Wang, H. Zhang, Y. Zhang, G. Wang and H. Zhao, Efficient Synthesis of Furfuryl Alcohol from H 2 -Hydrogenation/Transfer Hydrogenation of Furfural Using Sulfonate Group Modified Cu Catalyst, ACS Sustain. Chem. Eng., 2017, 5, 2172–2180, DOI:10.1021/acssuschemeng.6b02343.
- N. S. Date, A. M. Hengne, K.-W. Huang, R. C. Chikate and C. V. Rode, Single pot selective hydrogenation of furfural to 2-methylfuran over carbon supported iridium catalysts, Green Chem., 2018, 20, 2027–2037, 10.1039/C8GC00284C.
- B. Wang, C. Li, B. He, J. Qi and C. Liang, Highly stable and selective Ru/NiFe 2 O 4 catalysts for transfer hydrogenation of biomass-derived furfural to 2-methylfuran, J. Energy Chem., 2017, 26, 799–807, DOI:10.1016/j.jechem.2017.04.008.
- B. Li, L. Li, H. Sun and C. Zhao, Selective Deoxygenation of Aqueous Furfural to 2-Methylfuran over Cu 0/Cu 2 O·SiO 2 Sites via a Copper Phyllosilicate Precursor without Extraneous Gas, ACS Sustain. Chem. Eng., 2018, 6, 12096–12103, DOI:10.1021/acssuschemeng.8b02425.
- M. J. Gilkey, P. Panagiotopoulou, A. V. Mironenko, G. R. Jenness, D. G. Vlachos and B. Xu, Mechanistic Insights into Metal Lewis Acid-Mediated Catalytic Transfer Hydrogenation of Furfural to 2-Methylfuran, ACS Catal., 2015, 5, 3988–3994, DOI:10.1021/acscatal.5b00586.
- P. Panagiotopoulou and D. G. Vlachos, Liquid phase catalytic transfer hydrogenation of furfural over a Ru/C catalyst, Appl. Catal., A, 2014, 480, 17–24, DOI:10.1016/j.apcata.2014.04.018.
- J. Chuseang, R. Nakwachara, M. Kalong, S. Ratchahat, W. Koo-amornpattana, W. Klysubun, P. Khemthong, K. Faungnawakij, S. Assabumrungrat, V. Itthibenchapong and A. Srifa, Selective hydrogenolysis of furfural into fuel-additive 2-methylfuran over a rhenium-promoted copper catalyst, Sustainable Energy Fuels, 2021, 5, 1379–1393, 10.1039/D1SE00036E.
- I. Gandarias, S. García-Fernández, I. Obregón, I. Agirrezabal-Telleria and P. L. Arias, Production of 2-methylfuran from biomass through an integrated biorefinery approach, Fuel Process. Technol., 2018, 178, 336–343, DOI:10.1016/j.fuproc.2018.05.037.
- S. Srivastava, G. C. Jadeja and J. Parikh, Copper-cobalt catalyzed liquid phase hydrogenation of furfural to 2-methylfuran: An optimization, kinetics and reaction mechanism study, Chem. Eng. Res. Des., 2018, 132, 313–324, DOI:10.1016/j.cherd.2018.01.031.
- Z. Fu, Z. Wang, W. Lin, W. Song and S. Li, High efficient conversion of furfural to 2-methylfuran over Ni-Cu/Al2O3 catalyst with formic acid as a hydrogen donor, Appl. Catal., A, 2017, 547, 248–255, DOI:10.1016/j.apcata.2017.09.011.
- Y. Deng, R. Gao, L. Lin, T. Liu, X.-D. Wen, S. Wang and D. Ma, Solvent Tunes the Selectivity of Hydrogenation Reaction over α-MoC Catalyst, J. Am. Chem. Soc., 2018, 140, 14481–14489, DOI:10.1021/jacs.8b09310.
- I. Shilov, A. Smirnov, O. Bulavchenko and V. Yakovlev, Effect of Ni–Mo Carbide Catalyst Formation on Furfural Hydrogenation, Catalysts, 2018, 8, 560, DOI:10.3390/catal8110560.
- Y. Wang, X. Feng, S. Yang, L. Xiao and W. Wu, Influence of acidity on the catalytic performance of Ni2P in liquid-phase hydrodeoxygenation of furfural to 2-methylfuran, J. Nanopart. Res., 2020, 22, 67, DOI:10.1007/s11051-020-04784-z.
- M. Zhou, H. A. Doan, L. A. Curtiss and R. S. Assary, Identification of Active Metal Carbide and Nitride Catalytic Facets for Hydrodeoxygenation Reactions, J. Phys. Chem. C, 2021, 125, 8630–8637, DOI:10.1021/acs.jpcc.1c02387.
- C. Wan, Y. N. Regmi and B. M. Leonard, Multiple Phases of Molybdenum Carbide as Electrocatalysts for the Hydrogen Evolution Reaction, Angew. Chem., 2014, 126, 6525–6528, DOI:10.1002/ange.201402998.
- R. B. Levy and M. Boudart, Platinum-Like Behavior of Tungsten Carbide in Surface Catalysis, Science, 1973, 181, 547–549, DOI:10.1126/science.181.4099.547.
- F. Toledo, I. T. Ghampson, C. Sepúlveda, R. García, J. L. G. Fierro, A. Videla, R. Serpell and N. Escalona, Effect of Re content and support in the liquid phase conversion of furfural to furfuryl alcohol and 2-methyl furan over ReOx catalysts, Fuel, 2019, 242, 532–544, DOI:10.1016/j.fuel.2019.01.090.
- X. Chang, A.-F. Liu, B. Cai, J.-Y. Luo, H. Pan and Y.-B. Huang, Catalytic Transfer Hydrogenation of Furfural to 2-Methylfuran and 2-Methyltetrahydrofuran over Bimetallic Copper-Palladium Catalysts, ChemSusChem, 2016, 9, 3330–3337, DOI:10.1002/cssc.201601122.
- B. Seemala, C. M. Cai, R. Kumar, C. E. Wyman and P. Christopher, Effects of Cu–Ni Bimetallic Catalyst Composition and Support on Activity, Selectivity, and Stability for Furfural Conversion to 2-Methyfuran, ACS Sustain. Chem. Eng., 2018, 6, 2152–2161, DOI:10.1021/acssuschemeng.7b03572.
- S. Srivastava, G. C. Jadeja and J. Parikh, A versatile bi-metallic copper–cobalt catalyst for liquid phase hydrogenation of furfural to 2-methylfuran, RSC Adv., 2016, 6, 1649–1658, 10.1039/C5RA15048E.
- L. A. Sousa, J. L. Zotin and V. Teixeira da Silva, Hydrotreatment of sunflower oil using supported molybdenum carbide, Appl. Catal., A, 2012, 449, 105–111, DOI:10.1016/j.apcata.2012.09.030.
- V. Briois, C. La Fontaine, S. Belin, L. Barthe, T. Moreno, V. Pinty, A. Carcy, R. Girardot and E. Fonda, ROCK: the new Quick-EXAFS beamline at SOLEIL, J. Phys.: Conf. Ser., 2016, 712, 012149, DOI:10.1088/1742-6596/712/1/012149.
- C. La Fontaine, L. Barthe, A. Rochet and V. Briois, X-ray absorption spectroscopy and heterogeneous catalysis: Performances at the SOLEIL's SAMBA beamline, Catal. Today, 2013, 205, 148–158, DOI:10.1016/j.cattod.2012.09.032.
- C. Lesage, E. Devers, C. Legens, G. Fernandes, O. Roudenko and V. Briois, High pressure cell for edge jumping X-ray absorption spectroscopy: Applications to industrial liquid sulfidation of hydrotreatment catalysts, Catal. Today, 2019, 336, 63–73, DOI:10.1016/j.cattod.2019.01.081.
- B. Ravel and M. Newville, ATHENA , ARTEMIS , HEPHAESTUS : data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541, DOI:10.1107/S0909049505012719.
- M. Newville, IFEFFIT : interactive XAFS analysis and FEFF fitting, J. Synchrotron Radiat., 2001, 8, 322–324, DOI:10.1107/S0909049500016964.
- D.-V. N. Vo and A. A. Adesina, Kinetics of the carbothermal synthesis of Mo carbide catalyst supported on various semiconductor oxides, Fuel Process. Technol., 2011, 92, 1249–1260, DOI:10.1016/j.fuproc.2011.02.012.
- B. Wang, C. Wang, W. Yu, Z. Li, Y. Xu and X. Ma, Effects of preparation method and Mo2C loading on the Mo2C_ZrO2 catalyst for sulfur-resistant methanation, Mol. Catal., 2020, 482, 110668, DOI:10.1016/j.mcat.2019.110668.
- S. Boullosa-Eiras, R. Lødeng, H. Bergem, M. Stöcker, L. Hannevold and E. A. Blekkan, Catalytic hydrodeoxygenation (HDO) of phenol over supported molybdenum carbide, nitride, phosphide and oxide catalysts, Catal. Today, 2014, 223, 44–53, DOI:10.1016/j.cattod.2013.09.044.
- H. Wang, Support effects on hydrotreating of soybean oil over NiMo carbide catalyst, Fuel, 2013, 81–87, DOI:10.1016/j.fuel.2013.04.066.
- C. G. Silva, F. B. Passos and V. T. da Silva, Influence of the support on the activity of a supported nickel-promoted molybdenum carbide catalyst for dry reforming of methane, J. Catal., 2019, 375, 507–518, DOI:10.1016/j.jcat.2019.05.024.
- J. Lee, Molybdenum carbide catalysts I. Synthesis of unsupported powders, J. Catal., 1987, 106, 125–133, DOI:10.1016/0021-9517(87)90218-1.
- A. Hanif, T. Xiao, A. P. E. York, J. Sloan and M. L. H. Green, Study on the Structure and Formation Mechanism of Molybdenum Carbides, Chem. Mater., 2002, 14, 1009–1015, DOI:10.1021/cm011096e.
- M. Rahman, A. Infantes-Molina, A. Boubnov, S. R. Bare, E. Stavitski, A. Sridhar and S. J. Khatib, Increasing the catalytic stability by optimizing the formation of zeolite-supported Mo carbide species ex situ for methane dehydroaromatization, J. Catal., 2019, 375, 314–328, DOI:10.1016/j.jcat.2019.06.002.
- T. Mo, J. Xu, Y. Yang and Y. Li, Effect of carburization protocols on molybdenum carbide synthesis and study on its performance in CO hydrogenation, Catal. Today, 2016, 261, 101–115, DOI:10.1016/j.cattod.2015.07.014.
- M. Abou Hamdan, S. Loridant, M. Jahjah, C. Pinel and N. Perret, TiO2-supported molybdenum carbide: An active catalyst for the aqueous phase hydrogenation of succinic acid, Appl. Catal., A, 2019, 571, 71–81, DOI:10.1016/j.apcata.2018.11.009.
- L. F. Sosa, P. M. De Souza, R. A. Rafael, R. Wojcieszak, V. Briois, L. R. Francisco, R. C. Rabelo-Neto, E. Marceau, S. Paul, F. S. Toniolo and F. B. Noronha, Study of the performance of SiO2-supported Mo2C and metal-promoted Mo2C catalysts for the hydrodeoxygenation of m-cresol, Appl. Catal., B, 2023, 331, 122720, DOI:10.1016/j.apcatb.2023.122720.
- Z.-Y. Ma, C. Yang, W. Wei, W.-H. Li and Y.-H. Sun, Surface properties and CO adsorption on zirconia polymorphs, J. Mol. Catal. A: Chem., 2005, 227, 119–124, DOI:10.1016/j.molcata.2004.10.017.
- M. E. Manríquez, T. López, R. Gómez and J. Navarrete, Preparation of TiO2–ZrO2 mixed oxides with controlled acid–basic properties, J. Mol. Catal. A: Chem., 2004, 220, 229–237, DOI:10.1016/j.molcata.2004.06.003.
- L. Yang, Z. Liu, Z. Liu, W. Peng, Y. Liu and C. Liu, Correlation between H-ZSM-5 crystal size and catalytic performance in the methanol-to-aromatics reaction, Chin. J. Catal., 2017, 38, 683–690, DOI:10.1016/S1872-2067(17)62791-8.
- X. Yu, B. Liu and Y. Zhang, Effect of Si/Al ratio of high-silica HZSM-5 catalysts on the prins condensation of isobutylene and formaldehyde to isoprene, Heliyon, 2019, 5, e01640, DOI:10.1016/j.heliyon.2019.e01640.
- S. K. Bej, C. A. Bennett and L. T. Thompson, Acid and base characteristics of molybdenum carbide catalysts, Appl. Catal., A, 2003, 250, 197–208, DOI:10.1016/S0926-860X(02)00664-6.
- C. Xu, E. Paone, D. Rodríguez-Padrón, R. Luque and F. Mauriello, Recent catalytic routes for the preparation and the upgrading of biomass derived furfural and 5-hydroxymethylfurfural, Chem. Soc. Rev., 2020, 49, 4273–4306, 10.1039/D0CS00041H.
- P. Nagaiah, C. V. Pramod, M. V. Rao, B. D. Raju and K. S. R. Rao, Liquid phase hydrogenation of furfural using 2-propanol over $$\hbox {ZrO}_{2}$$ ZrO 2, J. Chem. Sci., 2018, 130, 66, DOI:10.1007/s12039-018-1469-5.
- N. Scotti, F. Zaccheria, C. Bisio, C. Vittoni and N. Ravasio, Switching Selectivity in the Hydrogen Transfer Reduction of Furfural, ChemistrySelect, 2018, 3, 8344–8348, DOI:10.1002/slct.201801974.
- L. F. Sosa, P. M. De Souza, R. A. Rafael, R. Wojcieszak, E. Marceau, S. Paul, V. Briois, F. B. Noronha and F. S. Toniolo, Hydrodeoxygenation of Furfural over Unsupported, SiO 2 -supported or Metal-promoted Mo Carbides: Tunning the Selectivity between 2-Methylfuran and C 10 Furoins Diesel Precursors, ChemCatChem, 2023, e202300890, DOI:10.1002/cctc.202300890.
- G. Li, N. Li, S. Li, A. Wang, Y. Cong, X. Wang and T. Zhang, Synthesis of renewable diesel with hydroxyacetone and 2-methyl-furan, Chem. Commun., 2013, 49, 5727, 10.1039/c3cc42296h.
- A. Corma, O. de la Torre and M. Renz, Production of high quality diesel from cellulose and hemicellulose by the Sylvan process: catalysts and process variables, Energy Environ. Sci., 2012, 5, 6328, 10.1039/c2ee02778j.
- S. Li, N. Li, G. Li, A. Wang, Y. Cong, X. Wang and T. Zhang, Synthesis of diesel range alkanes with 2-methylfuran and mesityl oxide from lignocellulose, Catal. Today, 2014, 234, 91–99, DOI:10.1016/j.cattod.2014.01.028.
- Y.-J. Luo, Y.-H. Zhou and Y.-B. Huang, A New Lewis Acidic Zr Catalyst for the Synthesis of Furanic Diesel Precursor from Biomass Derived Furfural and 2-Methylfuran, Catal. Lett., 2019, 149, 292–302, DOI:10.1007/s10562-018-2599-6.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04256e |
| This journal is © The Royal Society of Chemistry 2024 |